

Abstract
Sensitization of adenylyl cyclase (AC) signaling has been implicated in a variety of neuropsychiatric and neurologic disorders including substance abuse and Parkinson's disease. Acute activation of Gαi/o-linked receptors inhibits AC activity, whereas persistent activation of these receptors results in heterologous sensitization of AC and increased levels of intracellular cAMP. Previous studies have demonstrated that this enhancement of AC responsiveness is observed both in vitro and in vivo following the chronic activation of several types of Gαi/o-linked receptors including D2 dopamine and μ opioid receptors. Although heterologous sensitization of AC was first reported four decades ago, the mechanism(s) that underlie this phenomenon remain largely unknown. The lack of mechanistic data presumably reflects the complexity involved with this adaptive response, suggesting that nonbiased approaches could aid in identifying the molecular pathways involved in heterologous sensitization of AC. Previous studies have implicated kinase and Gbγ signaling as overlapping components that regulate the heterologous sensitization of AC. To identify unique and additional overlapping targets associated with sensitization of AC, the development and validation of a scalable cAMP sensitization assay is required for greater throughput. Previous approaches to study sensitization are generally cumbersome involving continuous cell culture maintenance as well as a complex methodology for measuring cAMP accumulation that involves multiple wash steps. Thus, the development of a robust cell-based assay that can be used for high throughput screening (HTS) in a 384 well format would facilitate future studies. Using two D2 dopamine receptor cellular models (i.e. CHO-D2L and HEK-AC6/D2L), we have converted our 48-well sensitization assay (>20 steps 4-5 days) to a five-step, single day assay in 384-well format. This new format is amenable to small molecule screening, and we demonstrate that this assay design can also be readily used for reverse transfection of siRNA in anticipation of targeted siRNA library screening.
Keywords: Bioengineering, Issue 83, adenylyl cyclase, cAMP, heterologous sensitization, superactivation, D2 dopamine, μ opioid, siRNA
Introduction
An adaptive adenylyl cyclase (AC) signaling response known as heterologous- or super-sensitization was first discovered in the laboratory of Nobel Laureate, Dr. Marshall Nirenberg. Dr. Nirenberg proposed that the observed increased AC responsiveness following chronic δ opioid receptor activation was a mechanism involved in opiate tolerance and dependence1. In addition to chronic δ opioid receptor activation, this neuroadaptive response of AC signaling also occurs following persistent activation of several other Gαi/o-coupled receptors2. Notably, many of these receptors are associated with pain, neuropsychiatric and neurological disorders, and include μ/κ opioid, D2/4 dopamine, 5HT1A, and M2/4 muscarinic receptors2. In addition to Dr. Nirenberg's findings, a large body of evidence exists linking sensitization of AC signaling to chronic opioid receptor activation both in vitro and in vivo3-7. Sensitization of AC has also been associated with a variety of diseases involving D2-like dopamine receptors including schizophrenia and Parkinson's disease (for review see reference2). Despite the potential importance of sensitization, the precise mechanism(s) associated with persistent Gαi/o-coupled receptor activation that leads to increased AC responsiveness remains largely unknown.
These studies provide the rationale for examining the mechanisms for sensitization of adenylyl cyclase as an important neurobiological target. Likewise, the physiological relevance of AC signaling8 and the importance that the individual AC isoforms hold in this adaptive response should also be recognized2,9,10. In the context of our research, the general features associated with heterologous sensitization of the recombinant isoforms of AC parallel those characteristics described for studying the endogenous isoforms of AC. Specifically, previous research has found that the activation of Gαi/o proteins and subsequent release/rearrangement βγ subunits are important requirements for receptor induced sensitization of all AC isoforms. Additionally, several studies suggest that signaling from protein kinases and Gβγ subunits are involved in sensitization2,11-13. Individual ACs also display unique and distinct sensitization patterns12. For instance, persistent exposure of D2 receptors to agonists is associated with sensitization of AC1 and AC8 to Ca2+/calmodulin stimulation14,15, whereas the closely related AC3 is not sensitized2. AC2, AC4, and AC7 are closely related, however, only PKC-stimulated AC2 activity is robustly sensitized after prolonged exposure of D2 receptors to agonists7,14,16,17. Additionally, AC5 and AC6 show a marked degree of heterologous sensitization to Gas- and forskolin-stimulated cAMP accumulation following activation of D2 receptors14,18-20, but appear to differ in their requirement for Gβγ subunit-AC interactions21. Although most studies of AC sensitization have used model cell lines (e.g. HEK293 cells expressing individual AC isoforms), it appears that these findings translate to native neuronal cell models4,22. More recently, the effects of AC isoform selective small molecule inhibitors identified in HEK293 cells expressing AC isoforms can also be translated to in vivo behavioral studies23.
The lack of an identified molecular mechanism for heterologous sensitization likely reflects the complexity of the adaptive response as well as the unique regulatory properties of the individual AC isoforms12. Unraveling such complexity is further complicated by the use of cumbersome methodology that has limited academic investigators from employing unbiased approaches. For example, our previous mechanistic studies involved the use of continuously cultured cellular models using 24- and 48-well tissue culture format15. Cultured cells were typically grown for 48 hr and then subjected to agonist drug treatment (2-18 hr) followed by a series of cell washes and incubations (Figure 1). AC-isoform specific cAMP accumulation protocols were then employed followed by measurement of cAMP accumulation using a laborious and time consuming [3H]cAMP binding methodology15,24. The duration from start to finish for each assay generally required a total of four to five days from cell plating to data analysis (Figure 1). The application of new technologies and automation has led to marked enhancements for sensitization studies in the industrial and HTS center setting. For example, a group working with the National Center for Chemical Genomics reported a two day HTS assay procedure for identifying small molecule inhibitors of μ opioid receptor induced sensitization in 1,536-well format25.
