

Abstract
Mantle cell lymphoma (MCL) is an aggressive and incurable subtype of B-cell Non-Hodgkin’s lymphomas characterized by an initial response to first-line treatment with chemotherapy plus monoclonal antibodies followed by relapse and less responsiveness to further lines of treatment. Harnessing the immune system to elicit its exquisite specificity and long-lasting protection might provide sustained MCL immunity that could potentially eradicate residual malignant cells responsible for disease relapse. Here we show that genetic or pharmacologic disruption of Stat3 in malignant B-cells augments their immunogenicity leading to better activation of antigen-specific CD4+ T-cells and restoration of responsiveness of tolerized T-cells. The additional demonstration that in vivo treatment of MCL-bearing mice with a specific Stat3 inhibitor resulted in decreased Stat3 phosphorylation in malignant B-cells and anti-lymphoma immunity, points to Stat3 inhibition as an enticing strategy to overcome tolerance to tumor antigens and elicit a strong immunity against MCL and other B-cell malignancies.
Keywords: Stat3, anti-tumor immunity, B-cell lymphoma
Introduction
Previous studies in murine models of B-cell lymphoma indicate that generation of effective anti-lymphoma immunity requires: (1) Conversion of bone marrow (BM)-derived antigen presenting cells (APCs) from a non-inflammatory (or tolerogenic) status into inflammatory APCs that trigger effective T-cell responses1,2 and, (2) Augmentation of the antigen-presenting cell function of the malignant B-cells3. Therapeutic strategies endowed with the ability of fulfilling both requirements might lead not only to successful eradication of B-cell tumors but also to a long-lasting immunity, the latter a desirable effect for certain B-cell malignancies characterized by their high tendency to relapse.
Mantle cell lymphoma (MCL) is the prototype of a B-cell malignancy in which relapse is the major challenge to overcome. In spite of an good initial response to first-line treatment with chemotherapy plus monoclonal antibodies, almost all patients with MCL will eventually relapse, become less responsive to further lines of treatment and ultimately will succumb to their disease4,5. Given these sobering characteristics, MCL has one of the worst prognoses among all B-cell Non Hodgkin’s lymphomas (NHL) and, in 2011 this disease remains incurable6. As such, novel non-cross resistant treatment modalities capable of improving the response rate and more importantly, able of sustaining these responses are greatly needed for MCL patients.
Several lines of evidence point to manipulation of the immune system as an enticing non-cross resistant therapeutic strategy for MCL. The demonstration that immune cells are able to kill chemotherapy-resistant tumor cells7,8 together with the findings that T-cell responses are elicited in vaccinated MCL patients9 and, the encouraging responses observed in patients with relapsed/refractory MCL treated with immunomodulatory drugs (IMIDs)6,10,11, suggest that harnessing the immune system and in particular, eliciting its exquisite specificity and long-lasting protection, might lead to sustained immune responses in MCL12.
Given the above rationale, a significant effort has been devoted to identify molecular target(s) capable of influencing inflammatory pathways in the APC as well as in the malignant B-cell. Signal transducer and activators of transcription (STATs) are cytoplasmic transcription factors that are key mediators of cytokine and growth factor signaling pathways13. One of the members of the Stat family, Stat3, has emerged as a negative regulator of inflammatory responses in a variety of immune cells14–16. For instance, we have previously demonstrated that pharmacologic or genetic disruption of Stat3 in APCs resulted in diminished production of the anti-inflammatory cytokine IL-10, enhanced expression of co-stimulatory molecules, and increased release of pro-inflammatory mediators leading to augmentation of the function of these cells to effectively prime T-cells and restore the responsiveness of anergic CD4+ T-cells17. These observations prompted us to ask whether targeting Stat3 in malignant B-cells might also influence the immunogenicity/inflammatory status of these cells and whether such an effect might unleash effective antitumor immune responses in a murine model of MCL.
Materials and Methods
Mice
Six-to-eight-week-old male BALB/c (H-2d), C57BL/6 (H-2b) mice were obtained from the NIH (Frederick, Maryland). Male BALB/c severe combined immunodeficiency (SCID) or C57BL/6 SCID mice, aged 6 to 8 weeks, were purchased from Jackson Laboratories (Bar Harbor, ME). TCR transgenic mice expressing an αβ T-cell receptor specific for amino acids 110–120 from influenza hemagglutinin presented by I-Ed were a gift of H. von Boehmer18. TCR transgenic mice (OT-II) expressing an αβ TCR specific for peptide 323-339 from Ovalbumin (OVA) presented by MHC class II, I-Ab19 were provided by Dr. W. Heath (The Walter and Eliza Hall Institute of Medical Research, Victoria, Australia). All experiments involving the use of mice were performed in accordance with protocols approved by the Animal Care and Use Committees of the University Of South Florida College Of Medicine.
Tumor Cells
Murine A20 lymphoma cells (H-2d) and human JEKO MCL cells were obtained from ATCC (Rockville, MD). A20 lymphoma cells expressing HA (Hemagglutinin influenza) as a model tumor antigen were selected and grown in vitro as previously described20. FC-muMCL1 cell line (H-2b) was derived from a tumor explanted from a one year-old Bcl-1 transgenic mice injected with pristane intraperitoneally21. For in vivo tumor challenge experiments, cells were washed three times in sterile HBSS and then 1×106 A20 tumor cells or 5×106 FC-muMCL1 cells were injected into BALB/c or C57BL/6 mice respectively, in a total volume of 0.2 ml per mouse.
Reagents
LPS (Escherichia coli 055:B5, L-2880) was purchased from Sigma-Aldrich (St. Louis, MO). CPA-7 was provided by Dr. Said Sebti (Moffitt Cancer Center, Tampa, FL). CPA-7 was first reconstituted in DMSO for stock preparation (10mM), and then further diluted in RPMI 1640 for in vitro or in HBSS for in vivo use.
