

Abstract
Rare copy number variants (CNVs) disrupting ASTN2 or both ASTN2 and TRIM32 have been reported at 9q33.1 by genome-wide studies in a few individuals with neurodevelopmental disorders (NDDs). The vertebrate-specific astrotactins, ASTN2 and its paralog ASTN1, have key roles in glial-guided neuronal migration during brain development. To determine the prevalence of astrotactin mutations and delineate their associated phenotypic spectrum, we screened ASTN2/TRIM32 and ASTN1 (1q25.2) for exonic CNVs in clinical microarray data from 89 985 individuals across 10 sites, including 64 114 NDD subjects. In this clinical dataset, we identified 46 deletions and 12 duplications affecting ASTN2. Deletions of ASTN1 were much rarer. Deletions near the 3′ terminus of ASTN2, which would disrupt all transcript isoforms (a subset of these deletions also included TRIM32), were significantly enriched in the NDD subjects (P = 0.002) compared with 44 085 population-based controls. Frequent phenotypes observed in individuals with such deletions include autism spectrum disorder (ASD), attention deficit hyperactivity disorder (ADHD), speech delay, anxiety and obsessive compulsive disorder (OCD). The 3′-terminal ASTN2 deletions were significantly enriched compared with controls in males with NDDs, but not in females. Upon quantifying ASTN2 human brain RNA, we observed shorter isoforms expressed from an alternative transcription start site of recent evolutionary origin near the 3′ end. Spatiotemporal expression profiling in the human brain revealed consistently high ASTN1 expression while ASTN2 expression peaked in the early embryonic neocortex and postnatal cerebellar cortex. Our findings shed new light on the role of the astrotactins in psychopathology and their interplay in human neurodevelopment.
INTRODUCTION
Genomic studies driven by the recent advances in microarray and next-generation sequencing technology have begun to uncover the architecture of genetic risk for autism spectrum disorder (ASD) (1,2). Rapid implementation of these genome-wide screening methods in the clinical diagnostic and research settings has facilitated the identification of etiologic variants in some 15% of ASD cases (2). Particularly prominent among these genetic findings have been rare de novo and inherited copy number variants (CNVs) and single-nucleotide variants (SNVs) impacting genes encoding cell-adhesion and scaffolding proteins at the neuronal synapse including those from the neurexin (3–5), neuroligin (6), SHANK (7–10), contactin (11–14) and contactin-associated (14–16) protein families. The parallel discoveries of rare mutations affecting several of these and other synaptic genes in conditions such as schizophrenia and intellectual disability (ID) have highlighted the disruption of synaptic homeostasis as a key overarching etiologic factor underlying clinically diverse neurodevelopmental disorders (NDDs) (17–20).
In addition to disruption of synaptic pathways, dysfunction of proteins participating in embryonic neuronal migration has been linked to the etiology of several neurocognitive disorders (21). Notable examples include the disruption of key signaling molecules that stimulate neuronal migration such as BDNF deletions in patients with behavioral disorders (22), reelin (RELN) as a risk factor for several NDDs including ASD and schizophrenia (23), and the implication of neuregulin (NRG1) and its receptor ERBB4 in risk for schizophrenia (24). The NRG1/ERBB4 complex is a key facilitator of neuronal migration along radial glial fibers during cortical development of the cerebrum and cerebellum.
Another well-characterized molecule of critical functional relevance to glial-guided neuronal migration is the integral membrane protein astrotactin 1 (ASTN1), which forms adhesions between neurons and astroglia as a neuronal cell-surface antigen (25–27). Mouse Astn1 is highly expressed in migrating granule neuron cells in the cerebellum and also in other brain regions featuring formation of laminar structures via glial-guided neuronal migration including the cerebral cortex, hippocampus and olfactory bulb (28). Astn1 null mice exhibit impaired migration of cerebellar granule cells, smaller cerebellar size, reduced glial-neuron binding, abnormal Purkinje cell morphology and poorer balance and coordination in behavioral assays compared with wild-type (29). A second member of the astrotactin protein family, astrotactin 2 (ASTN2), has recently been found to interact with ASTN1 in the neuronal membrane and regulate its expression on the neuronal surface, thus mediating the formation and release of neuronal-glial adhesions during migration (30).
Rare CNVs affecting ASTN2 or both ASTN2 and TRIM32, a small gene nested within an intron of ASTN2 and transcribed from the opposite strand, at the 9q33.1 locus were the most intriguing findings in our recent genome-wide rare CNV scan for shared risk factors between ASD and ADHD (31). These rare genetic events were significantly enriched in individuals from the ADHD and ASD cohorts (exonic CNVs in 5/597 probands) (Supplementary Material, Fig. S1) compared with a collection of 2357 population-based controls, in which they were absent. Other genome-wide scans have also detected very rare exonic CNVs at the ASTN2/TRIM32 locus in a handful of individuals with diverse neurodevelopmental diagnoses (Supplementary Material, Fig. S1) including 3 with ASD (32), 2 with schizophrenia (one patient also had epilepsy) (33), 2 with Tourette syndrome (34), 10 with ID (35,36) and 1 with bipolar disorder (37). All of these CNVs impacted one or more exons of ASTN2, while a subset also encompassed TRIM32. There have been no reports to date of mutations at the ASTN1 locus at 1q25.2.
The intriguing preliminary human genetic findings and the well-established functions of the astrotactins in mammalian brain development highlight ASTN1 and ASTN2 as promising candidate risk genes for NDDs. We exploited the availability of massive clinical microarray databases to screen systematically for novel mutations affecting these two genetic loci. We sought to elucidate their prevalence and role in human psychopathology, investigate their patterns of transmission and penetrance, and delineate their associated clinical phenotype.
RESULTS
Rare CNV findings at ASTN2/TRIM32 and ASTN1 regions
We examined microarray data from 89 985 individuals referred for postnatal genetic testing across 10 different sites, including 64 114 NDD subjects (Table 1 and see Materials and Methods). We identified three individuals (two deletions and one duplication) with exonic CNVs overlapping ASTN1 and 58 individuals with CNVs impacting exons of ASTN2 (Figs 1 and 2, Table 2; Supplementary Material, Fig. S2). One individual with an exonic ASTN2 deletion (patient 18) was obtained from the DECIPHER database and was not included in the CNV counts and enrichment analysis, since it was not part of data from the 10 molecular diagnostic sites. The exonic ASTN2 CNVs used in the analyses included 46 deletions (patients 1–17 and 19–47 in Fig. 1 and Table 2) and 12 duplications (patients 25 and 48–58 in Supplementary Material, Fig. S2 and Table 2). Except for patient 3, who possessed a whole gene deletion completely overlapping all ASTN2 transcript isoforms, all other individuals had partial deletions or duplications of ASTN2. One individual was seen to possess both a deletion and duplication at the ASTN2 locus (patient 25). Twenty seven of 46 of the deletions but none of the duplications also affected TRIM32. There were no exonic CNVs in the clinical dataset that impacted only TRIM32 without simultaneously affecting one or more exons of ASTN2. However, such CNVs could have gone undetected due to being smaller than the resolution of the microarray platforms.
Table 1.
Clinical case cohorts
| Cohorta | Total no. of cases | Total no. exonic ASTN1 CNVsb | Total no. exonic ASTN2 CNVsb | No. of NDD individuals (males/females) | No. of exonic ASTN2 CNVs in NDD individuals |
|---|---|---|---|---|---|
| Alberta Children's Hospital | 1619 | 0 | 1 (1 loss) | 1170 (675/495) | 1 (1 loss) |
| BBGRE | 14 847 | 2 (1 loss, 1 gain) | 3 (3 losses) | 9650 (6486/3164) | 1 (1 loss) |
| Boston Children's Hospital | 7320 | 1 (1 loss) | 8 (6 losses, 2 gains) | 6623 (4152/2471) | 6 (5 losses, 1 gain) |
| Credit Valley Hospital | 3552 | 0 | 2 (1 loss, 1 gain) | 3098 (2055/1043) | 2 (1 loss, 1 gain) |
| Hospital for Sick Children | 7411 | 0 | 5 (5 losses) | 4863 (3267/1596) | 5 (5 losses) |
| Italian diagnostic labsc | 6626 | 0 | 6 (3 losses, 3 gains) | 5568 (3272/2296) | 6 (3 losses, 3 gains) |
| Mayo Clinic | 19 131 | 0 | 7 (6 losses, 1 gain) | 11 208 (7282/3926) | 6 (5 losses, 1 gain) |
| Odense University Hospital | 551 | 0 | 2 (2 losses) | 289 (182/107) | 2 (2 losses) |
| Signature Genomics | 26 973 | 0 | 17 (13 losses, 4 gains) | 19 690 (11 617/8073)d | 13 (11 losses, 2 gains) |
| The Centre for Applied Genomicse | 1955 | 0 | 7 (6 losses, 1 gain) | 1955 (1450/505) | 7 (6 losses, 1 gain) |
| Total | 89 985 | 3 (2 losses, 1 gain) | 58 (46 losses, 12 gains) | 64 114 (40 438/23 676) | 49 (40 losses, 9 gains) |
BBGRE, brain and body genetic resource exchange (http://bbgre.org); NDD, neurodevelopmental disorders.
aTen different molecular diagnostic sites that contributed clinical microarray data for this study. Further descriptions are available in the following references: Chen et al. (97) for Boston Children's Hospital data, Hodge et al. (53) for Mayo Clinic data, Rosenfeld et al. (98) for Signature Genomics data and Ahn et al. (99) for BBGRE data. The microarray platforms utilized at each site and their corresponding number of probes within ASTN1 and ASTN2 are summarized in Supplementary Material, Table S9.
bAll CNVs in the clinical cohorts <6 Mb that overlapped one of more exons of ASTN1 or ASTN2 were included in the counts above.
cItalian cohort includes data from individuals tested at five different molecular diagnostic sites: Cremona, Pavia, San Giovanni Rotondo, Tor Vergata and Troina.
dSex distribution of the Signature Genomics cohort was extrapolated from that found in a sampling cross-section of the data by Ernst et al. (22).
eThe Centre for Applied Genomics cohort includes 415 Canadian individuals with ADHD (Lionel et al. (31)) genotyped on the Affymetrix 6.0 (n = 248) and the Affymetrix CytoScan HD (n = 167), 174 individuals with OCD genotyped on the Illumina Omni2.5M-quad and 1366 Canadian individuals with ASD (Sato et al. (7)) genotyped on one of the following microarray platforms: Affymetrix 6.0, Agilent 1M, Illumina 1M or Affymetrix CytoScan HD.
Figure 1.

Exonic deletions found at the ASTN2/TRIM32 locus in clinical and control cohorts. Exonic deletions identified in 46 of 89 985 cases and 19 of 44 085 controls are depicted. Filled red bars represent deletions detected in individuals with NDD phenotypes. Empty red bars denote deletions in cases without known NDD phenotypes (from available clinical information) and in controls. Shaded gray region denotes the critical region defined by deletions that disrupt multiple isoforms of ASTN2. Numbers adjacent to the bars are the randomized sample identifiers of individuals with the deletions and correlate with information in Table 2, Supplementary Material, Tables S1 and S3. Gender information was not available for the two control individuals marked with * at the bottom of the figure. Dashed purple lines intersect deletions that overlap exons shared by multiple ASTN2 isoforms and dashed green lines intersect those affecting only the long isoform. Dashed vertical black line intersects deletions that overlap an exon of TRIM32. Genomic locations and coordinates are based on hg18 (NCBI36). Information about genes and transcript isoforms was obtained from the RefSeq database. The three transcript isoforms of ASTN2 possessing different numbers of exons are depicted including the long isoform (NM_014010) and two shorter isoforms (NM_198186 and NM_001184735). The three other shorter isoforms of the gene (NM_198187, NM_198188 and NM_001184734) have the same number and location of exons as NM_198186 but differ slightly in the length of their first and terminal exons and UTRs.
