

Abstract
BACKGROUND
The association between hepatitis B virus (HBV) mutations and hepatocarcinogenesis were reported in the literature. Preference for G over C in the leading DNA strand has been reported to account for the asymmetry in nucleotide (nt) composition. The aim of this study was to analyze the complete genome sequence and compositional asymmetry of HBV in different stages of hepatitis B.
METHODS
Full genome sequencing of 24 patients with chronic hepatitis B, some of whom also had cirrhosis and hepatocellular carcinoma (HCC) was performed. Mutations analysis was implemented in a comparison with a HBV genotype D reference from an international DNA database. CpGProD, a web-based application, was used to evaluate CG content and predict CpG islands.
RESULTS
All strains were 3182 base pairs (bp) in length, except for two cases of HCC in which 9 and 21 nt, respectively, were deleted in preS2. The genetic relatedness of these isolates was 97%-100%. There were common CpG-rich regions in all 24 isolated full genome sequences, however a strong negative GC skew for forming a CpG island in the minus strand were exhibited in overlap with enhancer I in three HCC patients, a cirrhotic patient and three with chronic hepatitis.
CONCLUSION
The high percentage of sequence identity between HBV isolates in our patients demonstrates that genomic factors, except for genotype, are involved in hepatocarcinogenesis. Variations in GC content which were caused by a different spectrum of mutations may affect DNA compositional asymmetry and epigenetic modification of HBV DNA in HCC.
Keywords: Hepatitis B virus (HBV), Hepatocellular carcinoma (HCC), HBV genome CpG
INTRODUCTION
Accumulation of naturally occurring mutations in the hepatitis B virus (HBV) genome may be related to the development of hepatocellular carcinoma (HCC).1 The genome of HBV has for four partially overlapping open reading frames (ORFs). HBV replication seems to form the basis of strand asymmetries in transcription-induced mutations. It is replicated via a greater-than-genome-length RNA intermediate known as pregenomic RNA (pgRNA) which is very delicate until paired with the newly synthesized strand.2- 4
Two parameters used for strand asymmetry are the GC skew, (G - C)/(G + C), and the TA skew, (T - A)/(T + A), where G, C, T and A represent the frequencies of the four nucleotides (nt) in the strand under study.5 A peak in the GC skew at the transcription start site (TSS) can explain the pattern of CpG dinucleotides and the phenomenon of higher mutability of unprotected cytosines.6 Unmethylated CpG Oligodeoxynucleotides in specific sequence locations (CpG motifs) of HBV which has been proposed to provide signals that activate innate immune responses.7 Unmethylated CpGs cause activation of macrophages and natural killer cells, which induce release of cytokines including IL-12, IL-6 and IFN-gamma, and polyclonal stimulation of B-lymphocytes.8, 9 However, decreased production of viral proteins and DNA have been described from the methylated form of the HBV genome in artificially-infected hepatocytes.10 Therefore, the approach of DNA methylation is either for cloak immune detection of CpG sites or to silence the HBV genome transcription in self-defense.7
Analysis of HBV DNA sequences by characterization of two parameters, AT-skew and the GC-skew, helps to gain a better understanding of epigenetic evolution of HBV and its immunostimulatory activities. Genomic analysis of HBV also clarifies the genetic variations of HBV for specific amino acid substitutions. Therefore, in this study, we aimed to investigate the nt compositions of the full genome sequence of HBV in different stages of chronic hepatitis B for the frequency of CpG dinucleotides and the incidence of mutations in different ORFs.
MATERIALS AND METHODS
Patients
Full genome analysis of HBV was performed on 24 samples of HbsAg-positive patients who attended the Hepatitis Clinic at Shariati Hospital, Tehran University of Medical Sciences. Samples were from 16 men and 8 women whose mean age was 44 years (Table 1). Cirrhosis was defined as the presence of ascites and/or esophageal varices and/or splenomegaly, together with low serum albumin, prolonged prothrombin time and thrombocytopenia at enrollment. HCC was diagnosed by the presence of a lesion with typical dynamic imaging characteristics on a triple phase CT scan with or without an elevated level of serum AFP.