Transfection of tumor cells
A20 B-cells were transfected with either a dominant negative variant of Stat3, Stat3β22,23 or a mutant form of Stat3, Stat3c, that is constitutively activated without tyrosine phosphorylation24. Transfections were performed according to the manufacturer’s instructions (Bio-Rad). Briefly, A20 B-cells were harvested and washed with cold PBS then resuspended at the concentration of 1×107/0.3 ml in PBS and transferred into an electroporation cuvette. Then, 15 mcg of either GFP, Stat3β GFP DNA, or PBS was added and cells were subjected to a high-voltage electrical pulse of defined magnitude and length as per manufacturer’s instructions. A similar procedure was followed to transfect A20 cells with a Stat3c expression vector or with a control pcDNA3 empty vector. Inhibition of Stat3 in JEKO human MCL was accomplished with siRNA specific for Stat3 using Amaxa Nucleofector methodology as per manufacturer’s protocol (Dharmacon).
Isolation of B-cells from tumor
Mice were sacrificed and tumor nodules were carefully dissected from their livers. Tumors were gently mashed in tissue culture plates using a plunger. Then cells were transferred to a conical tube and washed twice in RPMI 1640. Cells were cultured for 3 hours at 37°C, 5% CO2 and floating cells were collected for further experiments and analyses.
Immunoblotting
Whole-cell lysates were prepared using modified RIPA lysis buffer. 50mcg of protein was subjected to 7% SDS-PAGE and transferred onto PVDF (Millipore) membranes and incubated overnight with primary antibodies, then followed by a secondary antibody (Pierce) and proteins were visualized with a Chemiluminescent Detection kit (Pierce). Primary antibodies against phospho-Stat3 (Tyr705), phospho-AKT, and phospho-p42/44 MAPK were purchased from Cell Signaling Technology (Cambridge, MA, USA). Total Stat3 and total AKT antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
In vitro and in vivo pharmacologic inhibition of Stat3
CPA-7 is a platinum-containing compound that disrupts Stat3 DNA binding activity, but not Stat5 nor Stat1 in malignant cells25. For in vitro studies, FC-muMCL1 cells were treated with CPA-7 alone (31.25 to 1000nM) or in combination with LPS (2mcg/ml) and their ability to present cognate peptide to antigen-specific CD4+ T-cells was determined as described under antigen presentation studies. For in vivo studies, FC-muMCL1 or A20 tumor bearing mice were given CPA-7 intravenously at the dose of 5 mg/kg every 3 days as previously described26.
In vivo generation of tolerized CD4+ T-cells
Briefly, 2.5 ×106 CD4+ transgenic T-cells specific for an MHC class II epitope of influenza hemagglutin (HA) were injected intravenously (iv) into A20HA lymphoma bearing mice. Twenty-one days after T-cell transfer, animals were sacrificed and tolerized T-cells were re-isolated from their spleens as previously described20. Cytokine production by re-isolated clonotypic CD4+ T-cells in response to HA-peptide110-120 presented by A20 B-cells was determined as described under antigen presentation studies.
For induction of antigen-specific T-cell tolerance in H-2b tumor bearing mice, a similar experimental approach was utilized, the only difference being that 1×106 anti-OVA CD4+ transgenic T cells (OT-II) were transferred into animals bearing an OVA-expressing tumor (B16OVA). Fourteen days after T-cell transfer, animals were sacrificed and tolerized OT-II cells were re-isolated from their spleens17. Cytokine production by OT-II cells in response to OVA-peptide323-339 presented by FC-muMCL1 cells was determined as described under antigen-presentation studies.
In vitro antigen-presentation studies
A20 or FC-muMCL1 cells (1×105/well) were cultured with 5×104 purified naïve or tolerized antigen-specific CD4+ T cells in the presence or not of cognate peptide (either synthetic HA peptide110-120 SFERFEIFPKE for studies with A20 B-cells or OVA peptide323-339, ISQAVHAAHAEINEAGR for studies with FC-muMCL1 cells). After 48 hours, supernatants were collected and stored at −70°C until assayed for IL-2 and IFN-γ production by ELISA (R&D Systems, Minneapolis, MN). Values for T-cells cultured in media alone were routinely less than 10% of the values for antigen-stimulated T-cells.
Flow Cytometric Analysis
FC-muMCL1 cells were stained with PE anti-CD5 (53-7.3, BD Bioscience), PE-Cy7 anti-CD19 (1D3, BD Bioscience) and FITC anti-Cyclin D1 (DCS-6, Millipore) antibodies. Fifty-thousand gated events were collected on a FACSCALIBUR (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Statistical Analysis
A 2-way analysis of variance (ANOVA) was used to evaluate the magnitudes of cytokine production by clonotypic T-cells. Differences in survival were assessed with the log-rank test.
Results
Manipulation of Stat3 signaling in malignant B-cells influences their antigen-presenting function and T-cell responsiveness in vitro
First, we asked whether genetic manipulation of Stat3 in A20 lymphoma B-cells could influence their intrinsic antigen-presenting capabilities and the responsiveness of antigen-specific CD4+ T-cells. To inhibit Stat3 in A20 cells we used a dominant negative variant of Stat3, Stat3β22. A20 cells were transfected with Stat3β (A20-Stat3β) and then cultured in vitro with syngeneic naïve CD4+ T-cells specific for a MHC class II restricted epitope of influenza hemagglutinin (HA) in the presence -or not- of cognate HA-peptide. As shown in Fig. 1 (left panel), clonotypic T-cells encountering HA-peptide on A20-Stat3β cells displayed an enhanced production of IL-2 (Fig. 1A) and IFN-γ (Fig. 1B) relative to those T-cells encountering cognate peptide on either non-transfected (None), mock-transfected (Mock) or GFP-transfected (GFP) A20 B-cells.
Figure 1. Disruption of Stat3 in malignant B-cells augments their antigen-presenting function.

A20 B-cells were left untransfected, mock transfected or transiently transfected with GFP vector or Stat3β GFP expression vector. Then, 1×105 transfected cells as well as non-transfected or mock transfected cells were incubated with either 5×104 naïve anti-HA CD4+ T-cells (left panel) or with 5×104 tolerized anti-HA CD4+ T-cells isolated from the spleen of A20HA-bearing mice (right panel), in the presence of 12.5 mcg of HA peptide110-120 SFERFEIFPKE. After 48 hours, supernatants were collected and IL-2 (A, C) and IFN-γ (B, D) production by antigen-specific CD4+ T-cells were determined by ELISA. Values represent mean ± S.E of triplicate cultures and are representative of three independent experiments (*p statistically significant for the difference in cytokine production between treatment with STAT3β and GFP).