Figure 2.
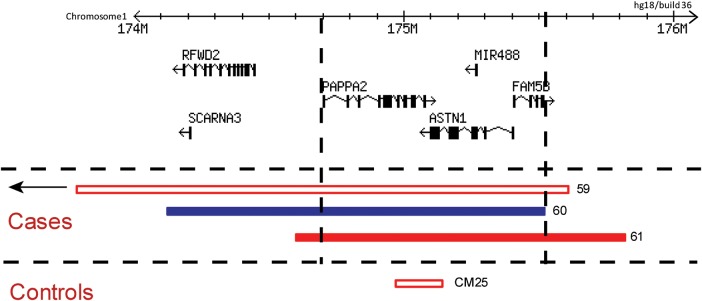
Exonic CNVs found at the ASTN1 locus in clinical and control cohorts. Red and blue bars represent deletions and duplications, respectively, that overlap ASTN1. Empty bars denote CNVs in cases without known NDD phenotypes (from available clinical information) and in controls. Dashed black lines outline the common region of overlap shared among the three CNVs detected in the clinical case dataset. Genomic locations and coordinates are based on hg18 (NCBI36). Information about genes and transcript isoforms was obtained from the RefSeq database.
Table 2.
Genetic and clinical information for individuals with rare CNVs of interest
| Case no. | Sex | Sitea | CNV coordinates (hg18) | CNV size | CNV | NDD phenotypesb |
|---|---|---|---|---|---|---|
| 1 | M | TCAG | chr9:117 954 428–118 356 243 | 401 816 | Loss | ADHD, Anx, LD, SD |
| 2 | M | OUH | chr9:118,055 333–118 646 904 | 591 572 | Loss | DD, MD |
| 3 | M | ACH | chr9:118 069 649–119 679 670 | 1 610 022 | Loss | Chiari I malformation |
| 4 | M | MC | chr9:118 130 121–119 029 857 | 899 737 | Loss | Hydrocephalus, Mac |
| 5 | M | HSC | chr9:118 164 272–118 358 705 | 194 434 | Loss | ADHD, ASD, DD, LD, Mic, MD, OCD, SD |
| 6 | M | SG | chr9:118 291 060–118 661 674 | 370 615 | Loss | Anx, ASD, LD, Mac, MD, OCD, SD |
| 7 | M | SG | chr9:118 327 395–118 595 433 | 268 039 | Loss | ASD, DD |
| 8 | M | CVH | chr9:118 342 936–118 685 436 | 342 501 | Loss | DD, seizures |
| 9 | M | BCH | chr9:118 358 646–118 459 563 | 100 918 | Loss | Anx, DD, Mac |
| 10 | M | ITA | chr9:118 358 837–118 728 270 | 369 434 | Loss | ASD, SD |
| 11 | M | HSC | chr9:118 390 436–118 524 432 | 133 997 | Loss | DD, ID, LD, MD, SD |
| 12 | M | SG | chr9:118 395 767–118 520 501 | 124 735 | Loss | ADHD, SD, CNS disorder |
| 13 | M | SG | chr9:118 395 767–118 595 433 | 199 667 | Loss | Hydrocephalus |
| 14 | M | TCAG | chr9:118 407 129–118 523 510 | 116 382 | Loss | Anx, ASD, SD |
| 15 | M | SG | chr9:118 421 170–118 683 092 | 261 923 | Loss | Behavioral problems |
| 16 | M | SG | chr9:118 430 585–118 569 556 | 138 972 | Loss | DD, MD, seizures, SD |
| 17 | M | SG | chr9:118 430 585–118 610 907 | 180 323 | Loss | DD, MD, SD |
| 18 | M | DEC | chr9:118 440 935–118 584 415 | 143 481 | Loss | ID, Mac |
| 19 | M | MC | chr9:118 459 294–118 616 407 | 157 114 | Loss | Chronic static encephalopathy |
| 20 | M | BCH | chr9:118 469 713–118 524 312 | 54 600 | Loss | DD, LD, tics |
| 21 | M | TCAG | chr9:118 479 893–118 627 637 | 147 745 | Loss | ADHD, Anx, OCD, tics |
| 22 | M | TCAG | chr9:118 480 042–118 570 447 | 90 406 | Loss | ADHD, ASD, Mac, OCD, SD, seizures |
| 23 | M | TCAG | chr9:118 481 308–118 654 031 | 172 724 | Loss | Anx, OCD |
| 24 | M | BCH | chr9:118 488 204–118 558 274 | 70 071 | Loss | ASD, seizures |
| 25 | M | BCH | chr9:118 488 204–118 700 657 | 212 454 | Loss | – |
| chr9:118 814 591–118 867 559 | 52 969 | Gain | ||||
| 26 | M | TCAG | chr9:118 493 276–118 670 608 | 177 333 | Loss | ADHD, Anx, LD |
| 27 | M | BBG | chr9:118 497 759–118 661 673 | 163 915 | Loss | – |
| 28 | M | MC | chr9:118 502 294–118 616 407 | 114 114 | Loss | ADHD, Anx, ASD, Mac, SD |
| 29 | M | SG | chr9:118 530 143–118 569 556 | 39 414 | Loss | Mic |
| 30 | M | MC | chr9:118 541 180–118 685 465 | 144 286 | Loss | Dizziness, dysgraphia, migraines |
| 31 | M | BCH | chr9:118 572 937–118 637 250 | 64 314 | Loss | DCD, DD, MD, SD |
| 32 | M | ITA | chr9:118 580 317–118 814 591 | 234 275 | Loss | ASD, structural brain anomaly |
| 33 | M | SG | chr9:118 580 891–118 630 518 | 49 628 | Loss | DD |
| 34 | M | HSC | chr9:118 616 347–118 907 058 | 290 712 | Loss | Anx, ASD, hydrocephalus, LD, Mac, MD, structural brain anomaly |
| 35 | M | SG | chr9:118 620 063–118 781 101 | 161 039 | Loss | ASD |
| 36 | M | BCH | chr9:118 743 266–118 991 977 | 248 712 | Loss | ASD, DD |
| 37 | M | ITA | chr9:118 840 027–118 935 027 | 95 001 | Loss | Behavioral problems, ID, OCD, SD |
| 38 | M | SG | chr9:118 874 947–119 109 618 | 234 672 | Loss | – |
| 39 | F | SG | chr9:118 199 243–118 248 950 | 49 708 | Loss | ADHD, DD, Mic, SD |
| 40 | F | BBG | chr9:118 202 805–118 227 359 | 24 555 | Loss | MD, plagiocephaly, SD |
| 41 | F | HSC | chr9:118 202 811–118 459 563 | 256 753 | Loss | Mac |
| 42 | F | HSC | chr9:118 390 436–118 524 432 | 133 997 | Loss | ID, MD, SD, seizures |
| 43 | F | SG | chr9:118 481 080–118 610 907 | 129 828 | Loss | – |
| 44 | F | BBG | chr9:118 497 759–118 661 673 | 163 915 | Loss | – |
| 45 | F | OUH | chr9:118 608 198–118 669 889 | 61 692 | Loss | ADHD, MD, SD |
| 46 | F | MC | chr9:118 728 240–118 992 036 | 263 797 | Loss | – |
| 47 | F | MC | chr9:118 829 818–118 890 615 | 60 798 | Loss | Septo-optic dysplasia |
| 48 | M | TCAG | chr9:118 263 609–118 308 641 | 45 033 | Gain | Anx, ASD, MD, seizures |
| 49 | M | BCH | chr9:118 899 840–121 893 169 | 2 993 330 | Gain | ADHD, ASD, DD, LD |
| 50 | M | ITA | chr9:118 916 018–119 875 655 | 959 638 | Gain | ASD, ID, LD, Mic, MD, SD |
| 51 | M | ITA | chr9:118 934 968–119 903 304 | 968 337 | Gain | Anx, ID, LD, MD, SD |
| 52 | M | MC | chr9:118 934 968–121 071 351 | 2 136 384 | Gain | Mac |
| 53 | M | SG | chr9:118 982 145–121 054 922 | 2 072 778 | Gain | Encephalocele |
| 54 | M | SG | chr9:118 982 145–121 054 922 | 2 072 778 | Gain | LD |
| 55 | M | SG | chr9:118 991 976–119 281 150 | 289 175 | Gain | – |
| 56 | M | ITA | chr9:119 042 541–119 297 292 | 254 752 | Gain | ADHD, ID, LD, MD, Mic, SD |
| 57 | M | SG | chr9:119 115 694–119 500 725 | 385 032 | Gain | – |
| 58 | F | CVH | chr9:118 899 870–120 282 623 | 1 382 754 | Gain | Craniosynostosis, ID |
| 59 | F | BBG | chr1:169 964 282–175 595 424 | 5 631 143 | Loss | – |
| 60 | M | BBG | chr1:174 117 247–175 518 085 | 1 400 839 | Gain | Anx, ASD, LD, Mac, MD |
| 61 | M | BCH | chr1:174 591 306–175 817 067 | 1 225 762 | Loss | Agenesis of corpus callosum, DD, Mic, seizures |
aMolecular diagnostic testing site of origin of the case: ACH, Alberta Children's Hospital; BBG, Brain and Body Genetic Resource Exchange (BBGRE); BCH, Boston Children's Hospital; CVH, Credit Valley Hospital; DEC, DECIPHER database; HSC, The Hospital for Sick Children; ITA, Italian diagnostic labs; MC, Mayo Clinic; OUH, Odense University Hospital; SG, Signature Genomics; TCAG, The Centre for Applied Genomics.
bNDD trait abbreviations: ADHD, attention deficit hyperactivity disorder; Anx, anxiety; ASD, autism spectrum disorder; CNS, central nervous system; DCD, developmental coordination disorder; DD, developmental delay; ID, intellectual disability; LD, learning disability; Mac; macrocephaly; MD, motor delay; Mic; microcephaly; NDD; neurodevelopmental disorder; OCD, obsessive compulsive disorder; SD, speech delay.‘–’ indicates non-NDD cases (no NDD terms were present in their reasons for referral for genetic testing).
We were able to determine inheritance for 20 of the exonic ASTN2 deletions (Supplementary Material, Table S1). The deletions were inherited from the mother in 10 (50%) and from the father in 8 (40%) individuals. In two individuals (10%), the deletions arose de novo (patients 6 and 36). Although caution is required given the small numbers involved, this observed rate of de novo deletions in the cases (2/20) was significantly higher (one-sided binomial test P = 0.017) than the expected genome-wide background rate of 1% for de novo deletions in the general population. The latter rate was derived from findings in control individuals by previous work (38–40), which used microarrays of similar resolution to those in this study. There have also been recent reports of de novo ASTN2 deletions in individuals with ID (35,36). Inheritance testing was performed for 7 of the individuals with exonic duplications. These events were maternally inherited in five (71%) individuals and paternally inherited in the other two (29%). Both deletions at the ASTN1 locus were found to be de novo, and the duplication was seen to be paternally inherited.