Table 1. Demographic characteristics of 24 patients with chronic HBV infection .
| Clinical status | No (%) |
Gender (male:female) |
*Age (years) |
|
| Chronic | HBeAg positive | 13 (54) | 9:4 | 30±11 |
| hepatitis | HBeAg negative | 6 (25) | 3:3 | 42±13 |
| Cirrhosis | 1 (4) | 1:0 | 65 | |
| HCC | 4(17) | 3:1 | 60±7.6 |
*Mean±SD
Amplification of full length of HBV
HBV DNA was extracted from 200 μL of serum using the semi-automated Roche Magna Pure System (version 2.1 Roche Branchberg, NJ) according to the manufacturer’s instruction and amplified using the PicoMaxxTM High Fidelity PCR System from Stratagene for the full length of the genome. PCR reactions were set up based on 20 μl DH2O, 0.5 of each p1 and p2 primer (100 ng/ml), 4 μl DNA template and 25 μl of PicoMaxxTM.
Forward P1 (nt 421-441: CCGGAAAGCTTGAGCTCTTCTTTTTCACCTCTGCCTAATCA) and reverse P2 (nt 425-406: CCGGAAAGCTTGAGCTCTTCAAAAAGTTGCATGGTGCTGG) primers for amplification of full length HBV genomes were adapted from Gunther et al.11 The amplification was performed for the appropriate number of cycles using the following cycling program: denaturation at 94°C for 40 sec, annealing at 60°C for 1 min, elongation at 72°C for 4 min, with an increment of 5 sec/cycle for 40 cycles.
The PCR products of the full genome were purified with ultra clean columns (MO BIO Laboratories, USA) according to the manufacturer’s specification and sequenced by using several primers and the Big Dye Terminator Cycle Sequencing Ready Reaction Kit Version 3.1 (Applied Biosystems, Foster City, CA, USA).
The nt position of the primers’ nomenclature according to HPBADR1CG12 on the HBV genome were as follows: SeqP1 (nt 421-441) TTTTTCACCTCTGCCTAATCA; SeqP2 (nt 425-406) AAAAAGTTGCATGGTGCTGG; 865 (nt 864-883) TTCGGAGTGTGGATTCGCAC; OS2 (nt 2798-2817) TCTCTGACATACTTTCCAAT; JM (nt 1676-1696) TTGGGGTGGAGCCCTCAGGCT; 1798 (nt 1799-1820) CCAACTGCATGGCCTGAGGATG; 903 (nt 900-925) GTTGATAAGATAGGGGCATTTGGTGG; OSX1 (nt 2628-2648) TTTTCT TTTGTCTTTGGGTAT; and OS1 (nt 1408-1430) GCCTCATTTTGTGGGTCACCATA.
Identification of amino acid mutations
All ORFs for the HBV genome were studied for amino acid mutations and substitutions. The polymerase and overlapping S region were studied together as a single frame whereas core and X ORFs were studied separately. They were compared and evaluated according to the database of the Victorian Infectious Disease Reference Laboratory, Melbourne, Australia.
Measuring of HBV genome GC content average
Web-based application CpGProD6 have been proposed to report the distribution of CpG regions. This program produces graphic visualizations of the GC% and the O/E ratio that allows ease of viewing CpG island information. CpGProD searches for all the CGIs (CpG islands) along the sequence query. The average values for G+C frequency and CpGo/e ratio are calculated by using a window of 500 nt in CpGProD and window moving along the sequence by steps of one nt.
