

Abstract
A newly identified lethal form of hereditary sensory and autonomic neuropathy (HSAN), designated HSAN-VI, is caused by a homozygous mutation in the bullous pemphigoid antigen 1 (BPAG1)/dystonin gene (DST). The HSAN-VI mutation impacts all major neuronal BPAG1/dystonin protein isoforms: dystonin-a1, -a2 and -a3. Homozygous mutations in the murine Dst gene cause a severe sensory neuropathy termed dystonia musculorum (dt). Phenotypically, dt mice are similar to HSAN-VI patients, manifesting progressive limb contractures, dystonia, dysautonomia and early postnatal death. To obtain a better molecular understanding of disease pathogenesis in HSAN-VI patients and the dt disorder, we generated transgenic mice expressing a myc-tagged dystonin-a2 protein under the regulation of the neuronal prion protein promoter on the dtTg4/Tg4 background, which is devoid of endogenous dystonin-a1 and -a2, but does express dystonin-a3. Restoring dystonin-a2 expression in the nervous system, particularly within sensory neurons, prevented the disorganization of organelle membranes and microtubule networks, attenuated the degeneration of sensory neuron subtypes and ameliorated the phenotype and increased life span in these mice. Despite these improvements, complete rescue was not observed likely because of inadequate expression of the transgene. Taken together, this study provides needed insight into the molecular basis of the dt disorder and other peripheral neuropathies including HSAN-VI.
INTRODUCTION
Hereditary sensory and autonomic neuropathies encompass a diverse group of inherited disorders of the peripheral nervous system, characterized by progressive sensory neuron degeneration and varying degrees of autonomic dysfunction (1). The genetic spectrum of hereditary sensory and autonomic neuropathies (HSANs) covers both autosomal dominant and autosomal recessive forms. Autosomal dominant traits typically present in the second and third decade of life with marked sensory involvement and little autonomic and variable motor involvement, while autosomal recessive forms typically show an early onset pattern (i.e. congenital or during childhood) with overt sensory and autonomic dysfunction (2). Based on age of onset, mode of inheritance and predominant clinical features, a classification system was devised representing HSAN types I–V (3). Over the past 15 years, 12 disease-causing HSAN alleles have been identified, albeit the pathological mechanisms in over two-thirds of HSAN patients remain unresolved. As such, understanding the functional implications of known or novel genetic defects is urgently needed to better understand the molecular basis of these disorders.
A recent study has identified deleterious homozygous mutations in the BPAG1/DST gene that impart a frame-shift mutation resulting in a newly described hereditary autonomic sensory neuropathy in four infants (4). The dystonin protein is exceptionally large (>600 kDa) and is capable of interacting with all cytoskeletal filaments. The frame-shift mutation, which reduces dystonin-a transcript expression, starts at Glu4995 and leads to a loss of the C-terminus, which harbors the microtubule-binding domain (MTBD), a domain common to all major dystonin-a isoforms (5). The patients' clinical features are reminiscent of familial dysautonomia (also known as HSAN-III, or Riley-Day syndrome), although the course and outcome of disease are more severe and are ultimately fatal. This newly reported dystonin-related neuropathy was termed HSAN-VI and shares clinical features reminiscent of those seen in dystonia musculorum (dt) mice. Indeed, both human patients and dt mice suffer from limb contractures and dysautonomia.
The dt phenotype is characterized as a loss of limb coordination beginning between 11 and 14 days postnatal (6,7). As the disease progresses, which it does rapidly, limb movement becomes more uncoordinated, while writhing and twisting of the trunk become increasingly prominent features. Among the pathological features displayed by dt mice, degeneration is most apparent in large and medium-sized proprioceptive primary sensory afferents of the dorsal root ganglion (DRG) and cranial nerves (8–10).
In both humans and rodents, the DST gene is characterized as having tissue-specific promoters and an abundance of exons that are alternatively spliced yielding differentially expressed transcripts (11). Within the nervous system, three prominently large (>600 kDa) neuronal dystonin isoforms exist (schematically represented in Fig. 1A), dystonin-a1, -a2, and -a3, each of which have distinct cellular localizations and are capable of interacting with microtubules (MTs) and actin filaments (12,13). Each isoform is endowed with a functional N-terminal actin binding domain, a plakin domain, a spectrin repeat-containing rod domain in the middle of the molecule and a MTBD at the C-terminus (14). Isoform uniqueness is achieved through alternative splicing of the first 5′ exons. Dystonin-a1 encodes a short N-terminal domain that includes an actin-binding domain, while dystonin-a2 possess a transmembrane domain capable of interacting with various organelle membranes, and dystonin-a3 harbors a myristoylation domain, aiding in anchoring to the plasma membrane (12,13).
Figure 1.

Generation of the PrP-dystonin-a2 transgenic mice. (A) Schematic representation of the N-terminal regions of the major dystonin neuronal isoforms (dystonin-a1, -a2 and -a3). The actin binding domain (ABD) of both dystonin-a1 and -a2 contains a pair of calponin homology sites, while dystonin-a3 contains a single calponin homology site. The plakin domain is common to all three isoforms. The differentiating feature of all dystonin-a isoforms lies in the initial N-terminal segment. Dystonin-a2 harbors a highly conserved transmembrane (TM) domain, which aids its localization to the membrane of organelles. Dystonin-a3 contains a myristoylation motif that localizes it to the plasma membrane. The C-termini of these proteins contain a MTBD, consisting of EF hands, a Gas2-related domain and a glycine–serine–arginine rich domain (data not shown). (B) Schematic representation of the PrP-dystonin-a2 cDNA construct used to generate transgenic mice. The construct harbors full-length dystonin-a2 cDNA in frame with a myc/his tag under the regulation of a strong neuronal promoter [prion protein promoter (PrP)]. (C) Validation of PrP-dystonin-a2 transgene expression in F11 sensory neurons. Antigenic labeling of the PrP-dystonin-a2 myc/his tag (using anti-c-myc) 48 h post-transfection in F11 sensory neurons produced a perinuclear/cytoplasmic-staining pattern expected for the dystonin-a2 isoform. Cells were counterstained with DAPI to label the nuclei (scale bar = 20 μm). (D) Schematic representation of the location of oligonucleotide primers used to amplify the endogenous Dst gene and the PrP-dystonin-a2 transgene. Arrows indicate the position of primers used to amplify between exons 7 and 8 of the Dst gene. Amplification of the endogenous locus yields a 318 bp fragment due to the presence of an intron, whereas amplification of the PrP-dystonin-a2 transgene gives rise to a 220 bp product. (E) PCR screening of the F0 generation indicates five offspring positive for the PrP-dystonin-a2 transgene (220 bp). Founder lines 542 and 559 were bred and established for further analysis.
While the dystonin-a isoforms mediating disease in HSAN-VI remain enigmatic, loss-of-function of dystonin-a1 and -a2, but not dystonin-a3, in the murine nervous system is causal in the dtTg4/Tg4 disorder (15,16). As such, numerous studies and speculations have been invested into parsing apart the biological functions of individual dystonin-a isoforms. Initial inspection of dystonin-a transcripts found dystonin-a2 to be the more unique isoform and least likely to be compensated for by any related protein (e.g. microtubule actin crosslinking factor and plectin) (16). Moreover, dystonin-a2 expression predominated in sensory ganglia—the most severely affected tissue of dt mice—and poorly expressed in non-neuronal tissues (17). These initial descriptions of the dystonin-a2 variant suggested that it might be the principle mediator of dt pathogenesis (16,18). Support for this notion was later corroborated through isoform-specific loss-of-function analyses in cell culture model systems. Indeed, depletion of dystonin-a2 perturbs membrane organization [endoplasmic reticulum (ER) and Golgi apparatus] and transport flux through the secretory pathway (19,20).
In a separate study exploring the individual roles of dystonin-a1 and -a2 isoforms in mediating dt sensory neuron degeneration, dystonin-a2 was determined to be the principle initiator of neuronal degeneration (20). Silencing dystonin-a2 in neurons in culture elicited distinct neurodegenerative pathologies, including Ca2+ dyshomeostasis, unfolded protein response induction, caspase activation and apoptosis. In addition to these aberrations, ER structural integrity was also compromised, presumably through uncoupling of cytoskeletal filaments, dystonin-a2 and ER membranes. Taken together, these studies suggest that dystonin-a2 is the chief contributor in the demise of dt sensory neurons. Nevertheless, dystonin-a1 is also involved in fundamental neuronal processes, including anterograde and retrograde trafficking (21,22). As disturbance of these processes are well-known to underlie many neurological diseases (23), including HSANs (1), dystonin-a1 likely also remains an important contributor in dt pathogenesis.
To determine the dystonin-a isoform mediating dt pathogenesis, and to gain insight into viable pathogenetic mechanisms underlying HSAN-VI, we generated and characterized transgenic mice expressing dystonin-a2 under the nervous system-specific prion protein promoter (PrP-dystonin-a2). These transgenic mice were subsequently crossed onto the dtTg4 background, producing homozygous (PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4) transgenic mice. We find restoring dystonin-a2 expression within the nervous system greatly diminishes the severity of the dt disorder—due, in part, to prolonged survival of specific sensory neuron subtypes—and significantly extends life span. Despite these improvements, complete rescue was not observed likely because of inadequate expression of the transgene.