Other rare CNV findings in the individuals with exonic variants at ASTN2 or ASTN1 were inspected and categorized (Supplementary Material, Table S1) based on American College of Medical Genetics guidelines for CNV interpretation (41). Of the 46 individuals with ASTN2 exonic deletions, only one had another CNV that could be classified as ‘Pathogenic’ or ‘Uncertain clinical significance: likely pathogenic (UCS-LP)’. Male patient 16 had a 2.5 Mb microdeletion at the 22q11.2 Velocardiofacial syndrome locus. None of the 12 individuals with ASTN2 duplications had other CNVs in the pathogenic or UCS-LP categories. One of the individuals with a de novo deletion overlapping ASTN1 (patient 61) had an additional de novo deletion on chromosome 1 in the UCS-LP category.
Exonic deletions affecting multiple isoforms of ASTN2 and/or TRIM32 are significantly enriched in NDD cases
The majority of the individuals with exonic ASTN2 CNVs (40/46 with deletions and 9/12 with duplications) belonged to the case subset of 64 114 with NDD phenotypes (Table 2). We observed significant enrichment of ASTN2 exonic deletions (P = 0.01; OR = 2.691; 95% CI = 1.134–7.767) in the NDD cases compared with the 25 871 non-NDD cases in the clinical cohort but not for exonic duplications (P = 0.531; OR = 1.211; 95% CI = 0.302–6.954). Two of the three individuals with CNVs overlapping ASTN1 presented with NDD phenotypes (Table 2).
Upon inspection of microarray data from 44 085 control individuals (Supplementary Material, Table S2 and Materials and Methods), we discovered 18 exonic deletions and five duplications at the ASTN2 locus, one deletion exonic solely to TRIM32 and one deletion affecting ASTN1 (Figs 1 and 2, Supplementary Material, Fig. S2 and Table S3). The frequency of exonic ASTN2 duplications in controls did not differ significantly from that of either the NDD or non-NDD case cohorts (Table 3). The relative frequency of exonic ASTN2 deletions in NDD cases compared with controls was above the threshold for significance (P = 0.084). In a secondary analysis, we observed a strong enrichment of deletions around the 3′ end of ASTN2 (Fig. 1) that disrupted multiple transcript isoforms of ASTN2 in NDD cases versus controls (P = 0.002; OR = 3.714; 95% CI = 1.41–12.356) but not in non-NDD cases versus controls (Table 3). To test the robustness of this secondary analysis, we performed a permutation-based multiple test correction. After permuting 70 000 times the case–control labels of the 64 114 NDD cases and 44 085 controls, we found only 133 of 70 000 permutations with an FET P-value of ≤0.002, corresponding to a type I error estimate of 0.0019 that is almost identical to the real test P-value. Considering the expanded set of tests for losses and gains overlapping the multiple isoform or long isoform region or any of the two (2 × 3 = 6 tests), we found 457 of 70 000 permutations with at least one test with FET P-value ≤ 0.002, corresponding to a multiple test permutation-corrected association P-value of 0.007. This indicates that the significant enrichment we observe is robust to the type of multiple tests that were performed. Deletions near the 5′ end of ASTN2 that affected only the long ASTN2 transcript isoform (NM_014010) were not enriched in NDD or non-NDD cases versus controls (Table 3). Deletions affecting TRIM32 were significantly enriched in NDD cases (P = 0.019; OR = 2.636; 95% CI = 1.043–7.916) but not in non-NDD cases compared with controls (Table 3).
Table 3.
Results of CNV enrichment analysis of ASTN2/TRIM32 locus
| CNV type | Total NDD dataset |
Male NDD |
Female NDD |
Total non-NDD dataset | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NDD cases (N = 64 114) | Controls (N = 44 085) | P-valuea | NDD males (N = 40 438) | Male controls (N = 14 953)b | P-valuea | NDD females (N = 23 676) | Female controls (N = 18 218)b | P-valuea | Non-NDD cases (N = 25 871) | Controls (N = 44 085) | P-valuea | |
| Exonic ASTN2 CNVs | 49 | 23 | 0.079 | 42 | 13 | 0.349 | 7 | 7 | 0.778 | 8 | 23 | 0.933 |
| Exonic ASTN2 gains | 9 | 5 | 0.463 | 8 | 3 | 0.657 | 1 | 1 | 0.811 | 3 | 5 | 0.619 |
| Exonic ASTN2 losses | 40 | 18 | 0.084 | 34 | 10 | 0.328 | 6 | 6 | 0.773 | 6 | 18 | 0.927 |
| Exonic losses affecting only long ASTN2 isoform | 13 | 13 | 0.876 | 11 | 9 | 0.976 | 2 | 3 | 0.883 | 4 | 13 | 0.924 |
| Exonic losses affecting multiple ASTN2 isoforms | 27 | 5 | 0.002 | 23 | 1 | 0.005 | 4 | 3 | 0.64 | 2 | 5 | 0.798 |
| Exonic losses affecting TRIM32c | 23 | 6 | 0.019 | 22 | 2 | 0.024 | 1 | 3 | 0.964 | 4 | 6 | 0.54 |
NDD, neurodevelopmental disorders.
aP-values are from one-sided Fisher's exact test. Values in bold are significant at threshold of P < 0.05.
bGender information was available for 33 171 control individuals (Supplementary Material, Table S2), and this subset was used for the sex-specific enrichment analysis.
cAll deletions which affected TRIM32 in the cases also overlapped exon(s) of ASTN2. One of the exonic TRIM32 deletions in the controls (CF14) did not overlap an ASTN2 exon.
The functional impact of two independent deletions affecting multiple ASTN2 isoforms on ASTN2 expression was tested in lymphoblast cell lines from six individuals with such deletions including patients 14 and 22 (Supplementary Material, Fig. S3A). Expression was significantly lower in ASTN2 deletion carriers (Supplementary Material, Fig. S3B) compared with expression levels in nine individuals with two copies of ASTN2.
Exonic deletions affecting multiple isoforms of ASTN2 and/or TRIM32 are significantly enriched in male NDD but not in female NDD individuals
We observed a difference in sex-specific frequencies for ASTN2 exonic deletions among the NDD cases, with an excess of such events in male cases compared with female cases (two-tailed P = 0.003; OR = 3.32; 95% CI = 1.377–9.672), but not for duplications (P = 0.168; OR = 4.684; 95% CI = 0.628–207.702). We did not see a sex-specific difference in deletion frequencies among the controls. The sex-specific difference was also observed for the deletions at the 3′ end of ASTN2 (Fig. 1) that disrupted multiple transcript isoforms of the gene. Such deletions were enriched in the male NDD cases compared with male controls (P = 0.005; OR = 8.509; 95% CI = 1.381–350.06). On the contrary, the frequency of these events in female NDD cases did not differ relative to female controls (Table 3). We tested the robustness of the enrichment in male cases using a permutation-based multiple test correction. After permuting 70 000 times the case–control labels of the individuals with sex information, we found only 668 of 70 000 permutations with an FET P-value of ≤0.005. This corresponds to a multiple-test corrected P-value (type I error estimate) of 0.009 and provides evidence that such deletions are indeed NDD risk factors in male individuals.
We specifically tested the significance of the effect of sex with respect to the NDD case versus control association results. The logistic regression model interaction analysis (interaction coefficient P-value: 0.094) and the Tarone's test for odds ratio heterogeneity (P-value: 0.061) supported a trend for the interaction of sex with NDD phenotype in individuals with deletions affecting multiple ASTN2 isoforms. These results were observed to be robust upon reassessment after randomly re-assigning sex to subjects within each of the case and control cohorts: logistic regression (768/70 000 permutations ≤ 0.094; multiple test corrected P-value = 0.011) and Tarone's test (4877/70 000 permutations ≤ 0.061, multiple test corrected P-value = 0.069). We additionally confirmed these trends by re-testing the Fisher's exact test for enrichment of deletions affecting multiple isoforms in males (4045/70 000 permutations ≤ 0.005; multiple test corrected P-value = 0.058), showing that the sex label permutation (in cases and controls separately) decreases the significance of the association. Overall, these results suggest sex as a key modifier of the association of 3′-terminal ASTN2 deletions to NDD risk, but an expanded cohort is required to conclusively prove this trend.
Rare SNV findings at ASTN2/TRIM32 and ASTN1
Screening of additional Canadian ASD individuals by whole-exome sequencing (WES) and whole-genome sequencing (WGS) identified several rare (present at <1% frequency in the 1000 genomes data) missense coding variants (Supplementary Material, Tables S4–S6) at the three genes of interest: ASTN1 (seven SNVs in eight individuals), ASTN2 (five SNVs in five individuals) and TRIM32 (one SNV in one individual). No nonsense or frame-shift mutations were detected in the three genes. Seventy-seven percent of the 13 SNVs were predicted to be damaging by at least one of either the SIFT or PolyPhen software programs. However, these variants were all found to be inherited from parents with no reported ASD phenotypes (Supplementary Material, Table S4–S6). There were no de novo or loss-of-function mutations reported at ASTN1, ASTN2 or TRIM32 in published exome sequencing data from ASD individuals (42–45).
In addition, we accessed the NIH National Heart, Lung and Blood Institute (NHLBI)-ESP database to evaluate the presence of loss-of-function mutations in TRIM32 and ASTN2. TRIM32 has one stop-gain (rs199664043) supported only by 1 of 13 005 alleles, which affects only 41 of 653 amino acids. ASTN2 has only one putative splice variant, also supported only by 1 of 13 005 alleles, potentially affecting the last two coding exons of isoform NM_198188.2, but not the other isoforms. We can conclude that loss-of-function SNVs disrupting the gene products of TRIM32 and ASTN2 are extremely rare and almost never observed.
Clinical phenotypes observed in individuals with rare CNVs of interest
The reasons for referral for genetic testing, together with more detailed clinical phenotypes when available, were obtained (Supplementary Material, Table S1) for the 61 individuals with exonic CNVs at the ASTN2/TRIM32 and ASTN1 loci (Table 2). Given the enrichment of exonic ASTN2 deletions in cases relative to controls, we examined the clinical features of individuals with such CNVs for phenotypic trends. The major commonality among these individuals was some form of an NDD phenotype, which was observed in 87% of ASTN2 deletion cases (n = 41). The most common NDD diagnoses were speech–language delay (n = 18), ASD (n = 12), ADHD (n = 9), generalized developmental delay (DD) (n = 9), anxiety (n = 9), obsessive compulsive disorder (OCD) (n = 6) and learning disability (n = 8) (Table 2). Gross motor delay was present in 12 cases and fine motor delay in 6 cases. In addition, a wide range of dysmorphic features were present in ASTN2 deletion carriers, although macrocephaly was the only feature found to be common in more than 10% of the cases (n = 7). Phenotypic information was available for 12 (8 mothers and 4 fathers) of the 17 parents with exonic ASTN2 deletions. Seventy-five percent of the paternal carriers and 50% of the maternal carriers reported some form of neurodevelopmental trait (primarily anxiety, depression, learning disabilities, dyslexia and in some cases formal adult diagnoses of ASD or ADHD), though usually milder than those seen in the probands.
