

Abstract
Varied stresses to cells can lead to a repression in translation by triggering phosphorylation of eukaryotic translation initiator factor 2α (eIF2α), which is central to a process known as the integrated stress response (ISR). PKR-like ER-localized eIF2 kinase (PERK), one of the kinases that phosphorylates eIF2α and coordinates the ISR, is activated by stress occurring from the accumulation of misfolded or unfolded proteins in the endoplasmic reticulum (ER). Mutant Cu/Zn superoxide dismutase (mtSOD1) is thought to cause familial amyotrophic lateral sclerosis (FALS) because it misfolds and aggregates. Published studies have suggested that ER stress is involved in FALS pathogenesis since mtSOD1 accumulates inside the ER and activates PERK leading to phosphorylated eIF2α (p-eIF2α). We previously used a genetic approach to show that haploinsufficiency of PERK significantly accelerates disease onset and shortens survival of G85R mtSOD1 FALS transgenic mice. We now show that G85R mice that express reduced levels of active GADD34, which normally dephosphorylates p-eIF2α and allows recovery from the global suppression of protein synthesis, markedly ameliorates disease. These studies emphasize the importance of the ISR, and specifically the PERK pathway, in the pathogenesis of mtSOD1-induced FALS and as a target for treatment. Furthermore, the ISR may be an appropriate therapeutic target for sporadic ALS and other neurodegenerative diseases since misfolded proteins have been implicated in these disorders.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease characterized by the selective loss of motor neurons (MNs). Approximately 10% of ALS cases are familial (known as FALS) with an autosomal dominant inheritance pattern, and ∼20% of FALS cases are caused by mutant Cu/Zn superoxide dismutase (mtSOD1) (reviewed in 1). Compelling evidence suggests that mtSOD1 causes FALS through a toxic gain in function rather than a loss in function; however, the nature of the toxicity is not well-defined. The presence of mtSOD1 aggregates as a characteristic feature of the neuropathology of FALS as well as the role of misfolded proteins in the pathogenesis of many neurodegenerative diseases have suggested that accumulation and aggregation of misfolded mtSOD1 is fundamental to the mutant protein's toxicity and leads to the death of MNs.
Varied stresses to cells can trigger phosphorylation of eukaryotic translation initiator factor 2α (eIF2α) on serine 51 by one of four known kinases, which is central to a process known as the integrated stress response (ISR). PKR-like ER-localized eIF2 kinase (PERK), one of the kinases that phosphorylates eIF2α and coordinates the ISR, is activated by stress occurring from the accumulation of misfolded or unfolded proteins in the endoplasmic reticulum (ER). The PERK pathway is one of three that constitutes the unfolded protein response (UPR), and the most rapidly activated arm. Although phosphorylated eIF2α (p-eIF2α) represses most translation, it promotes translation of selected genes and transcription factors that can enhance protein folding and lead to ER-associated degradation (ERAD) of the misfolded protein by the ubiquitin-proteasomal system (UPS) following retrotranslocation into the cytosol. For example, p-eIF2α induces translation of ATF4, a transcription factor that activates transcription of cytoprotective genes, including those involving chaperone function, the maintenance of redox homeostasis and protein degradation (2). ATF4 activates transcription of growth arrest and DNA damage-inducible protein (GADD34) and CCAAT enhancer-binding homologous protein (CHOP). GADD34 is a stress-inducible regulatory subunit of a phosphatase complex that quickly dephosphorylates p-eIF2α, allowing recovery from the global suppression of protein synthesis. CHOP expression can lead to cellular apoptosis in certain cell types if the UPR fails to compensate for the misfolding, for example, if the ER stress is sustained and excessive. The UPR involves activation of two other ER-resident stress sensors besides PERK: activating transcription factor 6 (ATF6) and inositol-requiring transmembrane kinase/endonuclease-α (IRE1α) (3). ATF6 and IRE1α/XBP1 activation by the UPR upregulates transcription of multiple genes, including genes important in protein quality control.
Although SOD1 is primarily cytosolic, mtSOD1, and to a lesser extent wild type (wt) SOD1, is also present in the secretory pathway (4–7). These observations have drawn attention to the possible role of ER stress in FALS (reviewed in 8,9), especially since ER stress has been implicated in the pathogenesis of another autosomal dominantly inherited form of MN disease caused by vesicle-associated membrane protein-associated protein B (10,11)—as well as other neurodegenerative diseases (12) (reviewed in 13). There are a number of studies that have demonstrated involvement of ER stress in mtSOD1-induced cell death in vitro (14–18), as well as in tissues from FALS transgenic mice (7,19,20) and from patients with FALS and sporadic ALS (21). Of note, studies have shown that mtSOD1 binds Derlin-1, a transmembrane ER protein involved in the translocation of misfolded proteins from the ER to the cytosol (17). In addition, overexpression of Derlin-1 decreases mtSOD1-induced cell death in vitro (22). All pathogenic mtSOD1s, but not human wtSOD1 or mutants of SOD1 that are not thought to be pathogenic, are reported to have an epitope that binds Derlin-1 and is recognized by a monoclonal antibody (23). These studies have suggested that the UPR is overwhelmed in FALS and therefore leads to toxicity and apoptosis, and that enhancing the UPR may ameliorate disease.
Of interest, recent data have demonstrated abnormalities in the structure of the SOD1 protein or its activity in the central nervous system of patients with sporadic ALS in addition to those with mtSOD1-induced FALS (24–29). Furthermore, a role for ER stress in the pathogenesis of sporadic ALS is supported by the presence of aggregates of misfolded TDP-43 in all cases of sporadic ALS as well as cases of non-mtSOD1-linked FALS (30). Recent studies demonstrated that C. elegans and zebrafish that express mtTDP-43 and mtFUS exhibit MN toxicity (31) that was thought to be related to oxidative stress in the ER (32). Four drugs were found to be protective against this toxicity, and all of them targeted the UPR (32). Therefore, treatment approaches that target the UPR may be effective in sporadic ALS as well as FALS. Indeed, these therapies may have a role in the treatment of a number of neurodegenerative diseases in which misfolded proteins have been implicated (9).
