

Abstract
To identify the genes responsible for yield related traits, and heterosis, massively parallel signature sequencing (MPSS) libraries were constructed from leaves, roots and meristem tissues from the two parents, ‘Nipponbare’ and ‘93-11’, and their F1 hybrid. From the MPSS libraries, 1–3 million signatures were obtained. Using cluster analysis, commonly and specifically expressed genes in the parents and their F1 hybrid were identified. To understand heterosis in the F1 hybrid, the differentially expressed genes in the F1 hybrid were mapped to yield related quantitative trait loci (QTL) regions using a linkage map constructed from 131 polymorphic simple sequence repeat markers with 266 recombinant inbred lines derived from a cross between Nipponbare and 93-11. QTLs were identified for yield related traits including days to heading, plant height, plant type, number of tillers, main panicle length, number of primary branches per main panicle, number of kernels per main panicle, total kernel weight per main panicle, 1000 grain weight and total grain yield per plant. Seventy one QTLs for these traits were mapped, of which 3 QTLs were novel. Many highly expressed chromatin-related genes in the F1 hybrid encoding histone demethylases, histone deacetylases, argonaute-like proteins and polycomb proteins were located in these yield QTL regions. A total of 336 highly expressed transcription factor (TF) genes belonging to 50 TF families were identified in the yield QTL intervals. These findings provide the starting genomic materials to elucidate the molecular basis of yield related traits and heterosis in rice.
Introduction
Rice is one of the most important cereal crops feeding half of the worlds' population. Because of the increasing population and reduction of arable lands for rice production, improving grain yield is one of the most important goals of rice breeding programs [1], [2]. The genetic basis of yield and its component traits are complex, and controlled simultaneously by QTLs that are sensitive to environmental changes [3]–[5]. Hybrid rice where F1 plants are used has provided the highest yield potential in comparison with inbred cultivars. Since the 1970's hybrid rice has been widely cultivated in China and is now being extended to United States and worldwide.
Rice yield is either directly or indirectly affected by various yield related traits including days to heading [DTH], plant height [PHT], lodging [LOG], tiller angle [PTY], numbers of tillers [NOT], number of primary branches per panicle [NOB], total kernel weight per panicle [KWP], number of kernels per panicle [NOK], panicle length [PLE], 1000 grain weight [TGW] and total yield per plant [TYP]. Heading date is important to rice breeders because it affects adaptation of plants to various crop seasons and cultivation areas [6]. Heading date is regulated by a complex gene network consisting of a series of genetic factors [7]. Many genes that control heading date have been identified by QTL analysis [8]–[11]. Some of the important QTLs, Hd1, Hd3a and Ehd1 involved in heading date were cloned [12]–[14]. In addition, genes influencing heading date, plant height and rice yield like Ghd7 and Ghd8 were also cloned [15], [16]. A major plant height gene, the semi-dwarf gene sd1 was responsible for the green revolution in rice [17]. Some major QTLs for grain shape and 1000 grain weight such as GS3, GW2 and qSW5/GW5 were fine mapped and cloned [2], [18], [19]. The QTL Gn1a influencing the number of kernels per panicle was isolated by a map-based cloning strategy [1]. In addition, QTL controlling grain weight, gw8.1 and gw9.1 [20], [21] and number of spikelets per panicle, GPP1, gpa7 and SPP3b/TGW3b, were recently fine mapped [22]–[24]. In spite of hundreds of QTL mapping studies in rice for yield related traits, few of them have been isolated. Most of the genes either cloned or fine mapped so far belong to major QTLs, and the genes located in the minor QTL regions have not been fully explored.
In hybrids, novel patterns of gene action resulting from the combination of allelic variants are thought to be responsible for heterosis [25]–[28]. Dominance [29], over-dominance [30], [31], or epistasis [32], [33] were used to explain heterosis. For example, indica x japonica crosses show maximum heterosis compared to any other combination between other subspecies [34]. Gene expression and QTL analysis provide an avenue for identifying candidate genes for heterosis [35]. Several genomic approaches have been employed in rice and many genes underlying yield related traits have been identified [1], [2], [18], [19], [36], [37], [38]. For example, plant height is related to synthesis of sucrose phosphate synthase [SPS] [39], and phytohormones such as gibberellin and brassinolide [40], [41]. Further, large-scale transcriptome profiling has been used to identify the genes related to heterosis in crop plants such as rice [42]–[44], maize [45] and wheat [46]. Using a cDNA microarray consisting of 9198 expressed sequence tags [ESTs], gene expression profiles from an elite hybrid rice Shanyou 63, its parents [Zhenshan 97 and Minghui 63] revealed patterns of expressed genes may be associated with heterosis at three stages of young panicle development [42]. In addition, differentially expressed genes related to heterosis were identified in the super hybrid rice LYP9 compared to its parents [93–11 and PeiAi64S] using microarray and SAGE technologies [44], [45]. The root transcriptomes of the super-hybrid rice variety Xieyou 9308 and its parents were analyzed at tillering and heading stages for identification of candidate genes for heterosis [46] using RNA sequencing technology [RNA-Seq].