In previous studies we have demonstrated that adoptive transfer of naïve anti-HA transgenic CD4+ T-cells into mice bearing A20 B-cell lymphoma expressing HA as a model tumor antigen (A20HA) resulted in the induction of antigen-specific CD4+ T-cell tolerance. In this system, re-isolated T cells from lymphoma bearing mice were found to be anergic by their failure to be primed in vivo as well as by their diminished IL-2 and IFN-γ production in response to in vitro re-stimulation with cognate HA-peptide20. However, as shown in Fig. 1 (right panel) in vitro incubation of these same tolerant T-cells (re-isolated from A20HA lymphoma bearing mice) with A20-Stat3β lymphoma cells resulted in restoration of T-cell responsiveness to cognate HA-antigen. Indeed, presentation of HA-peptide by A20-Stat3β triggered IL-2 (Fig. 1C) and IFN-γ production by tolerant CD4+ T-cells (Fig. 1D). In sharp contrast, tolerant T-cells encountering HA-antigen on non-transfected, mock-transfected or GFP-transfected A20 B-cells remained unresponsive.
Given the above findings, we next asked whether an opposite effect would be observed when Stat3 is over-expressed in malignant B-cells. A20 cells were therefore transfected with Stat3c, a mutant form of Stat3 that is constitutively activated without tyrosine phosphorylation24. Unlike naïve anti-HA CD4+ T-cells that produce IL-2 and IFN-γ in response to cognate antigen presented by control A20 B-cells (Fig. 2A–B, non-transfected or PC-DNA transfected), CD4+ T-cells cultured with A20-Stat3c cells were rendered unresponsive given their minimal production of IL-2 (Fig. 2A, Stat3c) and inability to produce IFN-γ in response to cognate antigen (Fig. 2B, Stat3c). Taken together, our results demonstrate that genetic manipulation of Stat3 in A20 B-cells influences their antigen-presenting capabilities and points to Stat3 inhibition as an enticing approach to augment the immunogenicity of B-cell lymphomas and overcome T-cell anergy.
Figure 2. Increased Stat3 activity in malignant B-cells inhibits antigen-specific CD4+ T-cell responses.
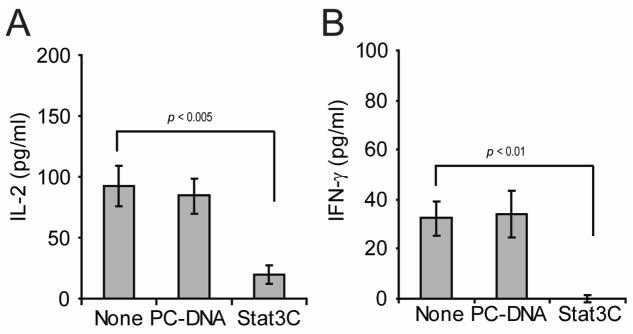
A20 B-cells were transiently transfected with either pcDNA3 empty vector or Stat3c expression vector. Then, 1×105 transfected cells as well as non-transfected cells were incubated with 5×104 naïve anti-HA CD4+ T-cells in the presence or not of 12.5 mcg of HA peptide. After 48 hours, supernatants were collected and IL-2 (A) and IFN-γ (B) production were determined by ELISA. Values represent mean ± S.E of triplicate cultures and are representative of three independent experiments (*p statistically significant for the difference in cytokine production between none and Stat3C).
Pharmacologic inhibition of Stat3 in Mantle cell lymphoma
Given the above results we next asked whether pharmacologic inhibition of Stat3 could also influence the immunogenicity of malignant B-cells, and in particular, of MCL cells. CPA-7, a platinum-containing compound, has been previously shown to disrupt Stat3 DNA binding activity, but not Stat5 nor Stat1, in malignant cells25. To address the selectivity of this agent in MCL cells, we first determined whether CPA-7 affects the Stat3 signaling pathway with minimal or absent off-target effects in treated cells. As such, we asked whether the phenotype displayed by JEKO MCL cells in which Stat3 has been knocked down using specific Stat3 siRNA could be recapitulated in cells treated with CPA-7 in vitro. As shown in Figure 3, phospho-Stat3 was diminished in JEKO cells subjected to either approach. Of note, CPA-7 induced a dose dependent inhibition of phospho-Stat3 (data not shown) without any effect on other signaling pathways such as phospho-MAPK or phospho-AKT in MCL cells even at 30μM, which is manifold over that used in subsequent in vitro assays. This effect mimicked results using Stat3 siRNA.
Figure 3. Comparison of the effects of CPA-7 treatment and Stat3 siRNA inhibition upon human MCL cells.

JEKO cells were transiently transfected with Stat3-specific siRNA (Stat3 siRNA) or non-targeting control (Control). In parallel, JEKO cells were treated or not with CPA-7 (30μM) for 24 hours. Cells were harvested and protein extracts were obtained and subjected to western blot using antibodies against p-Stat3, Stat3, p-MAPK, MAPK, p-Akt and Akt. Shown is a representative experiment of two with similar results.
Recently, Smith and collaborators have developed a murine model of MCL by injecting pristane intraperitoneally (ip) into one year-old Bcl-1 transgenic mice (Eμ-cyclin D1). In these animals, the pattern of disease consists of diffuse adenopathy, splenomegaly, bone marrow infiltration as well as lung, kidney and periportal hepatic infiltration. Analysis of tumor explants revealed malignant B-cells that co-express cyclin D1, CD20, and CD5, but lack expression of CD23, findings reminiscent of human MCL21. A cell line, FC-muMCL1, has been derived from one of these lymphoma explants and phenotypic analysis confirmed that they express cyclin D1, CD19 and CD5 (Fig. 4A). Furthermore, all C57BL/6 mice challenged with 5×106 FC-muMCL1 given either sc (black circle) or ip (open circle) developed tumors (Fig. 4B). In mice challenged sc, tumor nodules developed by day 21. Intra-peritoneal injection of MCL cells resulted in the development of ascites by day 30 and at necropsy we found enlarged mesenteric and retroperitoneal lymph nodes as well as tumor nodules in the peritoneum and small bowel (data not shown).
