

Abstract
Nuclear factor E2-related factor 2 (Nrf2) is a pivotal transcription factor in the defense against oxidative stress. Here we provide evidence that activation of the Nrf2 pathway reduces the levels of phosphorylated tau by induction of an autophagy adaptor protein NDP52 (also known as CALCOCO2) in neurons. The expression of NDP52, which we show has three antioxidant response elements (AREs) in its promoter region, is strongly induced by Nrf2, and its over-expression facilitates clearance of phosphorylated tau in the presence of an autophagy stimulator. In Nrf2 knockout mice, phosphorylated and sarkosyl-insoluble tau accumulates in the brains concurrent with decreased levels of NDP52. Moreover, NDP52 associates with phosphorylated tau from brain cortical samples of Alzheimer disease cases, and the amount of phosphorylated tau in sarkosyl-insoluble fractions is inversely proportional to that of NDP52. These results suggest that NDP52 plays a key role in autophagy-mediated degradation of phosphorylated tau in vivo.
In Alzheimer disease (AD), tau becomes aberrantly phosphorylated and oligomerized which likely contributes to neuronal dysfunction and death1–3. The mechanism involved in clearing pathological, phosphorylated tau has not yet been fully defined. Given that tau plays a central role in the pathogenesis of AD2, 3, there is a growing interest in understanding the processes by which pathological, phosphorylated tau is removed from neurons and subsequently developing interventions that result in the selective removal of the pathological tau as a therapeutic approach for the treatment of AD2, 4.
To date, several reports have suggested that misfolded or phosphorylated tau could be degraded through the proteasome5–8. On the other hand, some studies found that tau was not degraded by the proteasome, and unexpectedly observed decreases in the levels of tau in the presence of proteasome inhibitors9, 10. In addition to degradation by the proteasome, several groups have presented data indicating that tau can be turned over by the autophagy-lysosome pathway11–17. The triage process directing specific forms of tau to a given degradation pathway is not completely understood and an area of interest.
A family of autophagy adaptor proteins which contain LC3-interacting regions (LIRs) has been identified and include p62/sequestosome1 (SQSTM1), neighbor of BRCA1 gene 1 (NBR1) and nuclear dot protein 52 (NDP52)18, 19. The most well-studied member of this group is p62/SQSTM1, a multifunctional protein that acts as a scaffold for signaling protein complexes as well as being an adaptor for protein trafficking, aggregation and degradation20. p62/SQSTM1 also plays an important role in regulating the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway21, as well as being upregulated by Nrf222. Genetic inactivation of p62/SQSTM1 in mice leads to an accumulation of hyperphosphorylated tau23. NBR1 was shown to be involved in the autophagy targeting of ubiquitylated substrates24 and defective ribosomal products (DRiPs)25, and co-localized with phosphorylated tau together with p62/SQSTM1 in the muscle fiber aggregates of sporadic inclusion-body myositis26. NDP52 has been identified primarily in peripheral tissues as an innate immune autophagy receptor for ubiquitylated bacteria, thus coordinating their destruction by the autophagy pathway27, 28. NDP52 also functions as an autophagic adaptor for the selective degradation of Toll/IL-1 receptor homology domain-containing adaptor inducing IFN-β (TRIF) and TNF receptor-associated factor 6 (TRAF6) in the periphery29. Recently, it was suggested that NDP52 participates in autophagosome maturation by docking with myosin VI together with T6BP and optineurin30. However, a biological role for NDP52 in neurons or in mediating the degradation of tau has not been examined.
Oxidative stress has been considered as one of the early pathogenic events during disease progression in the brains of AD patients31. One of the defense mechanisms against oxidative stress as well as other stressors is the activation of the Nrf2 pathway32. Nrf2 acts by binding to the consensus antioxidant response element (ARE) sequence following heterodimerization with Maf, leading to the expression of genes involved in protein degradation like p62/STSQM122, as well as cytoprotective genes including heme oxygenase 1 (HO1), NAD(P)H:quinine oxidoreductase 1 (NQO1) and glutathione peroxidase (GPx)32. Although activation of Nrf2 improved learning and memory retention in a mouse model of AD33, 34, there is no study on its role in the clearance of pathological, phosphorylated tau.
Here, we show that in Nrf2 knockout mice the levels of phosphorylated tau and the levels of sarkosyl-insoluble tau are increased. We also demonstrate that NDP52 is a Nrf2 responsive gene and a major, downstream facilitator of Nrf2-mediated phosphorylated tau degradation by autophagy.
Results
Nrf2 activation promotes degradation of phosphorylated tau
To examine whether Nrf2 is involved in the degradation of phosphorylated tau, we examined the levels of phosphorylated tau in the hippocampal tissues of Nrf2 knockout (−/−) mice. As shown in Fig. 1a, b and Supplementary Fig. 1, 2.9 and 2.7 fold increases of tau phosphorylated at Ser262/Ser356 [12E8] and at Ser396/Ser404 [PHF1], respectively, were observed in Nrf2 (−/−) mice in comparison to wild-type mice. In addition, the level of total tau also showed a 1.8 or 2.1 fold increase by the use of a polyclonal tau or the Tau5 antibody, respectively. When the presence of phosphorylated tau in the hippocampal region was immunohistochemically evaluated, phosphorylated tau staining with the 12E8 or PHF1 antibodies was readily detected in the hippocampal region of Nrf2 (−/−) mice, whereas almost none was apparent in wild-type mice (Fig. 1c, d). Further, the amount of sarkosyl-insoluble tau was 5.3 fold increased in Nrf2 (−/−) mice compared with wild-type mice (Fig. 2). These results suggest that Nrf2 is involved in the degradation of pathologic, phosphorylated tau.
Figure 1. Phosphorylated tau accumulates in the hippocampus of Nrf2 (−/−) mice.
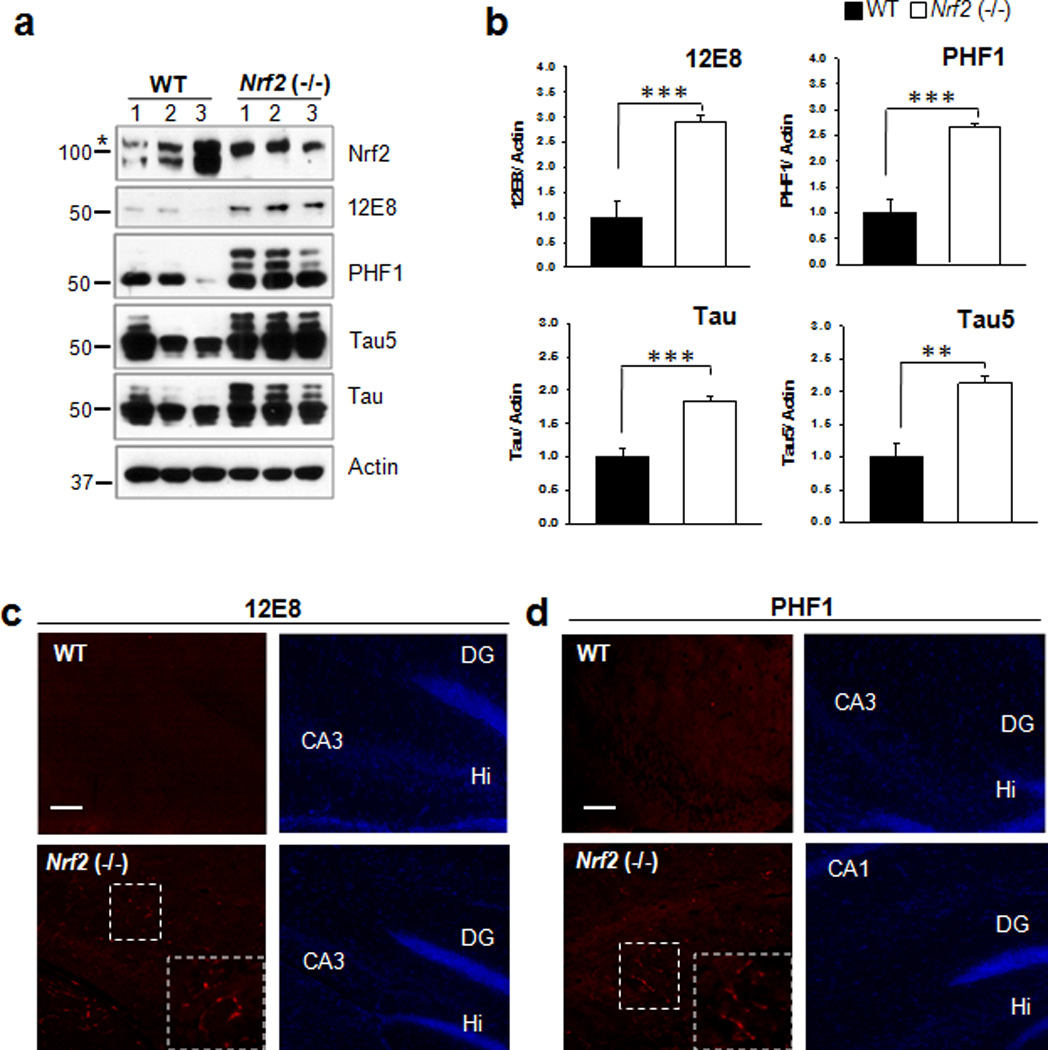
(a) Hippocampal tissues obtained from Nrf2 (−/−) (6 months old, 1 male and 2 female) or wild-type (11 months old, 2 male, and 1 female) mice were immunoblotted with a Nrf2 antibody, 12E8, PHF1, a monoclonal to total tau (Tau5) or a polyclonal to total tau (Tau) antibody. The asterisk to the left of the Nrf2 blot designates the position at which a non-specific band migrated. The relative molecular masses (kD) are indicated to the left of each blot. (b) Bar graphs represent the relative optical density of phosphorylated or total tau in the hippocampal tissues. n=6 (These data are based on the mouse hippocampi immunoblotted in Supplementary Fig. 1).. Data shown are mean±SE and were analyzed using Student’s t test. (**, p<0.01; ***, p<0.001) (c, d) Left panels show brain sections stained for tau phosphorylated at Ser262/Ser356 (12E8) (c) and Ser396/Ser404 (PHF1) (d). The area of delineated by dotted lines is enlarged and shown in the right corner of the image. Right panels shows the same slice stained with DAPI. Scale bar = 50 µm. DG, dentate gyrus; Hi, dentate hilar area.
Figure 2. Sarkosyl-insoluble tau accumulates in the hippocampus of Nrf2 (−/−) mice.