Both positive and negative heterosis can be employed in breeding depending on target traits, In general, positive heterosis is desirable for yield, and negative heterosis of growth duration is useful for earliness [47]–[49]. In F1 hybrid, the combination of allelic variants results in novel patterns of gene action possibly leading to heterosis [45], [49], [50]. Genetic variation, epistatic interaction, epigenetic modification and small- RNA-directed gene regulation were also shown to be related to heterosis [30], [33], [51]–[53]. Expression of transcription factors [TFs] and polymorphic cis-regulatory elements in the promoters of related genes in hybrids play an important role in heterotic gene expression and heterosis in rice [54]. Recently, gene expression profiling in Arabidopsis suggests that the genes involved in the circadian rhythm such as MYB-like transcription factors were associated with heterosis [55]. However, the molecular mechanism of either positive or negative heterosis remains poorly understood. For investigating heterosis at the transcriptome level EST library sequencing, microarray hybridization and serial analysis of gene expression [SAGE] have been used in crop plants, However, these technologies have drawbacks, such as low throughput, high cost, low sensitivity, cloning bias, high background signal, and pre-determined probe requirements [43], [44], [56]. Deep sequencing technologies including llumina's Massively Parallel Signature Sequencing [MPSS], Sequencing By Synthesis [SBS], RNA-Seq and pyrosequencing which offer large sequencing output with lower cost, have been widely applied in studying transcriptomes in plants and animals [47], [57]. The MPSS and SBS tags are short cDNA tags or digital gene expression tags, which are mainly derived from the 3′ regions of a transcript. These are deep sequencing methods previously used in rice and Arabidopsis [58], [59]. These tag- or sequence-based technologies determine the expression level of a gene by counting the precise abundance of a specific transcript in a library [25], [58], [59].
An inter-subspecific F1 hybrid was developed from a cross between Nipponbare [japonica] and 93-11[indica]. The genomes of both parents were completely sequenced [60]–[62]. Nipponbare is a rice cultivar developed in Japan [63]. Cultivar 93-11 is an elite parental line used in developing several super hybrid rice such as LYP9, YLY7, YLY1 etc. in China [44]. Thus, the F1 hybrid produced in this study provides a unique opportunity to investigate the molecular basis of yield related traits. The objectives of the present study were to 1) evaluate yield related traits and determine the transcription profiles of leaves, roots and meristems of the two parents, Nipponbare and 93-11] and their F1 hybrids using MPSS technology; 2) determine their commonly and specifically expressed genes; and 3) to map differentially expressed transcripts onto a genetic map and analyze their potentials for rice heterosis.
Results
Phenotyping and transcriptome sequencing of hybrid and their parents
F1 hybrid plants, RILs and the parents Nipponbare and 93-11 were phenotyped for yield traits (Table 1, Table S1). The F1 hybrid showed longer DTH [124 days], increased PHT [134 cm], narrower PTY [1], increased NOT [48], slightly shorter PLE [20.5], decreased number of NOK [16], decreased KWP [0.42], intermediate TGW [26.4] and decreased TYP [5.9] compared to the inbred parents (Figure S1). In this study, DTH, PHT and NOT in the F1 hybrid plants were greater than in the parents. PTY, PLE, NOK, KWP, and TYP in the F1 hybrid were less than the parents.