In a previous study (33), we used a genetic approach to clarify the importance of the UPR in mtSOD1-induced ALS. We found that G85R mtSOD1 transgenic mice that are haploinsufficient for PERK had accelerated disease, presumably because of a relative decrease in eIF2α phosphorylation. In the present study, we tested whether enhanced eIF2α phosphorylation would ameliorate mtSOD1-induced disease. In order to carry this out, G85R FALS transgenic mice were crossed with mice that express GADD34ΔC, a mutation that interferes with the normal function of GADD34 and confers resistance to cell death induced by ER stress (34). We predicted that compared with G85R mice, G85R/GADD34+/ΔC mice would have a decrease in misfolded proteins (because enhanced eIF2α phosphorylation would augment translation repression and lead to upregulation of ATF4, with subsequent induction of genes important in chaperone activity, degradation machinery, redox homeostasis and apoptosis). Our results show that G85R/GADD34+/ΔC mice have a remarkably delayed disease onset, delayed early phase of disease and prolonged survival compared with control littermate G85R mice. The data indicate the importance of the ER stress in the pathogenesis of mtSOD1-induced ALS, and the ISR as therapeutic target.
RESULTS
G85R/GADD34+/ΔC transgenic mice have a prolongation in disease onset, longer early phase of disease and longer survival than G85R transgenic mice
In order to determine the effect of modulating ER stress by elevating the level of p-eIF2α in mtSOD1-induced FALS, we crossed G85R transgenic mice (35) with GADD34+/ΔC mice (34). GADD34+/ΔC mice have an inactive allele that encodes a truncated GADD34 protein that lacks the phosphatase domain. GADD34ΔC/ΔC mice have a normal phenotype, although they display increased resistance to ER stress (34,36). Compared with control cells, GADD34+/ΔC fibroblasts have elevated p-eIF2α levels following tunicamycin treatment (Supplementary Material, Fig. S1). The G85R/GADD34+/ΔC transgenic mice were compared with G85R littermate mice with respect to the following previously defined clinical parameters (37): onset of disease, defined as peak weight before a decline; early phase of disease, defined as the period from peak weight until loss of 10% of maximal weight; late phase of disease, defined as the time from 10% loss in weight until death (when a mouse is unable to right itself within 20 s after being put on its back).
The mean onset of disease for the G85R/GADD34+/ΔC transgenic mice (n = 19) was significantly later than for G85R mice (n = 20) (389.1 ± 87.5 versus 304.2 ± 30.7 days, P < 0.001) (Fig. 1A). In addition, there was a prolongation in the duration of the early phase of disease in G85R/GADD34+/ΔC mice versus G85R mice (25.1 ± 6.8 versus 17.6 ± 3.3 days, P < 0.001) (Fig. 1B and D). There was no statistically significant difference in the duration of the late phase of disease (16.5 ± 3.6 versus 15.6 ± 3.6 days, P > 0.05) (Fig. 1E). As expected, survival of G85R/GADD34+/ΔC mice was significantly prolonged compared with G85R mice (430.7 ± 94.0 versus 337.4 ± 29.4 days, P < 0.001) (Fig. 1C).
Figure 1.

Plots of disease onset (A), the end of early disease (B), and survival (C) as well as diagrams of the duration (with standard deviation) of the early phase (D) and late phase (E) of disease in G85R/GADD34+/ΔC (n = 19) versus G85R transgenic mice (n = 20). In the Y axis of (A)–(C), 1 refers to the total number of animals. ** P < 0.001.
G85R/GADD34+/ΔC transgenic mice have less pathology, total mtSOD1 and mtSOD1 aggregation compared with similarly aged G85R transgenic mice
In order to compare the pathology of the different groups of mice, we examined the anterior horn of the lumbar spinal cord from G85R/GADD34+/ΔC, G85R and non-transgenic littermate control mice at varied ages. G85R transgenic mice exhibited MN loss (i.e. fewer Nissl staining cells), astrocytosis [more prominent glial fibrillary acid protein (GFAP)-positive cells] and microgliosis (more abundant Iba-positive cells) at end stage of disease at 345 days, compared with non-transgenic controls (Fig. 2A). In contrast, G85R/GADD34+/ΔC mice had little if any pathology at 345 days (Fig. 2A); however, by end stage of disease at ∼420 days, G85R/GADD34+/ΔC mice showed similar pathological changes to those seen in end-stage G85R mice. A quantitative study (Fig. 2B) showed that G85R/GADD34+/ΔC mice at 345 days had only a slight loss in the number of MNs compared with control mice, while G85R mice had a very significant decrease. There was a similar significant decrease in MNs as well as a similar astrocytosis and microgliosis at end stage in G85R and G85R/GADD34+/ΔC mice (Fig. 2B). The findings in the anterior horn of control GADD34+/ΔC mice were similar to those of non-transgenic controls (data not shown).
Figure 2.

Neuropathological and immunohistochemical studies of the anterior horn of the lumbar spinal cord of G85R, G85R/GADD34+/ΔC and littermate non-transgenic mice at varying ages. Sections of the anterior horn of the lumbar spinal cord were stained with: (A) Nissl for MNs, anti-GFAP antibody for astrocytes and anti-Iba1 antibody for microglia. (B) Bar diagrams show mean ± standard deviation calculated by counting cells in 15–20 sections from the lumbar spinal cord anterior horn of four mice from each of the groups (G85R, G85R/GADD34+/ΔC and littermate non-transgenic mice) at varying ages. The scale bar = 50 µm. ** P < 0.001.
The lumbar spinal cord was examined for SOD1 aggregation by immunohistochemistry. Abundant mtSOD1 aggregates were present in cells with MN morphology and their cell processes in G85R mice at 345 days (Fig. 3A). In contrast, there were few if any aggregates of mtSOD1 in G85R/GADD34+/ΔC mice at 345 days. Also of interest was the finding of fewer aggregates in G85R/GADD34+/ΔC mice at end stage at ∼420 days compared with G85R mice at end stage (Fig. 3B). Aggregated SOD1 was also examined on western blots of electrophoresed lumbar spinal cord homogenates that were not treated with β-mercaptoethanol prior to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE). A representative western blot (Fig. 3C), which is overexposed to display high molecular weight SOD1 forms, shows that when similar amounts of protein were loaded, the lumbar spinal cord homogenates from G85R mice at 345 days had much greater amounts of SOD1 monomer and high molecular weight protein bands that immunostained with anti-human SOD1 when compared with G85R/GADD34+/ΔC mice at 380 and 400 days. A representative western blot (Fig. 3D) shows that there is a statistically significant increase in the amount of total SOD1 homogenates (treated with β-mercaptoethanol prior to SDS–PAGE) in the lumbar spinal cord of G85R mice at 345 days (end stage) compared with G85R/GADD34+/ΔC mice at 380 days (P < 0.001), 400 days (P < 0.001) and 420 days (end stage) (P < 0.05) (Fig. 3E). The results suggest that the reduced level of GADD34 in G85R/GADD34+/ΔC mice led to a decrease in the amount of aggregated and total SOD1.