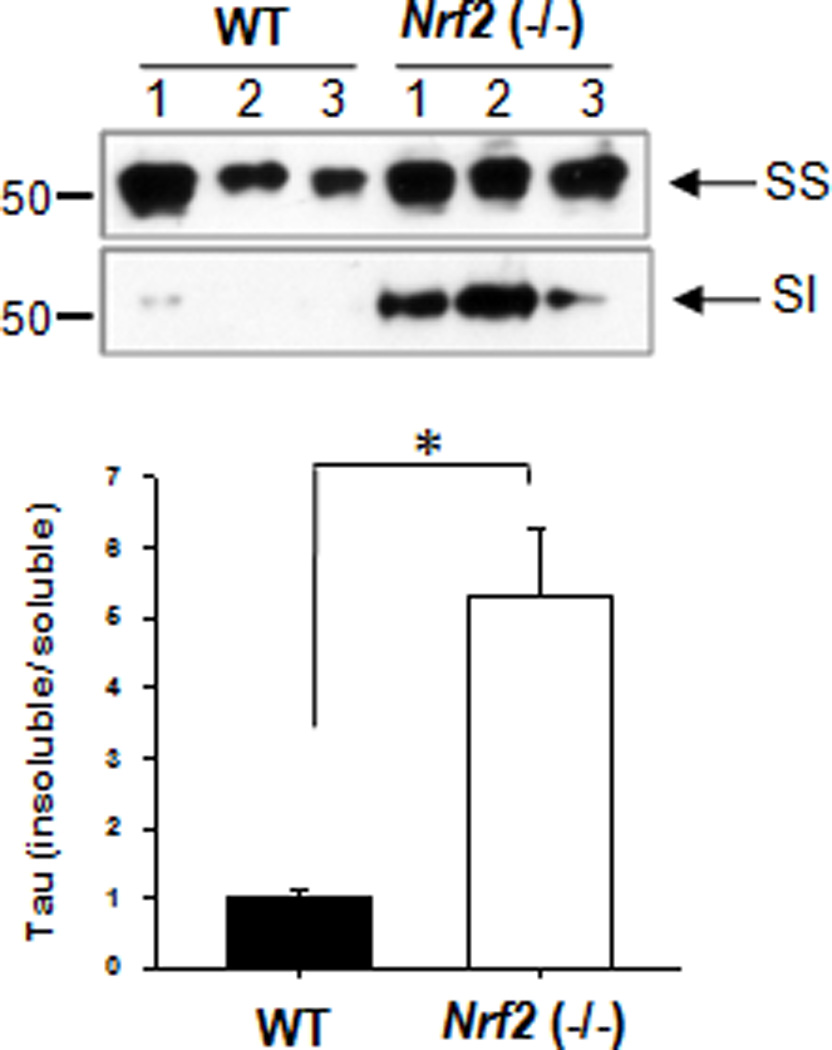
Cortex tissues obtained from Nrf2 (−/−) (6 months old 1 male; 2 female) or wild-type (11 months old 2 male; 1 female) mice were separated into sarkosyl-soluble (SS) and insoluble (SI) fractions as described in the Methods. The level of total tau was analyzed by immunoblotting using a monoclonal antibody to total tau (Tau5). The relative molecular masses (kD) are indicated to the left of each blot. Bar graph represents the relative optical density of sarkosyl-insoluble tau normalized with that of sarkosyl-soluble tau. n=3. Data shown are mean±SE and were analyzed using Student’s t test. (*, p<0.05)
To further investigate whether Nrf2 activity is involved in the degradation of phosphorylated tau, primary rat cortical neurons and immortalized CN1.4 mouse cortical cells that inducibly express wild-type tau14 were treated with sulforaphane (10 µM, SFN), a Nrf2 activator (Supplementary Fig.2a, b). Twenty-four hours after treatment with SFN the levels of tau phosphorylated at Ser262/Ser356 [12E8] were reduced 30% and 70% in primary cortical neurons and CN1.4 mouse cortical cells, respectively. In the case of tau phosphorylated at Ser396/Ser404 [PHF1], 50% and 43% reductions were observed, respectively (Fig. 3). Treatment with 10 µM of SFN did not affect the viability of neurons (Supplementary Fig. 2d, e), indicating that the reduction of phosphorylated tau was not due to increased cell death in the presence of SFN. In addition, when primary rat cortical neurons were treated with 50 µM tert-butylhydroquinone (tBHQ), another Nrf2 activator (Supplementary Fig. 2c), a significant reduction of tau phosphorylated at Ser262/Ser356 [12E8] and Ser396/Ser404 [PHF1] was also observed as shown in Supplementary Fig. 3a, b, c. Thus, these results suggest that Nrf2 activation facilitates the degradation of phosphorylated tau.
Figure 3. Nrf2 activation enhances the degradation of phosphorylated tau in neurons.
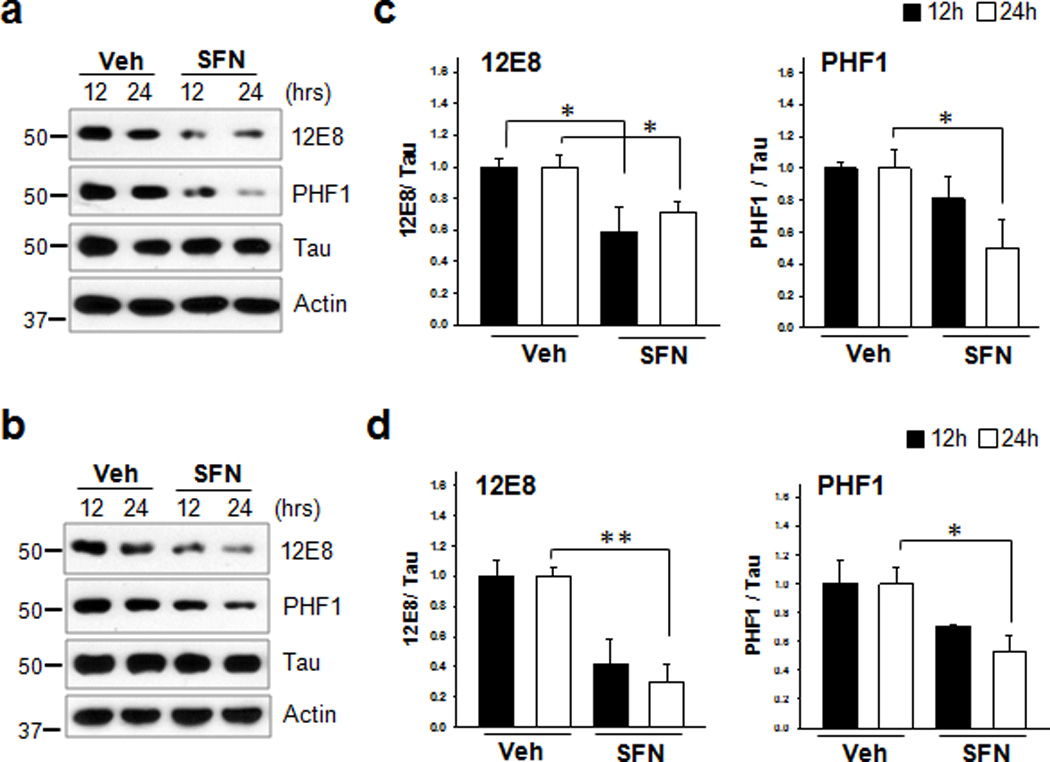
(a) Primary cortical neurons (DIV 5) were treated with sulforaphane (10 µM, SFN) for 12 h or 24 h. (b) Stably transfected CN1.4 cortical neurons were maintained in the presence of doxycycline (1 µg/ml) to induce the expression of tau for 24 h, and subsequently treated with SFN for 12 h or 24 h. The levels of tau phosphorylated at Ser262/Ser356 and Ser396/Ser404 were analyzed by immunoblotting using the 12E8- and PHF1 antibodies, respectively. Tau was detected with a polyclonal phospho-independent tau antibody (Tau). The relative molecular masses (kD) are indicated to the left of each blot. (c, d) Bar graphs represent the relative optical density of phosphorylated tau normalized with that of total tau. Data shown are mean±SE of three independent experiments and were analyzed using Student’s t test. (*, p<0.05; **, p<0.01)
Tau phosphotransferases are not different in Nrf2(−/−) mice
The Ser/Thr-Pro sites of tau have been proposed to be phosphorylated by such kinases as cyclin-dependent kinase 5 (Cdk5), glycogen synthase kinase 3β (GSK3β), and extracellular signal regulated kinase 2 (ERK2), while the KXGS motif (the 12E8 epitope) in the microtubule-binding domains is likely phosphorylated by microtubule-affinity regulating kinases (MARKs)1. Given the results, it is possible that the increased levels of phosphorylated tau could result from increased kinase activity in Nrf2 (−/−) mice. Thus, we performed in vitro kinase assays using mouse brain lysates and found there was no significant difference in kinase activity between Nrf2 (−/−) and wild-type mice (Fig. 4a and Supplementary Fig. 4). It also can be speculated that in Nrf2 (−/−) mice there may be decreased activity of protein phosphatase 2A (PP2A), since PP2A is known to be the primary phosphatase involved in the dephosphorylation of tau35, 36. When the activity of protein phosphatases was assayed using mouse brain lysates, there was no difference in the activity of PP2A, between Nrf2 (−/−) and wild-type mice (Fig. 4b). These results strongly support the conclusion that the increased levels of phosphorylated tau in Nrf2 (−/−) mouse brain does not result from alterations in the activities of kinases or phosphatases.
Figure 4. There is no significant difference in tau kinase and phosphatase activities between wild-type and Nrf2 (−/−) mice.
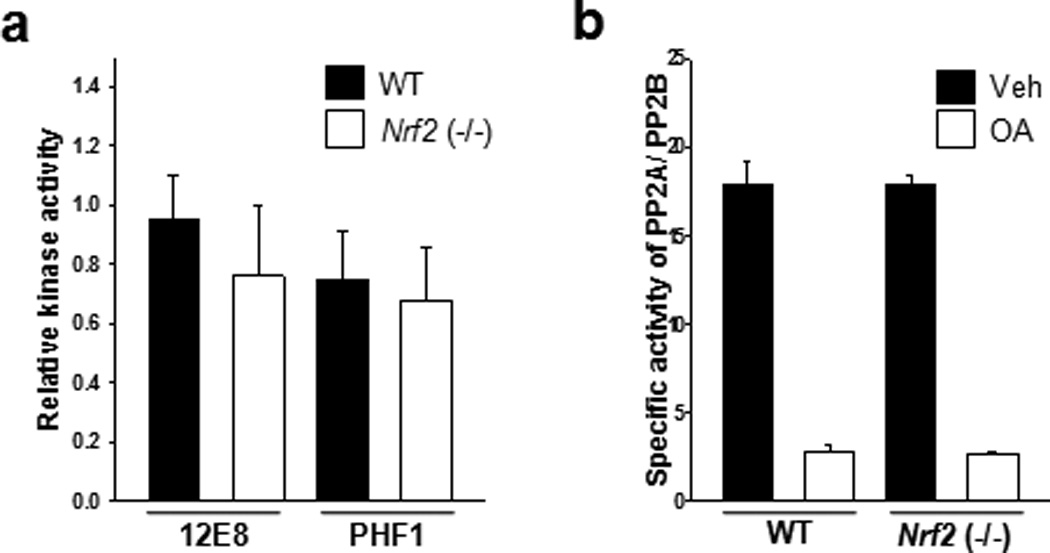
Mouse brains were obtained from wild-type (11 months old, 1 male; 5 months old, 1 female; 12 months old, 2 female) and Nrf2 (−/−) (10 months old, 2 male; 2 female) mice. (a)Mouse brain tissues were homogenized in the lysis buffer, and 10 µg of lysates was incubated with 2 µg of GST-tau protein at 37°C for 30 min in the presence or absence of 1 mM ATP. The levels of tau phosphorylated at Ser262/Ser356 and Ser396/Ser404 were analyzed by immunoblotting using a 12E8- and PHF1-specific antibody, respectively. GST-tau was detected with a monoclonal GST antibody (GST-tau) (Supplementary Fig. 4). Bar graph of the relative optical density of phosphorylated tau normalized to actin. Data shown are mean±SE of duplicated independent experiments and were analyzed using Student’s t test. (b) Mouse brain tissues were homogenized in the phosphatase storage buffer, and phosphatase activity in the lysates was quantitated using the Serine/Threonine Phosphatase Assay System (Promega) by measuring the dephosphorylation of a phospho-peptide, RRA(pT)VA in the presence or absence of okadaic acid (OA, 20 nM). PP2A activity was defined as the activity inhibited by the addition of okadaic acid to the phosphatase reaction mixture. n=4. Data shown are mean±SE of three independent experiments and were analyzed using Student’s t test.
Autophagy mediates the degradation of phosphorylated tau
To investigate the contribution of the proteasome and autophagy pathways to the degradation of phosphorylated tau, we examined the levels of phosphorylated tau in CN1.4 cortical cells14 using inhibitors. Following withdrawal of doxycycline, tau (and phospho-tau) levels decreased as expected. Treatment with the autophagy inhibitor, 3-methyladenine (3-MA, 10 mM) for 24 h following the withdrawal of doxycycline inhibited the degradation of phosphorylated tau and total tau (Fig. 5a). In contrast, treatment with the proteasome inhibitors epoxomicin (12.5 nM) or MG132 (7 µM) increased tau degradation (Fig. 5a). These data are in agreement with a recent study that showed proteasome inhibition in primary cortical neurons increased autophagy and tau clearance, and that 3-MA reduces tau clearance15. Further, addition of chloroquine (CQ, 50 µM) or bafilomycin A1 (BaF A1, 10 nM)37 to media devoid of doxycycline attenuated the degradation of phosphorylated tau and total tau (Fig. 5b). Also, the knockdown of beclin-1, a key molecule of the autophagy pathway, using siRNA resulted in an accumulation of tau (Fig. 5c). Interestingly, SFN causes an increase in autophagy in neurons (Supplementary Fig. 5, 6), thus the SFN (10 µM) induced reduction in phosphorylated tau levels (Fig. 3) is likely due to increased autophagic clearance. Indeed, the SFN-induced degradation of tau was significantly attenuated by CQ (Fig. 5d, e). These data strongly indicate that the autophagy pathway plays a major role in the degradation of phosphorylated tau.