Table 1. Summary statistics of phenotypic performance of recombinant inbred lines, F1 hybrids and their parents Nipponbare and 93-11 (Parenthesis indicate averages).
| Trait | Stuttgart, AR-2009 | Stuttgart, AR-2010 | Stuttgart, AR-2011 | Stuttgart, AR-2012 | Beaumont, TX-2009 | Beaumont, TX-2010 | |||||||||||||
| NPB | 93-11 | RILs | NPB | 93-11 | RILs | NPB | 93-11 | RILs | F1 hybrids | NPB | 93-11 | RILs | NPB | 93-11 | RILs | NPB | 93-11 | RILs | |
| Number of days to heading | 84–87 (85.5) | 86–88 (87) | 68–131 (85.8) | 90–91 (90.3) | 84–86 (85.2) | 63.6-117-6 (80.1) | 96–99 (97.7) | 74–78 (75.2) | 61.7–105 (72.8) | 124 | 88.3–91.7 (90.1) | 92.7–102 (96.36) | 68–111.6 (83.6) | 80–81 (79.8) | 85–86 (85.5) | 67–137 (92.2) | 79 (79) | 87–100 (94.3) | 69.5–111.5 (87.8) |
| Plant height (cm) | 86–103 (93.5) | 99–120 (111.4) | 60–168 (106.9) | 88–98 (93.3) | 124–136 (130) | 67.33–152 (108.6) | 70–78 (73.3) | 118–128 (123.1) | 64–154 (104.3) | 134 | 84–94 (90) | 98–118 (108) | 54–146 (102.6) | 79.5–82.5 (82) | 100.5–105-5 (103) | 66.7–153.7 (98.5) | 70–84 (76) | 84–90 (86.8) | 51.88–128.6 (83.4) |
| Lodging status | 0 (0) | 0 (0) | 0–8 (0.3) | 0 (0) | 0 (0) | 0–3 (0.03) | - | - | - | - | - | - | - | 0 | 0 | 0–3 (0.04) | 0 | 0 | 0–0.33 (0.002) |
| Tiller angle (plant type) | 3 (3) | 3 (3) | 1–7 (3.8) | 3 (3) | 3 (3) | 1–7 (3.4) | 3 (3) | 3 (3) | 1–7 (3.8) | 1 | 3 (3) | 3(3) | 1–6 (3.1) | 1–3 (2) | 3–5 (4) | 1–7 (2.8) | 1–3 (2) | 3 (3) | 1–7 (2.9) |
| Number of tillers per plant | 21–29 (25.1) | 18–25.3 (21.8) | 8.3–81 (30.6) | - | - | - | 27–41 (38.4) | 29–44 (36.5) | 22–62 (36.6) | 48 | 19–34.3 (25.8) | 18–23 (19.75) | 10.5–48.4 (26.8) | 29–42 (37.7) | 35–39 (36.7) | 15.7–70.7 (37.2) | - | - | - |
| Main panicle length (cm) | 21.3–22.2 (21.9) | 24–25 (24) | 14.3–32.5 (22.2) | - | - | - | 20–22 (21.5) | 22–24 (22.5) | 15.7–29.2 (21.3) | 20.5 | 20–23.2 (21.7) | 23.8–26.5 (25) | 15.6–31 (23) | 20–21 (20.4) | 24.5–26.7 (25.7) | 16.5–32.5 (22.4) | - | - | - |
| Number of branches in main panicle | 9.7–11 (10.3) | 10–12.3 (11.1) | 7–15.7 (10.6) | - | - | - | - | - | - | - | 8.7–10.3 (9.5) | 10–12 (11) | 6.5–17.5 (10) | 10–12 (11) | 10–12 (11.33) | 8–18 (11.6) | - | - | - |
| Number of grains per main panicle | 92.7–113.3 (105.8) | 165.3–191 (179.3) | 10–272.7 (126.2) | - | - | - | 27–41 (35) | 152–192 (177.9) | 2.8–176.4 (76.9) | 16 | 98.3–138 (117.2) | 68.3–230 (160.2) | 19.5–305.6 (112.5) | 93–105 (99.3) | 180–201 (189) | 28.7–248.7 (114.9) | - | - | - |
| Total kernel weight per panicle | 2.6–3.1 (2.93) | 5.8–6.2 (6) | 0.2–7.4 (3.6) | - | - | - | 0.9–1.1 (1.05) | 5.1–5.5 (5.2) | 0.067–5 (1.97) | 0.42 | 2–3.5 (2.8) | 3.3–6.6 (5.4) | 0.4–7 (2.8) | 2.4–2.6 (2.5) | 5.2–5.8 (5.4) | 1.1–5.4 (3.08) | - | - | - |
| 1000 grain weight (grams) | 27.1–28.1 (27.8) | 32.5–34.8 (33.4) | 11.1–43.4 (27.8) | - | - | - | 24–29 (26) | 26.8–29.3 (28.2) | 15.9–39.7 (25) | 26.4 | 24.2–25.7 (25.3) | 25.4–28.7 (27) | 16.5–40.2 (25.2) | 25–26 (24.8) | 28–29 (28.8) | 15.2–33.2 (24.8) | - | - | - |