Alternative transcription start site of ASTN2 shorter isoforms is of recent evolutionary origin
Given the location-dependent penetrance observed for the ASTN2 exonic deletions, we assessed the average nucleotide conservation of all exons in ASTN2 (Fig. 3A) and TRIM32 across vertebrates, placental mammals and primates (Fig. 3B). Exons with coding sequence were well-conserved across all vertebrate species. Interestingly, the conservation was much lower between humans and other vertebrates for the exons unique to the shorter isoforms (exons 1B and 5C in Fig. 3A). Even though this pattern was partially driven by the UTR regions of these unique exons, their conservation was also lower than the conservation in the UTRs present in the long isoform or in TRIM32 (Supplementary Material, Fig. S4). Additionally, we did not find any evidence from EST databases for the presence of shorter 3′-terminal Astn2 isoforms in mouse. These observations suggest that the unique exons of the shorter isoforms (and the alternative transcription site in exon 1B) are of more recent evolutionary origin and are potentially derived in or just prior to the primate lineage. In support of this claim, both exons 1B and 5C contain transposable elements. Recent research has revealed the ability of transposable elements to contribute novel regulatory elements and thus give rise to new transcript isoforms (46).
Figure 3.

Relative expression of ASTN2 transcript isoforms and exon conservation. (A) Schematic presentation of the ASTN2 transcript isoforms. Red arrows denote location of the primers used for the RT–PCR and/or qRT–PCR assays (Supplementary Material, Table S10). The ‘*’ symbol denotes exons with variable length in different isoforms. (B) Conservation profile of ASTN2 and TRIM32 exons across vertebrates, placental mammals and primates. (C) The expression of ASTN2 transcript isoforms (long, shorter and all) in eight different human brain regions. ACTB was used as a control gene. (D) Quantification of ASTN2 isoform expression levels in triplicate from adult brain and fetal brain RNA samples by qRT–PCR (standard curve method). The expression was normalized using ACTB as a housekeeping gene, and the expression ratio is relative to the expression from all ASTN2 isoforms (mean ratio ± standard deviation). The results were replicated using GAPDH as a housekeeping gene.
Although there was no evident difference in the average nucleotide conservation between the coding exons of ASTN2, the amino acid alignment of eight ASTN2 protein orthologs revealed that the C-terminal end exhibits a greater degree of conservation compared with the rest of the protein (Supplementary Material, Fig. S5 and S6). This section of the protein corresponds to the 3′-terminal region of the gene exhibiting the strongest enrichment of exonic deletions in male NDD cases (Fig. 1) and encodes the fibronectin type III domain (Supplementary Material, Fig. S6).
Expression of ASTN2 transcript isoforms in the human brain
The expression profile of ASTN2 and its different isoforms in the human brain has not been described previously. Therefore, we performed reverse transcriptase–PCR (RT–PCR) analysis of the ASTN2 isoforms in human brain samples using primers with locations as depicted in Figure 3A. We detected abundant expression for primers amplifying exons present in all isoforms (Fig. 3C, ASTN2 all) and for exons specific to the long ASTN2 isoform (NM_014010) (Fig. 3C, ASTN2 long isoform) in all the brain regions. The shorter transcripts (NM_198187, NM_198188, NM_001184734 and NM_001184735) were expressed at a lower level (Fig. 3C, ASTN2 shorter isoforms). We did not detect any expression when using primers specific for NM_198186. In addition, we measured the mRNA expression ratios of the isoforms in whole brain and fetal brain and observed a change in the ratios between the developmental stages. In the fetal brain, the shorter isoforms accounted for ∼40% of the total ASTN2 expression, which decreased to 20% in the adult brain (Fig. 3D). This suggests a functional role for the shorter isoforms during early development.
Spatiotemporal expression patterns of ASTN1, ASTN2 and TRIM32 in human brain
We analyzed the pattern of ASTN2, ASTN1 and TRIM32 gene level expression in different brain regions during human development by utilizing the comprehensive gene expression data from the BrainSpan database (47). Overall, ASTN2 is expressed at a moderate level throughout development and exhibits an increase in the late prenatal period and during postnatal development in many of the brain regions (Fig. 4A). The highest level of ASTN2 expression was observed in the cerebellar cortex (CBC), where the expression increase during infancy and early childhood is in concordance with the expression pattern previously reported in mice (30). Genes exhibiting similar expression patterns in the CBC (as quantified by Pearson correlation, Supplementary Material, Table S7) were more enriched in gene ontology (GO) terms such as synaptic transmission and plasticity (Supplementary Material, Fig. S7A and Table S7). Interestingly, during the prenatal period, ASTN2 has a dynamic spike in the expression at around 12–13 gestational weeks in the neocortical regions (frontal cortex, parietal cortex and occipital cortex) (Fig. 4A). This expression pattern is enriched in neuronal development GO terms such as ‘axonogenesis’ and ‘neuron differentiation’ (Supplementary Material, Fig. S7B and Table S7). The dynamic expression pattern of ASTN2 together with the biological annotation of genes with similar expression patterns suggests that ASTN2 could have an important role in both prenatal and postnatal brain development.
Figure 4.

Expression profiles of ASTN2 and ASTN1 across human brain development. The gene level expression profiles of (A) ASTN2 and (B) ASTN1 across developmental time points in nine regions of the human brain; amygdala (AMY), cerebellar cortex (CBC), diencephalon (DIE), frontal cortex (FC), hippocampus (HIP), occipital cortex (OC), parietal cortex (PC), temporal cortex (TC) and ventral forebrain (VF).
In contrast to ASTN2, ASTN1 has a high and steady level of expression across different brain regions suggesting a constant and fundamental role in human brain development (Fig. 4B). The expression pattern of TRIM32 differs from the astrotactins and is marked by high expression during early prenatal development, a decrease after birth followed by an increase during adolescence in all the brain regions studied (Supplementary Material, Fig. S8). Similar to ASTN2, TRIM32 has highest levels of expression in the CBC.
DISCUSSION
By leveraging information from multiple diagnostic laboratories to aid in the clinical interpretation of rare variants, we detected specific enrichment of exonic ASTN2/TRIM32 deletions in NDD cases compared with either non-NDD cases or with controls. Follow-up clinical characterization revealed a spectrum of NDD diagnoses, with ASD, ADHD, OCD and speech and language delay being the most common. A similarly diverse range of phenotypic outcomes from deletions at a single locus have been reported before for NRXN1 (48–50), GPHN (51), MBD5 (52,53), and other regions (19,54), which could reflect some shared genetic risk for different NDDs, consistent with overlap of clinical phenotypes often observed across diagnostic boundaries in psychiatry (55–57). In addition to the heterogeneity of their clinical presentation, ASTN2/TRIM32 deletions exhibit reduced penetrance, as demonstrated by their predominantly inherited nature and their presence in a small number of control individuals. SNPs within ASTN2 have been highlighted by recent genome-wide association studies (GWAS) in risk for ADHD (58), schizophrenia (59,60), bipolar disorder (60), migraine without aura (61), cognitive decline and reduced hippocampal volume (62). These reports of common risk factors at this gene complement our rare variant findings and lend support to the etiological contributions of inherited ASTN2 variants to a range of neurodevelopmental conditions. While several of the NDD risk gene discovery efforts to date have focused on de novo events, the role of inherited rare variants has been less thoroughly explored. It is likely that a combination of both de novo and inherited risk factors contribute to the genetic architecture of different NDDs. The presence of deletions in controls might also reflect the absence of stringent psychiatric screening of the control individuals or could arise from false-positive calls, since experimental validation was not possible due to unavailability of DNA from control individuals. Importantly, we note that the relatively lower resolution of the clinical microarray platforms compared with those used for the controls (Supplementary Material, Fig. S9) could imply an underestimation of smaller CNVs at ASTN2, ASTN1 and TRIM32 in the patient cohorts.
The striking enrichment in the NDD cases compared with the controls of exonic deletions at the 3′ end of ASTN2 (corresponding to multiple ASTN2 isoforms) defines a critical region for pathogenicity that has several functional implications. As suggested by our lymphoblast expression analysis, such deletions could disrupt the expression of all transcript isoforms of ASTN2 and potentially lead to complete haploinsufficiency of the protein. This may have more severe phenotypic consequences than those deletions affecting only the long transcript isoform. Similar trends of greater penetrance of deletions impacting multiple isoforms of a gene, relative to those overlapping a single isoform, have recently been observed at other loci such as NRXN1 (49,50,63), NRXN3 (5) and AUTS2 (64) in connection with risk for NDDs. Our findings also highlight the importance of the C-terminal end of the protein, which encodes the fibronectin III domain and was observed to be the most conserved part of the protein in our cross-species analysis. Interestingly, this domain is also found in other genes implicated in NDDs such as the contactins (13). In addition, our data suggest that the shorter transcript isoforms are of functional importance especially during early brain development. This claim is supported by our mRNA quantification assays in which the shorter isoforms account for 40% of the total expression of the gene in fetal brain. Our evolutionary conservation analyses suggest that the first exon shared by the shorter ASTN2 isoforms is of more recent evolutionary origin and could have a function specific to primates. There is no indication from previous functional work (30) or from mouse EST databases for existence of shorter 3′-terminal Astn2 transcript isoforms in the mouse. Taken together, the recent emergence of the shorter isoforms comprising a functionally important domain and their widespread expression in the human brain could indicate primate-specific involvement in neurodevelopment.
Several of the deletions at the 3′ end of ASTN2 also encompass TRIM32, a small two-exon gene nested within an intron of the long isoform of ASTN2, which is transcribed from the opposite strand to ASTN2. The first exons of TRIM32 and the ASTN2 shorter isoforms are only 39 bp apart. TRIM32 encodes an ubiquitin ligase and is expressed at high levels in a wide range of tissues, including the brain, muscle and skeletal tissue (65). Homozygous point mutations in this gene have been implicated in autosomal recessive disorders such as limb-girdle muscular dystrophy (66,67) and Bardet–Biedl syndrome (68). The latter is a heterogeneous multi-system disorder presenting with retinopathy, obesity and cognitive impairment, among other symptoms. TRIM32 expression levels in the mouse neocortex have been shown to determine the post-differentiation fates of neuronal stem cells (69) and the TRIM32 protein is a key regulator of neural differentiation (70). It is unlikely that disruption of TRIM32 alone, and not ASTN2, could be primarily responsible for the neurodevelopmental phenotypes observed in our study given the absence of such diagnoses in individuals with heterozygous TRIM32 missense mutations (66,68) and in a limb-girdle muscular dystrophy patient who possessed both a TRIM32 nonsense mutation and a deletion of the entire gene (67). The expression of ASTN2 is also notably higher than that of TRIM32 in the brain relative to other body tissues (33). Furthermore, we did not detect any deletions in the large clinical dataset that overlapped TRIM32 alone but did observe several deletions at both the 3′ and 5′ ends of ASTN2 that do not affect TRIM32. Given the very close genomic proximity of TRIM32 and ASTN2, and the finding that deletions impacting both genes exhibit stronger enrichment in cases versus controls than those affecting either gene alone, the most conservative interpretation of our data would suggest that the joint disruption of both genes potentially contributes to risk for a spectrum of NDD phenotypes.