Figure 5. Autophagy contributes to tau clearance in neuronal cells.
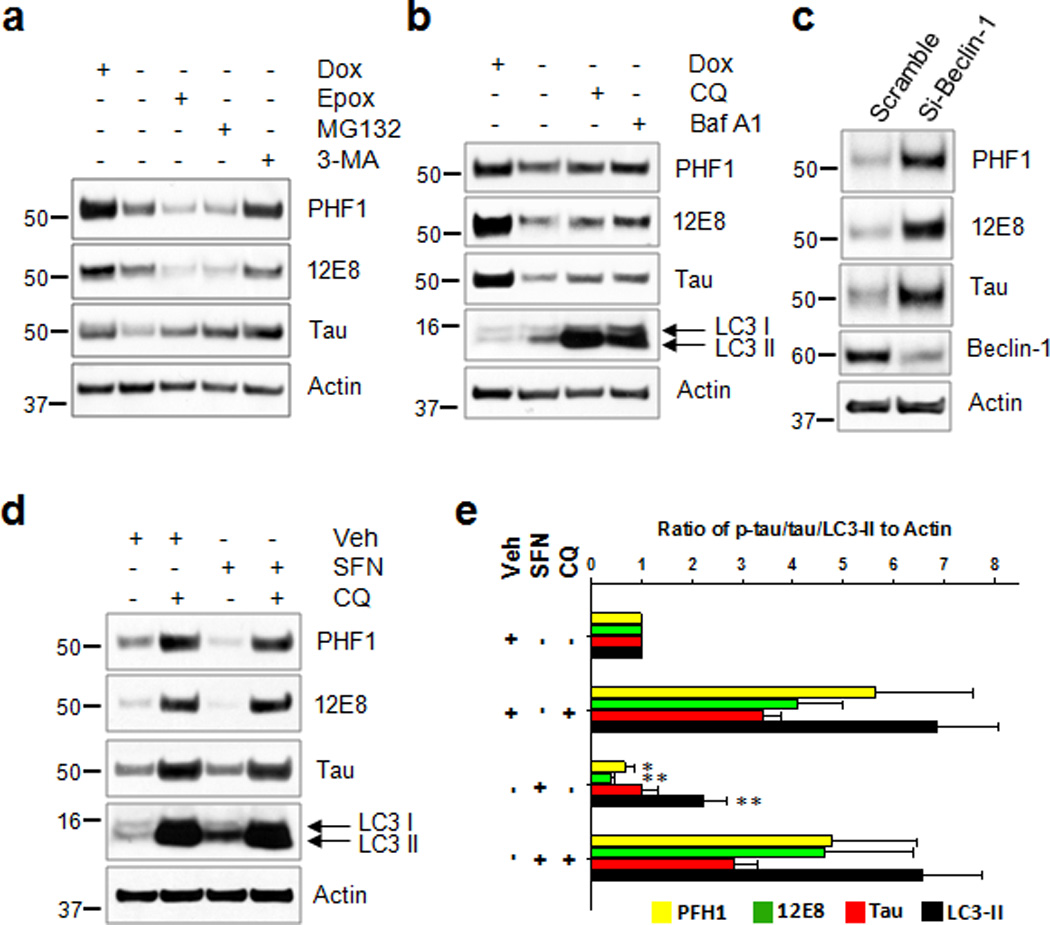
CN1.4 mouse cortical neurons stably expressing inducible tau were treated with doxycycline (Dox, 1 ug/ml) for 24 h. On the next day (a) the cells were washed twice with PBS, and the media were replaced with doxycycline-deficient media with either vehicle, epoxomicin (12.5 nM, Epox), MG132 (7 µM) or 3-methyladenine (10 mM, 3-MA) for 24 h. (b) Cells were treated with either chloroquine (50 µM, CQ) or bafilomycin A1 (10 nM, Baf A1) for 24 h. Samples in first lane were from cells maintained in the presence of Dox without medium exchange. (c) HEK293-TN cells were co-transfected with the plasmid expressing tau together either with siRNA of beclin-1 or scramble RNA as a control, which was kept for 48 h. (d) Stably transfected CN1.4 cortical neurons were maintained in the presence of doxycycline (1 µg/ml) to induce the expression of tau for 24 h, and subsequently treated with sulforaphane (10 µM, SFN) for 12 h. The culture media was exchanged for fresh media containing doxycycline (1 µg/ml) but no SFN after washing with PBS. The cells were then incubated for an additional 18 h in the absence or presence of chloroquine (50 µM, CQ). The levels of tau phosphorylated at Ser262/Ser356 and Ser396/Ser404 were analyzed by immunoblotting using the 12E8- and PHF1 antibodies, respectively. Tau, beclin-1 and LC3 were detected with the appropriate polyclonal antibodies. The relative molecular masses (kD) are indicated to the left of each blot. (e) Bar graphs represent the relative optical density of phosphorylated tau, tau or LC3-II normalized with that of actin. n=4. Data shown are mean±SE and were analyzed using Student’s t test. (*, p<0.05; **, p<0.01)
NDP52 expression is dependent on Nrf2
To elucidate the down-stream molecules involved in the Nrf2-mediated turnover of phosphorylated tau, we determined the transcript levels of the autophagy adaptor proteins p62/SQSTM1, NBR1 and NDP5218, 19 using qRT-PCR in the primary cortical neurons and CN1.4 cortical cells14 following treatment with SFN (10 µM) for 12 h. As shown in Fig. 6a and Supplementary Fig. 7, a 1.7 fold increase of p62/SQSTM1 mRNA was observed in the CN1.4 cortical cells, but not in the primary cultured neurons. NBR1 mRNA was not affected by SFN treatment in both cell types. However, in the case of NDP52 mRNA, expression was induced 2.4 and 2.6 fold in the primary cultured neurons and the CN1.4 mouse cortical cells, respectively following SFN treatment. Analysis of the transcript levels of these genes in the hippocampal tissues of Nrf2 (−/−) mice revealed that p62/SQSTM1 mRNA was significantly decreased 1.9 fold, compared to wild-type mice. By contrast, the decrease of NDP52 mRNA in the Nrf2 (−/−) mice was much more dramatic, as the level was 7.8 fold lower than in wild-type mice. This decrease was even greater than the decrease in the mRNA for heme oxygenase 1 (HO1) (~3 fold), a well-known Nrf2 down-stream gene (Fig. 6b). Protein levels of p62/SQSTM1 and NDP52, which showed a significant difference in the mRNA levels between Nrf2 (−/−) and wild-type mice, were also determined. In line with the results of the qRT-PCR, NDP52 protein was scarcely detected in the hippocampal tissues of Nrf2 (−/−) mice compared to those of wild-type mice (Fig. 6c and Supplementary Fig. 8c, d); however, no differences were observed in the protein levels of p62/SQSTM1 despite its decrease at the mRNA level (Fig. 6c).
Figure 6. NDP52 is induced by Nrf2 activation and its expression is significantly reduced in Nrf2 (−/−) mice.
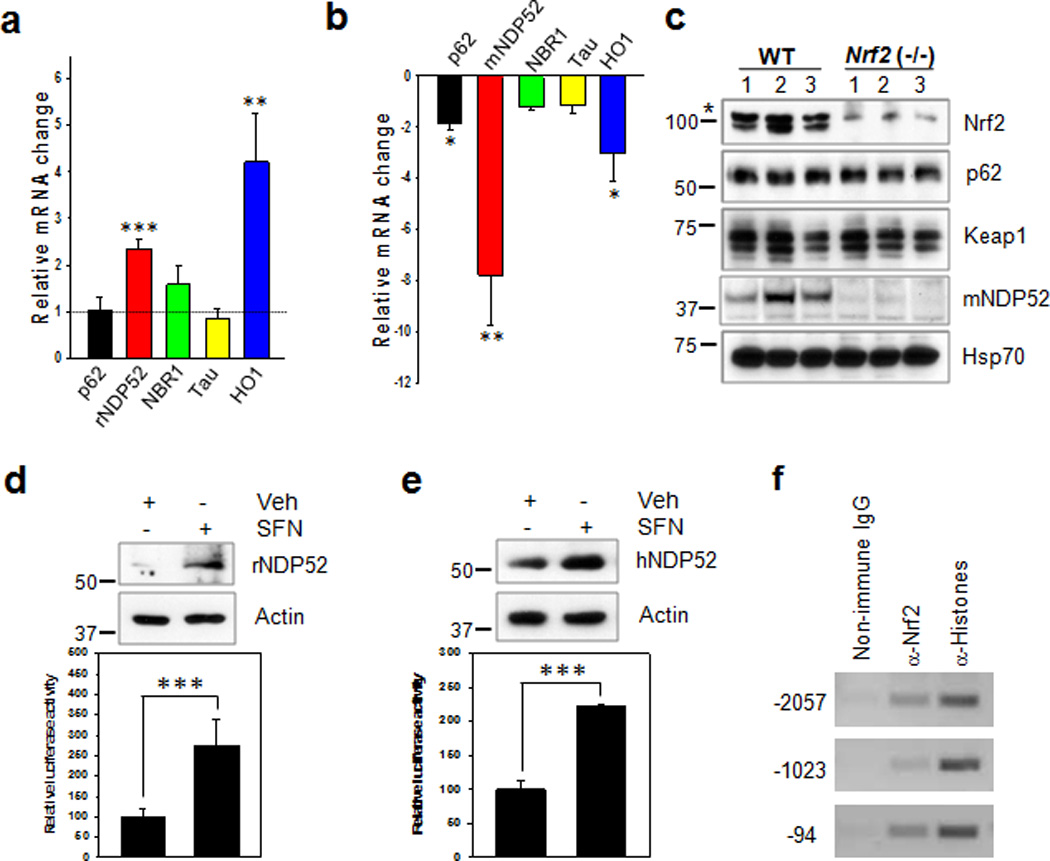
(a) Primary cortical neurons (DIV 5) were treated with sulforaphane (SFN, 10 µM) for 12 h, and the relative mRNA level of each gene was compared to that of cells not treated with SFN using qRT-PCR. (b) In hippocampal tissues from Nrf2 (−/−) (5 months old, 1 male; 11 months old 1 female; 5 months old, 1 female; 12 months old, 3 female) or wild-type (10 months old, 4 male; 2 female) mice, the relative mRNA level of each gene compared with that of wild-type mice. n=6. (c) The levels of p62/SQSTM1, Keap1, mouse NDP52 (mNDP52) and Hsp70 in Nrf2 (−/−) or wild-type mice were examined by immunoblotting. The asterisk on the Nrf2 panel indicates a non-specific band. (d, e: upper panel) Primary cortical neurons (DIV 5) (d) and SH-SY5Y cells (e) were treated with SFN for 24 h, and the expression levels of rat and human NDP52 (rNDP52 and hNDP52) examined by immunoblotting using anti-NDP52 antibody. The relative molecular masses (kD) are indicated to the left of each blot. (d, e: low panel) Primary cortical neurons (DIV 5) (d) and SH-SY5Y cells (e) transiently transfected with the pGL4.14/human NDP52 promoter (2,173 bps) luciferase reporter plasmid were treated with SFN for 24 h, and assayed for the luciferase activity. (f) SH-SY5Y cells were treated with SFN for 12 h, and cell lysates were used for the ChIP assay using anti-Nrf2 antibody as described in the Methods. Data shown are mean±SE and were analyzed using Student’s t test. (*, p<0.05; **, p<0.01; ***, p<0.001)
If the transcription of NDP52 is dependent on Nrf2 activation, there should be conserved antioxidant response elements (AREs) for Nrf2 in the promoter region of human NDP52. Interestingly, five putative, consensus cis-acting ARE elements were identified in the promoter DNA sequence (Fig. 7a). Therefore, we cloned the promoter region (2,137 bps) of human NDP52 gene from human genomic DNA. The protein levels of NDP52, as well as the luciferase activities of human NDP52 promoter, were significantly induced in both primary cultured neurons and SH-SY5Y cells in response to SFN (10 µM) treatment (Fig. 6d, e). In addition, the protein level of NDP52 increased in SH-SY5Y cells in response to tBHQ (50 µM) treatment (Supplementary Fig. 3d). To identify which of the consensus ARE sites in the NDP52 promoter are functional, we made several site-directed or deleted mutants of the putative ARE sites such that the Nrf2/Maf complex would be unable to bind. The luciferase activity of NDP52 promoter was abolished with the deletion of between −450 and +1 (HindIII), indicating this region is very important in the basal transcription of NDP52 gene (Fig. 7b). Nrf2-induced luciferase activity was completely abolished only when the three functional ARE sites (−2057, −1023 and −94) in the NDP52 promoter were mutated (Fig. 7c, d). The direct binding of Nrf2 to these ARE sites in vivo was confirmed with a ChIP assay (Fig. 6f). Together, the results suggest that the expression of NDP52 is strongly dependent on the activity of Nrf2 and mediated by the direct binding of Nrf2 to its promoter.