| Total grain yield per plant (grams) | 50.1–65.1 (56.6) | 97.2–126.1 (107.9) | 6–179.5 (74.5) | - | - | - | 30–46 (40.2) | 152–187 (155.6) | 3.9–155 (55) | 5.9 | 52.3–89.3 (71) | 78.6 (78.6) | 13.6–223.2 (79.8) | 52–73 (69.6) | 140–156 (152) | 27.3–149.5 (82.9) | - | - | - |
About 1.0 to 3.0 million 17-base MPSS signatures were obtained in the 21 libraries (Table 2, Table S2). These signatures were clustered and processed with reliability and significance filters as described by Meyers et al. [40]–[42] (Figure S2). To compare the expression levels across the libraries, the frequency of signatures in the individual libraries were normalized to one million [transcripts per million or TPM] [40]–[42]. The number of distinct signatures ranged from 14,127 to 28,621 in the MPSS libraries. The number of distinct genes identified using reliable and significance filtered signatures from 5,444 to 12,717 genes. About 56 to 87% of the signatures from Nipponbare matched to the Nipponbare genomic sequence. Similarly, about 77 to 87% of the signatures obtained from the 93-11 tissues matched to the 93-11 genomic sequence (Table S2). In F1, about 74 to 84% of signatures matched Nipponbare and 75 to 85% of signatures matched 93-11 (Table S2). The significant MPSS signatures from all 21 libraries were classified into seven classes based on their location on the annotated genes (Table S3).
Table 2. Characteristics of MPSS libraries of Nipponbare, 93-11 and F1 hybrid from leaves, roots and meristem tissues.
| Category | Nipponbare | 93-11 | F1 hybrid | ||||||
| Leaves∞ | Roots∞ | Meristems | Leaves∞ | Roots∞ | Meristems | Leaves∞ | Roots∞ | Meristems | |
| Total reads sequenced | 4940921 | 5293337 | 2,568,641 | 4902516 | 4319104 | 2,112,790 | 4687682 | 4642271 | 3,045,290 |
| Distinct | 60782 | 48206 | 28621 | 38478 | 34771 | 21836 | 44455 | 29119 | 24715 |
| Reliable* | 44971 | 38064 | 24183 | 31696 | 28109 | 17570 | 38082 | 24291 | 19945 |
| Significant* | 48688 | 38330 | 24305 | 33332 | 29195 | 19027 | 40037 | 24476 | 20056 |
| Reliable and Significant (≥4 TPM) | 35617 | 27282 | 18106 | 26697 | 22996 | 14081 | 32370 | 17289 | 15033 |
| 1–100 TPM | 57321 | 45257 | 26237 | 35346 | 31371 | 19695 | 41236 | 26983 | 22579 |
| 101–1,000 TPM | 3279 | 2804 | 2245 | 2973 | 3249 | 1962 | 3072 | 1958 | 1950 |
| 1,001–10,000 TPM | 190 | 151 | 134 | 169 | 158 | 166 | 161 | 184 | 176 |
| >10,000 TPM | 8 | 4 | 5 | 6 | 3 | 13 | 7 | 10 | 10 |
| #Distinct genes | 11902 | 11816 | 9360 | 10890 | 10186 | 6696 | 12717 | 8007 | 7591 |
| $ Genome match | 22786 (64%) | 21122 (77%) | 14936 (83%) | 20991 (79%) | 17756 (77%) | 10777 (77%) | 24717(76%) 24895 (77%) | 12855(74%) 13005 (75%) | 11763 (78%) 11857 (79%) |
Note: *- Number of signatures passed through reliability and significance filters as described by Meyers et al. [58]–[59]
#- Using all reliable and significance filter signatures.
$- Number and % of signatures match Nipponbare/93-11/both genomic sequences (≥4 TPM).
- Mean calculated from four leaf libraries are shown for leaf libraries. Similarly, the mean calculated from two root libraries are shown for root libraries (details of all the libraries can be found in Table S4).
Italics indicate- match to 93-11 genomic sequence.