We observed evidence for a sex-specific effect for the phenotypic expression of exonic deletions affecting ASTN2 or both ASTN2 and TRIM32 (Table 3). These mutations were significantly in excess in the males compared with the females within the NDD case cohort, whereas controls did not exhibit this difference. Deletions affecting the 3′ end of ASTN2 and/or TRIM32 were also significantly enriched in male NDD cases compared with male controls, whereas the difference in frequencies of such events between female cases and female controls was non-significant. These findings suggest greater penetrance in males relative to females of the NDD phenotypes linked to ASTN2/TRIM32 deletions, although a larger cohort is required to conclusively prove the significance of this sex-specific effect and the role of such deletions in females. Similar sex-specific effects have been recently reported for CNVs at other autosomal loci including SHANK1 deletions in ASD risk (7) and 16p13.11 deletions and duplications in NDD risk (71). The discovery of additional autosomal loci with male-biased penetrance (72), together with risk genes on the X chromosome (6,73), could help explain the molecular basis of the skewed sex ratios observed in the prevalence of NDDs such as ADHD, ASD and ID. Although the biological basis of this sex-modulated penetrance is unknown, theories involving sex hormones have been proposed (74). Interestingly, there is some evidence that ASTN2 could be regulated by estrogen (75).
The extreme rarity of exonic deletions affecting the ASTN1 gene is intriguing, especially in comparison with the relatively higher rate of such mutations at ASTN2. It is also striking that all the ASTN1 deletions in the cases were of de novo origin and overlapped multiple genes, in contrast to exonic ASTN2 deletions, which were predominantly of an inherited nature (only 10% of such deletions in families with parental DNA available for testing were de novo) and localized to ASTN2/TRIM32. These observations suggest stronger selective pressure against disruption of ASTN1, relative to ASTN2, and are consistent with results from functional characterization of the two proteins in the mouse brain. Although mouse ASTN1 and ASTN2 are both integral neuronal membrane proteins with similar domain structures, they have been shown to play non-redundant and complementary roles during neuronal migration (30). The C-terminus of mouse ASTN1 is exposed on the neuronal surface, enabling it to act as the inter-cellular linker molecule directly binding neurons to glia during migration. On the other hand, the C-terminus of ASTN2 was not detected on the neuronal surface. Rather than directly participating in neuron-glial adhesion, ASTN2 reportedly functions as a regulatory molecule by forming a complex with ASTN1 in the neuronal membrane, thus controlling ASTN1 surface expression levels and indirectly modulating the rate of neuronal migration. Presumably, the disruption of the key ligand enabling cerebellar neuron-glial binding would be more deleterious for neuronal migration relative to that of an indirect regulator of the process.
The differing functions of the two proteins are also reflected in the spatiotemporal mRNA expression profiles of ASTN1 and ASTN2. Both astrotactin genes are expressed in the developing human brain with highest overall expression during late prenatal development and early childhood. In contrast to ASTN1, which has a very static and high expression pattern during development across different regions, ASTN2 exhibits a wider range of expression levels suggesting region specific roles across different phases of human brain development. These findings, taken together with previous work (30), are in line with a more fundamental, constant role for ASTN1 protein, regulated at specific time points by varying ASTN2 protein levels.
Comparison of the human brain expression profile of ASTN2 with previous work in mice revealed both intriguing differences and similarities. The striking prenatal spike in ASTN2 expression in the neocortical regions, towards the end of the first trimester, has not been reported in the embryonic mouse brain (30). This finding might indicate transcriptional regulation patterns or additional functions of the protein and/or transcript isoforms specific to primates. Interestingly, this period in the developmental timeline of the human cerebral cortex is marked by extensive neuronal migration, increasing axonal outgrowth and the formation of the early synapses (47,76). Several genes previously implicated in risk for ASD (and also other NDDs) such as DOCK4 and NRCAM also exhibit increasing cortical expression around this time point (15,77). In concordance with the experimental work from mouse (30), we observe the highest ASTN2 expression in the cerebellar cortex shortly after birth. Several studies investigating the neuropathology of ASD (78,79) and ADHD (80–82) have consistently highlighted the cerebellum as a major region of interest. Reported neuroanatomical abnormalities include disrupted neuronal migration in the cerebellar cortex (79) as well as reduction in volume of the cerebellar vermis (83), which has been linked to repetitive and stereotyped behavior in ASD (84). Furthermore, the number of Purkinje cells, one of the main cell types in the cerebellum, has been found to be decreased by up to 50% in individuals with ASD (79). Interestingly, the Astn1 KO mouse has reduced cerebellar volume, slower neuronal migration rates and abnormal development of the Purkinje cells compared with wild type (29). Astn2 has also been shown to be very highly expressed in the cerebellum in general, and in the Purkinje cells in particular, during both embryonic and postnatal development (30).
While the role of the astrotactins in neuronal migration is well established, their potential involvement in other brain developmental processes remains to be elucidated. Our gene ontology analyses revealed that many of the genes with expression patterns similar to ASTN2 are involved in synapse-related biological processes. The presence of both astrotactins at high levels in the postnatal and adult brain, well after the completion of most neuronal migration, also suggests that ASTN2 and ASTN1 might potentially act together as a receptor system for synapse guidance in addition to their role in neuronal migration. Interestingly, recent studies show that genes important for embryonic neuronal migration such as PAFAH1B1 (LIS1) and RELN also participate in guiding and maintaining the synapses in postnatal brain development (23,85). CNTNAP2, another ASD risk gene, encodes a neuronal trans-membrane protein with important roles in diverse processes including neuron-glia interactions during migration, maintenance of the connectivity and synchronization of neuronal circuits and the clustering of K+ channels in myelinated axons (86).
Our data emphasize the need to characterize rare CNVs (and other genetic variants) in the context of large case and control cohorts, in order to extract meaningful genotype and phenotype data necessary for proper clinical genetic interpretation. For the ASTN2/TRIM32 locus, we show that a neurodevelopmental phenotype ensues preferentially in male patients when deletions of ASTN2 impact all its transcript isoforms. Functional dissection of the influences gender has on the ASTN2 isoforms during brain development may also inform on new treatment strategies in psychopathology.
MATERIALS AND METHODS
Clinical case cohorts
The clinical case cohorts utilized for this study are summarized in Table 1. These comprise a total of 89 985 postnatal patient samples submitted for clinical microarray testing to 10 different molecular diagnostic centers in Canada, Denmark, Italy, the UK and the USA. The reasons for referral for clinical microarray testing were systematically assessed at each of the study sites and the number of NDD cases was tabulated based on the presence of one or more of the following phenotypes: ADHD, ASD, behavioral disorders, cognitive impairment, developmental delay, ID, learning disability, macrocephaly, microcephaly, neurological disorders, OCD, psychoses, seizures and speech/language disorders. The remaining patients in the clinical cohorts, whose reasons for referral for genetic testing did not contain any of the NDD terms listed above, were counted as non-NDD cases. The gender composition of each clinical dataset was also tabulated in order to test for sex-biased effects. Individuals with CNVs that spanned one or more exons of the ASTN2/TRIM32 (Fig. 1) or ASTN1 (Fig. 2) genetic loci at 9q33.1 and 1q25.2, respectively, were included in the analysis and are listed in Table 2. To avoid bias in the CNV burden analysis by inclusion of large-scale multigenic chromosomal abnormalities, CNVs >6 Mb in the clinical dataset were excluded. Independent validation was performed for 71% (44/62) of the CNVs presented in Table 2 for which DNA was available, using one of the following methods: quantitative PCR (qPCR), multiplex ligation-dependent probe amplification (MLPA), fluorescence in situ hybridization (FISH) or by a second microarray platform (Supplementary Material, Table S1). All attempted assays revealed true positive CNVs. To obtain standardized clinical information from the individuals with the CNVs of interest, a phenotype checklist was sent to referring physicians for completion, based on physical and/or psychological examination, as well as review of the subject's medical history. The frequencies of specific features were tabulated (Supplementary Material, Table S1). In addition to the clinical cohorts described above, we inspected the DECIPHER database (https://decipher.sanger.ac.uk) for individuals with exonic deletions and duplications, smaller than 6 Mb, that overlapped ASTN2 (Supplementary Material, Table S8). We were able to obtain additional phenotype details from one individual (patient 18), who possessed an exonic ASTN2/TRIM32 deletion. Clinical information from this individual was included in the phenotype summary but this case was not counted in the CNV counts and enrichment analysis described in the Results since it was not part of data from the 10 molecular diagnostic sites. This study was approved by the Research Ethics Board at the Hospital for Sick Children, Toronto.
Control cohorts
For the purpose of statistical testing of findings in the case cohorts, we compiled exonic CNV findings at the ASTN2/TRIM32 and ASTN1 regions in high resolution microarray data from 44 085 individuals from population-based control cohorts and studies of individuals with disorders unrelated to NDDs such as diabetes (Supplementary Material, Table S2). These included individuals from different control datasets analyzed by us (14,31,51,87–90), published genome-wide CNV data (91,92) and published data from controls that were inspected for CNVs affecting ASTN2 (33,34). Gender information was available for 33 171 control individuals (Supplementary Material, Table S2), and this subset was used for the sex-specific enrichment analysis. Fisher's one-sided exact test was used to test for enrichment of CNVs in cases versus non-NDD cases and versus controls with a significance threshold of P < 0.05. A challenge in combining data across multiple case and control cohorts is the heterogeneity of microarray platforms and the resulting differences in probe coverage. We compiled probe numbers from the different microarray platforms featured among the clinical and control datasets in the ASTN2/TRIM32 and ASTN1 regions (Supplementary Material, Table S9). The array platforms used for the control dataset had much higher probe densities on average, both genome wide and in the specific regions of interest, than those constituting the clinical dataset (Supplementary Material, Table S9 and Fig. S9). The higher resolution of the control CNV dataset relative to the cases decreases the likelihood of spurious enrichment findings driven by false negatives in the controls and provides a conservative estimate of the significance and effect size of our findings.
Mutation screening of ASTN1, ASTN2 and TRIM32 from ASD exome sequencing data
Exons and splice sites of ASTN1, ASTN2 and TRIM32 were inspected for single-nucleotide changes and small indels in next-generation WES and WGS data from Canadian individuals (of European ancestry) with ASD (Supplementary Material, Tables S4–S6). ASTN1 was screened in 338 individuals, while ASTN2 and TRIM32 were inspected in 182 individuals. The subjects screened for mutations in ASTN2 and TRIM32 by WES and WGS were distinct from those in whom the two genes were inspected using Sanger sequencing in our previous study (31). Data generation and SNV analysis were performed as previously described for the WES (51) and WGS (93) data. Rare SNVs detected in the WES and WGS data that were present at <1% frequency in the 1000 genomes data (94) were confirmed by bidirectional Sanger sequencing. Inheritance testing of such variants was also conducted when parental DNA was available. We also examined the published results of four recent ASD exome sequencing studies (42–45) for variants at ASTN1, ASTN2 and TRIM32. In addition, we accessed the NHLBI exome sequencing database to evaluate the presence of loss-of-function mutations in TRIM32 and ASTN2.