Figure 4. Phenotypic and functional characteristics of FC-muMCL1 cells.

(A) Expression of Cyclin D1, CD19 and CD5 by FC-muMCL1 cells (open histogram). Gray histogram: Isotype control. (B) In vivo growth of FC-muMCL1 tumors. C57BL/6 mice were injected either sc in the right leg (closed circles) or ip (open circles) with 5×106 FC-muMCL1 cells. Five mice were included in each group and they were inspected three times a week for the development of tumor nodules (sc model) or abdominal girth (ip model). Shown is a representative experiment of two with similar results. (C–D). Antigen-presenting function of MCL cells. FC-muMCL1 cells (1×105 cells/well) were treated with LPS (2 mcg/ml), LPS + increasing concentrations of CPA-7, or left untreated (Media) for 24 hours. Then, cells were washed and plated with either 5×104 naïve anti-OVA CD4+ T-cells/well or tolerized anti-OVA CD4+ T-cells isolated from mice bearing an OVA-expressing tumor together with 3mcg/ml of OVA peptide323-339. Forty-eight hours later supernatants were collected and the production of IL-2 and IFN-γ by naïve T-cells (C) and the production of IFN-γ by tolerized T-cells (D) were determined by ELISA. Shown is a representative experiment of three independent experiments with similar results (*p statistically significant for the difference in cytokine production between treatment).
Next, we asked whether FC-muMCL1 cells can present cognate antigen to antigen-specific CD4+ T-cells in vitro and if so, whether this intrinsic APC function can be enhanced by CPA-7. Given the background of FC-muMCL1 cells (H-2b), we assessed their ability to present ovalbumin (OVA) peptide to transgenic CD4+ T-cells expressing an αβTCR specific for OVA-peptide323-33919. First, anti-OVA CD4+ T-cells encountering cognate antigen on untreated MCL cells (OVA peptide) or in LPS-treated MCL cells (LPS+OVA) produced IL-2 (Fig. 4C, left) and IFN-γ (Fig. 4C, right) indicative of the ability of murine MCL cells to present antigen to CD4+ T-cells in vitro. This intrinsic APC function was further enhanced following exposure of MCL cells to LPS in the presence of increasing concentrations of Stat3 inhibitor. Indeed, CPA-7-treated MCL cells triggered an increased production of IL-2 and IFN-γ by CD4+ T-cells (Fig 4C: CPA-7+LPS). Similarly, in vitro culture of CPA-7-treated human JEKO cells with allogeneic human peripheral blood mononuclear cells also resulted in enhanced IL-2 and IFN-γ production by T-cells as determined by ELISPOT assay (data not shown).
Finally, tolerized anti-OVA CD4+ T-cells cultured in vitro with CPA-7-treated FC-muMCL1 cells regained their ability to produce IFN-γ in response to cognate OVA-peptide (Fig. 4D, CPA-7+LPS). In contrast, tolerized T-cells encountering OVA-peptide on untreated MCL cells (OVA) or in cells treated with LPS+OVA peptide (in the absence of CPA-7) remained unresponsive. Taken together, treatment of FC-muMCL1 cells with the Stat3 inhibitor CPA-7 augments their immunogenicity resulting in enhanced activation of naïve CD4+ T-cells and restoration of responsiveness of tolerized CD4+ T-cells.
In vivo inhibition of Stat3 in B-cell lymphomas
Next we determined whether CPA-7 inhibits Stat3 signaling in malignant B-cells in vivo. Previous studies have shown that CPA-7 induces in vivo antitumor responses when used at the dose of 5 mg/kg given iv every 3 days16. We therefore injected 5×106 FC-muMCL1 cells ip into C57BL/6 mice and twenty-one days later, animals were treated (or not) with 5 mg/kg of CPA-7 given iv on days +21, +24 and +27. Two days later (day +29), animals were sacrificed and tumor nodules were dissected from their livers. As shown in Figure 5A, a decrease in phospho-Stat3 expression was observed in malignant B-cells isolated from MCL-bearing mice treated with CPA-7. No such effect was observed in untreated tumor bearing mice or animals treated with vehicle control. Of note, less tumor burden was observed in CPA-7 treated mice. These results prompted us to determine the in vivo anti-MCL effect of CPA-7 in a larger group of animals. C57BL/6 mice were challenged with 5×106 FC-muMCL1 cells given sc. Half of the mice received vehicle control and the other half received CPA-7 (5mg/kg/iv every three days, starting on day +5 after tumor challenge). Unlike untreated MCL-bearing mice, which rapidly developed tumors (Fig. 5B, solid line), mice treated with CPA-7 had a significant delay in MCL tumor growth (Fig. 5B, dashed line). Similarly, BALB/c mice challenged with 1×106 A20 lymphoma cells sc and then treated with CPA-7 (same dose and frequency as in the MCL model) rejected this B-cell tumor (Fig. 5C, dashed line). No such rejection was observed in A20 B-cell lymphoma bearing mice given vehicle control (Fig. 5C, solid line). Therefore, in vivo treatment of lymphoma bearing mice with CPA-7 resulted in decreased Stat3 phosphorylation in malignant B-cells and a strong anti-lymphoma effect.
Figure 5. In vivo treatment with CPA-7 results in decreased Stat3 phosphorylation in malignant B-cells and an anti-lymphoma effect.