Commonly and specifically expressed genes
Based on the results of a Venn diagram, we analyzed the differences that existed in gene expression among leaves, roots and meristem tissues. The similarities between any two genotypes [Nipponbare, 93-11 and F1 hybrid] were established based on the number of similar genes expressed between any two genotypes, and the number of genotype specifically expressed genes. The number of genes expressed in all three tissues [leaves, roots and meristems] in Nipponbare, 93-11 and the F1 hybrid were identified. A total of 7,812, 5,181 and 4,009 genes were commonly expressed in leaves, roots and meristem tissues, respectively. The number of commonly expressed genes was higher than specifically expressed genes in all 3 tissues (Figure 1).
Figure 1. Identification of commonly and specifically expressed genes in Nipponbare, 93-11 and F1 hybrid tissues.
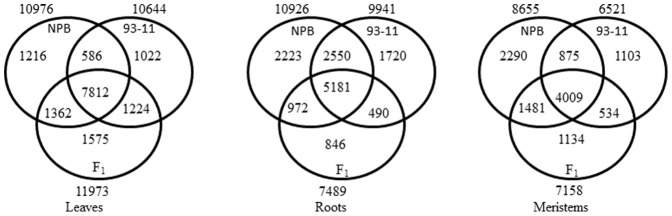
Signatures matched to both Nipponbare and 93-11 genomes were used to avoid sequence bias in the identification of commonly and specifically expressed genes among Nipponbare, 93-11 and F1 hybrid.
The expression levels of Nipponbare and 93-11 were compared with their F1 hybrid and the differentially expressed signatures were classified into eight expression patterns: above high parent level [AHPL], high parent level [HPL], mid parent level [MPL], low parent level [LPL], below low parent level [BLPL], transcripts specifically expressed in F1 and absent in parents [SEF1], transcripts expressed in either one of the parents and F1 [EOPF1], transcripts expressed in either of the parents but absent in F1 [EPAF1]. To observe which gene expression patterns were highly represented in the F1 hybrid, the gene expression patterns, including AHPL, HPL, MPL, LPL, BLPL and SEF1 were compared in leaves, roots and meristem tissues (Table S4). AHPL and SEF1 represent a majority of the gene expression pattern in leaves, roots and meristems (Figure 2). In leaves, roots and meristems combined AHPL and SEF1 expression patterns represented 58%, 48% and 54%, respectively. AHPL expression patterns represented 22%, 17%, and 17% while SEF1 expression patterns represented 36%, 31% and 37% in leaves, roots and meristems, respectively. These findings suggest that novel patterns of gene action thought to be involved in heterosis resulting in allelic variants from the parents and the hybrid.
Figure 2. Distribution of ‘gene expression level categories’ in F1 hybrid's leaf, root and meristem tissues.

The gene expression levels in leaf, root and meristem tissues of F1 hybrid were measured in comparison to their parents Nipponbare and 93-11. The differentially expressed signatures in F1 hybrid belonging to expression patterns: above high parent level (AHPL), high parent level (HPL), mid parent level (MPL), low parent level (LPL), below low parent level (BLPL) and transcripts expressed specifically in F1 and absent in parents (SEF1) are represented here.
Mapping differentially expressed genes
To better understand mechanisms underlying positive and negative heterosis in F1 hybrids, a linkage map was constructed using 131 polymorphic simple sequence repeat [SSR] markers with 266 RILs derived from a cross between Nipponbare and 93-11. Phenotypes of yield related traits were evaluated in 6 field studies [Stuttgart-2009, Stuttgart-2010, Stuttgart-2011, Stuttgart-2012, Beaumont-2009 and Beaumont-2010] (Table 1). Transgressive segregation was observed among the RILs for yield related traits (Table 1). Obvious transgressive segregation was observed for PTY [1]–[7], LOG [0–8], PHT [60–168 cm], NOT [8.3–81], NOK [10–272.7], TGW [11.1–43.4 g] and TYP [6–179.5 g] compared to other traits evaluated (Table 1; Figure S1; Table S1).
Seventy-one QTLs above the significant threshold level for yield related traits were mapped (Figure 3; Table 3; Table S5). A total of three novel QTLs related to PTY [2] and KWP [1] were identified. No QTL above the significant threshold level was found for LOG. We selected the QTLs which showed mean LOD scores of ≥5 for a given trait for identification of differentially expressed genes located in the 71 mapped QTLs (Table S6). Based on the physical location of SSR markers on the chromosomes, the differentially expressed genes between flanking markers were identified. Expression patterns for the differentially expressed genes in a particular QTL were listed in Table S6 (Table S6).