ASTN2 expression analysis using RT–PCR and quantitative reverse transcriptase–PCR
The expression of ASTN2 mRNA was analyzed by RT–PCR from a panel of eight different human brain RNA samples (adult whole brain, adult cerebellum, adult caudate nucleus, adult amygdala, adult hippocampus, adult corpus callosum, adult thalamus and fetal whole brain purchased from BD Biosciences, Clontech and AMSBIO). In addition, expression was measured in lymphoblast cell lines from six individuals with ASTN2 deletions and nine individuals with two copies of ASTN2 (detailed description in Supplementary Material, Fig. S3). For all the RNA samples, cDNA was synthesized using Superscript III First strand Synthesis Supermix (Invitrogen, Carlsbad, CA, USA) with 1 µg of poly (A+) or total DNase I treated RNA as a template. RT–PCR was performed under standard PCR conditions using 10 ng of cDNA as template. Eight primer pairs were used to amplify different transcripts of ASTN2 (Supplementary Material, Table S10). In total, ASTN2 has six different isoforms which include the long isoform (NM_014010) and shorter isoforms (NM_198186, NM_198187, NM_198188, NM_001184734 and NM_001184735). To quantify the expression ratio between the transcript isoforms, we used five suitable amplicons for quantitative reverse transcriptase–PCR (qRT–PCR) (Supplementary Material, Table S10). The qRT–PCR assay was performed using Brilliant III SYBR® Green PCR Master Mix (Agilent, Santa Clara, CA, USA) in a total reaction volume of 15 μl, containing 5 ng of cDNA templates. The reactions were amplified using the Mx3005P qPCR system (Agilent, Santa Clara, CA, USA). The expression ratios were calculated after determining the exact quantities of each isoform using the standard curve method, normalized against ACTB or GAPDH expression levels.
Nucleotide and amino acid conservation analysis of ASTN2
The nucleotide conservation scores for each base present in all ASTN2 and TRIM32 exons were computed using the PhyloP program based on alignment between 46 vertebrate species including 23 placental mammals and eight primates. The average score was calculated for each exon and for the unique UTRs. The amino acid sequences of 1:1 ASTN2 orthologs from eight species were downloaded from Ensembl, and the multiple sequence alignment was carried out using ClustalW2. After the alignment, the amino acid conservation was quantified using Scorecons server (95). To check for the presence of the shorter ASTN2 isoforms in mouse, we screened different databases including AceView, Ensembl and FANTOM.
Spatiotemporal expression analysis of ASTN2, ASTN1 and TRIM32 in the human brain
To analyze the expression pattern of ASTN2, ASTN1 and TRIM32 during human brain development, we utilized expression data from the BrainSpan database (www.brainspan.org) (47). This dataset contains extensive transcriptome profiles for 16 brain regions from 41 individuals. The age range of the subjects spans from 8 postconception weeks to 40 years (56–13 720 postconception days). Full sample information is available on the BrainSpan website. The data were quantile normalized and the gene level expression was averaged across donors for each time point in the following regions: the frontal cortex (FC), parietal cortex (PC), temporal cortex (TC), occipital cortex (OC), hippocampus (HIP), amygdala (AMY), ventral forebrain (VF), diencephalon (DIE) and cerebellar cortex (CBC). The expression levels of ASTN1, ASTN2 and TRIM32 in the different brain regions were plotted across age range using the R package ggplot2. Smooth curve lines were computed by loess using a span of 0.4. The BrainSpan dataset was also used to identify genes with expression patterns similar to that of ASTN2, and these genes were used for the gene set enrichment analysis (90,96) (Supplementary Material, Table S7).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by grants from the University of Toronto McLaughlin Centre, NeuroDevNet, Genome Canada and the Ontario Genomics Institute, the Canadian Institutes for Health Research (CIHR), National Institutes of Health (MH095867), the Canadian Institute for Advanced Research, the Canada Foundation for Innovation, the Government of Ontario, Autism Speaks and The Hospital for Sick Children Foundation. A.C.L. was supported by a NeuroDevNet doctoral fellowship. K.T. holds a postdoctoral fellowship from the Swedish Research Council. S.W.S. holds the GlaxoSmithKline-CIHR Chair in Genome Sciences at the University of Toronto and The Hospital for Sick Children. D.T., D.S.C. and T.M.W. are US military service members and this work was prepared as part of their official duties. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Army, Department of the Navy, Department of Defense, nor the US Government. Title 17, USC § 105 provides that ‘Copyright protection under this title is not available for any work of the U.S. Government.’ Title 17, USC § 101 defines a US Government work as a work prepared by a military service member or employee of the US Government as part of that person's official duties. Control datasets were obtained, along with permission for use, from the database of Genotypes and Phenotypes (dbGaP) found at http://www.ncbi.nlm.nih.gov/gap through accession numbers phs000143.v1.p1 (Starr County Health Studies’ Genetics of Diabetes Study), phs000091.v2.p1 (GENEVA NHS/HPFS Diabetes study), phs000169.v1.p1 (Whole Genome Association Study of Visceral Adiposity in the HABC Study), phs000303.v1.p1 (Genetic Epidemiology of Refractive Error in the KORA Study), phs000404.v1.p1 (COGEND; The Genetic Architecture of Smoking and Smoking Cessation) and phs000086.v2.p1 (DCCT-EDIC Clinical Trial and Follow-up of Persons with Type 1 Diabetes). The Starr County Health Studies Genetics of Diabetes Study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and the NIDDK Central Repositories. Support for the GWAS of Gene and Environment Initiatives in Type 2 Diabetes was provided through the NIH Genes, Environment and Health Initiative [GEI] (U01HG004399). The human subjects participating in the GWAS derive from The Nurses' Health Study and Health Professionals' Follow-up Study and these studies are supported by National Institutes of Health (NIH) grants CA87969, CA55075 and DK58845. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by the Gene Environment Association Studies, GENEVA Coordinating Center (U01 HG004446) and the National Center for Biotechnology Information. Support for genotyping, which was performed at the Broad Institute of MIT and Harvard, was provided by the NIH GEI (U01HG004424). Support for the ‘CIDR Visceral Adiposity Study’ was provided through the Division of Aging Biology and the Division of Geriatrics and Clinical Gerontology, National Institute on Aging. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by Health ABC Study (HABC) Investigators. The KORA dataset was obtained from the NEI Refractive Error Collaboration (NEIREC) Database, support for which was provided by the National Eye Institute. Support for genotyping of the COGEND samples, which was performed at the Center for Inherited Disease Research (CIDR), was provided by 1 X01 HG005274-01. Assistance with genotype cleaning of the COGEND samples, as well as with general study coordination, was provided by the Gene Environment Association Studies (GENEVA) Coordinating Center (U01HG004446). Support for the collection of COGEND datasets and samples was provided by the Collaborative Genetic Study of Nicotine Dependence (COGEND; P01 CA089392) and the University of Wisconsin Transdisciplinary Tobacco Use Research Center (P50 DA019706, P50 CA084724). The DCCT-EDIC Research Group is sponsored through research contracts from the National Institute of Diabetes, Endocrinology and Metabolic Diseases of the NIDDK and the NIH. The contents of this article are solely the responsibility of the authors and do not necessarily represent the official views of the NIDDK or the NIH.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the patients and their families, clinicians and diagnostic lab personnel who participated in this study. We also thank Dr Maria Tropeano, the CHOP CNV team and members of Dr Evan Eichler's group for sharing gender information for the control datasets.
Conflict of Interest statement. J.A.R. is an employee of Signature Genomic Laboratories, a subsidiary of PerkinElmer, Inc. E.H. and P.S.E. are employees of Population Diagnostics, Inc. S.W.S. is on the Scientific Advisory Board of Population Diagnostics, Inc and is a founding scientist of YouNique Genomics, both of which could use data from this study.
REFERENCES
- 1.Scherer S.W., Dawson G. Risk factors for autism: translating genomic discoveries into diagnostics. Hum. Genet. 2011;130:123–148. doi: 10.1007/s00439-011-1037-2. [DOI] [PubMed] [Google Scholar]
- 2.Devlin B., Scherer S.W. Genetic architecture in autism spectrum disorder. Curr. Opin. Genet. Dev. 2012;22:229–237. doi: 10.1016/j.gde.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Szatmari P., Paterson A.D., Zwaigenbaum L., Roberts W., Brian J., Liu X.Q., Vincent J.B., Skaug J.L., Thompson A.P., Senman L., et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gauthier J., Siddiqui T., Huashan P., Yokomaku D., Hamdan F., Champagne N., Lapointe M., Spiegelman D., Noreau A., Lafrenière R., et al. Truncating mutations in NRXN2 and NRXN1 in autism spectrum disorders and schizophrenia. Hum. Genet. 2011;130:563–573. doi: 10.1007/s00439-011-0975-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaags A.K., Lionel A.C., Sato D., Goodenberger M., Stein Q.P., Curran S., Ogilvie C., Ahn J.W., Drmic I., Senman L., et al. Rare deletions at the neurexin 3 locus in autism spectrum disorder. Am. J. Hum. Genet. 2012;90:133–141. doi: 10.1016/j.ajhg.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jamain S., Quach H., Betancur C., Rastam M., Colineaux C., Gillberg I.C., Soderstrom H., Giros B., Leboyer M., Gillberg C., et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 2003;34:27–29. doi: 10.1038/ng1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sato D., Lionel A.C., Leblond C.S., Prasad A., Pinto D., Walker S., O'Connor I., Russell C., Drmic I.E., Hamdan F.F., et al. SHANK1 deletions in males with autism spectrum disorder. Am. J. Hum. Genet. 2012;90:879–887. doi: 10.1016/j.ajhg.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berkel S., Marshall C.R., Weiss B., Howe J., Roeth R., Moog U., Endris V., Roberts W., Szatmari P., Pinto D., et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat. Genet. 2010;42:489–491. doi: 10.1038/ng.589. [DOI] [PubMed] [Google Scholar]