This program was performed in order to recognize the mammalian promoter regions, and to identify CpG islands in large genomic sequences and to shows the predicted probability over a TSS chart.6 For any existing species i, the amount of GC3 divergence among n genes since the placental ancestor is indicated by Di,anc and measured as:
where GC3ki is the GC3 observed for gene k, species i, while GC3kanc is the estimated ancestral GC3 for gene k. Overlapping windows with a G+C frequency above 0.5 and a CpGo/e value greater than 0.6 are grouped together to form the CGIs. To determine whether sequences are compositionally stationary, compositional divergence was calculated for among-species pairwise comparisons of each gene according to the method of Gillespie.13
RESULTS
Sequence analysis of the full HBV genome
A total of 3182 base pairs (bp) of the HBV genome was sequenced for 24 individuals. Patients’ demographic data are presented in Table 1. HBV strains in this study were defined as genotype D in comparison with reference strain X02496.1. These isolates were closely related to each other and according to the McDonald-Kreitman test for the ratio of replacement changes within species polymorphism and between-species divergence, the genetic relatedness of these isolates revealed 97%-100% neutral sequence similarity. The mean inter-genotypic divergence was 1.9%. The divergence to the D genotype of isolates retrieved from the Genbank was 3.9%. The distance to the other unrelated genotypes was 7.5%-16%. The lowest divergence observed was for genotype E, and the highest was for the H genotype.
Frequency of mutations in different open reading frames (ORFs)
a) Amino acid mutations of polymerase in overlap with HBs
Polymerase mutations at rtV173L plus rtL180M and rtM204V which have been mapped to the YMDD motif were selected in four patients who received lamivudine treatment. The full-length S gene (including preS1, preS2 and S regions) comprised 1170 bp and 382 amino acids in all patients except for two HCC patients. Amino acid mutations in the full length of the surface gene are shown in Figure 1 . One of the HCC patients showed an M1I mutation (methionine to isoleucine at position 1) and a deletion of three amino acids at position 14-16 in the preS2 region. In another HCC patient, a deletion of seven amino acids at positions 16-22 were observed. Amino acid mutations, P120T, M133T, and S143L were located within the “a“ determinant (codon 110-146) of HBsAg in four patients. Mutations of amino acid residues N40S and L49R that coincided with HLA class-I restricted to CTL epitopes were observed in two patients. Mutations of the S gene that overlapped with polymerase were selected in four patients who received lamivudine. The mutations at rtV173L plus rtL180M and rtM204V which has been mapped to the YMDD motif of polymerase also mapped in the S region as sE164D and sI195M (Figure 1). Amino acids arginine (position 122) and lysine (position 160) which determine the d/y and w/r subtypes of HBsAg, and all cysteine residues of the “a“ determinant were conserved.
Fig 1 .

Amino acid mutation in the envelope gene of HBV. Positions of mutations in deduced amino acid residue(s?) are indicated by vertical lines. The consensus residue of genotype D is shown in the first line. Mutations that affect the determinant region are indicated by a shaded box and mutation associated with a resistance to lamivudine are in bold.
b) Core promoter and core gene
The length of the C ORF gene was 639 bp with no deletions or insertions. Mutations were common in basal core promoter (BCP) sequences. A double mutation (A1762T and G1764A) was found in four patients with HCC, four e antigen-negative, and three HBeAg-positive chronic hepatitis B patients. All patients with HCC and cirrhotic patients harbored the T1753C mutation. A mutation at G1896 A was detected in 9 of 11 patients with e antigen-negative chronic hepatitis B. The direct repeat 1 (DR1) sequences at positions 1824 to 1834 were conserved in all patients. Amino acid substitutions of cS21F/T, cN92T, cM93A, cP130Q, cT147C, cV149I, and cR151Q in the cytotoxic T lymphocyte (CTL) epitopes of core protein and cS181P mutation were detected in HCC patients.
c) X gene
The X genes consisted of 465 nt and 154 amino acids in all patients. The DR2 sequence at positions 217 to 227 was conserved. Mutations A1762T and G1764A in the core promoter correspond to amino acid mutations L130 M and V131I of the X-protein along with the I127T mutation due to a mutation at position T1753C were detected in patients with HCC.