The present article describes our efforts to develop an HTS assay for studies of heterologous sensitization using technologies that are available at most academic research institutions. This strategy was accomplished by incorporating the use of cryopreserved cells from cell models heterologously expressing the D2 dopamine receptor in combination with endogenous or individual recombinant adenylyl cyclase isoforms (CHO-D2L or HEK-AC6/D2L). To improve our throughput, we redesigned our 48 well sensitization assay (ca. >20 steps over 4-5 days) to a five-step, single day assay in 384-well format that was essentially "mix and read". The new format uses a commercially available homogenous time resolved fluorescence (HTRF) assay to measure cAMP accumulation in intact cells with a multi mode plate reader. The assay is robust and amenable to small molecule screening, and can be effectively applied to screen for inhibitors of heterologous sensitization. In addition, we provide data that allows the use of this assay with reverse transfection of siRNA for targeted or genome wide siRNA library screening with only a minor modification to the general approach.
Protocol
1. Expansion and Cryopreservation of Assay Ready Cells
Culture CHO-K1-DRD2L (CHO-D2L) cells on a 15 cm2 cell culture dish in Ham's F12 media supplemented with 1.0 μM L-glutamine, 800 μg/μl G418, 300 μg/μl hygromycin, 100 u/μl penicillin, 100 μg/μl streptomycin, and 10% fetal bovine serum (FBS).
Incubate the cells at 37 °C in a humidified incubator with 5% CO2 until the cells are 90-95% confluent. Wash the cells with 10 μl Phosphate Buffered Saline (PBS), and harvest the cells by adding 3 μl of cell dissociation buffer for 5 min at 37 °C. Resuspend the cells using 12 μl of the culture media and count cells using trypan blue exclusion.
Centrifuge the cell suspension at 500 x g for 5 min at room temperature. Aspirate the supernatant and resuspend the cell pellet in 5 μl of freezing media (10% dimethyl sulfoxide, 90% FBS). Dilute the cell suspension to achieve the desired cell concentration (e.g. 1-20 x 106 cells/μl).
Aliquot 1.0 μl of cell solution to each cryovial. Incubate the cryovials in a cell freezing container at -80 °C overnight. Transfer the cryovials to a liquid N2 tank for long term storage.
2. Plating Assay Ready Cells (and Reverse Transfection Option)
Rapidly thaw a frozen cryovial of cells in a 37 °C water bath. Once cells are thawed, transfer the cells to a 15 μl conical tube containing 9 μl of Opti-MEM, and mix by inverting the tube 3-5x.
Centrifuge the cells at 500 x g for 5 min at room temperature. Aspirate the supernatant and resuspend the cells in 1 μl Opti-MEM.
Count the cells using trypan blue exclusion to determine cell viability and dilute the viable cells as necessary (e.g. 3 x 105 cells/μl concentration) in Opti-MEM. Plate 10 μl/well of cells in a tissue culture treated 384-well plate using a multichannel pipette.
Make serial dilutions of cAMP in Opti-MEM to generate a standard curve for estimating cAMP production by the cells (according to the manufactures recommendations - see Section 7). Add 10 μl/well of the cAMP standards to the plate. Note: a single standard curve can be prepared on a separate plate or blank wells on one of the assay plates.
Centrifuge the plate at 100 x g for 15 sec at room temperature, and incubate at 37 °C in a humidified incubator with 5% CO2 for 1 hr.
3. Reverse siRNA Transfection Option*
This section is optional and relevant to Figure 3.
Prepare a solution of siRNA in RNase-free ddH2O (e.g. 0.4 pmol/μl). Add 5 μl to individual wells of 384-well plate, and centrifuge the plate at 100 x g for 15 sec at room temperature.
Dilute Lipofectamine 2000 in Opti-MEM by a factor of 0.006 (e.g. add 6 μl of Lipofectamine 2000 to 1,000 μl of Opti-MEM), and mix by pipetting up and down.
Incubate the Lipofectamine 2000/Opti-MEM solution for 5 min at room temperature. Add 5 μl of the diluted Lipofectamine 2000/Opti-MEM solution to individual wells of 384-well plate that already contains siRNA. Centrifuge the plate at 100 x g for 15 sec at room temperature.
Incubate the plate for 30 min at room temperature.
Plate assay ready cells as described above (Section 2). Centrifuge the plate at 100 x g for 15 sec at room temperature.
Return assay plate to humidified incubator at 37 °C (5% CO2) for 48-96 hr as determined for targeted gene knock down (see discussion).
4. Small Molecule Screening
Dilute the drug of interest (e.g. small molecule inhibitors) in Opti-MEM to 6x the desired final concentrations. Serial dilutions can be completed using hand-held pipettes or a liquid handling station.
Add 2.5 μl/well of the test compound or buffer containing vehicle (e.g. DMSO) to the side of the wells using a multichannel pipette.
Centrifuge the plate at 100 x g for 15 sec at room temperature (incubation is optional).
5. Persistent Agonist Treatment
Prepare a 600 nM (i.e. 6 times the desired final concentration of 100 nM) solution of quinpirole in Opti-MEM.
Add 2.5 μl/well of the 600 nM quinpirole solution to the side of the wells using a multichannel pipette.
Centrifuge the plate at 100 x g for 15 sec at room temperature, and incubate at 37 °C in a humidified incubator with 5% CO2 for 2 hr.
6. Stimulation of cAMP Accumulation
During the 2 hr incubation, prepare the stimulation solution in Opti-MEM. The stimulation solution is comprised of 40 μM forskolin, 2 μM 3-isobutyl-1-methyxanthine (IBMX), and 4 μM spiperone (all concentrations in step 6 are 4x the desired final concentration).
Add 5 μl/well of the stimulation solution to the side of the wells using a multichannel pipette.
Centrifuge the plate at 100 x g for 15 sec at room temperature, and incubate at room temperature for 1 hr.
7. Quenching and Estimation of cAMP Accumulation
Production of cAMP by the cells is measured using the cAMP dynamic 2 kit according to manufacturer’s instructions. Briefly, reconstitute anti-cAMP-cryptate and cAMP-d2 in distilled water. Freeze aliquots at -20 °C for short term storage.
Dilute one aliquot of anti-cAMP-cryptate and one aliquot of cAMP-d2 separately in the lysis buffer according to the manufacturer’s instructions to make working solutions.