Figure 3.
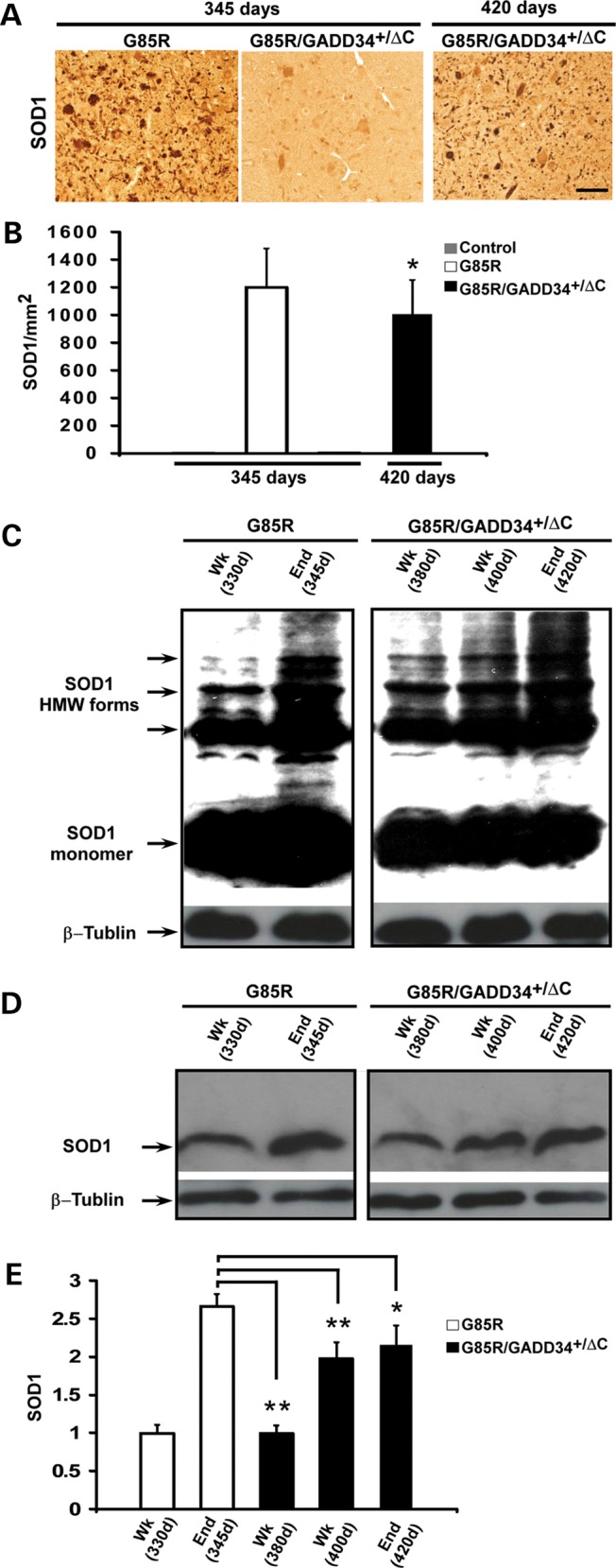
mtSOD1 in the anterior horn of the lumbar spinal cord of G85R, G85R/GADD34+/ΔC and littermate non-transgenic mice at varying ages. In this figure, ‘Wk’ refers to the early phase of disease, while ‘End’ refers to end stage; the approximate ages of the mice are noted. (A) Representative sections of the anterior horn of the lumbar spinal cord were stained with anti-human SOD1 antibody to identify aggregates. (B) Bar diagram shows the mean ± standard deviation of mtSOD1 aggregates/mm2. Note that there are so few aggregates in the anterior horn of control and G85R/GADD34+/ΔC mice at 345 days that the bars are not visible. The number of aggregates in G85R mice at end stage (345 days) was statistically significantly increased compared with G85R/GADD34+/ΔC mice at end stage (420 days). *P < 0.05. (C) Representative western blot of homogenates of the lumbar spinal cord of G85R and G85R/GADD34+/ΔC mice sacrificed at different times of disease, and processed as described in Materials and Methods. The samples were treated without β-mercaptoethanol before boiling and then loaded on 15% SDS polyacrylamide gels, electrophoresed, blotted and then immunostained using a human-specific anti-SOD1 antibody [to detect the monomeric and high molecular weight (HMW) forms] and an anti-β-tubulin antibody (as a loading control). (D) Representative western blot of homogenates of the lumbar spinal cord of G85R and G85R/GADD34+/ΔC mice that were harvested and processed as described above; however, the samples were diluted 1:5 and treated with β-mercaptoethanol before boiling to identify total SOD1. (E) Bar diagram shows total SOD1 in the lumbar spinal cord of G85R and G85R/GADD34+/ΔC mice at different ages and stages of disease. Each bar, which represents data from four animals, shows the mean ± standard deviation of the ratio of SOD1 signal to β-tubulin signal, displayed as a fraction of the value of the G85R mice at 330 days, which is set at 1. There was a statistically significant greater amount of total SOD1 in the lumbar spinal cord of G85R mice at 345 days (end stage) compared with G85R/GADD34+/ΔC mice at 380, 400 and 420 days (end stage). *P < 0.05, ** P < 0.001.