Figure 7. There are three functional ARE cis-elements in the human NDP52 promoter.
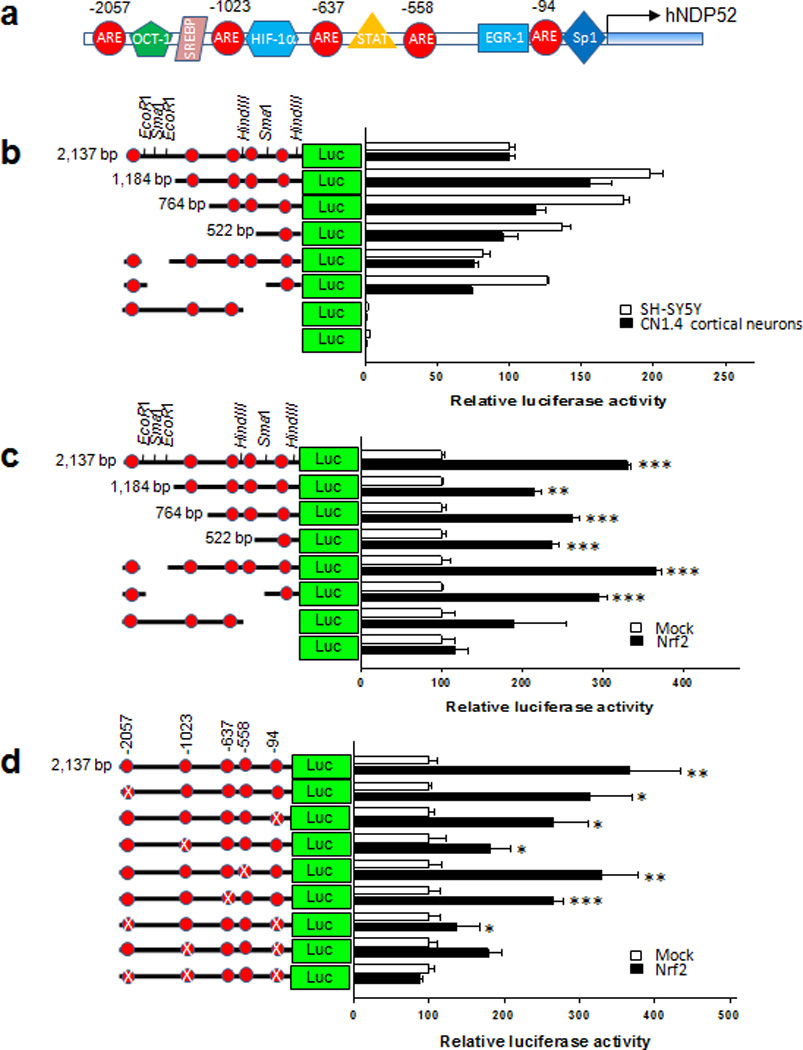
(a) Diagram shows five putative ARE cis-acting elements in human NDP52 promoter region (2,137 bps). Red ellipsoids indicate the putative ARE cis-acting elements. (b, c, d) The mutant forms of pGL4.14-Luc reporter plasmid with the human NDP52 promoter gene were prepared by digestion with specific restriction enzymes (e.g., HindIII, SmaI or EcoRI), by cloning of specific regions following PCR amplification, or with the QuikChange site-directed mutagenesis kit (Agilent Technologies). (b) CN1.4 cortical neurons and SH-SY5Y cells were transiently transfected with the reporter plasmids with the human NDP52 promoter (2,317 bps or deleted mutants), and luciferase activity assayed 24 h after transfection. (c) SH-SY5Y cells were co-transfected with the reporter plasmids with human NDP52 promoter (wild or deleted mutants) along with either the mock plasmid (pcDNA3.1) or the plasmid expressing Nrf2, and the luciferase activity was assayed 24 h after transfection. (d) SH-SY5Y cells were transiently co-transfected with the reporter plasmids with human NDP52 promoter or its mutant plasmid in which the putative ARE sequence was site-directly mutated (X), together with the mock plasmid (pcDNA3.1) or the plasmid expressing human Nrf2. The luciferase activity was assayed after 24 h. The induced luciferase activity with co-expression with Nrf2 was not observed in a mutant NDP52 promoter in which all three ARE sites (−2057, −1023 and −94) were site-directly mutated, indicating these three sites of five ARE cis-acting elements are functional. Data shown are mean±SD of three independent experiments and were analyzed using Student’s t test. (*, p<0.05; **, p<0.01; ***, p<0.001)
NDP52 facilitates the turnover of phosphorylated tau
NDP52 has an LC3 interacting region (LIR)28 and previous studies indicate that it functions as an autophagy adaptor recognizing ubiquitin-coated cytosolic bacteria or regulating Toll-like receptor signaling, but only in peripheral tissues27–29, 38. Therefore, we examined the co-localization of NDP52 with LC3 in both primary cortical neurons and CN1.4 naive cells following transient transfection. Expression of NDP52 along with tau in primary cultured neurons resulted in prominent, NDP52-postive punctae in the soma of the neurons (Supplementary Fig. 9a). All the NDP52 punctae overlapped with autophagic vesicles (AVs) as they were LC3-positive (Supplementary Fig. 9b, c and 10a, b). In addition, when HEK 293-TN cells were transiently transfected with GFP or GFP-LC3, endogenous NDP52 was co-immunoprecipitated with GFP-LC3, but not with GFP alone (Supplementary Fig. 11). The results indicate that NDP52 interacts with LC3 and likely plays a role in targeting client proteins to the autophagy pathway in neuronal cells.
To further investigate the role of NDP52 in facilitating the turnover of phosphorylated tau in primary cultured neurons, we used recombinant lentiviruses expressing sh-RNA for Nrf2 or NDP52. When primary cortical neurons were transduced with sh-Nrf2, the levels of tau endogenously phosphorylated at Ser262/Ser356 [12E8] and Ser396/Ser404 [PHF1] were 1.5 and 1.9 fold increased, respectively in comparison to what was observed in neurons transduced with sh-scramble (Fig. 8a, b). Transduction with sh-NDP52 resulted in 1.8 and 2.1 fold increases of tau phosphorylated at Ser262/Ser356 [12E8] and at Ser396/Ser404 [PHF1], respectively (Fig. 8a, b). A dramatic decrease in the levels of both wild-type tau and caspase-cleaved tau in HEK 293-TN cells was observed with co-expression of Nrf2 or NDP52 compared to the cells co-transfected with the mock plasmid, whereas co-expression of p62/SQSTM1 or the cochaperone Bag2, which has been reported to be involved in the degradation of phosphorylated tau7, 23, did not decrease tau levels (Supplementary Fig. 12a, b). In addition, a decrease in the intensity of endogenously phosphorylated tau immunostained with 12E8 or PHF1 antibody was observed in the primary neurons transduced with the recombinant lentivirus expressing NDP52 compared with those transduced with a control lentivirus in the presence of 150 mM trehalose, a disaccharide autophagy inducer15, 39 (Fig. 8c, d, e, f and Supplementary Fig. 12c, d). Together, these results suggest that NDP52 functions in facilitating the turnover of phosphorylated tau.
Figure 8. NDP52 plays an important role in the clearance of phosphorylated tau.