Add 10 μl/well of the anti-cAMP-cryptate working solution and 10 μl/well of the cAMP-d2 working solution to the 384-well plate using a multichannel pipette.
Centrifuge the plate at 100 x g for 15 sec at room temperature, and incubate at room temperature for 1 hr.
Read the plate in a fluorescence plate reader (auto scale setting for sensitivity) using an excitation of 337 nm, and measure the emissions at 620 nm and 665 nm per manufactures instructions.
Apply ratiometric analysis to assess cAMP standard curve per manufactures instructions. Using the resulting values, extrapolate the estimated cAMP accumulation in the test wells using data analysis software.
Representative Results
Part I. Developing a 384 well heterologous sensitization assay for identifying small molecule inhibitors using a commercially available cell model.
To study heterologous sensitization in a cell model, we made a number of improvements that enabled us to streamline the assay into a "mix and read" format. Several of the key modifications are highlighted below, and are described in more detail in the discussion. The first key step was modifying our cell culture from continuously cultured cells to cryopreserved cells26. We now routinely culture batches of cells that can be cryopreserved at concentrations between 1-20 x 106 cells/μl. For each respective assay, cell stocks are thawed, diluted, counted, and then plated directly in 384-well plates. Eliminating the need of a culture period and increasing the convenience of the assay. The format of our previous sensitization assay utilized 48-well tissue culture plates, and involved a number of wash, decant, transfer steps, and a filtration-based [3H]cAMP binding assay (Figure 1). Thus, this format for sensitization was not amenable to high throughput applications. Therefore, we exerted significant efforts to streamline the assay first to a 96-well and then a 384-well format amendable to high throughput screening. The optimization involved elimination of wash steps and evaluation of multiple types of culture/assay plates, varying cell densities, and different incubation periods. We also explored several methodologies for cAMP detection and found that the well characterized HTRF cAMP technology used in the present protocol was highly reproducible, reliable, and extremely stable. This assay platform is based on immunocompetition between the cAMP produced by the cells and labeled cAMP (i.e. cAMP-d2) provided by the kit. We have now successfully developed and applied protocols for heterologous sensitization in 384-well format that are essentially "mix and read" (right panel Figure 1).
Our initial goal was to generate a high-throughput D2L receptor sensitization assay using commercially available CHO-D2L cells (human DRD2L, accession number: NM_000795) that endogenously express AC6 and AC727. Cryopreserved CHO-D2L cells were plated at 3,000 cells/well in a 384-well tissue culture plate, and were equilibrated for 1 hr at 37 °C in a humidified incubator. The plates were removed from the incubator and increasing concentrations of the D2 agonist quinpirole were added at room temperature. The cells were then incubated for 2 hr at 37 °C in a humidified incubator. Cyclic AMP accumulation was then initiated by the addition of 10 µM forskolin (a direct activator of AC). The cAMP accumulation buffer contained a phosphodiesterase inhibitor, isobutylmethylxanthine (IBMX), as well as the D2 antagonist, spiperone (1 µM, Ki for D,2L = 0.07 nM), in order to preclude residual D2 receptor activation during the cAMP accumulation period. The cells were incubated at room temperature for 1 hr. The assay was terminated by the addition of a lysis buffer containing the HTRF cAMP reagents. The studies revealed that a 2 hr pretreatment with quinpirole enhanced subsequent forskolin-stimulated cAMP accumulation in a dose-dependent manner consistent with heterologous sensitization (Figure 2A). The results of this experiment demonstrate that sensitization assays can be performed in a 384-well format. The new assay format reduced the number of steps from more than 20 to 4 steps without the need for wash or decant steps making it a "mix and read" assay (Figure 1).
The second series of experiments explored whether this new streamlined assay could be utilized to assess inhibitors of sensitization in the CHO-D2L model. Cryopreserved CHO-D2L cells were plated in a 384 well tissue culture plate at a density of 3,000 cells/well in Opti-MEM. Known D2 antagonists were added to block D2 receptor-induced sensitization. Opti-MEM containing 100 nM quinpirole (final concentration) was then added to a total volume of 15 μl, and the cells were incubated for 2 hr at 37 °C in a humidified incubator. Following the incubation, cAMP accumulation was initiated by the direct addition of forskolin (10 µM final concentration), and the assay was terminated and cAMP measured using HTRF. The initial results revealed that pretreatment with spiperone or haloperidol (prototypical D2 antagonists) completely prevented quinpirole induced sensitization of forskolin stimulated cAMP accumulation (Figure 2B). These results provide validation that this approach can be utilized to identify small molecule inhibitors of D2 receptor-induced sensitization. In a second experiment, we used this method to assess the potency of a series of D2 antagonists to test whether these compounds inhibit sensitization. Similar to the previous results, these studies demonstrated that the D2 antagonists inhibited agonist induced sensitization in a dose dependent manner (Figure 2C). The rank order of potency for inhibition of sensitization was consistent with their actions as D2 receptor antagonists and their previously described pharmacology at D2 receptors28.
Part II. Application of the 384-well sensitization assay for reverse transfection of siRNA in screening endeavors.
Our next objective was to develop a 384-well sensitization assay that can be used to conduct scalable reverse transfection siRNA library screening. Preliminary studies were performed using an HEK cell model that heterologously expresses both D2L dopamine receptors (rat DRD2L, accession number: NM_012547) and AC6 (rat ADCY6, accession number: NC_005120) (i.e. HEK-AC6/D2L cells). The first experiments assessed the ability to demonstrate agonist induced sensitization of AC in the cell line. Briefly, cryopreserved AC6/D2L cells were plated (1,000 cells/well) in a 384-well tissue culture plate. Opti-MEM containing 1 µM quinpirole (final concentration) was added to a total volume of 15 μl, and the cells were incubated for 2 hr at 37 °C in a humidified incubator. Following the incubation, cAMP accumulation was initiated by the addition of increasing concentrations of forskolin. The assay was terminated by the addition of lysis buffer containing the HTRF reagents. The results revealed that exposure of cells to the agonist quinpirole for 2 hr led to a marked enhancement of forskolin-stimulated cAMP accumulation (Figure 3A). The increased accumulation of cAMP at both 100 nM and 300 nM of forskolin was greater than 15-fold compared to vehicle-treated cells (Figure 3A).