RESULTS
Generation and characterization of the PrP-dystonin-a2/PrP-dystonin-a2 transgenic mouse model
Transgenic mice harboring full-length dystonin-a2 cDNA were generated and used in rescue experiments using dtTg4/Tg4 mice. Full-length dystonin-a2 cDNA was placed under the control of a strong neuronal promoter, the 3.5 kb mouse PrP promoter (Fig. 1B). In addition, a myc/his epitope tag was included in frame at the 3′ end to facilitate detection of the transgene product. The mouse PrP promoter was previously demonstrated to yield high expression in neuronal tissues of various transgenes (24). Although the PrP promoter can exhibit low expression in other cell types (e.g. astrocytes and skeletal muscle), we are mainly concerned with expression within neuronal tissues.
To assess whether the PrP-dystonin-a2 transgene is functional and the myc-tag can be detected via immunocytochemical labeling, immortalized F11 sensory neurons were transfected with the construct. The transgene protein product displays a characteristic perinuclear/cytoplasmic staining pattern aligning with cytoskeletal filaments, particularly MTs (Fig. 1C). This pattern is very similar to what we had previously shown (13,18). Purified PrP-dystonin-a2 cDNA transgene fragment was microinjected into the pronuclei of one-cell mouse embryos and transgenic founder lines were established thereafter. To identify mice harboring the transgene, a genotyping assay was developed. Amplification of endogenous DNA from non-transgenic mice produces a 318 bp DNA fragment from within the Dst gene, while amplification of the transgene DNA produces a 220 bp DNA fragment (Fig. 1D and E). From five transgenic founder mice generated, two lines (founder lines F542 and F559) were bred to establish independent strains. These transgenic lines appeared normal and were fertile.
PrP-dystonin-a2 mice express the transgene in a neuronal specific manner
Using primers specific to the myc-his tag, transgene expression was assessed in multiple neuronal tissues including DRG, spinal cord and brain for both PrP-dystonin-a2 transgenic lines (Fig. 2A). At P7 stage, all neuronal tissues from heterozygous mice exhibited PrP-dystonin-a2 transgene expression, while no expression was evident in tibialis anterior muscle (Fig. 2B and C). PrP-dystonin-a2 transgene expression was also assessed in P7 heterozygous tissues by immunohistochemistry. Using an anti-myc monoclonal antibody to assess protein expression (this tag is placed in frame with the dystonin-a2 isoform), a positive signal was detected in the cerebral cortex and cerebellum of both transgenic lines; however, transgene expression in DRG tissue sections was faint and difficult to discern in mice from line 542 (data not shown). For this reason, transgenic line 559 was used and bred to homozygosity to increase relative transgene expression levels. Encouragingly, brain (P10) and DRG (P10 and P58) tissue sections of F559 homozygous (PrP-dystonin-a2/PrP-dystonin-a2) mice display the transgene product (Fig. 3A). Indeed, robust PrP-dystonin-a2 transgene expression was seen throughout the sensory neurons of the DRG. In comparison, as expected, there was no signal in age-matched brain and DRG tissues taken from wild-type mice (Fig. 3A). To further assess the pattern of transgene product expression, we have examined both DRG sections and primary cultures of DRG neurons by confocal microscopy. The transgene product is largely localized to the perinuclear region of the cell bodies of sensory neurons in DRG sections (Fig. 4A and B). As well, we observe a similar pattern in the cell bodies of DRG neurons in primary culture (Fig. 4C). Again, this is a very similar pattern to what we had previously shown (13,18).
Figure 2.

The PrP-dystonin-a2 transgene is expressed in neuronal tissues of transgenic lines 559 and 542. (A) Schematic representation of the location of oligonucleotide primers for the amplification of PrP-dystonin-a2 derived transcripts. Primers amplify a 62 bp fragment that includes the myc/his tag coding sequence. (B and C) RT–PCR analysis of RNA derived from P7 neuronal tissues (DRG, spinal cord and brain) of heterozygous transgenic mice from lines 559 and 542 indicates PrP-dystonin-a2 transgene expression (PrP, +RT lanes). Transgene expression was not evident in tibialis anterior (TA) muscle of either transgenic line. Controls: P7 wild-type (WT) cDNA of neuronal tissues, PrP (no RT, minus reverse transcriptase), (+) and (−) cDNA of F11 sensory neurons transfected or non-transfected with PrP-dystonin-a2 construct, respectively. Actin mRNA amplification served as a positive control for RNA.
Figure 3.

Immunohistochemical staining demonstrating robust PrP-dystonin-a2 transgene expression in neuronal tissues. (A) Representative tissue sections from brain (P10) and DRGs (P10 and P58) of homozygous PrP-dystonin-a2/PrP-dystonin-a2 (PrP/PrP) transgenic mice. Immunohistochemical staining with anti-c-myc antibody produced a peri-nuclear/cytoplasmic staining pattern in P10 cortical brain tissue, while a strong peri-nuclear staining pattern was observed in P10 and P58 DRGs. As expected, DRGs from wild-type (WT) non-transgenic mice stained with anti-c-myc revealed no specific staining (scale bars = 20 μm). (B) Analysis of transgene expression in cultured DRG neurons. Again, as expected, there was no anti-c-myc staining in P10 dtTg4/Tg4 sensory neurons (left panel), whereas staining was present in large-, medium- and small-sized sensory neurons of P10 PrP-dystonin-a2 mice (right panel). Scale bar = 50 μm. Sections or cells were counterstained with DAPI to label the nuclei.
Figure 4.
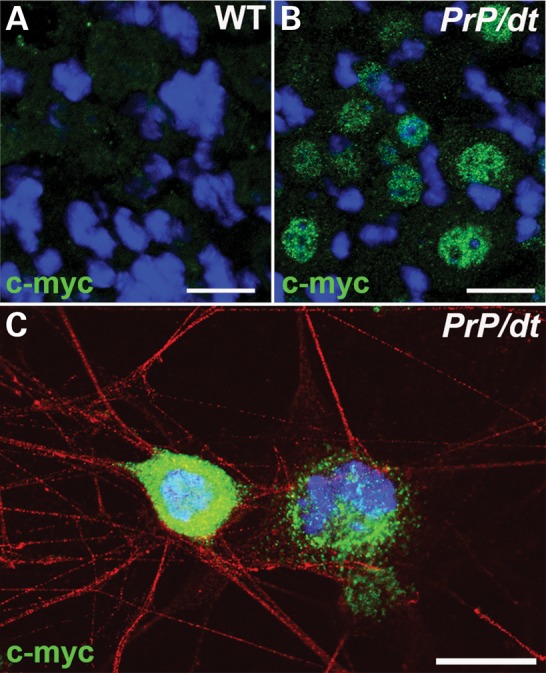
PrP-dystonin-a2 transgene product is expressed in DRG neurons and is organized into a perinuclear pattern. (A and B) Representative micrograph of anti-c-myc immunohistochemical labeling of P10 wild-type (WT) non-transgenic (A) and P10 PrP/PrP;dtTg4/Tg4 (B) DRG tissue sections. (C) Immunohistochemical staining with anti-c-myc antibody of P10 PrP/PrP;dtTg4/Tg4 sensory neurons in primary culture. PrP-dystonin-a2 expression is observed in the cell body in a perinuclear pattern. Images were captured by confocal microscopy. Scale bar = 20 μm.
The DRG contains distinct cell populations including proprioceptive large and medium-sized sensory afferents (muscle sensory) and small-sized (skin and visceral sensory) sensory neurons, each of which has specific physiological properties. Large- and medium-sized sensory neurons innervate muscle tissues and transmit proprioceptive and tactile information, whereas small-sized sensory neurons innervate and relay information from the skin and viscera. Dystonin-a is expressed throughout the DRG and not restricted to single subgroup (17). To ensure PrP-dystonin-a2 transgene was expressed in all sensory neuron subtypes, sensory neurons were cultured from P5 PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice for 5 days in vitro and immunolabeled with anti-c-myc. Different sized sensory neurons (small 100–400 μm, medium 400–700 μm and large 700–1200 μm) were categorized as previously described (10), using the Axiovision 4.6 software (Carl Zeiss) circumference-measuring tool. Anti-c-myc staining was replete throughout the PrP-dystonin-a2/PrP-dystonin-a2 sensory neuron subtypes, whereas no anti-myc labeling was observed in dtTg4/Tg4 sensory neurons (Fig. 3B).
To address how the levels of expression of dystonin-a2 from the transgene compared with endogenous dystonin-a2, we extracted RNA from the DRGs of P15 wild-type, dtTg4/Tg4 and PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice. We have performed real-time quantitative reverse transcription-polymerase chain reaction (RT–qPCR) and our analysis shows that dystonin-a2 transcripts are not detectable in dtTg4/Tg4 samples compared with the wild-type. Further, the level of dystonin-a2 transcripts in the PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 sample, although higher than the mutant alone, does not reach the same level as in the wild-type (Fig. 5).
Figure 5.
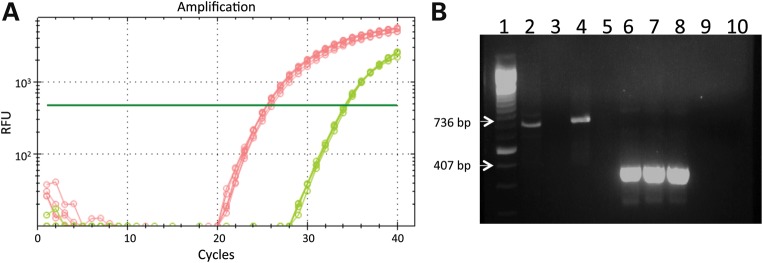
PrP-dystonin-a2 transgene transcript is expressed at lower levels than the endogenous dystonin-a2 transcript in DRGs from P15 mice. (A) Amplification curves from real-time RT–qPCR for dystonin-a2 (red—wild-type; green—PrP/PrP;dtTg4/Tg4; and dtTg4/Tg4—no detectable amplification). Curves shown represent n = 3 in technical triplicates. (B) Agarose gel electrophoresis of RT–qPCR products. Lane 1 = 100 bp ladder; lanes 2–4, products from dystonin-a2 mRNA amplification reactions (736 bp product) with lane 2 = wild-type, lane 3 = dtTg4/Tg4 and lane 4 = PrP/PrP;dtTg4/Tg4; lane 5 = blank; lanes 6–8, products from actin mRNA amplification reactions (407 bp product) with lane 6 = wild-type, lane 7 = dtTg4/Tg4 and lane 8 = PrP/PrP;dtTg4/Tg4; lane 9 = no template control (dystonin-a2 primers); lane 10 = no template control (actin primers).