G85R/GADD34+/ΔC transgenic mice have enhanced phosphorylation of eIF2α
The accumulation of misfolded protein activates PERK, which phosphorylates eIF2α leading to translational repression; however, there is enhanced translation of ATF4, which upregulates the expression of GADD34 and CHOP. GADD34 is part of a complex that rapidly dephosphorylates p-eIF2α. The GADD34+/ΔC mutation is predicted to augment the phosphorylation of eIF2α following ER stress. Figure 4A is a representative western blot showing immunostaining of p-eIF2α, ATF4 and CHOP. GADD34+/ΔC and non-transgenic littermate control mice showed no immunostaining of these proteins. p-eIF2α, ATF4 and CHOP were detected in all of the four G85R mice examined at end stage (∼345 days), but minimally seen (ATF4) or not seen (p-eIF2α and CHOP) in any of the four similarly aged G85R/GADD34+/ΔC mice. On the other hand, the amount of p-eIF2α and ATF4 at end stage at ∼420 days in G85R/GADD34+/ΔC mice was statistically significantly increased when compared with the amount seen in G85R mice at end stage at 345 days (Fig. 4B). These results suggest that G85R/GADD34+/ΔC mice are more protected early in the disease process from ER stress than G85R mice; however, at end stage, the enhanced phosphorylation of eIF2α ultimately fails to protect G85R/GADD34+/ΔC mice from mt SOD1 expression.
Figure 4.

Detection of markers of the ISR in G85R and G85R/GADD34+/ΔC mice. (A) Representative western blots from differently aged G85R, G85R/GADD34+/ΔC, GADD34+/ΔC and control non-transgenic littermate mice immunostained with antibodies to phosphorylated (p)-eIF2α, ATF4 and CHOP. Anti-β tubulin antibody was used as a loading control. ‘Pre’ refers to prior to disease onset, ‘Wk’ refers to the early phase of disease, while ‘End’ refers to end stage; the approximate ages of the mice are provided. (B) Bar diagrams showing quantitation of UPR markers from western blots of homogenates of the lumbar spinal cord from G85R (n = 4) and G85R/GADD34+/ΔC (n = 4) mice at end stage of disease. The mean ± standard deviation is shown. The value of the specific ISR marker detected in G85R mice was arbitrarily set as 1. * P < 0.05, ** P < 0.001.
DISCUSSION
ALS is a relentless paralyzing disease resulting from the death of MNs (reviewed in 1). Although effective treatment and cures are not available, there is increased understanding of certain features of its pathogenesis, primarily as a result of the study of FALS. Approximately 20% of FALS cases are caused by mutations in SOD1. The mechanism by which mtSOD1 kills MNs is not fully understood, but it appears to occur through a gain of function, rather than a loss of function (i.e. a loss of dismutase activity). mtSOD1 expression is associated with a number of abnormalities, including oxidative stress, protein aggregation, mitochondrial dysfunction and disruptions in axonal transport. Yet, the mechanisms by which mtSOD1 interferes with these cellular processes to cause FALS are poorly understood. One common pathological feature of FALS is the misfolding and aggregation of mtSOD1. Of note, abnormalities in the structure of wtSOD1 have also been reported in sporadic ALS (24–29). Furthermore, aggregates of other proteins, including TDP-43, have been identified in sporadic ALS (30).
Cells deal with unfolded and misfolded proteins using protein quality control systems. The quality control of secreted and membrane proteins is carried out in the ER by making use of the pathways of the UPR that can decrease protein synthesis and refold or degrade abnormally structured proteins. ER proteins that remain misfolded are cleared by the ERAD pathway, in which the misfolded proteins retrotranslocate to the cytoplasm for degradation by the UPS. When the UPS is overwhelmed, the autophagy-lysosomal pathway is activated (38). Eventually, if ER stress continues and protein quality control machinery exceeded, apoptosis and cell death can ensue. PERK reacts early to the presence of misfolded proteins, decreasing protein translation through phosphorylation of eIF2α. Despite this decrease in protein synthesis, ATF4 is selectively translated, activating transcription of cytoprotective genes (2).
The presumed importance of misfolded proteins in neurodegenerative diseases and the finding that mtSOD1 can be detected in the ER-Golgi pathway drew attention to the role of ER stress and the UPR in ALS. There are a number of studies demonstrating that tissues from FALS transgenic mice and human ALS patients have evidence of ER stress and activation of the ISR (reviewed in 39). MNs are especially vulnerable to ER stress because of the abundance of proteins synthesized and secreted in these exceptionally large, very metabolically active cells that possess huge amounts of associated membranes. The activation of the UPR may not only result from the presence of misfolded SOD1, but as a result of oxidative stress and axotomy (40).
We initially focused on the PERK pathway because it reacts early in the presence of misfolded proteins. In addition, the finding that treatment of mtSOD1 transgenic mice with salubrinal, which produces an elevation in p-eIF2α, ameliorates mtSOD1-induced disease supported the importance of PERK in FALS (19). Our previous study used a genetic approach to demonstrate the substantial effect of the PERK pathway on mtSOD1-induced FALS (33). G85R mice that were heterozygous for a PERK null mutation exhibited a prominent acceleration of disease, decreasing survival by ∼60 days. The data from this experiment demonstrated a less prominent increase in eIF2α phosphorylation in G85R/Perk+/− mice when compared with G85R mice. This relative decrease in eIF2α phosphorylation led to less suppression of protein synthesis, and therefore allowed for an accelerated and increased mtSOD1 aggregation that presumably overwhelmed the UPR earlier in the case of G85R/Perk+/− mice. These results prompted us to pursue the present genetic studies to test whether we would find a robust ameliorative effect on mtSOD1-induced disease by bolstering the effect of PERK.