Figure 3. Chromosomal locations of yield related QTL identified using RILs obtained from Nipponbare x 93-11 cross.

Seventy one QTL identified were mapped for yield related traits including days to heading (qdth), plant height (qpht), tiller angle (qpty), tiller number (qnot), panicle length (qple), number of primary branches per panicle (qnob), number of kernels per panicle (qnok), total kernel weight per panicle (qkwp), 1000 grain weight (qtgw) and total grain yield per plant (qtyp).
Table 3. List of previously published QTLs for yield and yield related traits identified in recombinant inbred line population (Nipponbare x 93-11) based on Gramene QTL database.
| Trait | Experiment location | Marker interval | Genetic distance (cM) | LOD peak position | Ch. | R2 (%) | Additive effect∧ | LOD threshold | LOD value | Avg. LOD score | No. of genes present | QTL name in this study | Published QTL |
| Days to heading | |||||||||||||
| 2009-Beaumont, TX | RM231–RM489 | 0–13.6 | 0.2 | 3 | 12.7 | −4.0 | 3.6 | 5.8 | 7.6 | 176 | qdth-1 # | QTL3, dth3.1, qFDN-3 | |
| 2009-Stuttgart, AR | RM231–RM489 | 0–13.6 | 0 | 3 | 10.2 | −3.0 | 3.6 | 11.2 | |||||
| 2010-Stuttgart, AR | RM231–RM489 | 0–13.6 | 4 | 3 | 11.3 | −3.5 | 3.6 | 10.5 | |||||
| 2010-Beaumont, TX | RM231–RM489 | 0–13.6 | 5 | 3 | 9.6 | −2.8 | 3.5 | 7.5 | |||||
| 2011-Stuttgart, AR | RM231–RM489 | 0–13.6 | 1 | 3 | 7.1 | −2.4 | 3.8 | 4.9 | |||||
| 2012-Stuttgart, AR | RM231–RM489 | 0–13.6 | 2.1 | 3 | 8.13 | −2.8 | 3.5 | 5.53 | |||||
| 2009-Stuttgart, AR | RM15766–RM15824 | 107.2–113.5 | 108.6 | 3 | 3.3 | −1.7 | 3.6 | 3.5 | 6.1 | 321 | qdth-2 # | QHd3c, Hd6, QHd3b | |
| 2010-Beaumont, TX | RM15824–RM6759 | 113.5–128.8 | 122.6 | 3 | 12.4 | −3.1 | 3.5 | 8.7 | |||||
| 2010-Stuttgart, AR | Con673–RM527 | 21–33.5 | 31.4 | 6 | 49.9 | 11.3 | 3.6 | 43.2 | 25.0 | 126 | qdth-5 # | Hd1, Hd-1 | |
| 2009-Beaumont, TX | Con673–RM527 | 21–33.5 | 30.5 | 6 | 21.0 | 8.6 | 3.6 | 8.4 | |||||
| 2010-Beaumont, TX | MRG2431–RM527 | 30.4–33.5 | 31.3 | 6 | 17.4 | 5.7 | 3.5 | 15.7 | |||||
| 2009-Stuttgart, AR | MRG2431–RM527 | 30.4–33.5 | 31.5 | 6 | 49.9 | 9.9 | 3.6 | 43.5 | |||||
| 2011-Stuttgart, AR | Con673–MRG2431 | 21–30.4 | 29.1 | 6 | 40.7 | 9.9 | 3.8 | 23.7 | |||||
| 2012-Stuttgar, AR | Con673–RM527 | 21–33.5 | 29 | 6 | 25.2 | 7.5 | 3.5 | 15.4 | |||||