These initial observations suggested that PrP-dystonin-a2/PrP-dystonin-a2 transgenic mice express the dystonin-a2 transgene throughout the nervous system, most importantly in all sensory neuron subtypes. PrP-dystonin-a2/PrP-dystonin-a2 mice are viable, fertile and phenotypically similar to wild-type mice.
Postnatal phenotypic characteristics of PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice
To characterize whether the PrP-dystonin-a2 transgene confers protection in dtTg4/Tg4 sensory neurons and ameliorates the dt phenotype, PrP-dystonin-a2/PrP-dystonin-a2 mice were crossed onto the dtTg4/Tg4 background. dtTg4/Tg4 mice heterozygous for the PrP-dystonin-a2 transgene mice died at approximately the same age as dtTg4/Tg4 mice (P21) and displayed poor locomotor coordination, unsteady gait and postural instability (data not shown). In comparison, homozygous PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice exhibited significant improvements in all these properties, as discussed below. This suggests that differences in dystonin-a2 protein levels in heterozygous versus homozygous transgenic mice likely influence the extent of rescue in dt mice.
The life span of dtTg4/Tg4 mice is ∼2–3 weeks (25). Here, we find the life span of PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice was significantly increased (mean age 55 days) compared with dtTg4/Tg4 mice (mean age 22 days, P < 0.0001) (Fig. 6A). Indeed, 60% of PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice assessed lived past P50, while 20% survived past 123 days (n = 10). Despite this increase in survival, PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 life span was compromised compared with control PrP-dystonin-a2/PrP-dystonin-a2 mice. In addition, PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice surviving past 120 days manifested additional phenotypes related to dysautonomia. Two mice (P123 and P126) showed blepharoptosis (drooping of the upper eyelid) and conjungtivitis (an early symptom of dry eye). While these dysautonomic features were not observable among the younger PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice, these features are occasionally seen in P20 dtTg4/Tg4 mice. This suggests that it takes a significantly longer period of time for PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice to manifest dysautonomic features compared with dtTg4/Tg4 mice.
Figure 6.

Postnatal characteristics of PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 transgenic rescue mice. The different groups analyzed were: control (PrP/PrP, n = 10), mutant (dtTg4/Tg4, n = 7) and transgenic rescue (PrP/PrP;dtTg4/Tg4, n = 10). (A) Kaplan–Meier survival curve analysis indicates a significant increase in lifespan of PrP/PrP;dtTg4/Tg4 mice (median lifespan, 55 days) when compared with dtTg4/Tg4 mice (median lifespan, 21 days) (****P < 0.0001). A significant decrease in lifespan was also observed between PrP/PrP;dtTg4/Tg4 mice and PrP/PrP mice (****P < 0.0001), indicating that although there was some rescue, it was not complete. (B) Postnatal growth curve analysis suggests that PrP/PrP;dtTg4/Tg4 mice have a progressive gain in weight between postnatal days P16 and P52, and plateau thereafter. PrP/PrP;dtTg4/Tg4 mice do not show comparable increases in weight to that of control PrP/PrP mice between P30 and P123 (**P < 0.01, ****P < 0.0001). (C) PrP/PrP;dtTg4/Tg4 mice show significant increases in weight compared with dtTg4/Tg4 mice at postnatal days P16 (**P < 0.01), P18 (**P < 0.01) and P20 (****P < 0.0001). This increase in weight at early postnatal days is comparable to PrP/PrP control mice. Further comparative weight analysis was not feasible for dtTg4/Tg4 mice as these mice die at ∼ P20. Two-way ANOVA, Bonferroni posttest, data are represented as mean ± SEM. (D) Performance analysis of PrP/PrP control, dtTg4/Tg4 and PrP/PrP;dtTg4/Tg4 mice in the pen test at various post-natal days between P23 and P123. PrP/PrP;dtTg4/Tg4 mice show a decreased latency to fall and as such are unable to effectively balance and/or grip to remain on the suspended pen compared with PrP/PrP control mice (****P < 0.0001). (E) PrP/PrP;dtTg4/Tg4 mice show improvements in balance and grip at early postnatal days (P18 and P20) compared with dtTg4/Tg4 mice (*P < 0.05). Despite this improvement, PrP/PrP control mice manifest superior balance and grip at P18 and P20 compared with PrP/PrP;dtTg4/Tg4 mice (***P < 0.001). Two-way ANOVA, Bonferroni posttest, data are represented as mean ± SEM. (F) Representative images of hind limb clasping during tail suspension, a hallmark dt phenotype. P15 dtTg4/Tg4 mice consistently displayed limb clasping (arrow), while P15 PrP/PrP;dtTg4/Tg4 mice manifested splayed hind limbs, reminiscent of wild-type or PrP/PrP control mice. Note that limb clasping was detectable in P21 PrP/PrP;dtTg4/Tg4 mice. (G) Representative examples of PrP/PrP (P20); dtTg4/Tg4 (P20) and PrP/PrP;dtTg4/Tg4 (P20) mouse paw prints used for gait analysis (red = front paws, red arrow; blue = back paws, blue arrows). Note the similar gait between P20 PrP/PrP and P20 PrP/PrP;dtTg4/Tg4 mice and the abnormal gait exhibited by P20 dtTg4/Tg4 mice.
From birth to P10, dtTg4/Tg4 mice are indistinguishable in size and phenotype from their wild-type littermates. At approximately 2 weeks of age, however, arrest in weight-gain and ambulating abnormalities is evident. We therefore assessed whether PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice show improved growth and ambulation. A progressive gain in weight in PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice was observed between postnatal days P16 and P52, albeit this progressive weight gain was not equivalent to PrP-dystonin-a2/PrP-dystonin-a2 control mice (Fig. 6B). In comparison with dtTg4/Tg4 mice, PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice showed a normal growth rate between P16 and P22, reminiscent of PrP-dystonin-a2/PrP-dystonin-a2 control mice (Fig. 6C). As dtTg4/Tg4 mice succumb to death at ∼P20, analyses between PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 and dtTg4/Tg4 mice could not be performed past this time point.
To assess whether the transgenic rescue mice have improved coordination, the mice were subjected to the pen test -- a test used to measure grip strength and coordination—and the duration of time the animal was able to stay on the pen was recorded. PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice had only minor improvements in this test at early time points compared with dtTg4/Tg4 mice and were unable to stay on the pen following the P24 time point (Fig. 6D and E). PrP-dystonin-a2/PrP-dystonin-a2 mice, on the other hand, performed normally. Despite poor performance on the pen test, PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice showed a delay in onset of hind limb clasping, a hallmark phenotypic feature of dtTg4/Tg4 mice (Fig. 6F). Indeed, dtTg4/Tg4 mice typically present this pathological reflex at P15, or earlier, whereas PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice only begin to show this phenotype at P21. This pathological reflex was non-existent in PrP-dystonin-a2/PrP-dystonin-a2 mice. Gait was assessed by using an in-house gait box as previously described (26). While P20 dtTg4/Tg4 mice exhibited an aberrant gait, the stride of P20 PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice was indistinguishable from that of P20 PrP-dystonin-a2/PrP-dystonin-a2 control mice (Fig. 6G). Taken together, restoring dystonin-a2 expression on the dtTg4/Tg4 background does improve features of the dt phenotype, albeit attenuation of the phenotype is transient.
The PrP-dystonin-a2 transgene delays sensory neuron degeneration in dt mice and improves neuromuscular junction maturation
The degeneration of dt sensory neurons is the primary contributor to the overt ataxia and dystonia observed in dt mice (17,27). We previously found the demise of dtTg4/Tg4 sensory neurons commences at P15, concomitant with the onset of dt pathogenesis and the dt phenotype (20). The PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mouse does not manifest impaired locomotion or dystonia at P15, suggesting delayed sensory neuron degeneration. To address this notion, quantitative TUNEL labeling on lumbar DRG tissue sections was performed.
As expected, P15 PrP-dystonin-a2/PrP-dystonin-a2 DRG tissue sections displayed a dearth of TUNEL-labeled neurons, while P15 dtTg4/Tg4 DRGs tissue sections exhibited a significant increase in TUNEL activity (Fig. 7B, C, F; P < 0.001). In comparison with dtTg4/Tg4 DRGs, P15 PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 DRGs showed a significant reduction in the number of TUNEL-positive sensory neurons (Fig. 7D and F; P < 0.001), suggesting that the PrP-dystonin-a2 transgene confers protection and delays the onset of neurodegeneration. As PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice eventually present ataxia, sensory neuron viability was assessed at a later time point. At P21, a significant increase in TUNEL-positive sensory neurons (P < 0.05) was observed compared with the P15 time point (compare Fig. 7D with 7E). Despite this increase, the percentage of dying cells was significantly less than that observed in P15 dtTg4/Tg4 sensory neurons (Fig. 7F, P < 0.001). These observations indicate that the aforementioned phenotypic improvements in PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice are due, in part, to increased viability in dtTg4/Tg4 sensory neurons. Furthermore, while the PrP-dystonin-a2 transgene confers protection in PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 sensory neurons, they still undergo degeneration, albeit at a slower rate.