Next, we used published siRNA methodologies29,30 to guide our optimization efforts which included an assessment of various reverse transfection conditions (e.g. transfection reagents, transfection efficiency, cell density, incubation time, etc.). Preliminary studies used siGloRed, an indicator of transfection, in combination with Lipofectamine 2000 to determine optimal reverse transfection conditions. The experiments indicated that 2-4 pmol siRNA/5 μl H2O combined with 0.03 μl Lipofectamine 2000/5 μl Opti-MEM provided a nearly 95% transfection efficiency at 72 hr without any observable toxicity in HEK cells (data not shown). We then examined the effect of Gαs siRNA on heterologous sensitization of forskolin stimulated cAMP accumulation in AC6/D2L cells. The Gas siRNA was proposed as a potential positive control because Gαs has been identified as a key component of heterologous sensitization for several AC isoforms (i.e. AC1, AC2, AC5, and AC6)19-21. Furthermore, the 15 fold sensitization signal in our AC6/D2L cell model (Figure 3A) provides a robust signal to noise window to assess the efficiency of reverse transfection. Using the conditions described above, we completed preliminary studies assessing Gαs siRNA as a positive control, and our results demonstrated a robust blockade (>90%) of sensitization (data not shown).
The dramatic siRNA-induced reduction in the sensitization signal provides an appropriate positive control for the siRNA screening process. To obtain a more quantitative assessment for siRNA screening of AC heterologous sensitization, we generated data in which a Z' factor was calculated for a series of test conditions using the AC6/D2L cells31. For this experiment, we combined 2 pmol Gαs siRNA, or the non targeting siRNA control, in 5 μl with 0.03 μl Lipofectamine 2000/5 μl Opti-MEM per well in a 384-well plate (total volume 10 μl). Cryopreserved cells were thawed, resuspended in Opti-MEM and then added to individual wells containing siRNA/Lipofectamine mixture using several cell densities (i.e. 500-5,000 cells/10 μl/well). The duration of quinpirole treatment (2 hr vs. 18 hr) as well as the final concentration of forskolin (100-300 nM) were also explored in our AC6/D2L sensitization assay. The results of these experiments revealed that a robust Z' factor was obtained in the following conditions: 750 cells/well, 72 hr transfection duration, 2 hr quinpirole treatment, and 300 nM forskolin to stimulate cAMP accumulation (Figure 3B). Under these conditions, we obtained a 15 fold sensitization response that was reduced by 94% in the presence of Gαs siRNA (Z' of 0.6 for Gαs siRNA vs. control siRNA).
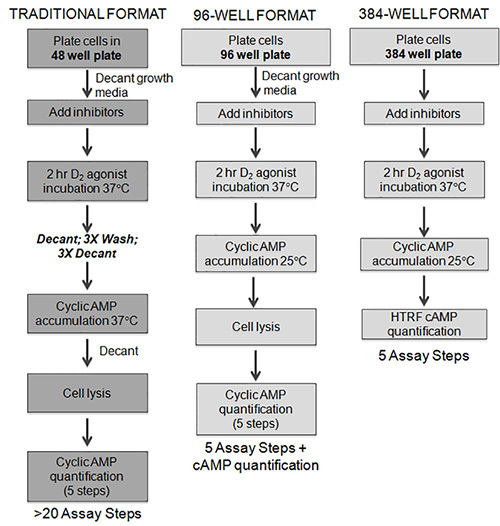 Figure 1. In the traditional format, cells are plated in a 48-well format and grown to confluence over 48 hr. The growth media is decanted, small molecule inhibitors added, followed by addition of the D2 agonist. After the 2 hr incubation, assay media is decanted and the cells are subjected to a series of wash/decant steps. Next, AC is stimulated and the cells are lysed. Cyclic AMP accumulation is then assessed using [3H] cAMP binding assay which is a 2 day assay requiring a filtration step and scintillation counting. While the 96 well format eliminated many of the wash and decant steps, we found that this format was not readily amenable to HTS or automation. The HTS format uses cryopreserved cells that are plated directly into 384-well plates. These cryopreserved cells are "assay ready" following a 1 hr equilibration. After this initial incubation period, small molecule inhibitors can be added followed by the addition of D2 agonist for a 2 hr incubation. AC is stimulated, and the cells are lysed using the HTRF detection reagents in the assay plate.
Figure 1. In the traditional format, cells are plated in a 48-well format and grown to confluence over 48 hr. The growth media is decanted, small molecule inhibitors added, followed by addition of the D2 agonist. After the 2 hr incubation, assay media is decanted and the cells are subjected to a series of wash/decant steps. Next, AC is stimulated and the cells are lysed. Cyclic AMP accumulation is then assessed using [3H] cAMP binding assay which is a 2 day assay requiring a filtration step and scintillation counting. While the 96 well format eliminated many of the wash and decant steps, we found that this format was not readily amenable to HTS or automation. The HTS format uses cryopreserved cells that are plated directly into 384-well plates. These cryopreserved cells are "assay ready" following a 1 hr equilibration. After this initial incubation period, small molecule inhibitors can be added followed by the addition of D2 agonist for a 2 hr incubation. AC is stimulated, and the cells are lysed using the HTRF detection reagents in the assay plate.