| 2010-Beaumont, TX | RM125–RM418 | 14.1–28.9 | 23 | 7 | 10.4 | −2.9 | 3.4 | 9.4 | 9.4 | 359 | qdth-6 | hd7c, Hd7, qHDD7-1, Hd4, qHD-7 | |
| 2009-Stuttgart, AR | RM1362–RM248 | 74.8–78.4 | 78 | 7 | 3.0 | 1.7 | 3.6 | 3.4 | 5.5 | 41 | qdth-7 # | qDTH-7, QTL7b | |
| 2010-Stuttgart, AR | RM1362–RM248 | 74.8–78.4 | 78 | 7 | 7.4 | 2.9 | 3.6 | 8.1 | |||||
| 2009-Beaumont, TX | RM1362–RM248 | 74.8–78.4 | 78 | 7 | 7.5 | 3.1 | 3.6 | 3.6 | |||||
| 2011-Stuttgart, AR | RM118–RM248 | 63.1–78.4 | 78 | 7 | 8.3 | 2.7 | 3.8 | 6.0 | |||||
| 2012-Stuttgart, AR | RM1362-RM248 | 74.8–78.4 | 78 | 7 | 8.4 | 2.9 | 3.5 | 6.3 | |||||
| Plant height | |||||||||||||
| 2009-Beaumont, TX | RM486–RM315 | 124.7–131.4 | 130.6 | 1 | 22.8 | 8.8 | 3.8 | 7.4 | 16.9 | 147 | qpht-1 # | Ph1, qph1.4, qPHT-1 | |
| 2009-Stuttgart, AR | RM486–RM315 | 124.7–131.4 | 131 | 1 | 32.0 | 12.4 | 3.5 | 21.4 | |||||
| 2010-Stuttgart, AR | RM486–RM315 | 124.7–131.4 | 130.6 | 1 | 29.2 | 9.7 | 3.6 | 22.1 | |||||
| 2010-Beaumont, TX | RM486–RM315 | 124.7–131.4 | 130.6 | 1 | 21.7 | 7.2 | 3.4 | 17.8 | |||||
| 2011-Stuttgart, AR | RM486–RM315 | 124.7–131.4 | 130.6 | 1 | 28.6 | 9.3 | 3.7 | 19.1 | |||||
| 2012-Stuttgart, AR | RM486–RM315 | 124.7–131.4 | 130.6 | 1 | 21.0 | 8.4 | 3.4 | 13.4 | |||||
| 2011-Stuttgart, AR | RM162–RM5371 | 55–63.8 | 62 | 6 | 7.0 | −5.3 | 3.7 | 5.0 | 5.0 | 81 | qpht-5 | Fh6-2, ph6.1, QPh6 | |
| 2010-Beaumont, TX | RM107–LJSSR1 | 66.3–77.1 | 69.4 | 9 | 7.7 | 4.2 | 3.4 | 6.1 | 5.1 | 137 | qpht-7 # | Fh9-2 | |
| 2009-Stuttgart, AR | RM107–LJSSR1 | 66.3–77.1 | 71.4 | 9 | 4.6 | 4.4 | 3.5 | 4.1 | |||||
| Tiller angle | |||||||||||||
| 2010-Stuttgart, AR | RM434–LJSSR1 | 48.3–77.1 | 70.2 | 9 | 48.6 | −1.1 | 3.5 | 36.6 | 31.80 | 477 | qpty-3 # | ta9, Ta, qTA-9a, ta9.1 | |
| 2009-Beaumont, TX | RM434–LJSSR1 | 48.3–77.1 | 66.3 | 9 | 37.9 | −1.6 | 4 | 13.2 | |||||
| 2010-Beaumont, TX | RM107–LJSSR1 | 66.3–77.1 | 69.6 | 9 | 58.8 | −1.2 | 3.6 | 45.6 | |||||
| 2009-Stuttgart, AR | RM107–LJSSR1 | 66.3–77.1 | 69.7 | 9 | 43.0 | −1.3 | 3.4 | 32.7 | |||||
| 2011-Stuttgart, AR | RM434–LJSSR1 | 48.3–77.1 | 61.6 | 9 | 50.9 | −1.5 | 3.7 | 25.2 | |||||
| 2012-Stuttgart, AR | RM107–LJSSR1 | 66.3–77.1 | 70.5 | 9 | 66.7 | −1.4 | 3.6 | 37.56 | |||||
| Tiller number | |||||||||||||
| 2010-Beaumont, TX | RM452–RM475 | 33.6–60.9 | 46.6 | 2 | 8.3 | 2.9 | 3.5 | 6.4 | 6.4 | 410 | qnot-2 | qNOT2-1, mtn2-1, tn2-1, qNOT2-1 | |