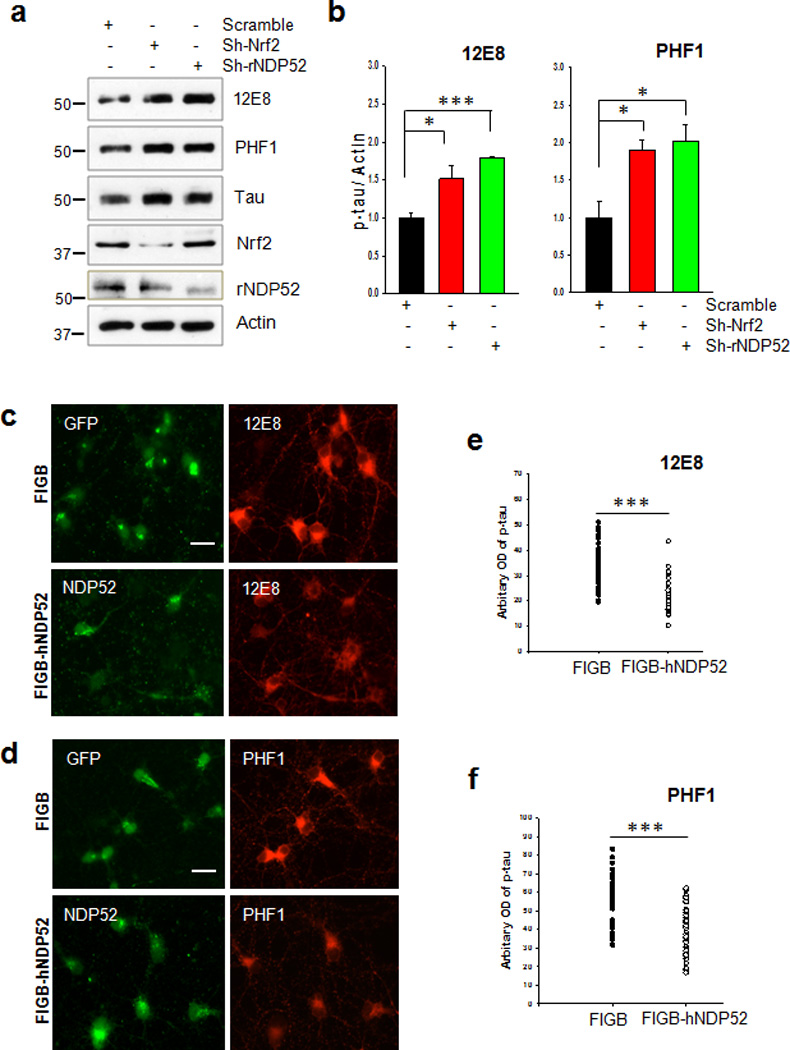
(a) Primary cortical neurons were transduced with lentivirus expressing sh-RNA for Nrf2 or rat NDP52 (rNDP52), or a scrambled sh-RNA at DIV 1, and maintained until DIV 6. The levels of tau phosphorylated at Ser262/Ser356 and Ser396/Ser404 were analyzed by immunoblotting using 12E8- and PHF1-specific antibodies, respectively. Total tau was detected with a polyclonal tau-specific antibody (Tau). The relative molecular masses (kD) are indicated to the left of each blot. (b) Bar graph of the relative optical density of phosphorylated tau normalized to actin. Data shown are mean±SE of three independent experiments and were analyzed using Student’s t test. (*, p<0.05; ***, p<0.001) (c, d) Primary cortical neurons were transduced with a control lentivirus (FIGB) or with one expressing human NDP52 (hNDP52) at DIV 1. To induce autophagy, trehalose (150 mM) was added at DIV 5 and the neurons incubated for 24 h (DIV 6). Primary cortical neurons were fixed with 4% PFA, and stained with the 12E8 or PHF1 antibodies. The optical density of tau phosphorylated at Ser262/Ser356 (12E8). Scale bar = 20 µm. (e) and Ser396/Ser404 (PHF1) (f) in the soma of approximately 30 neurons randomly chosen was analyzed with the ImageJ program. Data were analyzed using Student’s t test (***, p<0.001)
NDP52 interacts with tau through its SKICH domain
To determine how NDP52 may facilitate the clearance of tau, we performed co-immunoprecipitation studies using HEK 293-TN cell lysates transfected with GFP or GFP-tau to determine if NDP52 interacts with tau. Endogenous NDP52 protein co-immunoprecipitated with GFP-tau, but not with GFP alone (Supplementary Fig. 13), indicating that NDP52 associates with tau. Human NDP52 consists of an N-terminal skeletal muscle and kidney-enriched inositol phosphatase carboxyl homology (SKICH) domain (amino acids, 1–127), an intermediate coiled-coil (CC) domain (amino acids, 134–350) and a C-terminal Lin11, Is1-1 and Mec-3 (LIM)-like (LIM-L) domain (amino acids, 395–446)29. To investigate which domain of NDP52 is required for the interaction with tau, co-immunoprecipitation studies were carried out using lysates prepared from CN1.4 naive cortical cells co-transfected with GFP-tau along with different human NDP52 constructs (full length, ΔSKICH, coiled-coil (CC) and ΔLIM-L) as shown in Fig. 9a. Full length and the LIM-L domain-deleted construct of NDP52 co-immunoprecipitated with GFP-tau, however ΔSKICH and CC domain only constructs did not (Fig. 9b), suggesting that NDP52 interacts with tau through its SKICH domain. In CN1.4 mouse cortical cells14 endogenous mouse NDP52, which does not have the LIM-like domain (Fig. 9a), also co-immunoprecipitated with tau (which was induced by treatment with doxycycline (1 µg/ml)14) (Fig. 9c). The results strongly indicate that NDP52 selectively interacts with tau through the SKICH domain. In addition, overexpression of human and mouse NDP52 in CN1.4 cortical cells decreased the levels of phosphorylated tau, but that of ΔSKICH did not, supporting the involvement of the SKICH domain in its binding to phosphorylated tau (Fig. 9d and Supplementary Fig. 14). Further, knock-down of mouse NDP52 using siRNA prevented the SFN-induced reduction of phosphorylated tau (Fig. 9e). Together, the results suggest that NDP52, including mouse NDP52 which does not have the LIM-L domain, interacts with tau via its SKICH domain which is conserved in human, rat and mouse. It was previously reported that p62/SQSTM1 binding to non-ubiquitylated substrates facilitates autophagic clearance40. The LIM-L domain is known to bind specifically to ubiquitin chains of target molecules28. Considering the effect of mouse NDP52 on the degradation of phosphorylated tau, NDP52 likely plays a prominent role in the clearance of phosphorylated tau independent of ubiquitylation.
Figure 9. The SKICH domain of NDP52 interacts with tau.
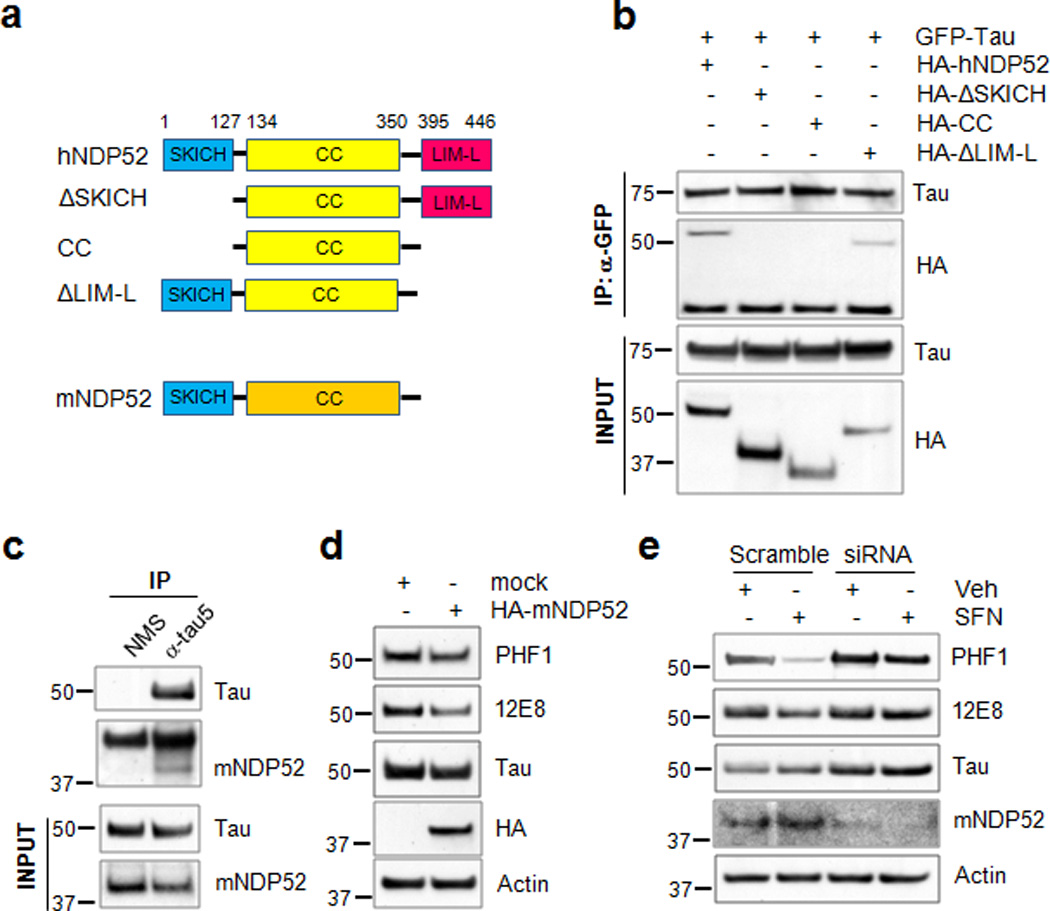
(a) Schematic diagram of human NDP52, the domain-deleted constructs and mouse NDP52. (b) Naïve cortical cells were co-transfected with the plasmids expressing HA-hNDP52 or HA-tagged domain-deleted mutants of human NDP52 together with GFP-tau. Twenty-four hours later cell lysates were prepared and transfected tau was immunoprecipitated and blots probed for both the HA-hNDP52 constructs and GFP-tau. Homogenates (input) used for co-immunoprecipitation were analyzed by immunoblotting using anti-HA or a polyclonal tau-specific antibody (Tau). (c) Stably transfected CN1.4 cortical neurons were maintained in the presence of doxycycline (1 µg/ml) to induce the expression of tau for 24 h. Homogenates from CN1.4 cortical neurons were used for the co-immunoprecipitation of mouse NDP52 (mNDP52) using a Tau5 antibody. Co-precipitation of mNDP52 was examined by immunoblotting using anti-NDP52 antibody. Input of the homogenates used for co-immunoprecipitation was analyzed by immunoblotting using anti-NDP52 or Tau antibody. (d, e) CN1.4 cortical neurons were transfected with the mock or HA-mNDP52 plasmid (d) or with mNDP52 siRNA or scramble RNA as a control (e). The cells transfected with siRNA or scramble RNA were treated with SFN (10 µM) for 24 h. The levels of tau phosphorylated at Ser262/Ser356 and Ser396/Ser404 were analyzed by immunoblotting using 12E8- and PHF1-specific antibodies, respectively. Total tau was detected with a polyclonal tau-specific antibody (Tau). The relative molecular masses (kD) are indicated to the left of each blot.
NDP52 decreases sarkosyl-insoluble tau in AD cases
We performed co-immunoprecipitation studies using lysates prepared from human AD cerebral cortex. Endogenous, human NDP52 protein co-immunoprecipitated when the phosphorylated tau antibodies 12E8 or PHF1 were used, but not when the phospho-independent Tau5 antibody was used (Fig. 10a, b). These results would suggest that NDP52 may more efficiently interact with phosphorylated forms of tau than with tau that is not pathologically phosphorylated.
Figure 10. The amount of sarkosyl-insoluble human tau is inversely proportional to that of human NDP52.
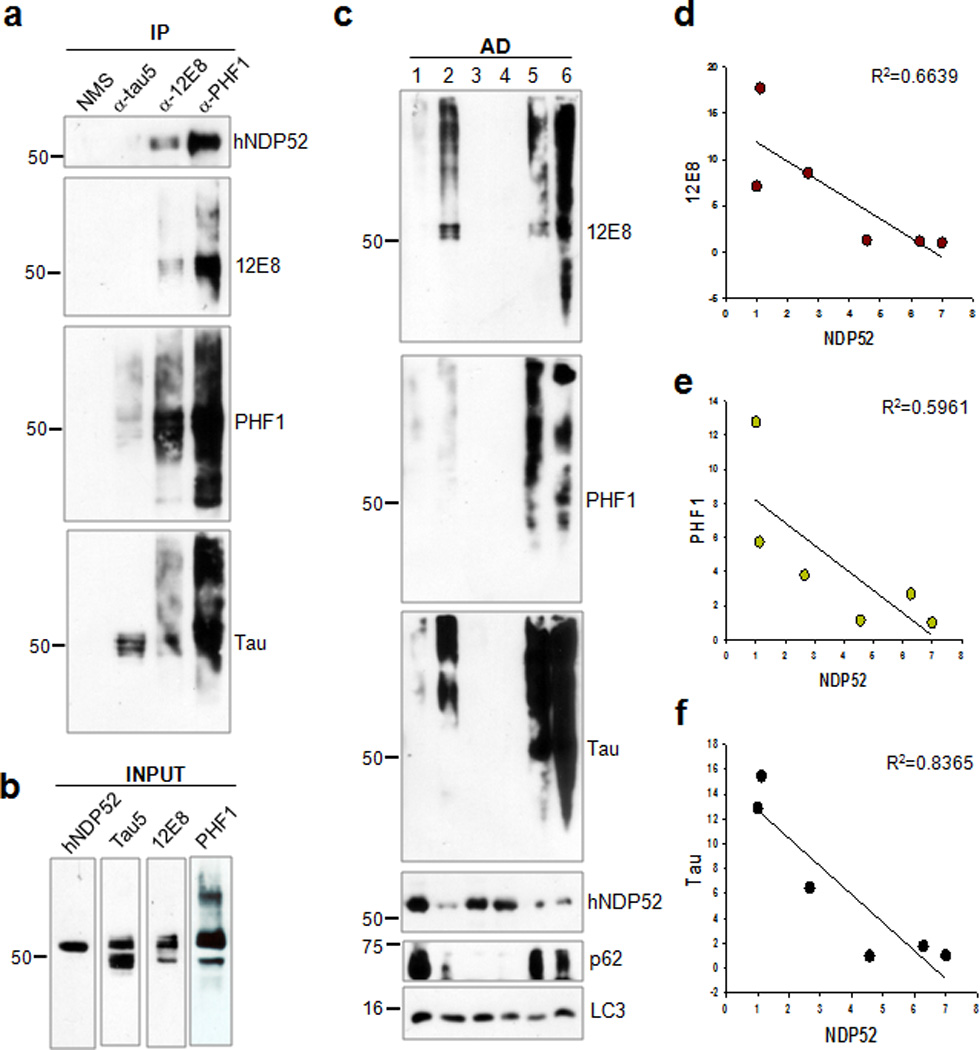
(a) Homogenates from cerebral cortical tissue of a diagnosed AD case were used for the co-immunoprecipitation of human NDP52 (hNDP52) using a Tau5, 12E8 or PHF1 antibody. (b) Homogenates used for co-immunoprecipitation in (a) were analyzed by immunoblotting using anti-NDP52, Tau5, 12E8 or PHF1 antibody. (c) Cerebral cortical tissues from diagnosed AD cases were separated into sarkosyl-soluble and insoluble fractions. The levels of total tau and tau phosphorylated at Ser262/Ser356 and Ser396/Ser404 in the sarkosyl-insoluble fractions were analyzed by immunoblotting using a 12E8, PHF1 and polyclonal to total tau (Tau) antibody, respectively. The levels of hNDP52, p62/SQSTM1 and LC3 were examined by immunoblotting. The relative molecular masses (kD) are indicated to the left of each blot. (d, e, f) The regression graphs show the relative amount of tau phosphorylated at Ser262/Ser356 (12E8) (d), at Ser396/Ser404 (PHF1) (e) or total tau (f) contrast to hNDP52 present in the sarkosyl-insoluble fractions.