The present study involves a cross between GADD34+/ΔC mice, which are resistant to ER stress (34), and G85R mice. Compared with G85R mice, G85R/GADD34+/ΔC mice had evidence of both a delayed accumulation of total (monomeric and high molecular forms of) mtSOD1 as well as a delayed formation of mtSOD1 aggregates. We suspect that the delayed accumulation of total mtSOD1 is related to a decrease in mtSOD1 synthesis when compared with G85R mice, as a result of enhanced phosphorylation of eIF2α. The delay in mtSOD1 aggregate formation in G85R/GADD34+/ΔC mice compared with G85R mice may be a result of less total mtSOD1 being available as a source of misfolded SOD1 and/or because of an enhanced expression of chaperone and cytoprotective genes due to selective translation of ATF4, which is downstream of p-eIF2α. Despite the expectation that G85R/GADD34+/ΔC mice would have enhanced phosphorylation of eIF2α, we found less p-eIF2α (and ATF4 and CHOP) in the lumbar spinal cords of G85R/GADD34+/ΔC mice compared with similarly aged G85R mice. We suspect that this decrease in ISR markers is a result of an initial enhanced phosphorylation of eIF2α in G85R/GADD34+/ΔC mice, which occurs in a rapid fashion, decreasing ER stress, followed by quick dephosphorylation of eIF2α. Presumably, the GADD34 mutation provides protection from ER stress, which lessens disease pathology. Compared with G85R/GADD34+/ΔC mice, G85R mice have more clinical disease, more pathology and more expression of stress proteins. However, as stress increases in G85R/GADD34+/ΔC mice as a result of the continuing accumulation of misfolded SOD1 and reactive oxygen species, the UPR finally becomes overwhelmed. Because of the mutation in GADD34, a greater amount of p-eIF2α was seen in G85R/GADD34+/ΔC mice compared with G85R mice at end stage.
G85R/GADD34+/ΔC mice had a very significant delay in the onset of disease and a prolongation in the early phase of disease resulting in a marked extension in survival, one of the longest rescues that has been observed. The delay in the onset of disease in G85R/GADD34+/ΔC mice is greater than that seen in G85R mice that have a knockdown of mtSOD1 in a specific neural cell type, including the MN, astrocyte, microglial cell and Schwann cell (41–44). We are presently testing whether mtSOD1/GADD34ΔC/ΔC mice will exhibit an even greater amelioration of disease.
The only longer rescue of mtSOD1-induced FALS was reported by Kang et al. (45) following a knockdown of mtSOD1 in oligodendrocyte precursor cells and oligodendrocytes in G37R mice. Kang et al. (45) found that in addition to MNs, oligodendrocytes die in ALS. Of note, oligodendrocytes like MNs are extremely vulnerable to ER stress (46). The sensitivity of oligodendrocytes to ER stress is due to the heavy demands placed on the secretory pathway by the synthesis and maintenance of myelin. Our results raise the possibility that the basis for mtSOD1 toxicity in oligodendrocytes may be ER stress.
The present findings and our previous publication (33) highlight the importance of the PERK pathway and the ISR in mtSOD1-induced FALS. The findings of abnormalities of structure of SOD1 along with the presence of ER stress in sporadic ALS suggest that the PERK pathway and the ISR may be effective targets for treatment of sporadic ALS as well as FALS. In addition, this therapeutic target may be an appropriate one for other neurodegenerative diseases, since misfolded proteins and ER stress have been implicated in these disorders (9,47,48).
Although our previously published genetic study as well as the present one demonstrated the ameliorative effect of bolstering the PERK pathway, there remain concerns about enhancing the ISR. PERK activation, which leads to CHOP expression, can be apoptotic (49) as well as protective (50,51) depending on the degree and chronicity of ER stress. In addition, the ISR can have unexpected effects, perhaps partly because the effects may be cell type-specific or because a large cascade of genes may be affected. Although disease in G85R mice worsened following decreased expression of PERK and was ameliorated following decreased GADD34 expression, prion disease neurodegeneration was exacerbated by inhibiting GADD34, and ameliorated by overexpressing GADD34 (52) and inhibiting PERK (53). In addition, decreased PERK expression ameliorated synaptic dysfunction in Alzheimer's disease (54). Furthermore, deletion of XBP-1, a component of the UPR, surprisingly extended rather than decreased survival of mtSOD1 mice (55); however, the effect of the XBP-1 deletion may have been related to its effect on the inflammatory response (56) rather than autophagy, as originally proposed (55). Even more complicated is a study that showed the XBP-1 stimulated retinal ganglion cell survival following axonal injury, although PERK-CHOP signaling led to retinal ganglion apoptosis (40,49). In summary, the powerful effects of the UPR and ISR indicate its potential for prominent effects on diseases involving misfolded proteins, but make one cautious because of its broad influence on gene expression and its different effects on neurodegenerative disease processes.
MATERIALS AND METHODS
Mice and breeding
G85R transgenic mice that carry G85R genomic sequence flanked by LoxP sites (and were prepared for other studies involving Cre/LoxP) have been previously described (35). These mice were crossed with GADD34ΔC/ΔC mice, which have a mutation that encodes a truncated GADD34 protein that lacks the phosphatase domain (34). All the mice were on a C57BL/6 background. The G85R/GADD34+/ΔC transgenic mice were identified by PCR using previously published primers for the detection of the SOD1 (35) and for GADD34 (forward, 5′-CCA GGA GAG AAG ACC AAG GGA CGT G-3′; reverse, 5′-AAG CCT TCG CCA TCT GCT TAT CCA G-3′) genes.
Assessment of disease phenotype
Clinical assessment
Mice were weighed every 2 days and clinically assessed as previously described in studies of FALS transgenic mice (57), including a different line of G85R transgenic mice (58): onset of disease was defined as peak weight before a decline; early phase of disease was the period from peak weight until loss of 10% of maximal weight; late phase of disease was the time from 10% loss in weight until death (when a mouse was unable to right itself within 20 s after being put on its back). In experiments monitoring disease parameters and survival, G85R/GADD34+/ΔC mice were compared with littermate G85R mice. Non-transgenic littermate and GADD34+/ΔC mice were used as controls.
Pathology
The two groups of mice that were evaluated clinically were also studied and compared with respect to pathology. The pathology of the spinal cord was evaluated as previously described (41,59).
Immunohistochemical evaluation
Nissl staining, GFAP antibody and Iba1 antibody staining have been described (44). SOD1 aggregation was determined by immunohistochemical staining using a rabbit antibody that recognizes the carboxyl end of mouse and human SOD1 (60), as previously described (41). Quantitation of MNs, astrocytes, activated microglia/macrophages, and SOD1 aggregation was carried out, using published methods (44), on 15 to 20 sections from the lumbar spinal cord anterior horn of 4 mice from each of the following groups: G85R mice at end stage of disease, non-transgenic littermate controls and G85R/GADD34+/ΔC mice at a similar age as well as at end stage. Cells were counted as MNs if they were large, located in the ventral horn, contained a distinct nucleus and nucleolus, and possessed at least one prominent process. The number of anti-GFAP immunoreactive astrocytes and anti-Iba1 activated microglia/macrophages per mm2 was calculated by using Image J software (NIH, Bethesda, MD, USA). The number of anti-SOD1 immunoreactive inclusions was counted by adjusting the Image J software to exclude very small immunoreactive material.