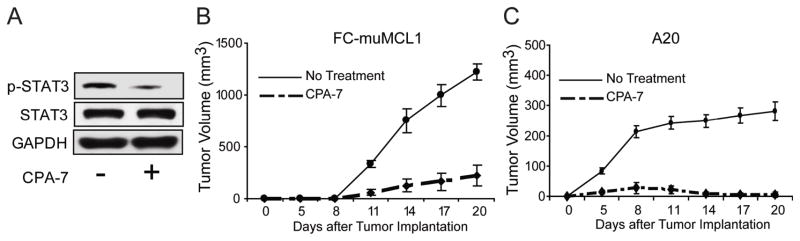
(A) 5×106 FC-muMCL1 cells were injected ip into C57BL/6 mice. Twenty-one days later animals were treated or not with 5 mg/kg of CPA-7 given iv every three days (days +21, +24 and +27). On day +29 animals were sacrificed and tumor nodules were carefully dissected from their livers. Malignant B-cells were then isolated and the expression of phopho-Stat3 was determined by western blot using an anti-p-Stat3 (Tyr705) antibody. Shown is a representative experiment of two independent experiments with similar results. (B) C57BL/6 mice (n=10) were challenged with 5×106 FC-muMCL1 cells given sc in the right leg. Half the mice were then treated with CPA-7 (5mg/kg) given IV every three days, starting on day +5 after tumor challenge (dashed line). The other half of the mice received vehicle control (solid line). Tumor volumes were calculated (LengthxWidthxWidthX1/2) from measurements made at times indicated. Two independent experiments were performed with similar results. (C) BALB/c mice (n=10) were challenged with 1×106 A20 lymphoma cells given sc in the right leg. Half the mice were treated with CPA-7 (dash line), and the other half with vehicle control (solid line) as indicated in B. Animals were monitored for the development of tumor nodules and tumor volumes measured on days indicated. Two independent experiments were performed with similar results.
The in vivo antitumor effect of CPA-7 requires an intact adaptive immune system
Previous studies have shown that disruption of Stat3 in malignant cells resulted in the induction of apoptosis27,28. As such, the in vivo antitumor effect observed in CPA-7 treated MCL bearing mice (Fig. 5) could be a reflection of a direct effect of this drug upon tumor cells themselves rather than immune effects triggered by Stat3 inhibition. To address this question, C57BL/6 SCID mice (Fig. 6A) or BALB/c-SCID mice (Fig. 6B) were challenged with 5×106 FC-muMCL1 cells or 1×106 A20 lymphoma cells given sc. respectively. Then, half the mice in each group were treated with CPA-7 (5mg/kg every three days, starting on day +5 after tumor challenge) and the other half received vehicle control. Unlike immunocompetent lymphoma bearing mice treated with CPA-7 in which a strong antitumor effect was clearly demonstrated (Fig. 5B–C), such an antitumor effect was not observed in immunodeficient animals treated with CPA-7. Indeed, no difference in the kinetics of tumor growth was observed among untreated or CPA-7 treated lymphoma-bearing mice (Fig 6A: MCL, Fig. 6B: A20). These results indicate that the antitumor effect of CPA-7 requires an intact adaptive immune system and points to the immunological rather than the non-immunological antitumor effects of this Stat3 inhibitor as playing a dominant role in its in vivo anti-lymphoma activity.
Figure 6. Similar kinetics of lymphoma growth in CPA-7 treated SCID mice.
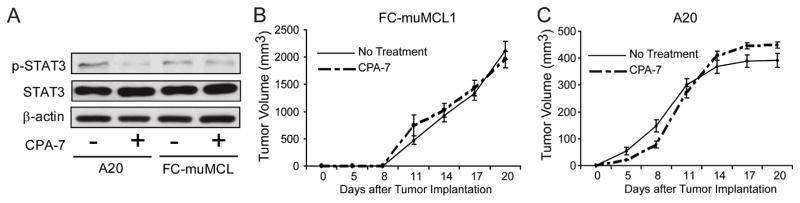
(A) C57BL/6 SCID and BALB/c SCID mice were treated as indicated in (B) and (C). Mice were sacrificed after 19 days of tumor challenge and tumor nodules were collected. Malignant B-cells were then isolated and the expression of phopho-Stat3 was determined by western blot using an anti-p-Stat3 (Tyr705) antibody. (B) C57BL/6 SCID mice (n=10) were challenged with 5×106 Fc-muMCL1 cells given sc. Half the mice were then treated with CPA-7 (dash line) (5mg/kg/iv given every three days, starting on day +5 after tumor challenge) and the other half received vehicle control (solid line). (C) BALB/c SCID mice (n=10) were challenged with 1×106 A20 cells given sc. Mice were then treated with CPA-7 (dash line) or received vehicle control (solid line) as indicated in A. Tumors were measured at times indicated. Two independent experiments were performed with similar results.
Discussion
In this study we have shown that genetic or pharmacologic disruption of Stat3 in malignant B-cells increased their immunogenicity leading to augmentation of antigen-specific CD4+ T-cell function and restoration of responsiveness of tolerized T-cells. These findings expand the previously known pro-inflammatory effects of Stat3 inhibition upon other immune cells such as BM-derived APCs17,29,30. This unique property of Stat3 inhibition to influence the inflammatory status of both the malignant B-cell as well as the APC points to pharmacologic inhibition of this signaling pathway as an appealing strategy to overcome tolerance to tumor antigens and elicit a strong antitumor immunity.
In the in vivo immune response against B-cell lymphomas, it is likely that both malignant cells themselves as well as BM-derived APCs present tumor antigens to antigen-specific CD4+ T-cells. B-cell lymphomas are the transformed counterparts of cells endowed with antigen-presenting capabilities. Normal B lymphocytes have long been known to interact with CD4+ T-cells during physiological immune responses in a process that involves presentation of peptide-MHC class II complexes, along with co-stimulatory signals to antigen specific T-cells31,32. Like normal B-cells, malignant B-cells also express major histocompatibility complex (MHC) class I and II molecules and low but inducible levels of adhesion and co-stimulatory molecules1,33,34. In spite of these intrinsic properties, it is quite paradoxical that B-cell malignancies fail to be eliminated in the very same compartment -lymph nodes- where tumor antigen-specific T-cell responses are initiated.