Cortical tissues from AD cases (Supplementary Table 1) were then separated into soluble, sarkosyl-soluble and sarkosyl-insoluble fractions. No correlation between the amount of phosphorylated tau and the amount of NDP52 was observed in the soluble and sarkosyl-soluble fractions. In contrast, the amount of total tau, as well as tau phosphorylated at Ser262/Ser356 [12E8] or a Ser396/Ser404 [PHF1], in the sarkosyl-insoluble fractions was inversely proportional to that of human NDP52 (Fig. 10c, d, e, f). This result indicates that NDP52 may prevent the aggregation of tau through the clearance of phosphorylated tau.
Discussion
Tau is normally an abundant soluble neuronal protein; however in AD tau becomes inappropriately phosphorylated and eventually accumulates as neurofibrillary tangles (NFTs)1–3. There is increasing evidence that the accumulation of soluble, but pathologically phosphorylated and oligomerized tau is likely a toxic event compromising neuronal function41–43. Hence the strategy of promoting the clearance of pathological tau in AD has been proposed as a therapeutic approach2, 4. In this context, understanding the mechanisms involved in selectively directing pathological, phosphorylated forms of tau to a degradative pathway is of significance. In this study, we found that NDP52, which is an autophagy adaptor that contains an LIR28, is induced by Nrf2 and may play an important role in directing tau, particularly phosphorylated tau, to the autophagic degradative pathway.
The expression of p62/SQSTM1 is induced by the activation of Nrf222. Since the accumulation of hyperphosphorylated tau was observed in the brains of p62/SQSTM1 (−/−) mice23, we postulated that p62/SQSTM1 might be involved in the Nrf2-mediated degradation of phosphorylated tau (Fig. 1, 3). Although a 1.9 fold decrease in the mRNA levels for p62/SQSTM1 was observed in Nrf2 (−/−) mice, the protein level of p62/SQSTM1 was not changed compared to wild-type mice (Fig. 6b, c). In addition, the protein levels of Keap1 (Kelch-like ECH associated protein 1), an adaptor protein for Cul3-based ubiquitin E3 ligase complex for Nrf2, showed no difference between Nrf2 (−/−) and wild-type mice (Fig. 6c), indicating that the activity of p62/SQSTM1 in Nrf2 (−/−) mice may not be different from that of wild-type mice. Although we could not exclude completely a role for p62/SQSTM1 as an autophagy adaptor for phosphorylated or misfolded tau in our models, our data strongly indicate that NDP52 is likely a primary autophagy adaptor that is facilitating degradation of phosphorylated tau.
Activation of Nrf2 in response to SFN treatment resulted in the induction of NDP52 at both the message and protein level in the neuronal models used in this study (Fig. 6a, d, e and Supplementary Fig. 7). In Nrf2 (−/−) mice the expression levels of p62/SQSTM1 and NDP52 were reduced 1.9 and 7.8 fold, respectively, indicating that the expression of NDP52 is more dependent on Nrf2 activity than that of p62/SQSTM1 (Fig. 6b). Since the degradation of Keap1 is dependent on the activity of p62/SQSTM121, it is plausible that Keap1, a cytosolic inhibitor of Nrf2, would accumulate in p62/SQSTM1 knockout mice, thus blocking the activation of Nrf2. Therefore, it can be speculated that the accumulation of phosphorylated tau observed in p62/SQSTM1 knockout mice23 could in part be due to a decreased expression of NDP52 by means of reduced Nrf2 activity.
A complex consisting of the E3 ligase CHIP (C terminus of Hsp70-interacting protein) in conjunction with several chaperones likely plays a role in maintaining tau in a functional conformation or directing it towards the degradation machinery8, 44–46. In this paradigm, inhibition of the chaperone function of Hsp90 results in CHIP/Hsp70 mediated ubiquitylation of tau, and subsequent degradation45, 47. Interestingly, Hsp90 inhibition did not affect the levels of tau phosphorylated at S262/S356 [12E8]47, and CHIP was unable to bind and ubiquitylate tau phosphorylated at S262/S356 [12E8] site as well8, indicating that tau phosphorylated at S262/S356 [12E8] site may not be recognized by this chaperone network. In this present study, by contrast, Nrf2 activation or NDP52 over-expression in neurons reduced the levels of tau phosphorylated at S396/S404 [PHF1] and/or S262/S356 [12E8] site(s) (Fig. 3, Fig. 8c, d, e, f, 9d and Supplementary Fig. 3a, b, c, 12). Therefore it can be suggested that the chaperone system and NDP52-mediated system may be complimentary in facilitating the clearance of different forms of tau.
Previously, NDP52 had been identified primarily in peripheral tissues as an innate immune autophagy receptor involved in the clearance of cytosolic bacteria27, 28, 38. Our results of these studies demonstrate for the first time that NDP52 is in the brain (Fig. 6, 10 and Supplementary Fig. 8) and expressed in neurons (Fig. 6, 8, 9), and is strongly regulated by Nrf2 (Fig. 6, 7 and Supplementary Fig. 8c, d). Further, we provide unique insight into NDP52 as an autophagy adaptor protein in the clearance of pathologic, phosphorylated tau in neurons (Fig. 8, 9, 10 and Supplementary Fig. 12). In previous studies, the hippocampal expression of Nrf2 or its activation with Nrf2 activators such as SFN, curcumin and α-lipoic acid in murine AD models significantly improved learning and memory retention compared to untreated animals33, 34. It is therefore plausible that NDP52 induced by Nrf2 could play a role in the improvement of AD symptoms through the clearance of pathological, phosphorylated tau. Thus, the pre-emptive activation of Nrf2 through the use of Nrf2 activators could be a possible therapeutic strategy for delaying the onset and slowing the progression of AD.
Methods
Plasmids and reagents
Tau5, PHF1 (phospho-Ser396/404) and 12E8 (phospho-Ser262/356) antibodies were kindly provided by Dr. L. Binder, Dr. P. Davies and Dr. P. Seubert, respectively. Polyclonal tau (A0024) antibody was purchased from Dako. Anti-Nrf2 (ab31163) antibody was obtained from Abcam. Anti-NDP52 (ab68588) antibody for human and rat NDP52 was purchased from Abcam, while the one (H00010241-B01P) used for mouse NDP52 was from Novus Biologicals48. Anti-p62/SQSTM1 (BML-PW9860), Keap1 (10503-2-AP), Hsp70 (ADI-SPA-757), GFP (11-814-460-001) and actin (MAB1501) antibodies were purchased from Biomol, Proteintech, Assay Designs, Roche and Chemicon, respectively. Anti-HA (2367), beclin-1 (3495), histone (12349) and Myc (9B11) (2276) were obtained from Cell Signaling Technology. The pEGFP-human NDP52 plasmid was generously provided by Dr J. Kendrick-Jones. The hemagglutinin (HA)-tagged human NDP52 (HA-hNDP52), SKICH domain-deleted HA-hNDP52 (ΔSKICH), LIM-like domain-deleted HA-hNDP52 (ΔLIM-L) and both SKICH and LIM-like domain-deleted HA-hNDP52 (CC) were gifts from by Dr. T. Into. The pcDNA3-Myc3-human Nrf2 plasmid was obtained from Addgene. The lentiviral vector expressing human NDP52 was constructed by subcloning the amplified gene into the AgeI and BamHI sites of the lentiviral expression plasmid, FIGB (a generous gift from Dr. C. Proschel). The lentiviral vectors expressing shRNA for scrambled, Nrf2 and NDP52 (Supplementary Table 2) were constructed by subcloning the cassette containing the H1 promoter and shRNA sequence from pSuper-GFP/neo (OligoEngine) into the HpaI and PacI sites of the lentiviral plasmid FG12 (Addgene). The vectors pVSV-G (encoding the viral envelope) and psPAX2 (encoding gag-pol) were from Addgene. The human promoter of NDP52 was amplified from human genomic DNA (Affymetrix), and cloned to the SacI and EcoRV sites of the pGL4.14-Luc reporter plasmid (Promega, E6691). The thymidine kinase (TK)-renilla plasmid (E224A) was from Promega. The antioxidant response element (ARE)-Luc reporter plasmid was kindly provided by Dr. M.O. Leonard. The plasmid expressing GFP-tau was constructed by subcloning the human tau gene into the HindIII and BamHI sites of the pEGFP-C1 plasmid (Clontech, 6084-1). The preparation of the plasmids expressing wild-type human tau (T4) and caspase-cleaved tau (T4C3) are described in the previous studies49, 50. In brief T4 was subcloned into the mammalian expression vector, pcDNA 3.1(−) (Invitrogen) at the EcoRI and KpnI sites. T4 in pcDNA 3.1(−) was then used as a template and PCR was performed with forward primer 5′-CGC GGA TCC GCG ATG GCT GAG CCC CGC CAG GAG TTC-3′ and reverse primer 5′-CTG CTC TAG AGC ATC AGT CTA CCA TGT CGA TGC TGC CGG TGG-3 to remove the last 20 amino acids of tau and subcloned into the BamHI and XbaI sites of pcDNA3.1(+). GFP-LC3 was prepared by using RNA extracted from naïve CN1.4 cells, creating cDNA by reverse transcription using Superscript III (Invitrogen, 12574030), followed by PCR and insertion into the EcoRI site of pEGFP-C1 as described previously14. YFP-Bag2 and Myc-p62/SQSTM1 were gifts from by Dr. K.S. Kosik and Dr. T. Johansen, respectively. The plasmid expressing HA-Bag2 was prepared by subcloning the mouse Bag2 gene into the KpnI and EcoRI sites of the pHM6 plasmid (Roche). The plasmids expressing mCherry-hNDP52 and mCherry–mNDP52 were constructed by subcloning human NDP52 and mouse NDP52 gene into BamHI and EcoRI sites and EcoRI and XhoI sites of the pmCherry plasmid (Clontech, 632524), respectively. All constructs were confirmed by sequencing. Sulforaphane (SFN, 574215), trehalose (625625), epoxomicin (324800) and bafilomycin A1 (196000) were purchased from Calbiochem. 3-methyladenine (3-MA, M9281) and chloroquine (C6628) were from Sigma.
Mice
The Nrf2 knockout (−/−) mice on a C57BL/6J background were generously supplied by Dr. M. Yamamoto51 via the RIKEN BioResource Center, Tsukuba, Japan. Wild-type C57BL/6J mice were purchased from Jackson laboratories. Information about the age and sex of the mice used are given in the appropriate figure legends. All animal protocols for this study were approved by the University Committee on Animal Research of the University of Rochester.