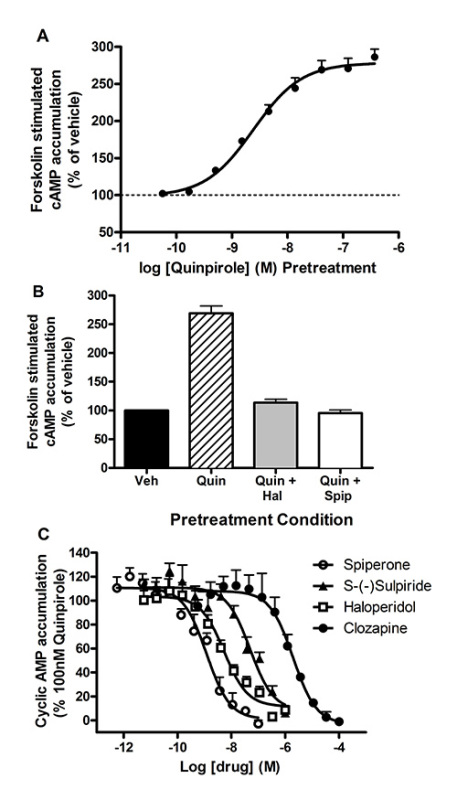 Figure 2. Cryopreserved CHO-D2L cells were plated directly into 384 well assay plates at 3000 cells/well and equilibrated for 1 hr. A.) Increasing concentrations of the D2 agonist, quinpirole, were added and cells were incubated for 2 hr at 37 °C in a humidified incubator. Cyclic AMP accumulation was stimulated with 10 µM forskolin (final concentration) in the presence of spiperone and IBMX for 1 hr at room temperature. The reaction was quenched using HTRF cAMP assay reagents (n=1, representative of at least 3 experiments). Data are expressed as a percent response of vehicle-treated cells. B.) CHO-D2L cells were pretreated in the absence (control) or the presence of the indicated D2 receptor antagonists (i.e. spiperone or haloperidol). Quinpirole (100 nM) was added, and the cells were incubated for 2 hr to induce sensitization. AC was stimulated with 10 μM forskolin (final concentration) for 1 hr at room temperature. Cyclic AMP was determined using HTRF. Data are expressed as a percent response of vehicle-treated cells. C.) CHO-D2L cells were pretreated in the absence (control = 100%) or the presence of increasing concentration of indicated D2 receptor antagonists. Quinpirole (100 nM) was added, and the cells were incubated for 2 hr to induce sensitization. AC was stimulated with 10 μM forskolin (final concentration) for 1 hr at room temperature. Cyclic AMP was determined using HTRF. Data are expressed as a percent of the quinpirole-induced sensitization response. Click here to view larger image.
Figure 2. Cryopreserved CHO-D2L cells were plated directly into 384 well assay plates at 3000 cells/well and equilibrated for 1 hr. A.) Increasing concentrations of the D2 agonist, quinpirole, were added and cells were incubated for 2 hr at 37 °C in a humidified incubator. Cyclic AMP accumulation was stimulated with 10 µM forskolin (final concentration) in the presence of spiperone and IBMX for 1 hr at room temperature. The reaction was quenched using HTRF cAMP assay reagents (n=1, representative of at least 3 experiments). Data are expressed as a percent response of vehicle-treated cells. B.) CHO-D2L cells were pretreated in the absence (control) or the presence of the indicated D2 receptor antagonists (i.e. spiperone or haloperidol). Quinpirole (100 nM) was added, and the cells were incubated for 2 hr to induce sensitization. AC was stimulated with 10 μM forskolin (final concentration) for 1 hr at room temperature. Cyclic AMP was determined using HTRF. Data are expressed as a percent response of vehicle-treated cells. C.) CHO-D2L cells were pretreated in the absence (control = 100%) or the presence of increasing concentration of indicated D2 receptor antagonists. Quinpirole (100 nM) was added, and the cells were incubated for 2 hr to induce sensitization. AC was stimulated with 10 μM forskolin (final concentration) for 1 hr at room temperature. Cyclic AMP was determined using HTRF. Data are expressed as a percent of the quinpirole-induced sensitization response. Click here to view larger image.
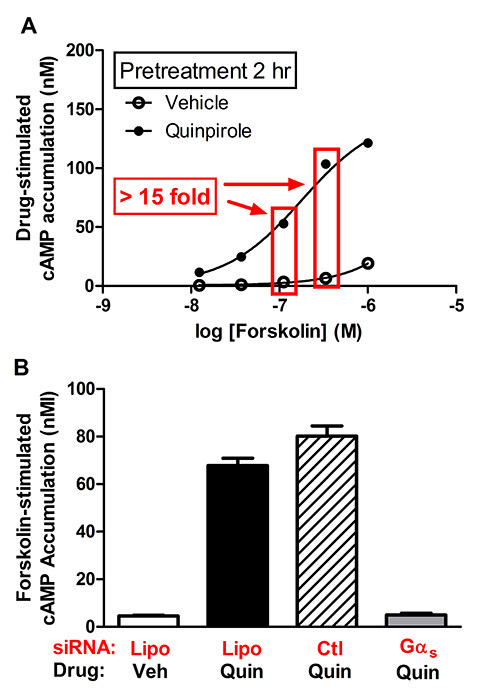 Figure 3. Cryopreserved HEK-AC6/D2L cells were plated at 1000 cells/well and equilibrated for 1 hr. A.) Cells were incubated with vehicle or 100 nM quinpirole for 2 hr in a humidified incubator. AC was stimulated with increasing concentrations of forskolin for 1 hr at room temperature. Cyclic AMP was determined using HTRF. B.) Reverse transfection of Gαs siRNA blocks sensitization in AC6/D2L cells. Gαs siRNA or the nontargeting siRNA control (2 pmol) was combined with 0.03 μl Lipofectamine2000 (Lipo) in a total volume of 10 μl, and the solution was added to individual wells in a 384-well plate. Cryopreserved AC6/D2L cells were then added for the reverse transfection and incubated for 70 hr at 37 °C in a humidified incubator. The D2 receptor agonist, quinpirole (1 μM final), or vehicle was added followed by a 2 hr incubation. Cyclic AMP accumulation was stimulated with 300 nM forskolin (final concentration) for 1 hr and measured using HTRF. Click here to view larger image.