- 9.Durand C.M., Betancur C., Boeckers T.M., Bockmann J., Chaste P., Fauchereau F., Nygren G., Rastam M., Gillberg I.C., Anckarsater H., et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moessner R., Marshall C.R., Sutcliffe J.S., Skaug J., Pinto D., Vincent J., Zwaigenbaum L., Fernandez B., Roberts W., Szatmari P., et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roohi J., Montagna C., Tegay D.H., Palmer L.E., DeVincent C., Pomeroy J.C., Christian S.L., Nowak N., Hatchwell E. Disruption of contactin 4 in three subjects with autism spectrum disorder. J. Med. Genet. 2009;46:176–182. doi: 10.1136/jmg.2008.057505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Daalen E., Kemner C., Verbeek N.E., van der Zwaag B., Dijkhuizen T., Rump P., Houben R., van 't Slot R., de Jonge M.V., Staal W.G., et al. Social responsiveness scale-aided analysis of the clinical impact of copy number variations in autism. Neurogenetics. 2011;12:315–323. doi: 10.1007/s10048-011-0297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burbach J.P., van der Zwaag B. Contact in the genetics of autism and schizophrenia. Trends Neurosci. 2009;32:69–72. doi: 10.1016/j.tins.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 14.Prasad A., Merico D., Thiruvahindrapuram B., Wei J., Lionel A.C., Sato D., Rickaby J., Lu C., Szatmari P., Roberts W., et al. A discovery resource of rare copy number variations in individuals with autism spectrum disorder. G3 (Bethesda) 2012;2:1665–1685. doi: 10.1534/g3.112.004689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pagnamenta A.T., Bacchelli E., de Jonge M.V., Mirza G., Scerri T.S., Minopoli F., Chiocchetti A., Ludwig K.U., Hoffmann P., Paracchini S., et al. Characterization of a family with rare deletions in CNTNAP5 and DOCK4 suggests novel risk loci for autism and dyslexia. Biol. Psychiatry. 2010;68:320–328. doi: 10.1016/j.biopsych.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bakkaloglu B., O'Roak B.J., Louvi A., Gupta A.R., Abelson J.F., Morgan T.M., Chawarska K., Klin A., Ercan-Sencicek A.G., Stillman A.A., et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 2008;82:165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramocki M.B., Zoghbi H.Y. Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature. 2008;455:912–918. doi: 10.1038/nature07457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Toro R., Konyukh M., Delorme R., Leblond C., Chaste P., Fauchereau F., Coleman M., Leboyer M., Gillberg C., Bourgeron T. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010;26:363–372. doi: 10.1016/j.tig.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 19.Guilmatre A., Dubourg C., Mosca A.L., Legallic S., Goldenberg A., Drouin-Garraud V., Layet V., Rosier A., Briault S., Bonnet-Brilhault F., et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch. Gen. Psychiatry. 2009;66:947–956. doi: 10.1001/archgenpsychiatry.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grant S.G. Synaptopathies: diseases of the synaptome. Curr. Opin. Neurobiol. 2012;22:522–529. doi: 10.1016/j.conb.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 21.Valiente M., Marin O. Neuronal migration mechanisms in development and disease. Curr. Opin. Neurobiol. 2010;20:68–78. doi: 10.1016/j.conb.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Ernst C., Marshall C.R., Shen Y., Metcalfe K., Rosenfeld J., Hodge J.C., Torres A., Blumenthal I., Chiang C., Pillalamarri V., et al. Highly penetrant alterations of a critical region including BDNF in human psychopathology and obesity. Arch. Gen. Psychiatry. 2012;69:1238–1246. doi: 10.1001/archgenpsychiatry.2012.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Folsom T.D., Fatemi S.H. The involvement of Reelin in neurodevelopmental disorders. Neuropharmacology. 2013;68:122–135. doi: 10.1016/j.neuropharm.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerjee A., Macdonald M.L., Borgmann-Winter K.E., Hahn C.G. Neuregulin 1-erbB4 pathway in schizophrenia: From genes to an interactome. Brain Res. Bull. 2010;83:132–139. doi: 10.1016/j.brainresbull.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fishell G., Hatten M.E. Astrotactin provides a receptor system for CNS neuronal migration. Development. 1991;113:755–765. doi: 10.1242/dev.113.3.755. [DOI] [PubMed] [Google Scholar]
- 26.Edmondson J.C., Liem R.K., Kuster J.E., Hatten M.E. Astrotactin: a novel neuronal cell surface antigen that mediates neuron-astroglial interactions in cerebellar microcultures. J. Cell Biol. 1988;106:505–517. doi: 10.1083/jcb.106.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stitt T.N., Hatten M.E. Antibodies that recognize astrotactin block granule neuron binding to astroglia. Neuron. 1990;5:639–649. doi: 10.1016/0896-6273(90)90218-5. [DOI] [PubMed] [Google Scholar]
- 28.Zheng C., Heintz N., Hatten M.E. CNS gene encoding astrotactin, which supports neuronal migration along glial fibers. Science. 1996;272:417–419. doi: 10.1126/science.272.5260.417. [DOI] [PubMed] [Google Scholar]
- 29.Adams N.C., Tomoda T., Cooper M., Dietz G., Hatten M.E. Mice that lack astrotactin have slowed neuronal migration. Development. 2002;129:965–972. doi: 10.1242/dev.129.4.965. [DOI] [PubMed] [Google Scholar]
- 30.Wilson P.M., Fryer R.H., Fang Y., Hatten M.E. Astn2, a novel member of the astrotactin gene family, regulates the trafficking of ASTN1 during glial-guided neuronal migration. J. Neurosci. 2010;30:8529–8540. doi: 10.1523/JNEUROSCI.0032-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lionel A.C., Crosbie J., Barbosa N., Goodale T., Thiruvahindrapuram B., Rickaby J., Gazzellone M., Carson A.R., Howe J.L., Wang Z., et al. Rare copy number variation discovery and cross-disorder comparisons identify risk genes for ADHD. Sci. Transl. Med. 2011;3 doi: 10.1126/scitranslmed.3002464. 95ra75. [DOI] [PubMed] [Google Scholar]
- 32.Glessner J.T., Wang K., Cai G., Korvatska O., Kim C.E., Wood S., Zhang H., Estes A., Brune C.W., Bradfield J.P., et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vrijenhoek T., Buizer-Voskamp J.E., van der Stelt I., Strengman E., Sabatti C., Geurts van Kessel A., Brunner H.G., Ophoff R.A., Veltman J.A. Recurrent CNVs disrupt three candidate genes in schizophrenia patients. Am. J. Hum. Genet. 2008;83:504–510. doi: 10.1016/j.ajhg.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernandez T.V., Sanders S.J., Yurkiewicz I.R., Ercan-Sencicek A.G., Kim Y.S., Fishman D.O., Raubeson M.J., Song Y., Yasuno K., Ho W.S., et al. Rare copy number variants in tourette syndrome disrupt genes in histaminergic pathways and overlap with autism. Biol. Psychiatry. 2012;71:392–402. doi: 10.1016/j.biopsych.2011.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bernardini L., Alesi V., Loddo S., Novelli A., Bottillo I., Battaglia A., Digilio M.C., Zampino G., Ertel A., Fortina P., et al. High-resolution SNP arrays in mental retardation diagnostics: how much do we gain? Eur. J. Hum. Genet. 2010;18:178–185. doi: 10.1038/ejhg.2009.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vulto-van Silfhout A.T., Hehir-Kwa J.Y., van Bon B.W., Schuurs-Hoeijmakers J.H., Meader S., Hellebrekers C.J., Thoonen I.J., de Brouwer A.P., Brunner H.G., Webber C., et al. Clinical significance of de novo and inherited copy-number variation. Hum. Mutat. 2013;34:1679–1687. doi: 10.1002/humu.22442. [DOI] [PubMed] [Google Scholar]
- 37.Grozeva D., Kirov G., Ivanov D., Jones I.R., Jones L., Green E.K., St Clair D.M., Young A.H., Ferrier N., Farmer A.E., et al. Rare copy number variants: a point of rarity in genetic risk for bipolar disorder and schizophrenia. Arch. Gen. Psychiatry. 2010;67:318–327. doi: 10.1001/archgenpsychiatry.2010.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu B., Roos J.L., Levy S., van Rensburg E.J., Gogos J.A., Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat. Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 39.Levy D., Ronemus M., Yamrom B., Lee Y.H., Leotta A., Kendall J., Marks S., Lakshmi B., Pai D., Ye K., et al. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron. 2011;70:886–897. doi: 10.1016/j.neuron.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 40.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A., et al. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kearney H.M., Thorland E.C., Brown K.K., Quintero-Rivera F., South S.T. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet. Med. 2011;13:680–685. doi: 10.1097/GIM.0b013e3182217a3a. [DOI] [PubMed] [Google Scholar]
- 42.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L., et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Neale B.M., Kou Y., Liu L., Ma'ayan A., Samocha K.E., Sabo A., Lin C.F., Stevens C., Wang L.S., Makarov V., et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O'Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D., et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.H., Narzisi G., Leotta A., et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jacques P.E., Jeyakani J., Bourque G. The majority of primate-specific regulatory sequences are derived from transposable elements. PLoS Genet. 2013;9:e1003504. doi: 10.1371/journal.pgen.1003504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang H.J., Kawasawa Y.I., Cheng F., Zhu Y., Xu X., Li M., Sousa A.M., Pletikos M., Meyer K.A., Sedmak G., et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duong L., Klitten L.L., Moller R.S., Ingason A., Jakobsen K.D., Skjodt C., Didriksen M., Hjalgrim H., Werge T., Tommerup N. Mutations in NRXN1 in a family multiply affected with brain disorders: NRXN1 mutations and brain disorders. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2012;159B:354–358. doi: 10.1002/ajmg.b.32036. [DOI] [PubMed] [Google Scholar]
- 49.Dabell M.P., Rosenfeld J.A., Bader P., Escobar L.F., El-Khechen D., Vallee S.E., Dinulos M.B., Curry C., Fisher J., Tervo R., et al. Investigation of NRXN1 deletions: clinical and molecular characterization. Am. J. Med. Genet. A. 2013;161:717–731. doi: 10.1002/ajmg.a.35780. [DOI] [PubMed] [Google Scholar]
- 50.Curran S., Ahn J.W., Grayton H., Collier D., Ogilvie C.M. NRXN1 deletions identified by array comparative genome hybridisation in a clinical case series – further understanding of the relevance of NRXN1 to neurodevelopmental disorders. Journal of Molecular Psychiatry. 2013;1:4. doi: 10.1186/2049-9256-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lionel A.C., Vaags A.K., Sato D., Gazzellone M.J., Mitchell E.B., Chen H.Y., Costain G., Walker S., Egger G., Thiruvahindrapuram B., et al. Rare exonic deletions implicate the synaptic organizer Gephyrin (GPHN) in risk for autism, schizophrenia and seizures. Hum. Mol. Genet. 2013;22:2055–2066. doi: 10.1093/hmg/ddt056. [DOI] [PubMed] [Google Scholar]
- 52.Talkowski M.E., Mullegama S.V., Rosenfeld J.A., van Bon B.W., Shen Y., Repnikova E.A., Gastier-Foster J., Thrush D.L., Kathiresan S., Ruderfer D.M., et al. Assessment of 2q23.1 microdeletion syndrome implicates MBD5 as a single causal locus of intellectual disability, epilepsy, and autism spectrum disorder. Am. J. Hum. Genet. 2011;89:551–563. doi: 10.1016/j.ajhg.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hodge J.C., Mitchell E., Pillalamarri V., Toler T.L., Bartel F., Kearney H.M., Zou Y.S., Tan W.H., Hanscom C., Kirmani S., et al. Disruption of MBD5 contributes to a spectrum of psychopathology and neurodevelopmental abnormalities. Mol. Psychiatry. 2013 doi: 10.1038/mp.2013.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Talkowski M.E., Rosenfeld J.A., Blumenthal I., Pillalamarri V., Chiang C., Heilbut A., Ernst C., Hanscom C., Rossin E., Lindgren A.M., et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Adam D. Mental health: On the spectrum. Nature. 2013;496:416–418. doi: 10.1038/496416a. [DOI] [PubMed] [Google Scholar]