Identification of CpG islands and analysis of HBV genome asymmetry
CpGProD platform generates a graphic visualization of CpG islands. We focused on GC content (GC%), O/E ratio and CpG island length design, as shown in Table 2. The prediction results were divided into four types of CpG island-related information: i) GC% charts, ii) O/E ratio charts, iii) the predicted probability of being over the TSS, and iv) the distribution of CpG in the predicted genome sequence position. Compositional asymmetry was assessed by calculating nt bias via two parameters, AT skew and GC skew, for each individual HBV genome.
Table 2. Comparison of nucleotide (nt) composition for characteristics of CpG .
| HBV complete sequence name | Number | Begin | End | Length (bp) | G+C frequency | CpGo/e ratio | Start-p | AT skew | GC skew |
Strand
(strand-p * ) |
| 104 | 1/2 | 961 | 1471 | 511 | 0.4990 | 0.6201 | 0.1240 | -0.1797 | -0.1922 | minus (0.6400) |
| 104 | 2/2 | 1789 | 2895 | 1107 | 0.5113 | 0.7667 | 0.3005 | -0.1978 | -0.0919 | plus (0.7255) |
| 105 | 1/2 | 952 | 1462 | 511 | 0.4990 | 0.6201 | 0.1240 | -0.1797 | -0.1922 | minus (0.6400) |
| 105 | 2/2 | 1777 | 2890 | 1114 | 0.5153 | 0.7510 | 0.2890 | -0.1889 | -0.0976 | plus (0.6895) |
| 111 | 1/2 | 964 | 1471 | 508 | 0.5059 | 0.6085 | 0.1207 | -0.1633 | -0.1984 | minus (0.6984) |
| 111 | 2/2 | 1815 | 2915 | 1101 | 0.5150 | 0.7320 | 0.2687 | -0.2285 | -0.0899 | plus (0.7918) |
| 112 | 1/1 | 1795 | 2865 | 1071 | 0.5089 | 0.7561 | 0.2835 | -0.1901 | -0.0899 | plus (0.7138) |
| 115 | 1/2 | 964 | 1471 | 508 | 0.5059 | 0.6066 | 0.1197 | -0.1873 | -0.1907 | minus (0.6158) |
| 115 | 2/2 | 1809 | 2899 | 1091 | 0.5188 | 0.7675 | 0.3058 | -0.2114 | -0.0777 | plus (0.7879) |
| 138 | 1/1 | 1810 | 2888 | 1079 | 0.5116 | 0.7698 | 0.3004 | -0.2182 | -0.0797 | plus (0.7959) |
| 140 | 1/1 | 1809 | 2888 | 1080 | 0.5111 | 0.7143 | 0.2470 | -0.2121 | -0.0870 | plus (0.7678) |
| 141 | 1/2 | 960 | 1469 | 510 | 0.4980 | 0.5941 | 0.1105 | -0.1719 | -0.2047 | minus (0.6964) |
| 141 | 2/2 | 1795 | 2895 | 1101 | 0.5123 | 0.7670 | 0.3010 | -0.1918 | -0.0851 | plus (0.7305) |
| 142 | 1/1 | 1809 | 2886 | 1078 | 0.5130 | 0.7243 | 0.2574 | -0.2229 | -0.0850 | plus (0.7926) |
| 143 | 1/1 | 1809 | 2888 | 1080 | 0.5130 | 0.7237 | 0.2570 | -0.2205 | -0.0903 | plus (0.7764) |
| 145 | 1/1 | 1795 | 2888 | 1094 | 0.5119 | 0.7324 | 0.2659 | -0.1910 | -0.0964 | plus (0.6977) |
| 146 | 1/1 | 1795 | 2888 | 1094 | 0.5110 | 0.7202 | 0.2538 | -0.2000 | -0.0912 | plus (0.7321) |
| 147 | 1/1 | 1811 | 2888 | 1078 | 0.5130 | 0.7243 | 0.2574 | -0.2152 | -0.0850 | plus (0.7785) |