Figure 7.
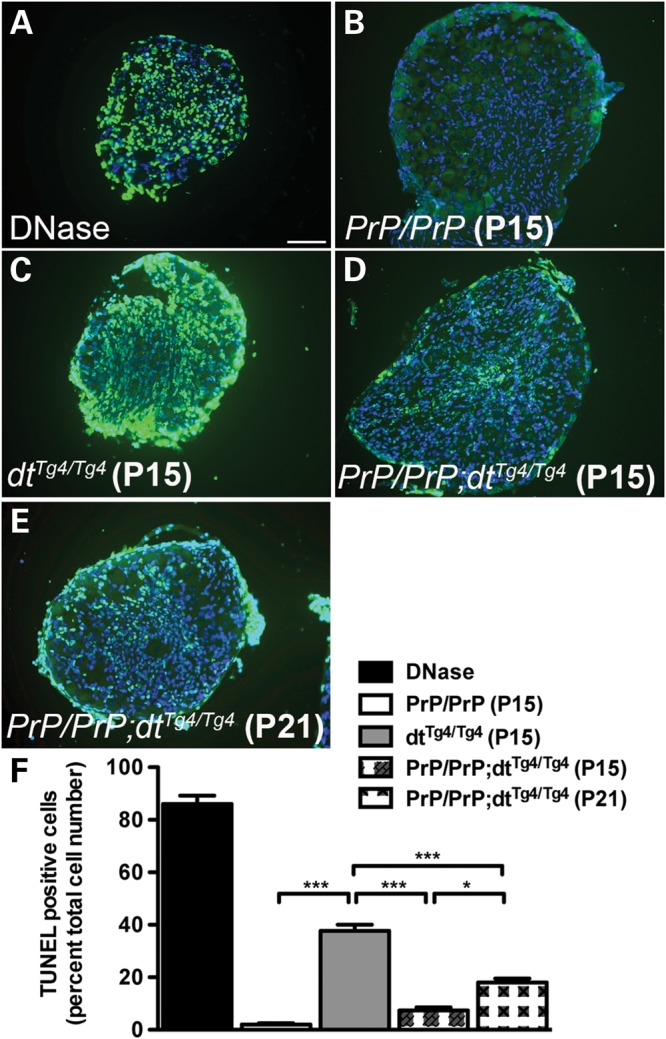
The PrP-dystonin-a2 transgene confers protection in dtTg4/Tg4 sensory neurons by delaying cellular demise. (A–E) Representative TUNEL labeling images of lumbar DRG tissue sections. (C) P15 dtTg4/Tg4 DRG tissue sections displayed significantly more TUNEL-positive cells (mean cell death, 37.6%) then P15 PrP/PrP;dtTg4/Tg4 (D) (mean cell death, 7.3%) and P21 PrP/PrP;dtTg4/Tg4 cells (E) (mean cell death, 18%). The decrease observed in TUNEL-positive cells for P15 PrP/PrP;dtTg4/Tg4 DRGs compared with P15 dtTg4/Tg4 was transient as P21 PrP/PrP;dtTg4/Tg4 DRGs showed an increase in cellular death. PrP/PrP control DRGs showed few TUNEL-positive cells, while DRG tissue sections that were DNase treated were replete with TUNEL-positive cells. (F) Quantification of the TUNEL analysis in DRG sections. ANOVA, post hoc Tukey, ***P < 0.001, *P < 0.05, n = 3/genotype. Scale bar = 20 μm.
To assess the impact of the transgene on the pathology in the axons, we have performed histopathology on dorsal root sections from P15 wild-type, dtTg4/Tg4 and PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice. Toluidine blue staining on thin sections was used to assess axon caliber. We have measured the axon caliber in at least 500 individual myelinated axons from cross-sections (n = 3 for each genotype). Note that there was no statistically significant difference in the axonal caliber between wild-type and dt. As well, there was no change in axon caliber after transgenic rescue of dt. Thus, this aspect of the morphology of axons is not affected in the dt mouse. As well, we have assessed the number of axonal swellings per dorsal root section (n = 3 for each genotype). Our quantification revealed that there were no axonal swellings observed in wild-type sections, 2 ± 1 axonal swellings in dt sections and 1.66 ± 1.53 axonal swellings in the rescued mice. Thus, this analysis revealed that axonal swellings were still present in the dorsal roots of rescued mice (Fig. 8). Interestingly, however, we did observe a relative increase in the density of axons in these mice, suggesting that axonal loss has been attenuated. Similarly, we have performed histopathology on muscles from P15 mice to assess neuromuscular junction (NMJ) and endplate maturity. In general, there was an increase in the number of immature endplates and NMJs in muscles from dtTg4/Tg4 mice compared with wild-type (Fig. 9, top and middle rows). By comparison, there was an improvement, albeit modest, in these defects in the PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice (Fig. 9, bottom rows). In particular, the incidence of neurofilament accumulation at the presynaptic site in PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice is reduced compared with dtTg4/Tg4 mice.
Figure 8.

Exogenous expression of dystonin-a2 partially rescues the number of myelinated axons in the dorsal sensory roots of dtTg4/Tg4 mice. Toluidine blue staining of transverse sections of dorsal sensory roots at P15. (A) Dorsal sensory roots from wild-type (WT) mice showing numerous myelinated axons (high magnification in A′). (B) Dorsal sensory roots from dtTg4/Tg4 mice are generally smaller than their wild-type counterparts, showing fewer axons, axons undergoing degeneration and axonal swellings (sa) (higher magnification in B′). (C) Dorsal sensory roots from PrP/PrP;dtTg4/Tg4 mice are generally larger than those from dtTg4/Tg4 mice, although they still remain smaller than wild-type dorsal roots. They display an increase in the number of myelinated axons when compared with dtTg4/Tg4 dorsal roots, but axonal swellings (sa) are still present (see higher magnification in C′). Scale bars = 50 μm (A–C) and 10 μm (A′–C′).
Figure 9.

Endplates from dtTg4/Tg4 mice are poorly developed and this defect is partially rescued in PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice. Representative photomicrographs showing NMJ endplate morphology in TA myofibers isolated from P15 wild-type (A–N), dtTg4/Tg4 (A′–N′) and PrP/PrP;dtTg4/Tg4 (A″–N″) mice. The endplates from dtTg4/Tg4 myofibers were less developed than those from wild-type (as indicated by asterisks and double asterisks). In addition to having an immature morphology, the endplates indicated by the double asterisks display neurofilament accumulation on the presynaptic side. Interestingly, both of these abnormal features were reduced in the PrP/PrP;dtTg4/Tg4 mice. Scale bar = 10 μm.
Finally, we have performed an analysis of muscle spindle morphology in 30 cross-sections of tibialis anterior muscles (n = 3 for each genotype) (Fig. 10). This analysis revealed that there was a significant degeneration of muscle spindles, regardless of their size, in dtTg4/Tg4 samples (Fig. 10D). Interestingly, this degeneration was substantially rescued in the PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice.
Figure 10.

PrP-dystonin-a2 transgene rescues muscle spindle degeneration in dtTg4/Tg4 mice. (A–C and A′–C′) Paraffin sections from the tibialis anterior muscle of P15 mice were stained with hematoxylin and eosin. (A and A′) Analysis of wild-type samples demonstrate a normal muscle spindle with its intrafusal muscle and its Ia afferent sensory nerve. (B and B′) Evidence of muscle spindle degeneration in dtTg4/Tg4 muscle at P15 (black arrow) and lack of detectable axons in the spindle (white arrow). (C and C′) Muscle spindle structure appears normal in PrP/PrP;dtTg4/Tg4 mice, with a normal encapsulated muscle spindle with its innervating sensory nerve shown. Scale bars = 10 μm. (D) Quantification of muscle spindle degeneration in tibialis anterior muscle at P15. There is a statistically significant difference between wild-type and dtTg4/Tg4 (***P < 0.001), wild-type and PrP/PrP;dtTg4/Tg4 (Prp/dt) (**P < 0.01), and between dtTg4/Tg4 and PrP/PrP;dtTg4/Tg4 (***P < 0.001) in the % of muscle spindles degenerating. (D′) Quantification of the number of muscle spindles by section of muscle. There is a statistically significant difference between wild-type and dtTg4/Tg4 (**P < 0.01). No significant difference was observed between wild-type and PrP/PrP;dtTg4/Tg4 in the average number of muscle spindles.
Dystonin-a2 protects proprioceptive sensory afferents and improves MT integrity
Various HSAN disorders are characterized by the demise of a specific sensory neuron subtype (1,28). Interestingly, while dystonin-a is expressed throughout different sensory neuron populations, large and medium proprioceptive sensory neurons are most affected by the dt disorder (8,10). Consequently, movement is severely impaired in dt mice. To address which population of sensory afferents the PrP-dystonin-a2 transgene protects, sensory neurons from PrP-dystonin-a2/PrP-dystonin-a2, PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4, and dtTg4/Tg4 mice were co-labeled with β-III tubulin (a neuronal marker) and caspase-3 (an apoptosis marker) (n = 3) (Fig. 11A–C). Furthermore, to differentiate between different-sized sensory neurons, soma circumference was measured and grouped by size as discussed above. Primary sensory neurons derived from P5 mice and cultured for 5 days in vitro (5 DIV) revealed a dearth of caspase-3 labeling for each genetic background (data not shown). As such, sensory neuron cultures were challenged with starvation media (serum-, glucose- and amino acid-free media) for 24 h following 5 DIV and cell viability was assessed thereafter. For cells to maintain viability following starvation, they activate cell-survival mechanisms (e.g. autophagy and robust intracellular transport).