Figure 3. Cryopreserved HEK-AC6/D2L cells were plated at 1000 cells/well and equilibrated for 1 hr. A.) Cells were incubated with vehicle or 100 nM quinpirole for 2 hr in a humidified incubator. AC was stimulated with increasing concentrations of forskolin for 1 hr at room temperature. Cyclic AMP was determined using HTRF. B.) Reverse transfection of Gαs siRNA blocks sensitization in AC6/D2L cells. Gαs siRNA or the nontargeting siRNA control (2 pmol) was combined with 0.03 μl Lipofectamine2000 (Lipo) in a total volume of 10 μl, and the solution was added to individual wells in a 384-well plate. Cryopreserved AC6/D2L cells were then added for the reverse transfection and incubated for 70 hr at 37 °C in a humidified incubator. The D2 receptor agonist, quinpirole (1 μM final), or vehicle was added followed by a 2 hr incubation. Cyclic AMP accumulation was stimulated with 300 nM forskolin (final concentration) for 1 hr and measured using HTRF. Click here to view larger image.
Discussion
In an effort to facilitate studies of heterologous sensitization, we have extensively modified our previous method achieving a streamlined "mix and read" format that is amendable to high-throughput screening and mechanism of action examination. The major modifications to our protocol can be summarized as follows: 1) the use of cryopreserved cells as "assay-ready" reagents; 2) the miniaturization of the assay into 384-well format; and 3) the utilization of the HTRF measurement of cAMP.
The adoption of cryopreservation for our cell based assays has greatly improved efficiency and reproducibility for our day to day experiments and screening endeavors. This approach is an industry standard, and can be readily translated to the academic research setting26. To improve cell culture efficiency, vials of cells are used as "off the shelf reagents" in that cell stocks are thawed, diluted, and then plated directly in 384-well plates for each assay. We previously incorporated cryopreserved cells in studies aimed at identifying antagonists of invertebrate G protein coupled receptors32,33. This methodology was advantageous in that the initial work was not performed in our laboratory and necessitated transporting cultured cells across campus which presented challenges and likely introduced variability. Utilizing our new approach, frozen cells can now be immediately thawed and diluted on site prior to the initiation of the assay. In addition to improving reproducibility, cryopreservation also eliminated the 48 hr time period from cell plating to assay execution (Figure 1). We have successfully applied the cryopreservation approach in stable and transient transfection studies using a variety of recombinant receptors and subtypes of ACs with functional readouts including cAMP accumulation and β-arrestin recruitment (Brust, T.F, Ejendal, K.F.K., and Watts, V.J. unpublished observations). Despite our positive experiences to date, researchers should verify that their receptor or target, and model cell system is compatible with cryopreservation prior to initiating any large scale studies.
A second modification that was important to our efforts to streamline the sensitization assay involved elimination of decant and wash steps (Figure 1). These changes were concurrent with our miniaturization of the assay to a 96-well format (Conley and Watts, unpublished observations). Specifically, a modified sensitization protocol was designed where cAMP accumulation was initiated by the direct addition of forskolin in assay buffer following D2 agonist incubation in the absence of any wash or decant steps (see middle panel Figure 1). Additionally, the cAMP accumulation buffer contained a relatively high concentration of spiperone (1 µM), a D2 antagonist that has a Ki value of 0.07 nM for D2 receptors15. This 100 fold excess concentration of spiperone results in complete D2 receptor occupation during the cAMP accumulation phase, thereby precluding residual D2 receptor activation by agonists (e.g. quinpirole) during the forskolin stimulated AC step15. This change eliminated the three decant and three wash steps following the initial agonist incubation. As part of the 96-well format development, we were also able to eliminate the final decant step prior to quenching and lysing the cells. This modification was accomplished by increasing the concentration of our lysing reagent (i.e. trichloroacetic acid; TCA) from 3-9% and adding the reagent directly to the cAMP accumulation buffer. The promising results of the modified 96 well sensitization assay provided support for the conversion of the sensitization assay to a 384-well format. Unfortunately, our previous methodology for quantifying cAMP was a laborious filtration-based [3H] cAMP protein binding assay (>5 steps). For example, the assay is generally completed over the course of two days to allow for filter drying and scintillation counting15,24. These drawbacks made the [3H] cAMP protein binding impractical to utilize for a high throughput 384-well format.
The third key development came through incorporation of methodologies for measuring and quantifying cAMP in a manner amenable to 384-well format. For example, we have significant experience using a cAMP Response Element (CRE)-luciferase-based reporter system for relative cAMP measurements in a 384-well format32,33. Although this approach has been successfully used by our laboratory for many studies of Gαs-coupled receptors, CRE-luc reporters are subject to a number of false positives and negatives during screening assays34. Additionally, our preliminary D2 sensitization studies with this approach were not encouraging in that we observed apparent feedback inhibition of the luciferase signal (data not shown). Therefore, we focused our efforts on the implementation of GloSensor cAMP technology by constructing and characterizing stable cell lines using both their "first" and "second" generation GloSensor vectors. The GloSensor is an engineered luciferase reporter containing a cAMP binding site within the luciferase protein35. Although the system is very useful for kinetic cAMP quantification, the method was not effective for measuring agonist-induced sensitization (Conley and Watts, unpublished observations). We also explored a BRET based cAMP biosensor36 for our studies as a cost effective means to assess cAMP accumulation. Unfortunately, the background noise associated with the transiently transfected biosensor limited our ability to adapt this method to our sensitization assay. Lastly, we evaluated several kits employing homogenous time resolved fluorescence (HTRF) as an approach to measure cAMP in 384-well format. We found that this established methodology was most compatible with academic laboratories and screening facilities in that they were robust, sensitive, stable, and easy to use. The primary disadvantage for academic investigators is the cost of the reagents, however bulk purchase of reagents and miniaturization to 384 well format makes the price more than competitive with other kits, including ELISA based assays and our previous "homemade" [3H] cAMP protein binding assay15.