| 148 | 1/1 | 1811 | 2888 | 1078 | 0.5130 | 0.7243 | 0.2574 | -0.2152 | -0.0850 | plus (0.7785) |
| 149 | 1/1 | 1811 | 2888 | 1078 | 0.5121 | 0.7130 | 0.2463 | -0.2281 | -0.0870 | plus (0.7978) |
| 151 | 1/1 | 1810 | 2885 | 1076 | 0.5121 | 0.7426 | 0.2736 | -0.2229 | -0.0853 | plus (0.7920) |
| 153 | 1/1 | 1809 | 2899 | 1091 | 0.5151 | 0.7499 | 0.2850 | -0.2136 | -0.0712 | plus (0.8061) |
| 156 | 1/2 | 964 | 1471 | 508 | 0.5079 | 0.6026 | 0.1186 | -0.1920 | -0.1938 | minus (0.6134) |
| 156 | 2/2 | 1795 | 2885 | 1091 | 0.5105 | 0.7239 | 0.2566 | -0.1948 | -0.0952 | plus (0.7099) |
| 159 | 1/1 | 1795 | 2885 | 1091 | 0.5105 | 0.7371 | 0.2689 | -0.2060 | -0.0880 | plus (0.7531) |
| 273 | 1/1 | 1809 | 2888 | 1080 | 0.5130 | 0.7091 | 0.2438 | -0.2205 | -0.0866 | plus (0.7847) |
| 317 | 1/2 | 964 | 1471 | 508 | 0.5059 | 0.6066 | 0.1197 | -0.1793 | -0.1907 | minus (0.6364) |
| 317 | 2/2 | 1792 | 2885 | 1094 | 0.5119 | 0.7460 | 0.2789 | -0.1910 | -0.0929 | plus (0.7077) |
| 327 | 1/1 | 1810 | 2895 | 1086 | 0.5129 | 0.7743 | 0.3070 | -0.2212 | -0.0736 | plus (0.8139) |
| 328 | 1/1 | 1809 | 2888 | 1080 | 0.5130 | 0.7237 | 0.2570 | -0.2205 | -0.0903 | plus (0.7764) |
| 334 | 1/1 | 1810 | 2888 | 1079 | 0.5116 | 0.7136 | 0.2467 | -0.2220 | -0.0870 | plus (0.7867) |
| HBV_A | 1/1 | 1853 | 2863 | 1011 | 0.5272 | 0.6734 | 0.2162 | -0.1632 | -0.0807 | plus (0.6773) |
| HBV_B | 1/1 | 1841 | 2947 | 1107 | 0.5285 | 0.7526 | 0.3008 | -0.1839 | -0.0530 | plus (0.7928) |
| HBV_C | 1/1 | 1826 | 2863 | 1038 | 0.5212 | 0.6714 | 0.2132 | -0.1710 | -0.0832 | plus (0.6888) |
| HBV-D | 1/1 | 1795 | 2870 | 1076 | 0.5093 | 0.7349 | 0.2641 | -0.2008 | -0.0730 | plus (0.7788) |
A common CpG island in the plus strand was characterized with moderate negative GC skew (0.0770–0.0960) at about nt 1795-2890, surrounding the ATG start site of HBs and polymerase genes in all 24 HBV full genome sequences. Interestingly, a CpG island located at about nt 950-1470 and was in overlap with enhancer I and the X-protein gene promoter was defined in three HCC patients, the cirrhotic patient, and three patients with chronic hepatitis. This region exhibited a strong negative GC skew (-0.1900 to –0.1980) in the minus strand that reflected a C to T mutation in the pgRNA. All the above results are shown in Figure 2 and Table 2.
Fig 2 .
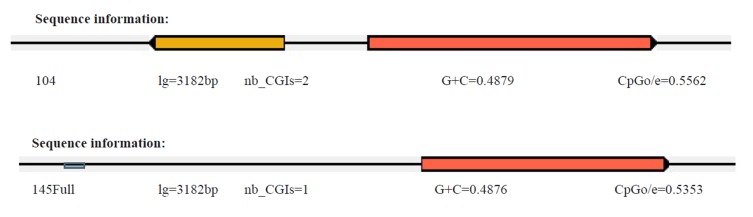
Visualization of the CpG island prediction results of two patients (104 with HCC and 145 with chronic hepatitis). GC% chart shows the GC content distribution and CpG nucleic and CpG island distribution in the input sequence.