Western blot
For experiments involving SOD1, homogenates of the spinal cord were processed using a method previously published (33). Briefly, 30 mg of fresh tissue was homogenized in 300 µl homogenizing buffer (50 mm HEPES, 1 mm EDTA, 100 mm iodoacetamide and 2.5% SDS, pH 7.2), incubated at 37°C for 1 h and then centrifuged at 20 000g for 15 min. Ten microgram of the protein of the supernatant of each sample was treated without β-mercaptoethanol before boiling and then loaded on to 15% SDS polyacrylamide gels for western blot analysis using a human-specific anti-SOD1 antibody (to detect monomeric SOD1 and high molecular weight forms) and anti-β-tubulin antibody (1:500, Developmental Studies Hybridoma Bank). A similar sample diluted 1:5 was treated with β-mercaptoethanol before boiling and then loaded on to 15% SDS polyacrylamide gels for western blot analysis, with overnight incubation at 4°C with a human-specific anti-SOD1 antibody (to detect total SOD1) and mouse anti-β-tubulin antibody. After washing, the rabbit antibody-stained blots were then incubated for 2 h at room temperature with horseradish peroxidase (HRP)-linked anti-rabbit IgG (1:3000, Cell Signaling), while the mouse antibody-stained blots were incubated with anti-mouse IgG (1:6000, Santa Cruz, CA, USA), followed by washing and then detection with an ECL-Plus detection kit (Amersham, NJ, USA). The signal intensity of human SOD1 (normalized to β-tubulin as a loading control) was estimated by densitometry, as previously published (35) using Image J (National Institute of Mental Health, Bethesda, MD, USA).
For experiments involving ISR markers, the lumbar spinal cord tissue from each mouse was collected and homogenized in 200 µl homogenizing buffer (20 mm Tris, pH 8.0, 150 mm NaCl, 1 mm EDTA, 0.5% Triton X-100, 0.5% SDS) containing a protease inhibitor cocktail. The homogenate was then centrifuged 20 000g for 15 min. Twenty micrograms of total protein from the supernatant were loaded on 10 or 15% SDS polyacrylamide gels for western blot analysis, as previously described. Immunostaining was carried out with overnight incubation at 4°C with the following antibodies: rabbit p-eIF2α (1:1,000, Cell Signaling Technology), rabbit CHOP (1:500, Santa Cruz), rabbit ATF4 (1:1,000, Santa Cruz) and mouse β-tubulin. After washing, the rabbit antibody stained blots were then incubated for 2 h at room temperature with HRP-linked anti-rabbit IgG (1:3000, Cell Signaling) while the mouse antibody stained blots were incubated with anti-mouse IgG (1:6000, Santa Cruz) followed by washing and then detection with an ECL-Plus detection kit. The signal intensity of ISR markers was estimated by densitometry using Image J.
Evaluation of eIF2α phosphorylation
Mouse embryonic fibroblasts were cultured from non-transgenic, GADD34+/ΔC and GADD34ΔC/ΔC mice. At the third passage, separate cultures in dishes from four embryos of each of the genotypes were incubated with 2 µg/ml of tunicamycin. The cells were harvested before incubation (zero time) and at 3, 4 and 6 h after incubation. The cell lysates were then processed for western blots using rabbit monoclonal anti-p-eIF2α (1:2,000, Epitomics).
Statistics
The data were statistically analyzed using a t-test or one-way ANOVA variance analysis with the Newman–Keuls multiple comparison test.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Muscular Dystrophy Association (#4346 to R.P.R.), the ALS Association (#1211 to R.P.R.) and the NIH (NS34939 to B.P.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Heather P. Harding and David Ron for providing us with the GADD34ΔC/ΔC mice.
REFERENCES
- 1.Rothstein J.D. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann. Neurol. 2009;65:S3–S9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- 2.Lewerenz J., Maher P. Control of redox state and redox signaling by neural antioxidant systems. Antioxid. Redox Signal. 2011;14:1449–1465. doi: 10.1089/ars.2010.3600. [DOI] [PubMed] [Google Scholar]
- 3.Ron D., Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 4.Urushitani M., Sik A., Sakurai T., Nukina N., Takahashi R., Julien J.P. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat. Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- 5.Turner B.J., Atkin J.D., Farg M.A., Zang D.W., Rembach A., Lopes E.C., Patch J.D., Hill A.F., Cheema S.S. Impaired extracellular secretion of mutant superoxide dismutase 1 associates with neurotoxicity in familial amyotrophic lateral sclerosis. J. Neurosci. 2005;25:108–117. doi: 10.1523/JNEUROSCI.4253-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Urushitani M., Ezzi S.A., Matsuo A., Tooyama I., Julien J.P. The endoplasmic reticulum-Golgi pathway is a target for translocation and aggregation of mutant superoxide dismutase linked to ALS. FASEB J. 2008;22:2476–2487. doi: 10.1096/fj.07-092783. [DOI] [PubMed] [Google Scholar]
- 7.Kikuchi H., Almer G., Yamashita S., Guegan C., Nagai M., Xu Z., Sosunov A.A., McKhann G.M., 2nd, Przedborski S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl Acad. Sci. USA. 2006;103:6025–6030. doi: 10.1073/pnas.0509227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nassif M., Matus S., Castillo K., Hetz C. Amyotrophic lateral sclerosis pathogenesis: a journey through the secretory pathway. Antioxid. Redox Signal. 2010;13:1955–1989. doi: 10.1089/ars.2009.2991. [DOI] [PubMed] [Google Scholar]
- 9.Matus S., Glimcher L.H., Hetz C. Protein folding stress in neurodegenerative diseases: a glimpse into the ER. Curr. Opin. Cell Biol. 2011;23:239–252. doi: 10.1016/j.ceb.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Kanekura K., Suzuki H., Aiso S., Matsuoka M. ER stress and unfolded protein response in amyotrophic lateral sclerosis. Mol. Neurobiol. 2009;39:81–89. doi: 10.1007/s12035-009-8054-3. [DOI] [PubMed] [Google Scholar]
- 11.Gkogkas C., Middleton S., Kremer A.M., Wardrope C., Hannah M., Gillingwater T.H., Skehel P. VAPB interacts with and modulates the activity of ATF6. Hum. Mol. Genet. 2008;17:1517–1526. doi: 10.1093/hmg/ddn040. [DOI] [PubMed] [Google Scholar]