Figure 11.

PrP-dystonin-a2 transgene imparts neuro-protection in small and medium caliber sensory neurons but not in large caliber sensory neurons. (A–C) Primary DRG sensory neuron cultures were established from P5 mice of various genetic backgrounds [PrP/PrP (A), PrP/PrP;dtTg4/Tg4 (B), dtTg4/Tg4 (C)], and cultured for 5 days in vitro. Cells were challenged with serum-free media for 24 h and antigenic labeling of β-III tubulin (neuronal marker) and the apoptotic marker caspase-3 was conducted thereafter. Note the accumulation of β-III tubulin in axons of dtTg4/Tg4 sensory neurons (arrowheads in C). (D) The average number of small (soma area, 100–400 μm2) and medium caliber sensory neurons (400–700 μm2) was not significantly different between genotypes. (E) A significant increase in caspase-3 staining was observed in small- and medium-sized dtTg4/Tg4 sensory neurons compared with PrP/PrP (***P < 0.001) and PrP/PrP;dtTg4/Tg4 (**P < 0.01), indicating the transgene confers protection in these cell types. (F) The average number of large caliber sensory neurons (700–1300 μm2) is significantly different between genotypes PrP/PrP and PrP/PrP;dtTg4/Tg4 (*P < 0.05); PrP/PrP and dtTg4/Tg4 (** P < 0.01). However, there was no difference between PrP/PrP;dtTg4/Tg4 and dtTg4/Tg4. (G) A significant increase in caspase-3 staining was observed in dtTg4/Tg4 large caliber sensory neurons compared with PrP/PrP large caliber sensory neurons (*P < 0.05). No significant difference in capase-3 staining was observed between PrP/PrP and PrP/PrP;dtTg4/Tg4 large caliber sensory neurons. Statistics: ANOVA, post hoc Tukey, n = 3/genotype. Scale bar = 20 μm.
In initial analysis, there was no difference in the average number of small and medium sensory neurons between PrP-dystonin-a2/PrP-dystonin-a2, PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4, and dtTg4/Tg4 cultures following 24 h serum-free treatment (Fig. 11D). However, a significant increase in caspase-3 labeling was noted in dtTg4/Tg4 small and medium sensory neurons compared with PrP-dystonin-a2/PrP-dystonin-a2 (P < 0.001) and PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 (P < 0.01) sensory neurons (Fig. 11E). Furthermore, no significant difference in capase-3 labeling was observed in small and medium sensory neurons between PrP-dystonin-a2/PrP-dystonin-a2 and PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 cultures (Fig. 10E). These observations suggest expression of the PrP-dystonin-a2 transgene on the dtTg4/Tg4 background imparts protection in small and medium sensory neurons.
Unlike small and medium sensory neurons, the average number of large sensory neurons following serum-free treatment was significantly different between genotypes. On average, there were 14 large sensory neurons per PrP-dystonin-a2/PrP-dystonin-a2 culture and significantly fewer in PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 (five large sensory neurons per culture, P < 0.05) and in the dtTg4/Tg4 culture (one large sensory neuron per culture, P < 0.01) (Fig. 11F). Coupled with this, 15% of large PrP-dystonin-a2/PrP-dystonin-a2 sensory neurons were capase-3 positive, while ∼52 and 100% of large PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 and dtTg4/Tg4 sensory neurons were capase-3 positive, respectively (Fig. 11G). These data indicate that although the PrP-dystonin-a2 transgene imparts slight protection to large sensory neurons, they remain vulnerable to dt pathogenesis.
In both HSAN-VI patients and dt mice, disorganized MT networks are thought to be a critical driver of disease pathogenesis (4,29,30). As such, we addressed whether the dystonin-a2 transgene improves MT network integrity. dtTg4/Tg4 cultures exhibited notable accumulations of β-III tubulin throughout axons (arrowheads, Fig. 11C), whereas accumulations of β-III tubulin were absent from the axons of PrP-dystonin-a2/PrP-dystonin-a2 and PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 sensory neurons (Fig. 11A and B). Similar accumulations seen herein have been observed in dt axons, and likely influence the bi-directional transport impairment observed in dt sensory neurons (31). We therefore conclude that the increased viability among small- and medium-sized PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 sensory afferents is mediated, in part, by improved MT organization.
Dystonin-a2 transgene preserves organelle integrity
The cytoskeleton and cytoskeletal stabilizing proteins are instrumental in organelle organization, movement and function (32–35). We previously demonstrated that dystonin-a2 loss of function leads to the structural disorganization of the ER and Golgi membranes through uncoupling of the cytoskeletal network (19,20,36). To address whether the PrP-dystonin-a2 transgene protects organelle integrity in PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice, we sought to evaluate the ultrastructure of control, dtTg4/Tg4 and PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 P5 sensory neurons (Fig. 12A–E). Representative EM micrographs of P5 WT sensory neurons displayed organized rough ER sheets (A) and Golgi stacks (A′) compared with the dilated rough ER sheets (B) and Golgi membranes (B′) observed in P5 dtTg4/Tg4 sensory neurons. Similar to control sensory neurons, we found P5 PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 sensory neurons were devoid of dilated ER (C) and Golgi (C′) membranes. Quantification of ER and Golgi membrane diameter indicated P5 control and PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 were comparable in size, and were significantly smaller in size compared with ER and Golgi membranes of dtTg4/Tg4 sensory neurons (Fig. 12D and E). These findings corroborate previous inferences that dystonin-a2 is critical in maintaining organelle structure. Moreover, as the PrP-dystonin-a2 transgene attenuates cellular demise, we conclude that stabilization of vital organelles is an important contributor to this cellular rescue.
Figure 12.

PrP-dystonin-a2 transgene prevents the alteration of ER and Golgi membranes. Representative electron micrographs show dilated ER and Golgi membranes (B and B′, respectively) in P5 dtTg4/Tg4 sensory neurons. Arrows depict organelles, whereas asterisks depict areas of dilation. In contrast to dtTg4/Tg4 sensory neurons, ER and Golgi membranes within PrP/PrP control (A and A′, respectively) and PrP/PrP;dtTg4/Tg4 (C and C′ respectively) sensory neurons do not display dilated membranes. (D and E) Quantitative analyses of ER and Golgi ultrastructure. ANOVA, post hoc Tukey, **P < 0.01, (5 cells examined per animal, n = 3/genotype).
DISCUSSION
Deleterious homozygous mutations in DST were recently reported to cause a dystonin-related neuropathy termed HSAN-VI. All three dystonin isoforms are impacted in HSAN-VI. The human and mouse DST gene are highly similar (16) and DST mutations in both humans and mice produce similar phenotypic features.
To gain a better understanding of the dystonin-a isoforms underlying both HSAN-VI and dt pathogenesis, we addressed whether dystonin-a2 expression in dtTg4/Tg4 mice is able to partially or completely rescue the dt phenotype and the underlying pathophysiology. We successfully generated transgenic mice expressing the PrP-dystonin-a2 transgene on the dtTg4/Tg4 background. Transgene expression was evident throughout neural tissues and was, most importantly, expressed throughout all sensory neuron subtypes. Of note, the transgene product was largely localized to the perinuclear region of the cell bodies of DRG sensory neurons. Increasing dystonin-a2 expression in neural tissues of dtTg4/Tg4 mice decreased disease severity and increased life span. We find these aforementioned improvements are due in part to improved MT and organelle integrity within proprioceptive sensory neurons, and as such, delay apoptosis of sensory neurons. Collectively, this study proposes dystonin-a2 loss of function is an important contributor to dt pathogenesis. Despite these improvements, complete rescue was not observed likely because of inadequate expression of the transgene.
Dystonin-a2 decreases disease severity and increases life span
By reintroducing dystonin-a2 expression within the nervous system of dtTg4/Tg4 mice, we extended life span to a mean age of 55 days coupled with a progressive gain in weight between P16 and P52 (Fig. 6). Why some rescue mice show prolonged survival compared with other rescue mice (e.g. 123 versus 46 days) is unclear. We suspect this variability in survival is due in part to disease modifiers and/or intrinsic genetic background differences. The PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice also showed a delay in the onset of hind limb clasping and exhibited a normal gait at P20 reminiscent of PrP-dystonin-a2/PrP-dystonin-a2 control mice. As hind limb clasping and aberrant gait in dt mice are provoked through the degeneration of proprioceptive sensory afferents, improvement in these areas suggests this subgroup of sensory neurons are transiently protected by dystonin-a2 transgenic expression. Qualitatively, PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice also display a milder form of dt phenotype (less limb incoordination, fewer dystonic postures and little to no writhing).
Dystonin-a2 is protective in subpopulations of sensory neurons
Although dystonin-a expression is widely distributed throughout DRG sensory afferents, the postnatal consequence of dystonin-a absence varies among sensory neuron subpopulations (8–10). The dt mutation provokes a very selective degeneration, mainly affecting large- and medium-sized sensory neurons which encompass group Ia and Ib (large), and group II (medium-sized) sensory afferents (8). These proprioceptive sensory afferents make up 20% of DRG neurons and innervate specialized sense organs within the musculature, including muscle spindles and Golgi tendon organs (37). These sensory organs are critical for posture and stability and goal-directed movements. Multiple studies found that the degeneration of muscle spindles is concomitant with phenotypic onset and that aberrations in spindle function are most likely the cause of both the ataxic and dystonic movements seen in dt mice (6,8,27). Delineating which dystonin-a isoform(s) is/are responsible for the demise of these populations is paramount, as it will not only narrow down the principle dystonin-a isoform, but also propose potential pathological mechanisms underlying the dt disorder and similar human diseases, like HSAN-VI.