Human samples
Human brain tissues (0.2 g) were homogenized in 1ml of ice-cold soluble buffer (0.1 M Mes [pH 7.0], 1 mM EDTA, 0.5 mM MgSO4, 1 M sucrose) containing 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF and 10 µg/ml each of aprotinin, leupeptin, and pepstatin. The homogenates were cleared by centrifugation at 50,000 × g for 20 min at 4°C, and the supernatants were collected as soluble fractions. To prepare the sarkosyl-insoluble fraction, the pellets were resuspended in RAB buffer (100 mM Mes [pH6.8], 10% sucrose, 2 mM EGTA, 0.5 mM MgSO4, 500 mM NaCl, 1 mM MgCl2, 10 mM NaH2PO4, 20 mM NaF) containing 1% N-lauroylsarcosine (Sarkosyl) with protease inhibitors, vortexed for 1 min at room temperature, incubated at 4°C overnight, and then centrifuged at 200,000×g for 30 min at room temperature. The supernatants were collected as sarkosyl-soluble fractions, and the pellets, sarkosyl-insoluble fractions, were resuspended in 1× SDS protein loading buffer and incubated in 95°C for 5 min. Pre-existing AD brain samples were obtained from the University of Rochester Medical Center Brain Bank. The protocol was designated as exempt (exemption #4) by the Institutional Review Board at the University of Rochester as it was a retrospective study of autopsy specimens that were pre-existing and de-identified.
Cell culture
Immortalized mouse cortical CN1.4 naïve cells52 (a gift from Dr. Campagnoni, UCLA) and CN1.4 cells that we engineered to inducibly express full-length tau in the presence of the tetracycline derivative doxycycline14 were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum (FBS), 0.1% gentamicin, 4 mM glutamine, and 10 units/ml penicillin and 100 units/ml streptomycin at 33°C in a humidified atmosphere containing 5% CO2. In this study, cells were treated with doxycycline at a concentration of 1 µg/ml for 24 h to induce tau expression. HEK293-TN and SH-SY5Y cells were grown in DMEM supplemented with 10% FBS, 10 units/ml penicillin and 100 units/ml streptomycin. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Primary cell culture and transduction
Primary cortical neuronal cultures from rat embryonic forebrain were prepared and maintained as described previously with modification53. In brief, whole brains were removed from E18 rats. The cortices were dissected, treated with 0.05% trypsin at 37°C for 30 min, and gently triturated with a fire polished glass Pasteur pipette. Dissociated cells were plated onto 6-well plates or glass-coverslips in 12-well plates coated with poly-D-lysine (40 µg/ml, Millipore, A-003-E) in MEM (Invitrogen, 11090) containing 5% FBS, 20 mM glucose, 2 mM glutamine in the incubator at 37°C in a humidified atmosphere containing 5% CO2. Five hours after plating, medium was replaced with Neurobasal medium (NBM, Invitrogen, 21103-049) supplemented with 0.5 mM glutamine and 2% B27 (Invitrogen, 17504-044). One-half of the medium was replaced with the fresh, complete NBM every 3 day. On DIV 1, 20 µl of virus solution was added directly to the media on the neurons cultured in 6-well plates or 10 µl of virus solution in 12-well plates, and the medium was replaced after 12 h. Experiments were performed on DIV 6–7.
Lentivirus production
HEK 293-TN cells (a generous gift from Dr. C. Proschel, University of Rochester) were transfected with the lentiviral plasmid/psPAX2/pVSV-G using Lipofectamine 2000 (Invitrogen, 11668-019). Twelve hours post-transfection, media was replaced with DMEM supplemented with 1% FetalcloneII, and the cells were incubated at 33°C in a humidified atmosphere containing 5% CO2. Seventy-two hours later the media was collected, spun at 800 × g for 10 min at room temperature and the resulting supernatant was then centrifuged at 60,000 × g for 2 h at 4°C. The pellet was resuspended in 500 µl sterile PBS containing 0.1% BSA per 100 mm dish, and the lentiviral solution was used immediately.
Protein kinase assay
Mouse brain tissue was homogenized in lysis buffer (50 mM Tris-HCl [pH 7.9], 100 mM NaCl, 10 mM MgCl2, 1 mM DTT) containing 0.1% NP-40 and protease inhibitors. Proteins were extracted on ice with periodic vortexing for 30 min, and lysates were cleared by centrifugation at 10,000 × g for 10 min at 4 °C. Ten µg of lysates was incubated with 2 µg of GST-tau protein at 37°C for 30 min in buffer consisting of 25 mM Tris-HCl [pH 7.4], 10 mM MgCl2, 1 mM EGTA, 1 mM DTT, 0.2 mM NaF, 0.1 mM Na3VO4, 1 µg/ml aprotinin, 10 µM dibutyryl-cAMP (Tocris, 1141) and 10 nM okadic acid without or with 1 mM ATP. The reaction was stopped by the addition of 1× SDS-sample loading buffer and boiling at 95°C for 5 min. Proteins were separated by SDS-PAGE and immunoblotted as described below.
Phosphatase assay
Phosphatase activity of mouse brain tissues was quantitated using the Serine/Threonine Phosphatase Assay System (Promega, V2460) by measuring the dephosphorylation of a phospho-peptide, RRA(pT)VA. Briefly, mouse brain tissues were homogenized in the phosphatase storage buffer (10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EDTA, I mM EGTA, 0.5% NP-40) containing protease inhibitors. Proteins were extracted on ice with periodic vortexing for 30 min, and lysates were cleared by centrifugation at 10,000 × g for 10 min at 4°C. Two hundred µl of supernatants was passed through Sephadex G-25 column to remove the residual phosphate in the supernatants. Two µl of samples was incubated with 100 µM of phosphopeptide (RRA(pT)VA) at 37 °C for 30 min in buffer consisting of 250 mM imidazole [pH 7.2], 1 mM EGTA, 50 mM MgCl2, 5 mM NiCl2, 250 µg/ml calmodulin and 0.1% β-mercaptoethanol without or with 20 nM okadaic acid. To terminate the reaction, 50 µl of Molybdate dye with additive mixture was added to the mixture. The mixture was incubated for 10 min at room temperature to allow color development. Absorbance was measured at A600 using the Synergy HT plate reader (BioTek). Phosphate released was determined by comparing absorbance to a standard phosphate curve. PP2A activity was defined as the activity inhibited by the addition of 20 nM okadaic acid to the phosphatase reaction mixture.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) was performed following the protocol from Upstate (Temecula, CA). Briefly, cells were treated with 1% formaldehyde at 37°C for 10 min to cross-link chromatin and lysed with SDS lysis buffer containing protease inhibitors. Then the cell extracts were sonicated to shear DNA and immunoprecipitated using a Nrf2 (1:100) or histone (1:100) antibody. Immunoprecipitates containing protein-DNA complexes were washed according to the instruction. In order to reverse cross-links 20 µl of 5 M NaCl was added to the eluates (500 µl) and heated at 65°C for 4 h. The eluates were then treated with 10 µl of Proteinase K (20 µg/ml) in a buffer containing 20 µl of 1M Tris-HCl (pH 6.5) and 10 µl of 0.5 M EDTA, and incubated at 45°C for 1 h. DNA was recovered by phenol/chloroform extraction and ethanol precipitation. To analyze ChIP products, PCR primers (Supplementary Table 3) were designed to flank ARE sequence in the human NDP52 promoter. PCR was performed with samples of ChIP DNAs for 35 cycles of denaturation at 97°C for 30 s, annealing at 56°C for 30 s, and extension at 72°C for 1 min.
siRNA transfection
For knock-down of endogenous mouse NDP52, mouse NDP52 specific siRNA or scrambled RNA as a control (Supplementary Table 4), was mixed with Oligofectamine (Invitrogen, 12252-011) in serum-free Opti-MEM medium for 15 min. The mixture was loaded onto CN1.4 cortical neurons14 following exchange of the culture medium for serum-free DMEM medium. After 4 h, FBS was added up to 5% and maintained at 33°C in a humidified atmosphere containing 5% CO2 for 24 h. The human beclin-1 specific siRNA (6222) was purchased from Cell Signaling Technology.
Transient transfection and luciferase assays
Cells were transiently transfected with human NDP52 promoter cloned to the reporter plasmid pGL4.14-Luc (Promega) together with the TK-renilla plasmid using Lipofectamine 2000 (Invitrogen). The total amount of DNA used for each well or plate was normalized with the relevant mock vectors. Luciferase activity was measured using the Luciferase assay system (Promega, E2920) and a Luminometer (Turner Designs). Transfection efficiency was normalized with renilla activity.
Immunoblotting
Cells were washed once with PBS and lysed with modified RIPA buffer (10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1 mM EGTA, 1% NP-40, 0.25% sodium deoxycholate, 0.1% SDS) containing 1 mM NaF, 1 mM Na3VO4, 1 mM PMSF and 10 µg/ml each of aprotinin, leupeptin and pepstatin. Proteins were extracted on ice with periodic vortexing for 30–40 min, and lysates were cleared by centrifugation at 10,000 × g for 10 min at 4 °C, and the supernatants were used for immunoblotting following boiling in 1× SDS-sample loading buffer for 5 min. For analysis samples (10–50 µg protein) were separated on 8%, 10% or 12% SDS-polyacrylamide gels followed by transfer to nitrocellulose membranes (Bio-Rad) and immunoblotting with the indicated antibodies: Tau5 (1: 25,000), PHF1 (1: 5,000), 12E8 (1: 5,000), polyclonal tau (1: 500,000), Nrf2 (1: 2,000), NDP52 (1: 2,000), mouse NDP52 (1: 500), p62/SQSTM1 (1: 5,000), Keap1 (1: 2,500), Hsp70 (1: 5000), GFP (1: 5,000), actin (1: 50,000), HA (1: 1,000), beclin-1 (1: 1,000) and Myc (9B11) (1: 1,000) antibodies. Blots were developed with chemiluminescence. All protein concentrations were determined using the BCA method (Sigma). Full blots are shown in Supplementary Fig. 15.
Immunoprecipitation
Human brain tissue (0.2 g) or cells were lysed in NP-40 lysis buffer (0.5% NP-40, 10 mM Tris-HCl [pH 8.0], 150 mM NaCl, 10 mM sodium pyrophosphate, 1 mM EDTA) containing 1 mM NaF, 1 mM Na3VO4, and protease inhibitors. Equal amounts of lysates were incubated with 2 µg of antibodies pre-conjugated with sheep anti-mouse magnetic beads (DYNAL, Invitrogen, 110-31) for 3 h on a rotational shaker at 4°C. After overnight incubation at 4°C, the beads were washed with three changes of NETN wash buffer (0.1% NP-40, 50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA), and boiled in 1× SDS-sample loading buffer for 5 min. Proteins were separated by SDS-PAGE and immunoblotted as described above.
Immunohistochemical staining
Brain slices (30 µm) prepared from mouse brains fixed with 4% paraformaldehyde were washed three times using PBS to remove cryoprotectant, and incubated in TBS containing 10% normal goat serum (NGS) and 0.4% Triton X-100 for 1 h at room temperature. To detect phosphorylated tau, the sections were incubated with a 12E8 (1:500) or PHF1 (1:600) antibody containing 5% NGS and 0.1% Triton X-100 in TBS (incubation solution) for overnight 4°C. On the next day, slices were incubated for 1 h at room with Alexa Fluro 594-conjugated secondary antibody (1:500, Invitrogen) followed by three washes with the incubation solution. For the immunostaining of primary cortical neurons grown on coverslips (18 mm), neurons were fixed with 4% paraformaldehyde in PBS, followed by a permeabilization with TBS containing 0.2% Triton X-100 for 30 min and a pre-blocking with the incubation solution containing 5% NGS and 0.1% Triton X-100 in TBS for 1 h at room temperature. To detect phosphorylated tau, coverslips were incubated overnight at 4°C with a 12E8 (1:1,000) or PHF1 (1:2,000) antibody in incubation solution, followed by incubation with the secondary antibody bound to Alexa 594 fluorophore (Invitrogen). The coverslips were mounted on the glass slides with ProLong® gold antifade reagent (Invitrogen, P36935) following three washes with TBS. Images were photographed using a flurorescence microscope (Carl Zeiss AX10).