Several factors might account for the failure of malignant B-cells to properly activate T-cells in vivo. First, their expression of MHC molecules, co-stimulatory molecules and/or adhesion molecules that participate in T-cell priming might not be sufficient to trigger a full T-cell activation. This “state” of partial T-cell activation, in the absence of additional signals capable of sustaining this initial response and/or in the presence of dominant suppressive mechanisms, might be followed instead by a state of T-cell anergy1,35. Second, among the suppressive mechanisms operative in the lymphoma microenvironment, one that has gained particular attention is the ability of BM-derived APCs to create a tolerogenic environment in the lymph node that favors T-cell anergy over T-cell activation36–38. It is plausible therefore that the combination of weak antigen-presenting capabilities of malignant B-cells together with cross-presentation of tumor antigens by non-inflammatory, immunosuppressive APCs2,39 would likely be conducive to T-cell unresponsiveness, a barrier that needs to be overcome if effective immunity against B-cell tumors is to be generated.
In the past several years, a number of therapeutic approaches have sought to improve the weak antigen-presenting capabilities of malignant B-cells mainly by genetically modifying these cells to enforce the expression of adhesion and costimulatory molecules as well as pro-inflammatory cytokines40–43. Other approaches have focused instead on the induction of inflammatory APCs as a strategy to improve cross-presentation of tumor antigens to antigen-specific T-cells3,44. Although each of these approaches induced productive immune responses in vivo, the duration and magnitude of these effects were transient and not strong enough to fully eradicate systemic lymphoma. A potential explanation for the limited success of these strategies is that they have targeted either the malignant B-cell or the APC, but not both, and as such they were unable to fully overcome tolerogenic mechanisms in cancer. Therefore, from a therapeutic perspective it would be desirable to find novel approaches with the dual ability of enhancing the antigen-presenting function of malignant B-cells and inducing inflammatory APCs displaying enhanced cross-presentation of tumor antigens to antigen-specific T-cells.
Inhibition of Stat3 signaling represents such a novel strategy given its known ability to influence the inflammatory status of the APC17,29,45 and as shown here, to also enhance the immunogenicity of malignant B-cells. Indeed, treatment of malignant B-cells with CPA-7, a novel Stat3 inhibitor, rendered these cells better activators of antigen-specific CD4+ T-cells and capable of restoring the responsiveness of tolerant T-cells isolated from lymphoma bearing mice. In addition to these in vitro effects, treatment of MCL-bearing mice with CPA-7 decreased Stat3 phosphorylation in tumor cells and resulted in effective antitumor immunity. Of note, the lack of antitumor activity in immunodeficient mice treated with CPA-7, points to the effects of Stat3 inhibition upon immune cells as being essential for effective lymphoma eradication in vivo.
Our demonstration that Stat3 inhibition is an effective strategy in a murine model of MCL provides the basis for evaluating the efficacy of this strategy in human MCL. The prior demonstration that Stat3 is constitutively activated in patients with this disease46,47 provides further support to targeting this signaling pathway in MCL. Of note, inhibition of Stat3 in tumor cells displaying aberrant activation of this pathway has been shown to result in “inflammatory death”, a process associated with release of pro-inflammatory mediators that could amplify ongoing antitumor immune responses also triggered by the effects of Stat3 inhibition upon APCs and other immune cells26,48. This pro-inflammatory environment generated by Stat3 inhibition is further enhanced by the inability of malignant B-cells and immune cells to produce IL-10 in the absence of intact Stat3 signaling15,17. Such a lack of production of IL-10 has the dual advantage of not only diminishing the generation of an immunosuppressive environment but also depriving malignant B-cells of an important survival factor49,50.
Taken together, the dual effects of Stat3 inhibition upon both the malignant B-cells as well as immune cells triggers a positive loop of pro-inflammatory events that likely generates an activating rather than a tolerogenic environment in the lymph nodes, which is ultimately conducive to effective anti-lymphoma immunity. Such an unique property of Stat3 inhibition makes this approach suitable for future evaluation in human MCL and other B-cell malignancies, either alone or as an adjuvant to therapeutic lymphoma vaccines.
Supplementary Material
Acknowledgments
We thank Moffitt’s Flow Cytometry Core and Animal Facility for technical assistance.
Grant Support
This work was supported by PHS grants CA134807 and CA087583 (EMS).
Footnotes
Disclosure of Potential Conflicts of Interest:
No potential conflicts of interest were disclosed.
References
- 1.Sotomayor EM, Borrello I, Rattis FM, Cuenca AG, Abrams J, Staveley-O’Carroll K, et al. Cross-presentation of tumor antigens by bone marrow-derived antigen-presenting cells is the dominant mechanism in the induction of T-cell tolerance during B-cell lymphoma progression. Blood. 2001;98(4):1070–1077. doi: 10.1182/blood.v98.4.1070. [DOI] [PubMed] [Google Scholar]