DISCUSSION
In the present study, the full-length genome sequence of HBV was analyzed in 24 patients. On the basis of 3182 nt sequences in the full length genome, the strains from different stages of hepatitis were closely related to each other. Previous studies of Iranian patients demonstrated that the distribution of HBV genotype D in different liver diseases and HBV genotype had no influence on clinical features of hepatitis.14, 15
It is revealed that variants with mutations and deletions in the preS2 region are frequent in patients who develop end stage liver disease or HCC.16, 17 Also mutations in the core promoter region may play a role in HBeAg clearance and the A1762T/G1764A mutations that are highly prevalent in chronic active hepatitis with long-standing liver disease, irrespective of the HBeAg statu.14 Asymmetric mutation pressures which occurred in replication supported the generation-time effect hypothesis18 in which molecular clock can explain HBV evolutionary in chronic hepatitis B and its effect on hepatocarcinogenesis. The presence of strong negative GC skew in the minus strand of HBV in HCC and cirrhotic patients leads to greater mutation pressure associated with greater differences in GC skew between the plus and minus strands. The computed GC content showed the existence of CpG-rich regions spanning the enhancer I and X-protein gene promoter. The latter might have a role in inducing expression of protein-x which is related to HCC.
Interestingly, some of the CpG sites in CpG islands of HBV were found to be methylated in HCC patients and hepatocyte samples, when checked for both the integrated or unintegrated HBV genomes.19 , 20DNA methylation
possibly is a self-defence mechanism of HBV to cloak itself from immune detection.7, 10
In conclusion, this study demonstrates that nt identity for HBV isolates varied from 97% to 100% in the Iranian population. However, HBV mutants were prevalent among patients with HCC. A negative GC skew in the minus strand of HBV can explain the mechanisms of evolution, such as natural selection and genetic drift, and generation of variations by mutations in developing liver disease in hepatitis B.
CONFLICT OF INTEREST
The authors declare no conflict of interest related to this work.
Please cite this paper as: Shokrgozar Z, Tayebi S, Minucheher Z, Mohamadkhani A. Hepatitis B Virus Genome Asymmetry in Hepatocellular Carcinoma: A Case-series Study. Middle East J Dig Dis 2012;4:150-7.
References
- 1.Liu S, Zhang H, Gu C, Yin J, He Y, Xie J. et al. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: a meta-analysis. J Natl Cancer Inst. 2009;101:1066–82. doi: 10.1093/jnci/djp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barone M, Spano D, D’Apolito M, Centra M, Lasalandra C, Capasso M. et al. Gene expression analysis in HBV transgenic mouse liver: a model to study early events related to hepatocarcinogenesis. Mol Med. 2006;12:115–23. doi: 10.2119/2006-00015.Barone. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fattovich G, Brollo L, Giustina G, Noventa F, Pontisso P, Alberti A. et al. Natural history and prognostic factors for chronic hepatitis type B. Gut. 1991;32:294–8. doi: 10.1136/gut.32.3.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mohamadkhani A, Montazeri G, Poustchi H. The Importance of Hepatitis B Virus Genome Diversity in Basal Core Promoter Region. Middle East J Dig Dis. 2011;3:13–9. [PMC free article] [PubMed] [Google Scholar]
- 5.Lobry JR. Asymmetric substitution patterns in the two DNA strands of bacteria. Mol Biol Evol. 1996;13:660–5. doi: 10.1093/oxfordjournals.molbev.a025626. [DOI] [PubMed] [Google Scholar]