Quantitative real-time PCR
For the synthesis of cDNA, total RNA was isolated using Trizol (Invitrogen) according to the manufacture’s protocol. cDNA was generated using 2 µg of RNA, oligo-dT and Superscript III (Invitrogen). Quantitative real-time PCR (qRT-PCR) was performed using the SYBR® Green real-time PCR master mix (Invitrogen, 4344463) on a Bio-Rad IQ5 Real-Time PCR Detection System. Each reaction consisted of 10 µL of cDNA product from the diluted reverse transcription reaction (x30), 2.5 µM of primers (forward and reverse, Supplementary Table 5), 12.5 µl of SYBR® Green real-time PCR master mix. The reactions were incubated in a 96-well plate at 95°C for 5 min, followed by 45 cycles of 95°C for 15 s, 60°C for 20 s and 72°C for 40 s. After the reactions were completed, the threshold was manually set and the threshold cycle (CT) was automatically recorded. The CT is defined as the fractional cycle number at which the fluorescence signal passes the fixed threshold. All reactions were run in three replicates for each sample.
Resazurin assay
The cell viability was determined by monitoring conversion of resazurin to a fluorescent product resorufin. Twenty four hours after treatment of neurons with sulforaphane (SFN) or tert-butylhydroquinone (tBHQ) in 24 well plates, resazurin at a final concentration of 400 µM was added to the media. Two hour after incubation, resorufin fluorescence was measured using the Synergy HT plate reader (BioTek) with excitation at 540 nm and emission at 590 nm. Results are presented as a percent of control cells.
RT-PCR
For the synthesis of cDNA, total RNA was isolated using Trizol (Invitrogen) according to the manufacture’s protocol. cDNA was synthesized using 2 µg of RNA with EcoDry premix (Clontech, 639543). Mouse NDP52 gene was amplified using the PCR premix (Bioneer, Korea) following addition of 1 µL of cDNA product and 1 µM of primers (forward and reverse, Supplementary Table 6) on a PCR machine (Applied Biosystems). The reaction tubes were incubated at 96°C for 5 min, followed by 35 cycles of 96°C for 30 s, 58°C for 30 s and 72°C for 45 s. The PCR products were analyzed by the agarose gel (1.2%) electrophoresis.
Supplementary Material
Acknowledgements
We thank Drs. Lester Binder (Michigan State University), Peter Davies (Albert Einstein College of Medicine), Peter Seubert (Neotope, Inc.), John Kendrick-Jones (MRC Laboratory of Molecular biology, UK), Martin O. Leonard (University College Dublin, Ireland), Christopher Proschel (University of Rochester), Terje Johansen (University of Tromso, Norway), Kenneth S. Kosik (University of California Santa Barbara), Dr. A. Campagnoni (University of California, Los Angeles) and Takeshi Into (Asahi University of School of Dentistry) for providing materials for this study. We thank Dr. Masayuki Yamamoto (Tohoku University, Japan) for providing Nrf2 knockout (−/−) mice via the RIKEN BioResource Center, Tsukuba, Japan. We thank Dr. Mahlon Johnson (University of Rochester Medical Center Brain Bank) for providing AD cerebral cortical samples. This work was supported by National Institutes of Health (NIH) grant NS076789.
Footnotes
Author contributions
C.J. and G.V.W.J. designed the experiments. S.G. performed the ChIP assay. S.P. made brain slices for the immunohistochemical staining. C.J. performed the experiments. Y.N.J. and I.R. provided critical review of the manuscript. C.J. and G.V.W.J wrote the manuscript.
Supplementary Information for this paper can be found at http://www.nature.com/
Naturecommunications
Competing financial interests: The authors declare no competing financial interests.
References
- 1.Jakob-Roetne R, Jacobsen H. Alzheimer's disease: from pathology to therapeutic approaches. Angew Chem Int Ed Engl. 2009;48:3030–3059. doi: 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- 2.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer's disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 3.Ittner LM, Gotz J. Amyloid-beta and tau--a toxic pas de deux in Alzheimer's disease. Nat Rev Neurosci. 2011;12:65–72. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 4.Gong CX, Grundke-Iqbal I, Iqbal K. Targeting tau protein in Alzheimer's disease. Drugs Aging. 2010;27:351–365. doi: 10.2165/11536110-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 5.Zhang JY, Liu SJ, Li HL, Wang JZ. Microtubule-associated protein tau is a substrate of ATP/Mg(2+)-dependent proteasome protease system. J Neural Transm. 2005;112:547–555. doi: 10.1007/s00702-004-0196-x. [DOI] [PubMed] [Google Scholar]
- 6.David DC, et al. Proteasomal degradation of tau protein. J Neurochem. 2002;83:176–185. doi: 10.1046/j.1471-4159.2002.01137.x. [DOI] [PubMed] [Google Scholar]
- 7.Carrettiero DC, Hernandez I, Neveu P, Papagiannakopoulos T, Kosik KS. The cochaperone BAG2 sweeps paired helical filament-insoluble tau from the microtubule. J Neurosci. 2009;29:2151–2161. doi: 10.1523/JNEUROSCI.4660-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dickey CA, et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delobel P, et al. Proteasome inhibition and Tau proteolysis: an unexpected regulation. FEBS Lett. 2005;579:1–5. doi: 10.1016/S0014-5793(05)01501-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feuillette S, et al. Tau is not normally degraded by the proteasome. J Neurosci Res. 2005;80:400–405. doi: 10.1002/jnr.20414. [DOI] [PubMed] [Google Scholar]
- 11.Hamano T, et al. Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci. 2008;27:1119–1130. doi: 10.1111/j.1460-9568.2008.06084.x. [DOI] [PubMed] [Google Scholar]
- 12.Berger Z, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–442. doi: 10.1093/hmg/ddi458. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi: 10.1093/hmg/ddp367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dolan PJ, Johnson GV. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem. 2010;285:21978–21987. doi: 10.1074/jbc.M110.110940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kruger U, Wang Y, Kumar S, Mandelkow EM. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 16.Khurana V, et al. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 2010;6:e1001026. doi: 10.1371/journal.pgen.1001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shimada K, et al. Long-term oral lithium treatment attenuates motor disturbance in tauopathy model mice: implications of autophagy promotion. Neurobiol Dis. 2012;46:101–108. doi: 10.1016/j.nbd.2011.12.050. [DOI] [PubMed] [Google Scholar]
- 18.Clague MJ, Urbe S. Ubiquitin: same molecule, different degradation pathways. Cell. 2010;143:682–685. doi: 10.1016/j.cell.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 19.Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi: 10.4161/auto.7.3.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salminen A, et al. Emerging role of p62/sequestosome-1 in the pathogenesis of Alzheimer's disease. Prog Neurobiol. 2012;96:87–95. doi: 10.1016/j.pneurobio.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Komatsu M, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 22.Jain A, et al. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285:22576–22591. doi: 10.1074/jbc.M110.118976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramesh Babu J, et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008;106:107–120. doi: 10.1111/j.1471-4159.2008.05340.x. [DOI] [PubMed] [Google Scholar]
- 24.Kirkin V, et al. A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell. 2009;33:505–516. doi: 10.1016/j.molcel.2009.01.020. [DOI] [PubMed] [Google Scholar]
- 25.Wenger T, et al. Autophagy inhibition promotes defective neosynthesized proteins storage in ALIS, and induces redirection toward proteasome processing and MHCI-restricted presentation. Autophagy. 2012;8 doi: 10.4161/auto.18806. [DOI] [PubMed] [Google Scholar]
- 26.D'Agostino C, Nogalska A, Cacciottolo M, Engel WK, Askanas V. Abnormalities of NBR1, a novel autophagy-associated protein, in muscle fibers of sporadic inclusion-body myositis. Acta Neuropathol. 2011;122:627–636. doi: 10.1007/s00401-011-0874-3. [DOI] [PubMed] [Google Scholar]
- 27.Mostowy S, et al. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J Biol Chem. 2011;286:26987–26995. doi: 10.1074/jbc.M111.223610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 29.Inomata M, Niida S, Shibata K, Into T. Regulation of Toll-like receptor signaling by NDP52-mediated selective autophagy is normally inactivated by A20. Cell Mol Life Sci. 2012;69:963–979. doi: 10.1007/s00018-011-0819-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tumbarello DA, et al. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol. 2012;14:1024–1035. doi: 10.1038/ncb2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanninen K, White AR, Koistinaho J, Malm T. Targeting Glycogen Synthase Kinase-3beta for Therapeutic Benefit against Oxidative Stress in Alzheimer's Disease: Involvement of the Nrf2-ARE Pathway. Int J Alzheimers Dis. 2011;2011:985085. doi: 10.4061/2011/985085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kensler TW, Wakabayashi N, Biswal S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu Rev Pharmacol Toxicol. 2007;47:89–116. doi: 10.1146/annurev.pharmtox.46.120604.141046. [DOI] [PubMed] [Google Scholar]
- 33.Kanninen K, et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2009;106:16505–16510. doi: 10.1073/pnas.0908397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hur W, Gray NS. Small molecule modulators of antioxidant response pathway. Curr Opin Chem Biol. 2011;15:162–173. doi: 10.1016/j.cbpa.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 35.Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci. 2005;22:1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 36.Sontag E, Nunbhakdi-Craig V, Lee G, Bloom GS, Mumby MC. Regulation of the phosphorylation state and microtubule-binding activity of Tau by protein phosphatase 2A. Neuron. 1996;17:1201–1207. doi: 10.1016/s0896-6273(00)80250-0. [DOI] [PubMed] [Google Scholar]
- 37.Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008;4:849–950. doi: 10.4161/auto.6845. [DOI] [PubMed] [Google Scholar]
- 38.Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–418. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem. 2007;282:5641–5652. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 40.Watanabe Y, Tanaka M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J Cell Sci. 2011;124:2692–2701. doi: 10.1242/jcs.081232. [DOI] [PubMed] [Google Scholar]
- 41.Steinhilb ML, et al. S/P and T/P phosphorylation is critical for tau neurotoxicity in Drosophila. J Neurosci Res. 2007;85:1271–1278. doi: 10.1002/jnr.21232. [DOI] [PubMed] [Google Scholar]
- 42.Berger Z, et al. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brunden KR, Trojanowski JQ, Lee VM. Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments. J Alzheimers Dis. 2008;14:393–399. doi: 10.3233/jad-2008-14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petrucelli L, et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum Mol Genet. 2004;13:703–714. doi: 10.1093/hmg/ddh083. [DOI] [PubMed] [Google Scholar]
- 45.Shimura H, Schwartz D, Gygi SP, Kosik KS. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J Biol Chem. 2004;279:4869–4876. doi: 10.1074/jbc.M305838200. [DOI] [PubMed] [Google Scholar]
- 46.Dickey CA, et al. Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho- and caspase-3-cleaved tau species. J Neurosci. 2006;26:6985–6996. doi: 10.1523/JNEUROSCI.0746-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dickey CA, et al. HSP induction mediates selective clearance of tau phosphorylated at proline-directed Ser/Thr sites but not KXGS (MARK) sites. FASEB J. 2006;20:753–755. doi: 10.1096/fj.05-5343fje. [DOI] [PubMed] [Google Scholar]
- 48.Watson RO, Manzanillo PS, Cox JS. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell. 2012;150:803–815. doi: 10.1016/j.cell.2012.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cho JH, Johnson GV. Glycogen synthase kinase 3 beta induces caspase-cleaved tau aggregation in situ. J Biol Chem. 2004;279:54716–54723. doi: 10.1074/jbc.M403364200. [DOI] [PubMed] [Google Scholar]
- 50.Cho JH, Johnson GV. Glycogen synthase kinase 3beta phosphorylates tau at both primed and unprimed sites. Differential impact on microtubule binding. J Biol Chem. 2003;278:187–193. doi: 10.1074/jbc.M206236200. [DOI] [PubMed] [Google Scholar]
- 51.Itoh K, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 52.Bongarzone ER, et al. Two neuronal cell lines expressing the myelin basic protein gene display differences in their in vitro survival and in their response to glia. J Neurosci Res. 1998;54:309–319. doi: 10.1002/(SICI)1097-4547(19981101)54:3<309::AID-JNR2>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 53.Kress GJ, Dineley KE, Reynolds IJ. The relationship between intracellular free iron and cell injury in cultured neurons, astrocytes, and oligodendrocytes. J Neurosci. 2002;22:5848–5855. doi: 10.1523/JNEUROSCI.22-14-05848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.