The representative work demonstrates the applicability of the 384-well sensitization assay to studies of D2 dopamine receptor induced sensitization of AC signaling in recombinant cell lines. In several preliminary studies, we have used this assay with minor modifications to assess D2 dopamine receptor and μ opioid receptor induced sensitization of several AC isoforms (Table 1). These studies highlight the general applicability of the 384-well assay to multiple cell lines, receptor subtypes, and AC isoforms. Those interested in developing similar assay formats would be encouraged to explore variables such as cell density, agonist pretreatment times, AC activation protocols/conditions, and HTRF cAMP detection methodology for their cellular models. The antagonist data presented herein demonstrate the ability of the assay to identify small molecule inhibitors of sensitization. The potency values are consistent with those determined with other functional assays28 as well as affinity (Ki) values obtained using radioreceptor binding assays (see: http://pdsp.med.unc.edu/pdsp.php). The present assay design could readily accommodate HTS efforts where compounds are delivered via a pintool or are preplated in assay ready plates. In addition to the sensitization assays described, the methodologies applied here should be applicable to other adaptive assays where multiple drugs/reagents are applied prior to an end point measure in 384-well format.
We have attempted to highlight important development steps for the entire 384-well assay, but also want to note that the development of the reverse transfection siRNA assays was particularly challenging. Our long term goal for these assays is to conduct a genome wide effort to identify the overlapping and unique genes involved in heterologous sensitization of AC isoforms. Using our initial siRNA assay as a guideline, we offer the following abbreviated suggestions for developing a siRNA reverse transfection assays: (1) examine transfection efficiency using ratios of an indicator such as siGlo and transfection reagent (e.g. Lipofectamine 2000); (2) determine the optimal seeding density (e.g. 500-5,000 cells/well); (3) explore transfection duration (e.g. 48-96 hr); and (4) assess appropriate assay conditions (e.g. drug treatments, end point measures, and robust controls) for achieving optimal signal (e.g. Z' factor analysis). Additional detail and more general information for the use of siRNA in screening endeavors can also be found in the previously referenced methods articles29,30. Those investigators who are interested in HTS efforts should also explore the adaptability of their assays to automation (e.g. pintool and robotics). In addition, other factors or controls for screening endeavors include formal assay validation, cell viability assays, and appropriate counter screens. For additional guidance, the interested reader would be encouraged to examine the NCGC Assay Guidance Manual available through NCATS at: http://www.ncbi.nlm.nih.gov/books/NBK53196/.
We have described our approach for streamlining a laborious multiday assay into an efficient "mix and read" assay that can be performed in a single day. The methodologies described here and a recent study of 1,280 compounds in μopioid receptor induced sensitization in 1,536-well format25 suggests that sensitization can now be readily interrogated using HTS. The assay described here is amenable to small molecule testing and can be applied to genetic approaches such as siRNA. Our assay format is focused on the adaptive response known as heterologous sensitization of AC; however, the general approach and methods can be widely applied to other cell based assays requiring multiple drug and reagent additions (e.g. desensitization and neuroprotection). The work reported here is easily accomplished within an academic setting using multichannel pipettes and a multi mode plate reader capable of reading the desired endpoint in 384-well format.
Table 1. Cellular models tested for agonist-induced sensitization.
| Receptor | AC Isoform | Cell line | Sensitization signal |
| D2L dopamine | Endogenous (AC6 and AC727) | CHO-D2L | 2-3 fold |
| D2L dopamine | Recombinant AC2, AC5, and AC6 | HEK 293 stably transfected | AC2 = 2-3 fold AC5 = 50 fold AC6 = 15 fold |
| μ opioid | Recombinant AC1, AC2, and AC5 | HEK 293 stably transfected | AC1 = 6-7 fold AC2 = 2-3 fold AC5 = 10-15 fold |
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to acknowledge Mr. Richard Zink and Todd Wiernicki for methodological training and assay guidance. We also thank John Paul Spence for careful reading and editorial suggestions. This work was supported by the National Institute of Health MH 060397, Brain and Behavior Research Foundation, and Eli Lilly and Company through the Lilly Research Award Program (LRAP).
References
- Sharma SK, Klee WA, Nirenberg M. Dual regulation of adenylate cyclase accounts for narcotic dependence and tolerance. Proc. Natl. Acad. Sci. U.S.A. 1975;72:3092–3096. doi: 10.1073/pnas.72.8.3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts VJ, Neve KA. Sensitization of adenylate cyclase by Galpha(i/o)-coupled receptors. Pharmacol. Ther. 2005;106:405–421. doi: 10.1016/j.pharmthera.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Christie MJ. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br. J. Pharmacol. 2008;154:384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan P, Jiang Z, Diamond I, Yao L. Up-regulation of AGS3 during morphine withdrawal promotes cAMP superactivation via adenylyl cyclase 5 and 7 in rat nucleus accumbens/striatal neurons. Mol. Pharmacol. 2009;76(3):526–533. doi: 10.1124/mol.109.057802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin RT, Lefkowitz RJ, Caron MG. m-Opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat. Rev. 2001;2:119–128. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- Avidor-Reiss T, Nevo I, Saya D, Bayewitch M, Vogel Z. Opiate-induced adenylyl cyclase superactivation is isozyme-specific. J. Biol. Chem. 1997;272:5040–5047. doi: 10.1074/jbc.272.8.5040. [DOI] [PubMed] [Google Scholar]