- 6.Ponger L, Mouchiroud D. CpGProD: identifying CpG islands associated with transcription start sites in large genomic mammalian sequences. Bioinformatics. 2002;18:631–3. doi: 10.1093/bioinformatics/18.4.631. [DOI] [PubMed] [Google Scholar]
- 7.Kaur P, Paliwal A, Durantel D, Hainaut P, Scoazec JY, Zoulim F. et al. DNA methylation of hepatitis B virus (HBV) genome associated with the development of hepatocellular carcinoma and occult HBV infection. J Infect Dis. 2010;202:700–4. doi: 10.1086/655398. [DOI] [PubMed] [Google Scholar]
- 8.Klinman DM, Yi AK, Beaucage SL, Conover J, Krieg AM. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci U S A. 1996;93:2879–83. doi: 10.1073/pnas.93.7.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R. et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–9. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 10.Hantz O, Parent R, Durantel D, Gripon P, Guguen-Guillouzo C, Zoulim F. Persistence of the hepatitis B virus covalently closed circular DNA in HepaRG human hepatocyte-like cells. J Gen Virol. 2009;90:127–35. doi: 10.1099/vir.0.004861-0. [DOI] [PubMed] [Google Scholar]
- 11.Günther S, Li BC, Miska S, Krüger DH, Meisel H, Will H. A novel method for efficient amplification of whole hepatitis B virus genomes permits rapid functional analysis and reveals deletion mutants in immunosuppressed patients. J Virol. 1995;69:5437–44. doi: 10.1128/jvi.69.9.5437-5444.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Günther S, Sommer G, Von Breunig F, Iwanska A, Kalinina T, Sterneck M. et al. Amplification of full-length hepatitis B virus genomes from samples from patients with low levels of viremia: frequency and functional consequences of PCR-introduced mutations. J Clin Microbiol. 1998;36:531–8. doi: 10.1128/jcm.36.2.531-538.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gillespie JH. Variability of evolutionary rates of DNA. Genetics. 1986;113:1077–91. doi: 10.1093/genetics/113.4.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poustchi H, Mohamadkhani A, Bowden S, Montazeri G, Ayres A, Revill P. et al. Clinical significance of precore and core promoter mutations in genotype D hepatitis B-related chronic liver disease. J Viral Hepat. 2008;15:753–60. doi: 10.1111/j.1365-2893.2008.00998.x. [DOI] [PubMed] [Google Scholar]
- 15.Sendi H, Mehrab-Mohseni M, Zali MR, Norder H, Magnius LO. T1764G1766 core promoter double mutants are restricted to Hepatitis B virus strains with an A1757 and are common in genotype D. J Gen Virol. 2005;86:2451–8. doi: 10.1099/vir.0.81023-0. [DOI] [PubMed] [Google Scholar]
- 16.Mun HS, Lee SA, Kim H, Hwang ES, Kook YH, Kim BJ. Novel F141L pre-S2 mutation in hepatitis B virus increases the risk of hepatocellular carcinoma in patients with chronic genotype C infections. J Virol. 2011;85:123–32. doi: 10.1128/JVI.01524-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chisari FV, Filippi P, Buras J, McLachlan A, Popper H, Pinkert CA. et al. Structural and pathological effects of synthesis of hepatitis B virus large envelope polypeptide in transgenic mice. Proc Natl Acad Sci U S A. 1987;84:6909–13. doi: 10.1073/pnas.84.19.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li WH, Ellsworth DL, Krushkal J, Chang BH, Hewett-Emmett D. Rates of nucleotide substitution in primates and rodents and the generation-time effect hypothesis. Mol Phylogenet Evol. 1996;5:182–7. doi: 10.1006/mpev.1996.0012. [DOI] [PubMed] [Google Scholar]
- 19.Guo Y, Li Y, Mu S, Zhang J, Yan Z. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J Med Virol. 2009;81:1177–83. doi: 10.1002/jmv.21525. [DOI] [PubMed] [Google Scholar]
- 20.Vivekanandan P, Thomas D, Torbenson M. Methylation regulates hepatitis B viral protein expression. J Infect Dis. 2009;199:1286–91. doi: 10.1086/597614. [DOI] [PMC free article] [PubMed] [Google Scholar]