Here, we show that dystonin-a2 is necessary in the maintenance of postnatal sensory neurons as its expression significantly delays the progressive degeneration seen in dtTg4/Tg4 mice (Fig. 7). Moreover, cultured primary sensory neurons from PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice revealed that the dystonin-a2 transgene confers protection among medium-sized sensory neurons (group II sensory afferents), but not large sensory neurons when challenged with serum-free media (Fig. 11). Why large sensory neurons are so vulnerable in the dt disorder is unclear. Previous studies have found large sensory neurons are more vulnerable to diseased conditions than small-sized neurons (38). Moreover, large- and medium-sized sensory neurons possess high-energy demands, and require significant trafficking capacity, and hence may be particularly vulnerable to vesicular and axonal transport defects. As transgene expression was replete throughout all sensory neuron subtypes (Fig. 3), we conclude that dystonin-a2 expression is critical for the survival of medium-sized sensory neurons. Although no rescue was seen in large sensory neurons, this does not imply dystonin-a2 expression is dispensable in this population of sensory neurons. It is likely that the level of transgene expression was not high enough for the survival of large sensory afferents.
PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice show partial, but not complete, improvements in both the pen test and gait analyses. This would imply that specific sensory nerves—likely large sensory afferents—are already dying or are not functioning optimally. Furthermore, the milder dt phenotype indicates that the progressive nature of dtTg4/Tg4 pathogenesis within proprioceptive sensory afferents is delayed.
There are several possibilities as to why PrP-dystonin-a2 transiently rescues medium-sized sensory afferents. The most likely reason is that expression of the dystonin-a2 transgene is not equivalent to that of endogenous dystonin-a2. However, expression of dystonin-a1, or a yet-to-be-determined dystonin-a isoform, may also be needed for a complete rescue of medium-sized sensory afferents.
To address how dystonin-a2 was mediating a partial rescue, we investigated the status of MT networks within our primary sensory neuron culture system. Dystonin-a has long been known to be involved in the maintenance of the neuronal cytoskeleton, particularly MTs (29,30). More recently, through interactions with the microtubule-associated protein-1B, dystonin-a2 was found to be necessary in maintaining the acetylation status of MTs, and as such, preserved optimal protein trafficking (19). Here, we find that the dystonin-a2 transgene is capable of maintaining the integrity of MT networks within dtTg4/Tg4 sensory axons. Indeed, we did not observe axonal accumulations of βIII-tubulin as seen in dtTg4/Tg4 sensory axons (Fig. 11C). In light of the aforementioned studies, improved MT stability is a conceivable explanation as to the delayed degeneration observed herein.
Coupled with improvements in MT stability was the ultrastructural preservation of ER and Golgi membranes in PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 sensory neurons (Fig. 12A–E). As organelle function is contingent upon structure, we conclude that preserved organelle structure enables organelles to fulfill their function and hence extend cellular viability. Previous studies demonstrated that dystonin-a2 mediates Golgi organization through the maintenance of MT acetylation (19). It is therefore likely that restoration of dystonin-a2 expression stabilizes MTs, which in turn contributes to the maintenance of Golgi membranes. Whether ER membranes are also organized via acetylated MT networks remains to be determined.
Taken altogether, both dystonin-a1 and dystonin-a2 are involved in fundamental biological functions within neurons, including vesicular trafficking and maintenance of MTs and organelle membranes. As similar biological processes are perturbed in both inherited and acquired peripheral neuropathies, our results should be of clinical interest. Moreover, results herein will provide needed biological insights into the newly identified dystonin-related HSAN-VI.
MATERIALS AND METHODS
Reagents
All chemicals were purchased through Sigma-Aldrich (St Louis, MO, USA) and all cell culture reagents were obtained from Invitrogen (Burlington, ON, Canada) except where indicated.
Ethics statement
All experimental protocols on mice were approved by the Animal Care Committee of the University of Ottawa. Care and use of experimental mice followed the guidelines of the Canadian Council on Animal Care.
Animals and cell culture
The dtTg4/Tg4, PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 and PrP-dystonin-a2/PrP-dystonin-a2 transgenic mice all shared the same mixed genetic background of CD1/C57BL6 and were used at pre-phenotype (P7) and phenotype stages (P15 and P58). The generation of the dtTg4/Tg4 line and characterization of the mutation have been described previously (15,25,39). The onset of phenotype was generally assessed by the appearance of clasping of hind limbs after picking the mice up by their tails. dtTg4/Tg4 mice were genotyped by PCR amplification of genomic tail DNA.
The dystonin-a2 cDNA was previously developed in the laboratory. In brief, dystonin-a2 cDNA was amplified from mouse brain RNA and cloned into the pEF1-myc/his vector (Invitrogen) encoding a C-terminal myc/his tag (13,18). The dystonin-a2-myc/his cDNA was thereafter cloned into the 3.5 kb PrP promoter and the expression vector pEF1myc/hisB (Invitrogen). The resulting transgene construct was microinjected into one-cell mouse embryos. Tail biopsies were obtained from potential founder mice, DNA was extracted and transgenic mice were identified by PCR amplification using sense oligo 5′ GTT TGC ACC AAT GCC TTC GC 3′ and antisense oligo 5′ GCC GGA CCT GAT AGA CAT GA 3′. These primers amplify a 318 bp fragment on the wild-type Dst gene and a 220 bp fragment from the PrP-dystonin-a2 transgene. Positive founder mice were bred with wild-type mice to establish two independent transgenic lines (founder lines 542 and 559). Heterozygous PrP-dystonin-a2/+ and homozygous PrP-dystonin-a2/PrP-dystonin-a2 mice were subsequently crossed to heterozygous dtTg4/+ mice, and the dtTg4 allele was thereafter bred to homozygosity producing both PrP-dystonin-a2/+;dtTg4/Tg4 mice and PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 mice. The genotype of offspring mice was confirmed by PCR.
F11 cells (a fusion of embryonic rat DRG cells with a mouse neuroblastoma cell line, kindly supplied by Dr Paul Albert, University of Ottawa) were maintained in DMEM + 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin/antimycotic. Cells were passaged at ∼70% confluency in 10 cm plastic Petri dishes and plated onto glass coverslips for use in immunofluorescence assays. Cell transfections were performed using Lipofectamine 2000 (Invitrogen), according to the manufacturer's directions.
Gait measurements
Gait assessment was performed using an in-house gait box as previously described (26). Both front and back paws were marked with red and blue ink, respectively. Mice were situated at the larger end of the gait apparatus with a light shone through to encourage the mice to walk toward the opposite end, leaving their imprints on a piece of white paper. Imprints were thereafter inspected and compared among different genotypes.
Pen test
Balance and strength were assessed using the pen test as described (40). Mice were placed on a suspended pen at different time points. The latency to fall from the pen was measured with a plateau of 30 s. At each time point, individual mice were placed three consecutive times.
RNA-isolation and reverse transcription-polymerase chain reaction analysis
All RNA used for experiments was extracted using the RNeasy® Mini Kit (Qiagen), as per the manufacturer's protocol. For RT–PCR analysis, RNA was isolated from transgenic P7, P15 and P21 tissues from the two PrP-dystonin-a2 founder lines (F542 and F559). To synthesize cDNA, equal amounts of RNA from brain, spinal cord, muscle and DRGs were reverse-transcribed in a standard reaction with MuLV reverse transcriptase (Invitrogen). PCR amplification using the sense oligo 5′ AAC AAA AAC TCA TCT CAG AAG AG 3′, and the antisense oligo 5′ ATG GTG ATG GTG ATG ATG AC 3′ (specific to the Myc/His cDNA), yielded a 62 bp fragment. Primers were chosen to flank the intronic region of the construct to selectively amplify the RNA transcript and prevent amplifying any contaminating genomic DNA. The reaction began with a 3 min incubation time at 94°C followed by 30 cycles of 45 s at 94°C, 45 s at 55°C, 1 min at 68°C, with a final extension time of 10 min at 68°C. Amplification of actin cDNA served as a control. The PCR products were electrophoresed on a 5% agarose gel containing ethidium bromide, and amplified fragments were visualized under UV transillumination. cDNAs encoding actin (control) derived from mouse were PCR amplified as previously described (15).
Quantitative RT–PCR
For the RT–qPCR analysis, RNA was isolated from the DRGs of P15 wild-type, dtTg4/Tg4 and PrP-dystonin-a2/PrP-dystonin-a2; dtTg4/Tg4 mice. A total of 1 μg of RNA was reverse transcribed to cDNA using the QuantiTect Reverse Transcription Kit (Qiagen) as per the manufacturer's protocol. RT–qPCR was performed on the Bio-Rad CFX Connect Real-time System using the following protocol: 95°C for 10 min, followed by 40 cycles of 95°C for 10 s and 59.5°C for 30 s. Each 25 μl reaction contained 1.0 μl of RT products, 12.5 μl SsoFast EvaGreen Supermix (Bio-Rad) and 0.1 μm forward and reverse primer for actin or 0.5 μm primers for dystonin-a2. Primers used for dystonin-a2 and actin have the following sequences from 5′ to 3′: dystonin-a2 sense oligo 5′ GAG GGC TGT GCT TCG GAT AG 3′, dystonin-a2 antisense oligo 5′ CAT CGT TTG CAC CAA TGC C 3′, actin sense oligo 5′ CCG TCA GGC AGC TCA TAG CTC TTC 3′ and actin antisense oligo 5′ CTG AAC CCT AAG GCC AAC CGT 3′. Amplification reactions were performed for each genotype using three biological replicates and each in technical triplicate. Separation of RT–qPCR products was done by electrophoresis on a 2% agarose gel with ethidium bromide, using representative samples from each genotype.