- Sadana R, Dessauer CW. Physiological roles for G protein-regulated adenylyl cyclase isoforms: insights from knockout and overexpression studies. Neurosignals. 2009;17:5–22. doi: 10.1159/000166277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, et al. Adenylyl cyclase type 5 (AC5) is an essential mediator of morphine action. Proc. Natl. Acad. Sci. U.S.A. 2006;103:3908–3913. doi: 10.1073/pnas.0508812103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts VJ. Adenylyl cyclase isoforms as novel therapeutic targets: an exciting example of excitotoxicity neuroprotection. Mol. Interv. 2007;7:70–73. doi: 10.1124/mi.7.2.6. [DOI] [PubMed] [Google Scholar]
- Beazely MA, Watts VJ. Regulatory properties of adenylate cyclases type 5 and 6: A progress report. Eur. J. Pharmacol. 2006;535:1–12. doi: 10.1016/j.ejphar.2006.01.054. [DOI] [PubMed] [Google Scholar]
- Ejendal KFK, Przybyla JA, Watts VJ. Chapter 10. In: Siehler S, Milligan G, editors. Adenylyl cyclase isoform-specific signaling of GPCRs. G Protein-Coupled Receptors: Structure, Signaling, and Physiology. Vol. 10. Cambridge University Chapter; 2010. pp. 189–217. [Google Scholar]
- Lin Y, Smrcka AV. Understanding molecular recognition by G protein betagamma subunits on the path to pharmacological targeting. Mol. Pharmacol. 2011;80:551–557. doi: 10.1124/mol.111.073072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cumbay MG, Watts VJ. Heterologous sensitization of recombinant adenylate cyclases by activation of D2 dopamine receptors. J. Pharmacol. Exp. Ther. 2001;297:1201–1209. [PubMed] [Google Scholar]
- Watts VJ, Neve KA. Sensitization of endogenous and recombinant adenylate cyclase by activation of D2 dopamine receptors. Mol. Pharmacol. 1996;50:966–976. [PubMed] [Google Scholar]
- Nevo I, et al. Regulation of adenylyl cyclase isozymes on acute and chronic activation of inhibitory receptors. Mol. Pharmacol. 1998;54:419–426. doi: 10.1124/mol.54.2.419. [DOI] [PubMed] [Google Scholar]
- Nevo I, Avidor-Reiss T, Levy R, Bayewitch M, Vogel Z. Acute and chronic activation of the m-opioid receptor with the endogenous ligand endomorphin differentially regulates adenylyl cyclase isozymes. Neuropharmacology. 2000;39:364–371. doi: 10.1016/s0028-3908(99)00155-0. [DOI] [PubMed] [Google Scholar]
- Beazely MA, Watts VJ. Activation of a novel PKC isoform synergistically enhances D2L dopamine receptor-mediated sensitization of adenylate cyclase type 6. Cell. Signal. 2005;17:647–653. doi: 10.1016/j.cellsig.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Vortherms TA, Nguyen CH, Berlot CH, Watts VJ. Using molecular tools to dissect the role of Gs in sensitization of AC1. Mol. Pharmacol. 2004;66:1617–1624. doi: 10.1124/mol.104.000166. [DOI] [PubMed] [Google Scholar]
- Watts VJ, Taussig R, Neve R, Neve KA. Dopamine D2 receptor-induced heterologous sensitization of adenylyl cyclase requires Gas: Characterization of Gas-insensitive mutants of adenylyl cyclase V. Mol. Pharmacol. 2001;60:1168–1172. doi: 10.1124/mol.60.6.1168. [DOI] [PubMed] [Google Scholar]
- Ejendal KF, Dessauer CW, Hebert TE, Watts VJ. Dopamine D(2) Receptor-Mediated Heterologous Sensitization of AC5 Requires Signalosome Assembly. J. Signal Transduct. 2012;2012:210324. doi: 10.1155/2012/210324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston CA, Beazely MA, Vancura AF, Wang JKT, Watts VJ. Heterologous sensitization of adenylate cyclase is protein kinase A-dependent in Cath.a differentiated (CAD)-D2L cells. J. Neurochem. 2002;82:1087–1096. doi: 10.1046/j.1471-4159.2002.01033.x. [DOI] [PubMed] [Google Scholar]
- Wang H, et al. Identification of an adenylyl cyclase inhibitor for treating neuropathic and inflammatory pain. Sci. Transl. Med. 2011;3:65ra3. doi: 10.1126/scitranslmed.3001269. [DOI] [PubMed] [Google Scholar]
- Nordstedt C, Fredholm BB. A modification of a protein-binding method for rapid quantification of cAMP in cell-culture supernatants and body fluid. Anal. Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- Xia M, et al. Inhibition of morphine-induced cAMP overshoot: a cell-based assay model in a high-throughput format. Cell. Mol. Neurobiol. 2011;31:901–907. doi: 10.1007/s10571-011-9689-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman GJ, de Roos JA, Blomenrohr M, Van Koppen CJ, Oosterom J. Cryopreserved cells facilitate cell-based drug discovery. Drug Discov. Today. 2007;12:521–526. doi: 10.1016/j.drudis.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Varga EV, et al. Identification of adenylyl cyclase isoenzymes in CHO and B82 cells. Eur. J. Pharmacol. 1998;348:R1–R2. doi: 10.1016/s0014-2999(98)00258-1. [DOI] [PubMed] [Google Scholar]
- Masri B, et al. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. U.S.A. 2008;105:13656–13661. doi: 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaker NG, et al. Functional genomic analysis of glioblastoma multiforme through short interfering RNA screening: a paradigm for therapeutic development. Neurosurg. Focus. 2010;28:E4. doi: 10.3171/2009.10.FOCUS09210. [DOI] [PubMed] [Google Scholar]
- Echeverri CJ, Perrimon N. High-throughput RNAi screening in cultured cells: a user's guide. Nat. Rev. Geneti. 2006;7:373–384. doi: 10.1038/nrg1836. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- Ejendal KF, et al. Discovery of antagonists of tick dopamine receptors via chemical library screening and comparative pharmacological analyses. Insect Biochem. Mol. Biol. 2012;42:846–853. doi: 10.1016/j.ibmb.2012.07.011. [DOI] [PubMed] [Google Scholar]
- Meyer JM, et al. A "genome-to-lead" approach for insecticide discovery: pharmacological characterization and screening of Aedes aegypti D(1)-like dopamine receptors. PLoS Negl.Trop. Dis. 2012;6:e1478. doi: 10.1371/journal.pntd.0001478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne N, Inglese J, Auld DS. Illuminating insights into firefly luciferase and other bioluminescent reporters used in chemical biology. Chem. Biol. 2010;17:646–657. doi: 10.1016/j.chembiol.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, et al. Novel genetically encoded biosensors using firefly luciferase. ACS Chem. Biol. 2008;3:346–351. doi: 10.1021/cb8000414. [DOI] [PubMed] [Google Scholar]
- Jiang LI, et al. Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem. 2007;282:10576–10584. doi: 10.1074/jbc.M609695200. [DOI] [PMC free article] [PubMed] [Google Scholar]