- 2.Rabinovich GA, Gabrilovich D, Sotomayor EM. Immunosuppressive Strategies that are Mediated by Tumor Cells. Annual Review of Immunology. 2007;25(1):267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horna P, Cuenca A, Cheng F, Brayer J, Wang HW, Borrello I, et al. In vivo disruption of tolerogenic cross-presentation mechanisms uncovers an effective T-cell activation by B-cell lymphomas leading to antitumor immunity. Blood. 2006;107(7):2871–2878. doi: 10.1182/blood-2005-07-3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howard OM, Gribben JG, Neuberg DS, Grossbard M, Poor C, Janicek MJ, et al. Rituximab and CHOP induction therapy for newly diagnosed mantle-cell lymphoma: molecular complete responses are not predictive of progression-free survival. J Clin Oncol. 2002;20(5):1288–1294. doi: 10.1200/JCO.2002.20.5.1288. [DOI] [PubMed] [Google Scholar]
- 5.Witzig TE. Current treatment approaches for mantle-cell lymphoma. J Clin Oncol. 2005;23(26):6409–6414. doi: 10.1200/JCO.2005.55.017. [DOI] [PubMed] [Google Scholar]
- 6.Martin P, Chadburn A, Christos P, Weil K, Furman RR, Ruan J, et al. Outcome of deferred initial therapy in mantle-cell lymphoma. J Clin Oncol. 2009;27(8):1209–1213. doi: 10.1200/JCO.2008.19.6121. Prepublished on 2009/02/04 as DOI JCO.2008.19.6121 [pii] [DOI] [PubMed] [Google Scholar]
- 7.Fuchs EJ, Bedi A, Jones RJ, Hess AD. Cytotoxic T cells overcome BCR-ABL-mediated resistance to apoptosis. Cancer Res. 1995;55(3):463–466. [PubMed] [Google Scholar]
- 8.Shtil AA, Turner JG, Durfee J, Dalton WS, Yu H. Cytokine-based tumor cell vaccine is equally effective against parental and isogenic multidrug-resistant myeloma cells: the role of cytotoxic T lymphocytes. Blood. 1999;93(6):1831–1837. [PubMed] [Google Scholar]
- 9.Neelapu SS, Kwak LW, Kobrin CB, Reynolds CW, Janik JE, Dunleavy K, et al. Vaccine-induced tumor-specific immunity despite severe B-cell depletion in mantle cell lymphoma. Nat Med. 2005;11(9):986–991. doi: 10.1038/nm1290. [DOI] [PubMed] [Google Scholar]
- 10.Kaufmann H, Raderer M, Wöhrer S, Püspök A, Bankier A, Zielinski C, et al. Antitumor activity of rituximab plus thalidomide in patients with relapsed/refractory mantle cell lymphoma. Blood. 2004;104(8):2269–2271. doi: 10.1182/blood-2004-03-1091. [DOI] [PubMed] [Google Scholar]
- 11.Habermann TM, Lossos IS, Justice G, Vose JM, Wiernik PH, McBride K, et al. Lenalidomide oral monotherapy produces a high response rate in patients with relapsed or refractory mantle cell lymphoma. Br J Haematol. 2009;145(3):344–349. doi: 10.1111/j.1365-2141.2009.07626.x. [DOI] [PubMed] [Google Scholar]
- 12.Zhou Y, Zhang L, Romaguera J, Delasalle K, Han X, Du X, et al. Immunotherapy in mantle cell lymphoma: anti-CD20-based therapy and beyond. Am J Hematol. 2008;83(2):144–149. doi: 10.1002/ajh.21036. [DOI] [PubMed] [Google Scholar]
- 13.Darnell JE., Jr STATs and gene regulation. Science. 1997;277(5332):1630–1635. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- 14.Sano S, Itami S, Takeda K, Tarutani M, Yamaquchi Y, Miura H, et al. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. Embo J. 1999;18(17):4657–4668. doi: 10.1093/emboj/18.17.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takeda K, Clausen BE, Kaisho T, Tusjimura T, Terada N, Förster I, et al. Enhanced Th1 activity and development of chronic enterocholitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity. 1999;10:39. doi: 10.1016/s1074-7613(00)80005-9. [DOI] [PubMed] [Google Scholar]
- 16.Kortylewski M, Jove R, Yu H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005;24(2):315–327. doi: 10.1007/s10555-005-1580-1. Prepublished on 2005/06/30 as DOI. [DOI] [PubMed] [Google Scholar]
- 17.Cheng F, Wang HW, Cuenca A, Huang M, Ghansah T, Brayer J, et al. A critical role for Stat3 signaling in immune tolerance. Immunity. 2003;19(3):425–436. doi: 10.1016/s1074-7613(03)00232-2. [DOI] [PubMed] [Google Scholar]
- 18.Kirberg J, Baron A, Jakob S, Rolink A, Karjalainen K, von Boehmer H. Thymic selection of CD8+ single positive cells with a class II major histocompatibility complex-restricted receptor. J Exp Med. 1994;180(1):25–34. doi: 10.1084/jem.180.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnden MJ, Allison J, Heath WR, Carbone FR. Defective TCR expression in transgenic mice constructed using cDNA-based alpha- and beta-chain genes under the control of heterologous regulatory elements. Immunol Cell Biol. 1998;76(1):34–40. doi: 10.1046/j.1440-1711.1998.00709.x. [DOI] [PubMed] [Google Scholar]
- 20.Staveley-O’Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, et al. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc Natl Acad Sci U S A. 1998;95(3):1178–1183. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith MR, Joshi I, Jin F, Al-Saleem T. Murine model for mantle cell lymphoma. Leukemia. 2006;20(5):891–893. doi: 10.1038/sj.leu.2404177. [DOI] [PubMed] [Google Scholar]
- 22.Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of Stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. 1999;10(1):105–115. doi: 10.1016/s1074-7613(00)80011-4. [DOI] [PubMed] [Google Scholar]
- 23.Niu G, Heller R, Catlett-Falcone R, Coppola D, Jaroszeski M, Dalton W, et al. Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer Res. 1999;59(20):5059–5063. [PubMed] [Google Scholar]
- 24.Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene [published erratum appears in Cell 1999 Oct 15;99(2):239] Cell. 1999;98(3):295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 25.Turkson J, Kim JS, Zhang S, Yuan J, Huang M, Glenn M, et al. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004;3(3):261–269. [PubMed] [Google Scholar]
- 26.Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11(12):1314–1321. doi: 10.1038/nm1325. [DOI] [PubMed] [Google Scholar]
- 27.Catlett-Falcone R, Dalton WS, Jove R. STAT proteins as novel targets for cancer therapy. Signal transducer an activator of transcription. Curr Opin Oncol. 1999;11(6):490–496. doi: 10.1097/00001622-199911000-00010. [DOI] [PubMed] [Google Scholar]
- 28.Niu G, Shain KH, Huang M, Ravi R, Bedi, Dalton WS, et al. Overexpression of a dominant-negative signal transducer and activator of transcription 3 variant in tumor cells leads to production of soluble factors that induce apoptosis and cell cycle arrest. Cancer Res. 2001;61(8):3276–3280. [PubMed] [Google Scholar]