- 56.Hofvander B., Delorme R., Chaste P., Nyden A., Wentz E., Stahlberg O., Herbrecht E., Stopin A., Anckarsater H., Gillberg C., et al. Psychiatric and psychosocial problems in adults with normal-intelligence autism spectrum disorders. BMC Psychiatry. 2009;9:35. doi: 10.1186/1471-244X-9-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Craddock N., Owen M.J. The Kraepelinian dichotomy – going, going...but still not gone. Br. J. Psychiatry. 2010;196:92–95. doi: 10.1192/bjp.bp.109.073429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lesch K.P., Timmesfeld N., Renner T.J., Halperin R., Roser C., Nguyen T.T., Craig D.W., Romanos J., Heine M., Meyer J., et al. Molecular genetics of adult ADHD: converging evidence from genome-wide association and extended pedigree linkage studies. J. Neural Transm. 2008;115:1573–1585. doi: 10.1007/s00702-008-0119-3. [DOI] [PubMed] [Google Scholar]
- 59.Glessner J.T., Reilly M.P., Kim C.E., Takahashi N., Albano A., Hou C., Bradfield J.P., Zhang H., Sleiman P.M., Flory J.H., et al. Strong synaptic transmission impact by copy number variations in schizophrenia. Proc. Natl. Acad. Sci. USA. 2010;107:10584–10589. doi: 10.1073/pnas.1000274107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang K.S., Liu X.F., Aragam N. A genome-wide meta-analysis identifies novel loci associated with schizophrenia and bipolar disorder. Schizophr. Res. 2010;124:192–199. doi: 10.1016/j.schres.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 61.Freilinger T., Anttila V., de Vries B., Malik R., Kallela M., Terwindt G.M., Pozo-Rosich P., Winsvold B., Nyholt D.R., van Oosterhout W.P., et al. Genome-wide association analysis identifies susceptibility loci for migraine without aura. Nat. Genet. 2012;44:777–782. doi: 10.1038/ng.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bis J.C., DeCarli C., Smith A.V., van der Lijn F., Crivello F., Fornage M., Debette S., Shulman J.M., Schmidt H., Srikanth V., et al. Common variants at 12q14 and 12q24 are associated with hippocampal volume. Nat. Genet. 2012;44:545–551. doi: 10.1038/ng.2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schaaf C.P., Boone P.M., Sampath S., Williams C., Bader P.I., Mueller J.M., Shchelochkov O.A., Brown C.W., Crawford H.P., Phalen J.A., et al. Phenotypic spectrum and genotype-phenotype correlations of NRXN1 exon deletions. Eur. J. Hum. Genet. 2012;20:1240–1247. doi: 10.1038/ejhg.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beunders G., Voorhoeve E., Golzio C., Pardo L.M., Rosenfeld J.A., Talkowski M.E., Simonic I., Lionel A.C., Vergult S., Pyatt R.E., et al. Exonic deletions in AUTS2 cause a syndromic form of intellectual disability and suggest a critical role for the C terminus. Am. J. Hum. Genet. 2013;92:210–220. doi: 10.1016/j.ajhg.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kudryashova E., Wu J., Havton L.A., Spencer M.J. Deficiency of the E3 ubiquitin ligase TRIM32 in mice leads to a myopathy with a neurogenic component. Hum. Mol. Genet. 2009;18:1353–1367. doi: 10.1093/hmg/ddp036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frosk P., Weiler T., Nylen E., Sudha T., Greenberg C.R., Morgan K., Fujiwara T.M., Wrogemann K. Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am. J. Hum. Genet. 2002;70:663–672. doi: 10.1086/339083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neri M., Selvatici R., Scotton C., Trabanelli C., Armaroli A., De Grandis D., Levy N., Gualandi F., Ferlini A. A patient with limb girdle muscular dystrophy carries a TRIM32 deletion, detected by a novel CGH array, in compound heterozygosis with a nonsense mutation. Neuromuscul. Disord. 2013;23:478–482. doi: 10.1016/j.nmd.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 68.Chiang A.P., Beck J.S., Yen H.J., Tayeh M.K., Scheetz T.E., Swiderski R.E., Nishimura D.Y., Braun T.A., Kim K.Y., Huang J., et al. Homozygosity mapping with SNP arrays identifies TRIM32, an E3 ubiquitin ligase, as a Bardet-Biedl syndrome gene (BBS11) Proc. Natl. Acad. Sci. USA. 2006;103:6287–6292. doi: 10.1073/pnas.0600158103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schwamborn J.C., Berezikov E., Knoblich J.A. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell. 2009;136:913–925. doi: 10.1016/j.cell.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sato T., Okumura F., Kano S., Kondo T., Ariga T., Hatakeyama S. TRIM32 promotes neural differentiation through retinoic acid receptor-mediated transcription. J. Cell Sci. 2011;124:3492–3502. doi: 10.1242/jcs.088799. [DOI] [PubMed] [Google Scholar]
- 71.Tropeano M., Ahn J.W., Dobson R.J., Breen G., Rucker J., Dixit A., Pal D.K., McGuffin P., Farmer A., White P.S., et al. Male-biased autosomal effect of 16p13.11 copy number variation in neurodevelopmental disorders. PLoS ONE. 2013;8:e61365. doi: 10.1371/journal.pone.0061365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Girirajan S., Rosenfeld J.A., Coe B.P., Parikh S., Friedman N., Goldstein A., Filipink R.A., McConnell J.S., Angle B., Meschino W.S., et al. Phenotypic heterogeneity of genomic disorders and rare copy-number variants. N. Engl. J. Med. 2012;367:1321–1331. doi: 10.1056/NEJMoa1200395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Noor A., Whibley A., Marshall C.R., Gianakopoulos P.J., Piton A., Carson A.R., Orlic-Milacic M., Lionel A.C., Sato D., Pinto D., et al. Disruption at the PTCHD1 Locus on Xp22.11 in Autism spectrum disorder and intellectual disability. Sci. Transl. Med. 2010;2 doi: 10.1126/scitranslmed.3001267. 49ra68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Werling D.M., Geschwind D.H. Sex differences in autism spectrum disorders. Curr. Opin. Neurol. 2013;26:146–153. doi: 10.1097/WCO.0b013e32835ee548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qin X.Y., Kojima Y., Mizuno K., Ueoka K., Muroya K., Miyado M., Zaha H., Akanuma H., Zeng Q., Fukuda T., et al. Identification of novel low-dose bisphenol a targets in human foreskin fibroblast cells derived from hypospadias patients. PLoS ONE. 2012;7:e36711. doi: 10.1371/journal.pone.0036711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pescosolido M.F., Yang U., Sabbagh M., Morrow E.M. Lighting a path: genetic studies pinpoint neurodevelopmental mechanisms in autism and related disorders. Dialogues Clin. Neurosci. 2012;14:239–252. [PMC free article] [PubMed] [Google Scholar]
- 77.Sakurai T. The role of NrCAM in neural development and disorders--beyond a simple glue in the brain. Mol. Cell Neurosci. 2012;49:351–363. doi: 10.1016/j.mcn.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 78.Hampson D.R., Gholizadeh S., Pacey L.K. Pathways to drug development for autism spectrum disorders. Clin. Pharmacol. Ther. 2012;91:189–200. doi: 10.1038/clpt.2011.245. [DOI] [PubMed] [Google Scholar]
- 79.Fatemi S.H., Aldinger K.A., Ashwood P., Bauman M.L., Blaha C.D., Blatt G.J., Chauhan A., Chauhan V., Dager S.R., Dickson P.E., et al. Consensus paper: pathological role of the cerebellum in autism. Cerebellum. 2012;11:777–807. doi: 10.1007/s12311-012-0355-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hart H., Radua J., Mataix-Cols D., Rubia K. Meta-analysis of fMRI studies of timing in attention-deficit hyperactivity disorder (ADHD) Neurosci. Biobehav. Rev. 2012;36:2248–2256. doi: 10.1016/j.neubiorev.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 81.O'Halloran C.J., Kinsella G.J., Storey E. The cerebellum and neuropsychological functioning: a critical review. J. Clin. Exp. Neuropsychol. 2012;34:35–56. doi: 10.1080/13803395.2011.614599. [DOI] [PubMed] [Google Scholar]
- 82.van Ewijk H., Heslenfeld D.J., Zwiers M.P., Buitelaar J.K., Oosterlaan J. Diffusion tensor imaging in attention deficit/hyperactivity disorder: a systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2012;36:1093–1106. doi: 10.1016/j.neubiorev.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 83.Anagnostou E., Taylor M.J. Review of neuroimaging in autism spectrum disorders: what have we learned and where we go from here. Mol. Autism. 2011;2:4. doi: 10.1186/2040-2392-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pierce K., Courchesne E. Evidence for a cerebellar role in reduced exploration and stereotyped behavior in autism. Biol. Psychiatry. 2001;49:655–664. doi: 10.1016/s0006-3223(00)01008-8. [DOI] [PubMed] [Google Scholar]
- 85.Sudarov A., Gooden F., Tseng D., Gan W.B., Ross M.E. Lis1 controls dynamics of neuronal filopodia and spines to impact synaptogenesis and social behaviour. EMBO Mol. Med. 2013;5:591–607. doi: 10.1002/emmm.201202106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Penagarikano O., Abrahams B.S., Herman E.I., Winden K.D., Gdalyahu A., Dong H., Sonnenblick L.I., Gruver R., Almajano J., Bragin A., et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marshall C.R., Noor A., Vincent J.B., Lionel A.C., Feuk L., Skaug J., Shago M., Moessner R., Pinto D., Ren Y., et al. Structural variation of chromosomes in autism spectrum disorder. Am. J. Hum. Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Silversides C.K., Lionel A.C., Costain G., Merico D., Migita O., Liu B., Yuen T., Rickaby J., Thiruvahindrapuram B., Marshall C.R., et al. Rare copy number variations in adults with tetralogy of Fallot implicate novel risk gene pathways. PLoS Genet. 2012;8:e1002843. doi: 10.1371/journal.pgen.1002843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Costain G., Lionel A.C., Merico D., Forsythe P., Russell K., Lowther C., Yuen T., Husted J., Stavropoulos D.J., Speevak M., et al. Pathogenic rare copy number variants in community-based schizophrenia suggest a potential role for clinical microarrays. Hum. Mol. Genet. 2013;22:4485–4501. doi: 10.1093/hmg/ddt297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pinto D., Pagnamenta A.T., Klei L., Anney R., Merico D., Regan R., Conroy J., Magalhaes T.R., Correia C., Abrahams B.S., et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature. 2010;466:368–372. doi: 10.1038/nature09146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V., et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shaikh T.H., Gai X., Perin J.C., Glessner J.T., Xie H., Murphy K., O'Hara R., Casalunovo T., Conlin L.K., D'Arcy M., et al. High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 2009;19:1682–1690. doi: 10.1101/gr.083501.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jiang Y.-h., Yuen Ryan K.C., Jin X., Wang M., Chen N., Wu X., Ju J., Mei J., Shi Y., He M., et al. Detection of Clinically Relevant Genetic Variants in Autism Spectrum Disorder by Whole-Genome Sequencing. Am. J. Hum. Genet. 2013;93:249–263. doi: 10.1016/j.ajhg.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Valdar W.S. Scoring residue conservation. Proteins. 2002;48:227–241. doi: 10.1002/prot.10146. [DOI] [PubMed] [Google Scholar]
- 96.Merico D., Isserlin R., Stueker O., Emili A., Bader G.D. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chen X., Shen Y., Zhang F., Chiang C., Pillalamarri V., Blumenthal I., Talkowski M., Wu B.L., Gusella J.F. Molecular analysis of a deletion hotspot in the NRXN1 region reveals the involvement of short inverted repeats in deletion CNVs. Am. J. Hum. Genet. 2013;92:375–386. doi: 10.1016/j.ajhg.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rosenfeld J.A., Coe B.P., Eichler E.E., Cuckle H., Shaffer L.G. Estimates of penetrance for recurrent pathogenic copy-number variations. Genet. Med. 2013;15:478–481. doi: 10.1038/gim.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ahn J.W., Dixit A., Johnston C., Ogilvie C.M., Collier D.A., Curran S., Dobson R.J. BBGRE: brain and body genetic resource exchange. Database. 2013 doi: 10.1093/database/bat067. (Oxford), doi:10.1093/database/bat067. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.