- 12.Uehara T. Accumulation of misfolded protein through nitrosative stress linked to neurodegenerative disorders. Antioxid. Redox Signal. 2007;9:597–601. doi: 10.1089/ars.2006.1517. [DOI] [PubMed] [Google Scholar]
- 13.Zhang K., Kaufman R.J. The unfolded protein response: a stress signaling pathway critical for health and disease. Neurology. 2006;66:S102–S109. doi: 10.1212/01.wnl.0000192306.98198.ec. [DOI] [PubMed] [Google Scholar]
- 14.Oh Y.K., Shin K.S., Yuan J., Kang S.J. Superoxide dismutase 1 mutants related to amyotrophic lateral sclerosis induce endoplasmic stress in neuro2a cells. J. Neurochem. 2008;104:993–1005. doi: 10.1111/j.1471-4159.2007.05053.x. [DOI] [PubMed] [Google Scholar]
- 15.Malhotra J.D., Miao H., Zhang K., Wolfson A., Pennathur S., Pipe S.W., Kaufman R.J. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl Acad. Sci. USA. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Atkin J.D., Farg M.A., Turner B.J., Tomas D., Lysaght J.A., Nunan J., Rembach A., Nagley P., Beart P.M., Cheema S.S., et al. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J. Biol. Chem. 2006;281:30152–30165. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- 17.Nishitoh H., Kadowaki H., Nagai A., Maruyama T., Yokota T., Fukutomi H., Noguchi T., Matsuzawa A., Takeda K., Ichijo H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008;22:1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tobisawa S., Hozumi Y., Arawaka S., Koyama S., Wada M., Nagai M., Aoki M., Itoyama Y., Goto K., Kato T. Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem. Biophys. Res. Commun. 2003;303:496–503. doi: 10.1016/s0006-291x(03)00353-x. [DOI] [PubMed] [Google Scholar]
- 19.Saxena S., Cabuy E., Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- 20.Wate R., Ito H., Zhang J.H., Ohnishi S., Nakano S., Kusaka H. Expression of an endoplasmic reticulum-resident chaperone, glucose-regulated stress protein 78, in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Acta Neuropathol. 2005;110:557–562. doi: 10.1007/s00401-005-1080-y. [DOI] [PubMed] [Google Scholar]
- 21.Ilieva E.V., Ayala V., Jove M., Dalfo E., Cacabelos D., Povedano M., Bellmunt M.J., Ferrer I., Pamplona R., Portero-Otin M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130:3111–3123. doi: 10.1093/brain/awm190. [DOI] [PubMed] [Google Scholar]
- 22.Mori A., Yamashita S., Uchino K., Suga T., Ikeda T., Takamatsu K., Ishizaki M., Koide T., Kimura E., Mita S., et al. Derlin-1 overexpression ameliorates mutant SOD1-induced endoplasmic reticulum stress by reducing mutant SOD1 accumulation. Neurochem. Int. 2011;58:344–353. doi: 10.1016/j.neuint.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 23.Fujisawa T., Homma K., Yamaguchi N., Kadowaki H., Tsuburaya N., Naguro I., Matsuzawa A., Takeda K., Takahashi Y., Goto J., et al. A novel monoclonal antibody reveals a conformational alteration shared by amyotrophic lateral sclerosis-linked SOD1 mutants. Ann. Neurol. 2012;72:739–749. doi: 10.1002/ana.23668. [DOI] [PubMed] [Google Scholar]
- 24.Ezzi S.A., Urushitani M., Julien J.P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007;102:170–178. doi: 10.1111/j.1471-4159.2007.04531.x. [DOI] [PubMed] [Google Scholar]
- 25.Gruzman A., Wood W.L., Alpert E., Prasad M.D., Miller R.G., Rothstein J.D., Bowser R., Hamilton R., Wood T.D., Cleveland D.W., et al. Common molecular signature in SOD1 for both sporadic and familial amyotrophic lateral sclerosis. Proc. Natl Acad. Sci. USA. 2007;104:12524–12529. doi: 10.1073/pnas.0705044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kabashi E., Valdmanis P.N., Dion P., Rouleau G.A. Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis? Ann. Neurol. 2007;62:553–559. doi: 10.1002/ana.21319. [DOI] [PubMed] [Google Scholar]
- 27.Bosco D.A., Morfini G., Karabacak N.M., Song Y., Gros-Louis F., Pasinelli P., Goolsby H., Fontaine B.A., Lemay N., McKenna-Yasek D., et al. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat. Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pokrishevsky E., Grad L.I., Yousefi M., Wang J., Mackenzie I.R., Cashman N.R. Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS ONE. 2012;7:e35050. doi: 10.1371/journal.pone.0035050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haidet-Phillips A.M., Hester M.E., Miranda C.J., Meyer K., Braun L., Frakes A., Song S., Likhite S., Murtha M.J., Foust K.D., et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011;29:824–828. doi: 10.1038/nbt.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mackenzie I.R., Bigio E.H., Ince P.G., Geser F., Neumann M., Cairns N.J., Kwong L.K., Forman M.S., Ravits J., Stewart H., et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann. Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- 31.Vaccaro A., Patten S.A., Ciura S., Maios C., Therrien M., Drapeau P., Kabashi E., Parker J.A. Methylene blue protects against TDP-43 and FUS neuronal toxicity in C. elegans and D. rerio. PLoS ONE. 2012;7:e42117. doi: 10.1371/journal.pone.0042117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vaccaro A., Patten S.A., Aggad D., Julien C., Maios C., Kabashi E., Drapeau P., Parker J.A. Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol. Dis. 2013;55:64–75. doi: 10.1016/j.nbd.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 33.Wang L., Popko B., Roos R.P. The unfolded response in familial amyotrophic lateral sclerosis. Hum. Mol. Genet. 2011;20:1008–1015. doi: 10.1093/hmg/ddq546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marciniak S.J., Yun C.Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H.P., Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang L., Deng H.X., Grisotti G., Zhai H., Siddique T., Roos R.P. Wild-type SOD1 overexpression accelerates disease onset of a G85R SOD1 mouse. Hum. Mol. Genet. 2009;18:1642–1651. doi: 10.1093/hmg/ddp085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Harding H.P., Zeng H., Zhang Y., Jungries R., Chung P., Plesken H., Sabatini D.D., Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol. Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- 37.Boillee S., Vande Velde C., Cleveland D.W. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 38.Pandey U.B., Nie Z., Batlevi Y., McCray B.A., Ritson G.P., Nedelsky N.B., Schwartz S.L., DiProspero N.A., Knight M.A., Schuldiner O., et al. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–863. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 39.Walker A.K., Atkin J.D. Stress signaling from the endoplasmic reticulum: a central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life. 2011;63:754–763. doi: 10.1002/iub.520. [DOI] [PubMed] [Google Scholar]