TUNEL labeling
Ten micrometer lumbar DRG tissue sections were dissected, washed in phosphate buffered saline (PBS) and permeabilized in ice cold 0.1% sodium citrate/0.1% Triton X-100 for 5 min followed by 2 min in 2:1 ethanol:acetic acid on ice. Samples were rinsed for 2 min in PBS and incubated for 1 h at 37°C with FITC-labeled dUTP in terminal deoxynucleotidyl transferase (TdT) buffer (30 mm Tris–HCl, pH 7.2, 140 mm sodium cacodylate and 1 mm cobalt chloride) and TdT according to the protocol provided by the manufacturer (Roche, Laval, QC, Canada). Negative controls included sections incubated with FITC-labeled dUTP in the absence of TdT. Cells were washed in PBS, mounted in fluorescent mounting media (Dako, Burlington, ON, Canada) and analyzed with a Zeiss Axiovert 200 m epifluorescent microscope equipped with an Axiocam HRM digital camera and Axiovision 4.6 software (Zeiss, Toronto, ON, Canada).
Primary culture of DRG neurons
Spinal columns were removed from P5 mice and transferred to a dissection microscope. Total DRGs were isolated per mouse, and subsequently digested for 15 min each with collagenase A (Roche) and papain (Worthington, Lakewood, NJ, USA) solutions. DRG neurons were dissociated with flame polished glass Pasteur pipettes and seeded onto 18 mm laminin-2 (Millipore, Billerica, MA, USA) coated cover slips at a density of 50 000 per 12-well cell culture vessel. Cells were cultured in DMEM with 10% FBS and 1% Pen/Strep and placed in a 37°C tissue culture incubator, under 8.5% CO2. The following day, media were changed to neuronal maintenance media (DMEM base, 0.5% FBS, 1% Glutamax, 16 µg/ml putrescine, 400 µg/ml thyroxine, 400 µg/ml triiodothyronine, 6.2 ng/ml progesterone, 5 ng/ml sodium selenite, 100 µg/ml bovine albumin serum, 5 µg/ml bovine insulin, 50 µg/ml holo-transferrin) supplemented with 200 ng/ml nerve growth factor and 1 mm 5-fluoro-2′-deoxyuridine. A three-quarter media change was carried out every other day. Cultures were fixed in 4% paraformaldehyde (PFA) before antigenic labeling. Primary antibodies used were anti-c-myc (1:800; Santa Cruz Biotechnology, Inc.), mouse polyclonal anti-βIII-tubulin (1:1000; Millipore) and rabbit polyclonal anti-cleaved caspase-3 (1:1000; Cell Signaling Technology, Beverly, MA, USA). Antibodies were diluted in antibody buffer (PBS, 0.3% Triton X-100, and 3% BSA). Where DAPI staining is indicated, samples were incubated in DAPI stain (0.2 µg/ml in PBS) for 10 min and washed three times in PBS for 5 min. Samples were mounted in fluorescent mounting media (Dako) and analyzed with a confocal microscope (LSM 510 meta) equipped with an EC Plan-Neofluar 40×/1.30 NA oil DIC M27 objective using Zen 8.0 software.
Subpopulations of sensory neurons were visualized with an epifluorescent microscope (Axiovert 200M; Carl Zeiss) under a ×20 objective (Achroplan 0.25) equipped with a digital camera (AxioCam HRm; Carl Zeiss). Using Axiovision 4.6 software (Carl Zeiss), the circumference-measuring tool was employed in determining sensory neuron size.
Immunohistochemistry
Tissues (cortical, cerebellum, DRG and muscle) were collected from at least three dtTg4/Tg4, PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4 and PrP-dystonin-a2/PrP-dystonin-a2 mice at multiple time points (P10, P15, P58) as previously described (18). Mice were anesthetized with tribromoethanol (Avertin) and perfused transcardially with 3 ml of PBS followed by 10 ml of 4% PFA in PBS. Samples were embedded in OCT compound (Sakura), and frozen in liquid nitrogen. Cryostat sections of 10 μm thickness were stored at −20°C before use. Primary antibodies used were rabbit monoclonal anti-Myc (1:100, Abcam). Secondary antibodies used were anti rabbit Alexa-488 (1:2000, Molecular Probes). Antibodies were diluted as mentioned above. Samples were mounted in fluorescent mounting media (Dako) and analyzed with a Zeiss Axiovert 200 m epifluorescent microscope equipped with an Axiocam HRM digital camera and Axiovision 4.6 software (Zeiss).
Axonal assessment in the dorsal roots and morphological analysis of endplates was performed essentially as described previously (41). The number of muscle spindles was counted in serial paraffin cross-sections of P15 tibialis anterior muscle of three mice per genotype stained with hematoxylin and eosin. Big and small spindles were examined for degeneration from 30 sections per muscle using light microscopy.
Cell preparation for transmission EM and morphometric analysis
P5 primary sensory neurons were cultured as described above, washed with PBS and trypsinized (0.25% trypsin/0.53 mm EDTA) for 5 min at 37°C. To obtain a suitable-sized pellet of sensory neurons, an n = 2 was collected and combined for each genotype (PrP-dystonin-a2/PrP-dystonin-a2 control, dtTg4/Tg4 and PrP-dystonin-a2/PrP-dystonin-a2;dtTg4/Tg4). Cells were centrifuged at 1000g for 5 min and supernatant removed. Neurons were fixed for 1 h at room temperature in Karnovsky's fixative (4% PFA, 2% glutaraldehyde and 0.1 m cacodylate in PBS, pH 7.4) and subsequently washed twice in 0.1 m cacodylate buffer. Neurons were post-fixed with 1% osmium tetroxide in 0.1 m cacodylate buffer for 1 h at room temperature, followed by 3 × 5 min washes in water. Cells were dehydrated twice for 20 min for each step in a graded series of ethanol from water through 30, 50, 70, 85 and 95% ethanol, and twice for 30 min in 100% ethanol (molecular sieves were used to dehydrate ethanol), followed by two washes for 15 min in 50% ethanol/50% acetone and twice for 15 min in 100% acetone. Neurons were infiltrated in 30% spurr/acetone for 15 h (overnight), then in 50% spurr/acetone for 6 h, and in fresh 100% spurr resin for overnight. Spurr was changed twice a day for 3 days at room temperature. All infiltration steps were performed on a rotator. Neurons were embedded in fresh liquid spurr epoxy resin and polymerized overnight at 70°C. Ultrathin sections (80 nm) of cells were collected onto a 200-mesh copper grid and let dry overnight. Grids were stained with 2% aqueous uranyl acetate and with Reynold's lead citrate. Sections were observed under a transmission electron microscope (Hitachi 7100). Approximately 10 EM micrographs at ×20 000 magnification were examined per genotype.
Statistical analysis
Data were analyzed using Student's t-test or factorial ANOVA. Following detection of a statistically significant difference in a given series of treatments by ANOVA, post hoc Dunnett's t-tests or Tukey tests were performed where appropriate. P-values under 0.05 were considered statistically significant (shown as *); P-values under 0.01, 0.001 or 0.0001 were considered highly statistically significant (shown as **, ***, ****, respectively).
FUNDING
This project was funded by a grant from the Canadian Institutes of Health Research (CIHR) to R.K. A.F. is supported by an Ontario Graduate Scholarship, R.W.O'M. is a recipient of a Frederick Banting and Charles Best CIHR Doctoral Research Award and R.K. is a recipient of a University Health Research Chair from the University of Ottawa.