- 29.Nefedova Y, Cheng P, Gilkes D, Blaskovich M, Beg AA, Sebti SM, et al. Activation of dendritic cells via inhibition of Jak2/STAT3 signaling. J Immunol. 2005;175(7):4338–4346. doi: 10.4049/jimmunol.175.7.4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005;65(20):9525–9535. doi: 10.1158/0008-5472.CAN-05-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark EA, Ledbetter JA. How B and T cells talk to each other. Nature. 1994;367(6462):425–428. doi: 10.1038/367425a0. [DOI] [PubMed] [Google Scholar]
- 32.Attanavanich K, Kearney JF. Marginal zone, but not follicular B cells, are potent activators of naive CD4 T cells. J Immunol. 2004;172(2):803–811. doi: 10.4049/jimmunol.172.2.803. [DOI] [PubMed] [Google Scholar]
- 33.Chaperot L, Plumas J, Jacob MC, Bost F, Molens JP, Sotto JJ, et al. Functional expression of CD80 and CD86 allows immunogenicity of malignant B cells from non-Hodgkin’s lymphomas. Exp Hematol. 1999;27(3):479–488. doi: 10.1016/s0301-472x(98)00059-9. [DOI] [PubMed] [Google Scholar]
- 34.Guilloux Y, Bai XF, Liu X, Zheng P, Liu Y. Optimal induction of effector but not memory antitumor cytotoxic T lymphocytes involves direct antigen presentation by the tumor cells. Cancer Res. 2001;61(3):1107–1112. [PubMed] [Google Scholar]
- 35.Adler AJ, Marsh DW, Yochum GS, Guzzo JL, Nigam A, Nelson WG, et al. CD4(+) T cell tolerance to parenchymal self-antigens requires presentation by bone marrow-derived antigen-presenting cells [In Process Citation] J Exp Med. 1998;187(10):1555–1564. doi: 10.1084/jem.187.10.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dave SS, Wright G, Tan B, Rosenwald A, Gascoyne RD, Chan WC, et al. Prediction of survival in follicular lymphoma based on molecular features of tumor-infiltrating immune cells. N Engl J Med. 2004;351(21):2159–2169. doi: 10.1056/NEJMoa041869. [DOI] [PubMed] [Google Scholar]
- 37.Zhou G, Lu Z, McCadden JD, Levitsky HI, Marson AL. Reciprocal changes in tumor antigenicity and antigen-specific T cell function during tumor progression. Journal of Experimental Medicine. 2004 doi: 10.1084/jem.20041240. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamper P, Bendix K, Hamilton-Dutoit S, Honore B, Nyengaard JR, d’Amore F. Tumor-infiltrating macrophages correlate with adverse prognosis and Epstein-Barr virus status in classical Hodgkin’s lymphoma. Haematologica. 2011;96(2):269–276. doi: 10.3324/haematol.2010.031542. Prepublished on 2010/11/13 as DOI haematol.2010.031542 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haabeth OA, Lorvik KB, Hammarstrom C, Donaldson IM, Haraldsen G, Bogen B, et al. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun. 2011;2:240. doi: 10.1038/ncomms1239. Prepublished on 2011/03/17 as DOI ncomms1239 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levitsky HI, Montgomery J, Ahmadzadeh M, Staveley-O’Carroll K, Guarnieri F, Longo DL, et al. Immunization with granulocyte-macrophage colony-stimulating factor- transduced, but not B7-1-transduced, lymphoma cells primes idiotype- specific T cells and generates potent systemic antitumor immunity. J Immunol. 1996;156(10):3858–3865. [PubMed] [Google Scholar]
- 41.Maric M, Chen L, Sherry B, Liu Y. A mechanism for selective recruitment of CD8 T cells into B7-1-transfected plasmacytoma: role of macrophage-inflammatory protein 1alpha. J Immunol. 1997;159(1):360–368. [PubMed] [Google Scholar]
- 42.Cantwell MJ, Wierda WG, Lossos IS, Levy R, Kipps TJ. T cell activation following infection of primary follicle center lymphoma B cells with adenovirus encoding CD154. Leukemia. 2001;15(9):1451–1457. doi: 10.1038/sj.leu.2402208. [DOI] [PubMed] [Google Scholar]
- 43.Briones J, Timmerman J, Levy R. In vivo antitumor effect of CD40L-transduced tumor cells as a vaccine for B-cell lymphoma. Cancer Res. 2002;62(11):3195–3199. [PubMed] [Google Scholar]
- 44.Evel-Kabler K, Song XT, Aldrich M, Huang XF, Chen SY. SOCS1 restricts dendritic cells’ ability to break self tolerance and induce antitumor immunity by regulating IL-12 production and signaling. J Clin Invest. 2006;116(1):90–100. doi: 10.1172/JCI26169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brayer J, Cheng F, Wang H, Horna P, Vicente-Suarez I, Pinilla-Ibarz J, et al. Enhanced CD8 T cell cross-presentation by macrophages with targeted disruption of STAT3. Immunol Lett. 2010;131(2):126–130. doi: 10.1016/j.imlet.2010.03.004. Prepublished on 2010/03/30 as DOI S0165-2478(10)00095-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lai R, Rassidakis GZ, Medeiros LJ, Leventaki V, Keating M, McDonnell TJ. Expression of STAT3 and its phosphorylated forms in mantle cell lymphoma cell lines and tumours. J Pathol. 2003;199(1):84–89. doi: 10.1002/path.1253. Prepublished on 2002/12/11 as DOI. [DOI] [PubMed] [Google Scholar]
- 47.Baran-Marszak F, Boukhiar M, Harel S, Laquiller C, Roger C, Gressin R, et al. Constitutive and B-cell receptor-induced activation of STAT3 are important signaling pathways targeted by bortezomib in leukemic mantle cell lymphoma. Haematologica. 2010;95(11):1865–1872. doi: 10.3324/haematol.2009.019745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4(2):97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 49.Madan R, Demircik F, Surianarayanan S, Allen JL, Divanovic S, Trompette A, et al. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J Immunol. 2009;183(4):2312–2320. doi: 10.4049/jimmunol.0900185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scuto A, Kujawski M, Kowolik C, Krymskaya L, Wang L, Weiss LM, et al. STAT3 inhibition is a therapeutic strategy for ABC-like diffuse large B-cell lymphoma. Cancer Res. 2011;71(9):3182–3188. doi: 10.1158/0008-5472.CAN-10-2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.