- 40.Hu Y., Park K.K., Yang L., Wei X., Yang Q., Cho K.S., Thielen P., Lee A.H., Cartoni R., Glimcher L.H., et al. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron. 2012;73:445–452. doi: 10.1016/j.neuron.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang L., Sharma K., Grisotti G., Roos R.P. The effect of mutant SOD1 dismutase activity on non-cell autonomous degeneration in familial amyotrophic lateral sclerosis. Neurobiol. Dis. 2009;35:234–240. doi: 10.1016/j.nbd.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang L., Pytel P., Feltri M.L., Wrabetz L., Roos R.P. Selective knockdown of mutant SOD1 in Schwann cells ameliorates disease in G85R mutant SOD1 transgenic mice. Neurobiol. Dis. 2012;48:52–57. doi: 10.1016/j.nbd.2012.05.014. [DOI] [PubMed] [Google Scholar]
- 43.Wang L., Gutmann D.H., Roos R.P. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum. Mol. Genet. 2011;20:286–293. doi: 10.1093/hmg/ddq463. [DOI] [PubMed] [Google Scholar]
- 44.Wang L., Grisotti G., Roos R.P. Mutant SOD1 knockdown in all cell types ameliorates disease in G85R SOD1 mice with a limited additional effect over knockdown restricted to motor neurons. J. Neurochem. 2010;113:166–174. doi: 10.1111/j.1471-4159.2010.06594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang S.H., Li Y., Fukaya M., Lorenzini I., Cleveland D.W., Ostrow L.W., Rothstein J.D., Bergles D.E. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 2013;16:571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin W., Popko B. Endoplasmic reticulum stress in disorders of myelinating cells. Nat. Neurosci. 2009;12:379–385. doi: 10.1038/nn.2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Colla E., Coune P., Liu Y., Pletnikova O., Troncoso J.C., Iwatsubo T., Schneider B.L., Lee M.K. Endoplasmic reticulum stress is important for the manifestations of alpha-Synucleinopathy in vivo. J. Neurosci. 2012;32:3306–3320. doi: 10.1523/JNEUROSCI.5367-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Naidoo N. The endoplasmic reticulum stress response and aging. Rev. Neurosci. 2009;20:23–37. doi: 10.1515/revneuro.2009.20.1.23. [DOI] [PubMed] [Google Scholar]
- 49.Roselli F., Caroni P. Life-or-death decisions upon axonal damage. Neuron. 2012;73:405–407. doi: 10.1016/j.neuron.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 50.Halterman M.W., Gill M., DeJesus C., Ogihara M., Schor N.F., Federoff H.J. The endoplasmic reticulum stress response factor CHOP-10 protects against hypoxia-induced neuronal death. J. Biol. Chem. 2010;285:21329–21340. doi: 10.1074/jbc.M109.095299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gow A., Wrabetz L. CHOP and the endoplasmic reticulum stress response in myelinating glia. Curr. Opin. Neurobiol. 2009;19:505–510. doi: 10.1016/j.conb.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreno J.A., Radford H., Peretti D., Steinert J.R., Verity N., Martin M.G., Halliday M., Morgan J., Dinsdale D., Ortori C.A., et al. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature. 2012;485:507–511. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Moreno J.A., Halliday M., Molloy C., Radford H., Verity N., Axten J.M., Ortori C.A., Willis A.E., Fischer P.M., Barrett D.A., et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013;5:206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- 54.Ma T., Trinh M.A., Wexler A.J., Bourbon C., Gatti E., Pierre P., Cavener D.R., Klann E. Suppression of eIF2alpha kinases alleviates Alzheimer's disease-related plasticity and memory deficits. Nat. Neurosci. 2013;16:1299–1305. doi: 10.1038/nn.3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hetz C., Thielen P., Matus S., Nassif M., Court F., Kiffin R., Martinez G., Cuervo A.M., Brown R.H., Glimcher L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23:2294–2306. doi: 10.1101/gad.1830709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwakoshi N.N., Pypaert M., Glimcher L.H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 2007;204:2267–2275. doi: 10.1084/jem.20070525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boillee S., Yamanaka K., Lobsiger C.S., Copeland N.G., Jenkins N.A., Kassiotis G., Kollias G., Cleveland D.W. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- 58.Lobsiger C.S., Boillee S., Cleveland D.W. Toxicity from different SOD1 mutants dysregulates the complement system and the neuronal regenerative response in ALS motor neurons. Proc. Natl Acad. Sci. USA. 2007;104:7319–7326. doi: 10.1073/pnas.0702230104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang L.J., Lu Y.Y., Muramatsu S., Ikeguchi K., Fujimoto K., Okada T., Mizukami H., Matsushita T., Hanazono Y., Kume A., et al. Neuroprotective effects of glial cell line-derived neurotrophic factor mediated by an adeno-associated virus vector in a transgenic animal model of amyotrophic lateral sclerosis. J. Neurosci. 2002;22:6920–6928. doi: 10.1523/JNEUROSCI.22-16-06920.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng H.X., Shi Y., Furukawa Y., Zhai H., Fu R., Liu E., Gorrie G.H., Khan M.S., Hung W.Y., Bigio E.H., et al. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc. Natl Acad. Sci. USA. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.