ACKNOWLEDGEMENTS
We are grateful to the Kothary laboratory for helpful discussions.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Rottier A., Baets J., Timmerman V., Janssens K. Mechanisms of disease in hereditary sensory and autonomic neuropathies. Nat. Rev. Neurol. 2012;8:73–85. doi: 10.1038/nrneurol.2011.227. [DOI] [PubMed] [Google Scholar]
- 2.Rotthier A., Baets J., De Vriendt E., Jacobs A., Auer-Grumbach M., Lévy N., Bonello-Palot N., Kilic S.S., Weis J., Nascimento A., et al. Genes for hereditary sensory and autonomic neuropathies: a genotype–phenotype correlation. Brain. 2009;132:2699–2711. doi: 10.1093/brain/awp198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dyck P.J., Thomas P.K., Griffin J.W., Low P.A., Poduslo J.F. Peripherial Neuropathy. Philadelphia: WB Saunders; 2005. [Google Scholar]
- 4.Edvardson S., Cinnamon Y., Jalas C., Shaag A., Maayan C., Axelrod F.B., Elpeleg O. Hereditary sensory autonomic neuropathy caused by a mutation in dystonin. Ann. Neurol. 2012;71:569–572. doi: 10.1002/ana.23524. [DOI] [PubMed] [Google Scholar]
- 5.Ferrier A., Boyer J.G., Kothary R. Cellular and molecular biology of neuronal dystonin. Int. Rev. Cell Mol. Biol. 2013;300:85–120. doi: 10.1016/B978-0-12-405210-9.00003-5. [DOI] [PubMed] [Google Scholar]
- 6.Duchen L.W., Strich S.J., Falconer D.S. Clinical and pathological studies of an hereditary neuropathy in mice (dystonia musculorum) Brain. 1964;87:367–378. doi: 10.1093/brain/87.2.367. [DOI] [PubMed] [Google Scholar]
- 7.Duchen L.W. Dystonia musculorum—an inherited disease of the nervous system in the mouse. Adv. Neurol. 1976;14:353–365. [PubMed] [Google Scholar]
- 8.Sotelo C., Guenet J.L. Pathologic changes in the CNS of dystonia musculorum mutant mouse: an animal model for human spinocerebellar ataxia. Neuroscience. 1988;27:403–424. doi: 10.1016/0306-4522(88)90277-1. [DOI] [PubMed] [Google Scholar]
- 9.al-Ali S.Y., al-Zuhair A.G. Fine structural study of the spinal cord and spinal ganglia in mice afflicted with a hereditary sensory neuropathy, dystonia musculorum. J. Submicrosc. Cytol. Pathol. 1989;21:737–748. [PubMed] [Google Scholar]
- 10.Carlsten J.A., Kothary R., Wright D.E. Glial cell line-derived neurotrophic factor-responsive and neurotrophin-3-responsive neurons require the cytoskeletal linker protein dystonin for postnatal survival. J. Comp. Neurol. 2001;432:155–168. doi: 10.1002/cne.1094. [DOI] [PubMed] [Google Scholar]
- 11.Suozzi K.C., Wu X., Fuchs E. Spectraplakins: master orchestrators of cytoskeletal dynamics. J. Cell Biol. 2012;197:465–475. doi: 10.1083/jcb.201112034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jefferson J.J., Leung C.L., Liem R.K.H. Dissecting the sequence specific functions of alternative N-terminal isoforms of mouse bullous pemphigoid antigen 1. Exp. Cell Res. 2006;312:2712–2725. doi: 10.1016/j.yexcr.2006.04.025. [DOI] [PubMed] [Google Scholar]
- 13.Young K.G., Pinheiro B., Kothary R. A Bpag1 isoform involved in cytoskeletal organization surrounding the nucleus. Exp. Cell Res. 2006;312:121–134. doi: 10.1016/j.yexcr.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Sun D., Leung C.L., Liem R.K. Characterization of the microtubule binding domain of microtubule actin crosslinking factor (MACF): identification of a novel group of microtubule associated proteins. J. Cell Sci. 2001;114:161–172. doi: 10.1242/jcs.114.1.161. [DOI] [PubMed] [Google Scholar]
- 15.Pool M., Boudreau Larivière C., Bernier G., Young K.G., Kothary R. Genetic alterations at the Bpag1 locus in dt mice and their impact on transcript expression. Mamm. Genome. 2005;16:909–917. doi: 10.1007/s00335-005-0073-4. [DOI] [PubMed] [Google Scholar]
- 16.Young K.G., Kothary R. Dystonin/Bpag1—a link to what? Cell Motil. Cytoskeleton. 2007;64:897–905. doi: 10.1002/cm.20235. [DOI] [PubMed] [Google Scholar]
- 17.Bernier G., Brown A., Dalpé G., De Repentigny Y., Mathieu M., Kothary R. Dystonin expression in the developing nervous system predominates in the neurons that degenerate in dystonia musculorum mutant mice. Mol. Cell Neurosci. 1995;6:509–520. doi: 10.1006/mcne.1995.0003. [DOI] [PubMed] [Google Scholar]
- 18.Young K.G., Kothary R. Dystonin/Bpag1 is a necessary endoplasmic reticulum/nuclear envelope protein in sensory neurons. Exp. Cell Res. 2008;314:2750–2761. doi: 10.1016/j.yexcr.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 19.Ryan S.D., Bhanot K., Ferrier A., De Repentigny Y., Chu A., Blais A., Kothary R. Microtubule stability, Golgi organization, and transport flux require dystonin-a2-MAP1B interaction. J. Cell Biol. 2012;196:727–742. doi: 10.1083/jcb.201107096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ryan S.D., Ferrier A., Sato T., O'Meara R.W., De Repentigny Y., Jiang S.X., Hou S.T., Kothary R. Neuronal dystonin isoform 2 is a mediator of endoplasmic reticulum structure and function. Mol. Biol. Cell. 2012;23:553–566. doi: 10.1091/mbc.E11-06-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J.-J., Ding J., Kowal A.S., Nardine T., Allen E., Delcroix J.-D., Wu C., Mobley W., Fuchs E., Yang Y. BPAG1n4 is essential for retrograde axonal transport in sensory neurons. J. Cell Biol. 2003;163:223–229. doi: 10.1083/jcb.200306075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J.J., Ding J., Wu C., Bhagavatula P., Cui B., Chu S., Mobley W.C., Yang Y. Retrolinkin, a membrane protein, plays an important role in retrograde axonal transport. Proc. Natl Acad. Sci. USA. 2007;104:2223–2228. doi: 10.1073/pnas.0602222104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Perlson E., Maday S., Fu M.-M., Moughamian A.J., Holzbaur E.L.F. Retrograde axonal transport: pathways to cell death. Trends Neurosci. 2010;33:335–344. doi: 10.1016/j.tins.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gavrilina T.O., McGovern V.L., Workman E., Crawford T.O., Gogliotti R.G., DiDonato C.J., Monani U.R., Morris G.E., Burghes A.H.M. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum. Mol. Genet. 2008;17:1063–1075. doi: 10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kothary R., Clapoff S., Brown A., Campbell R., Peterson A., Rossant J. A transgene containing lacZ inserted into the dystonia locus is expressed in neural tube. Nature. 1988;335:435–437. doi: 10.1038/335435a0. [DOI] [PubMed] [Google Scholar]
- 26.Bowerman M., Murray L.M., Beauvais A., Pinheiro B., Kothary R. A critical smn threshold in mice dictates onset of an intermediate spinal muscular atrophy phenotype associated with a distinct neuromuscular junction pathology. Neuromusc. Dis. 2012;22:263–276. doi: 10.1016/j.nmd.2011.09.007. [DOI] [PubMed] [Google Scholar]
- 27.Dowling J., Yang Y., Wollmann R., Reichardt L.F., Fuchs E. Developmental expression of BPAG1-n: insights into the spastic ataxia and gross neurologic degeneration in dystonia musculorum mice. Dev. Biol. 1997;187:131–142. doi: 10.1006/dbio.1997.8567. [DOI] [PubMed] [Google Scholar]
- 28.Axelrod F.B., Gold-von Simson G. Hereditary sensory and autonomic neuropathies: types II, III, and IV. Orphanet J. Rare Dis. 2007;2:39. doi: 10.1186/1750-1172-2-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dalpé G., Leclerc N., Vallée A., Messer A., Mathieu M., De Repentigny Y., Kothary R. Dystonin is essential for maintaining neuronal cytoskeleton organization. Mol. Cell Neurosci. 1998;10:243–257. doi: 10.1006/mcne.1997.0660. [DOI] [PubMed] [Google Scholar]
- 30.Yang Y., Dowling J., Yu Q.C., Kouklis P., Cleveland D.W., Fuchs E. An essential cytoskeletal linker protein connecting actin microfilaments to intermediate filaments. Cell. 1996;86:655–665. doi: 10.1016/s0092-8674(00)80138-5. [DOI] [PubMed] [Google Scholar]
- 31.De Repentigny Y., Deschenes-Furry J., Jasmin B.J., Kothary R. Impaired fast axonal transport in neurons of the sciatic nerves from dystonia musculorum mice. J. Neurochem. 2003;86:564–571. doi: 10.1046/j.1471-4159.2003.01861.x. [DOI] [PubMed] [Google Scholar]
- 32.Barr F.A., Egerer J. Golgi positioning: are we looking at the right MAP? J. Cell Biol. 2005;168:993–998. doi: 10.1083/jcb.200501088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Starr D.A. Communication between the cytoskeleton and the nuclear envelope to position the nucleus. Mol. Biosyst. 2007;3:583–589. doi: 10.1039/b703878j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bola B., Allan V. How and why does the endoplasmic reticulum move? Biochem. Soc. Trans. 2009;37:961–965. doi: 10.1042/BST0370961. [DOI] [PubMed] [Google Scholar]
- 35.Lin C.M., Chen H.J., Leung C.L., Parry D.A., Liem R.K. Microtubule actin crosslinking factor 1b: a novel plakin that localizes to the Golgi complex. J. Cell Sci. 2005;118:3727–3738. doi: 10.1242/jcs.02510. [DOI] [PubMed] [Google Scholar]
- 36.Ryan S.D., Ferrier A., Kothary R. A novel role for the cytoskeletal linker protein dystonin in the maintenance of microtubule stability and the regulation of ER-Golgi transport. BioArchitecture. 2012;2:2–5. doi: 10.4161/bioa.20302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mu X., Silos-Santiago I., Carroll S.L., Snider W.D. Neurotrophin receptor genes are expressed in distinct patterns in developing dorsal root ganglia. J. Neurosci. 1993;13:4029–4041. doi: 10.1523/JNEUROSCI.13-09-04029.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McIlwain D.L. Nuclear and cell body size in spinal motor neurons. Adv. Neurol. 1991;56:67–74. [PubMed] [Google Scholar]
- 39.Brown A., Bernier G., Mathieu M., Rossant J., Kothary R. The mouse dystonia musculorum gene is a neural isoform of bullous pemphigoid antigen 1. Nat. Genet. 1995;10:301–306. doi: 10.1038/ng0795-301. [DOI] [PubMed] [Google Scholar]
- 40.Willmann R., Dubach J., Chen K. Developing standard procedures for pre-clinical efficacy studies in mouse models of spinal muscular atrophy: report of the expert workshop “Pre-clinical testing for SMA”, Zürich, March 29-30th 2010. Neuromusc. Dis. 2011;21:74–77. doi: 10.1016/j.nmd.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 41.De Repentigny Y., Ferrier A., Ryan S.D., Sato T., Kothary R. Motor unit abnormalities in Dystonia musculorum mice. PLoS ONE. 2011;6:14. doi: 10.1371/journal.pone.0021093. [DOI] [PMC free article] [PubMed] [Google Scholar]