| 2009-Beaumont, TX | RM15824–RM514 | 113.5–145.5 | 131 | 3 | 13.7 | −4.0 | 3.9 | 5.0 | 5.0 | 583 | qnot-4 | tn3-4 | |
| 2010-Beaumont, TX | RM451–RM124 | 74.4–104 | 82.2 | 4 | 9.5 | −3.0 | 3.5 | 6.8 | 6.1 | 541 | qnot-5 # | tp4, mtn4, tn4-3 | |
| 2011-Stuttgart, AR | RM348–RM124 | 87.8–104 | 99.5 | 4 | 10.0 | −2.2 | 3.5 | 5.4 | |||||
| 2010-Beaumont, TX | RM125–RM418 | 14.1–28.9 | 18.9 | 7 | 7.9 | 2.8 | 3.5 | 5.8 | 5.1 | 515 | qnot-6 # | tp7, tn7.1, qTN-7-2 | |
| 2011-Stuttgart, AR | RM5711–RM125 | 0–14.1 | 10.9 | 7 | 8.2 | 2.0 | 3.5 | 4.5 | |||||
| 2010-Beaumont, TX | RM434–LJSSR1 | 48.3–77.1 | 70.8 | 9 | 9.7 | −3.1 | 3.5 | 7.3 | 6.3 | 477 | qnot-7 # | qNOT9-1 | |
| 2011-Stuttgart, AR | RM434–LJSSR1 | 48.3–77.1 | 67.2 | 9 | 9.2 | −2.2 | 3.5 | 5.9 | |||||
| 2012-Stuttgart, AR | RM107–LJSSR1 | 66.3–77.1 | 74.3 | 9 | 9.4 | −2.1 | 3.6 | 5.6 | |||||
| Panicle length | |||||||||||||
| 2009-Beaumont, TX | RM302–RM315 | 115.6–131.4 | 131 | 1 | 16.9 | 1.2 | 3.6 | 5.1 | 5.1 | 302 | qple-1 | pI1.1, qPL-1 | |
| 2011-Stuttgart, AR | RM450–RM166 | 97.5–126.4 | 115.2 | 2 | 7.7 | −0.7 | 3.6 | 5.1 | 5.1 | 504 | qple-2 | pI2a | |
| 2009-Stuttgart, AR | RM5711–RM2 | 0–23.9 | 16.3 | 7 | 7.9 | −0.8 | 3.5 | 5.3 | 6.7 | 515 | qple-5 # | QPI7, pI7.1 | |
| 2011-Stuttgart, AR | RM2–RM560 | 23.9–35.1 | 28.9 | 7 | 10.4 | −0.7 | 3.6 | 8.1 | |||||
| Number of branches | |||||||||||||
| 2009-Stuttgart, AR | RM452–RM475 | 33.6–60.9 | 44.6 | 2 | 11.3 | −0.5 | 3.5 | 6.3 | 6.3 | 410 | qnob-2 | qNRB-2-1 | |
| 2009-Beaumont, TX | RM149–RM5493 | 71.5–88.1 | 84.5 | 8 | 21.0 | 1.0 | 4.1 | 5.5 | 5.5 | 116 | qnob-4 | qNRB-8-1, qNRB-8-2 | |
| Number of grains per panicle | |||||||||||||
| 2009-Stuttgart, AR | RM341–RM475 | 52.9–60.9 | 62.2 | 2 | 8.6 | −13.3 | 3.8 | 6.5 | 6.5 | 54 | qnok-3 | gn2.1, sn2.1 | |
| Total grain wt. per panicle | |||||||||||||
| 2009-Stuttgart, AR | RM486–RM315 | 124.7–131.4 | 130.7 | 1 | 8.5 | 0.3 | 3.6 | 6.6 | 6.6 | 147 | qkwp-2 | Pdw1-1 | |
| 2009-Stuttgart, AR | RM341–RM5427 | 52.9–68.3 | 61.1 | 2 | 7.0 | −0.3 | 3.6 | 5.6 | 5.6 | 122 | qkwp-3 | Gw2 | |
| 2009-Beaumont, TX | RM5711–RM2 | 0–23.9 | 17.2 | 7 | 16.6 | −0.4 | 3.8 | 5.5 | 5.5 | 515 | qkwp-5 # | Pdw7 | |
| 2009-Stuttgart, AR | RM125–RM418 | 14.1–28.9 | 20.3 | 7 | 9.1 | −0.4 | 3.6 | 6.0 | |||||
| 2012-Stuttgart, AR | RM2–RM418 | 23.9–28.9 | 26.1 | 7 | 7.7 | −0.3 | 3.3 | 5.0 | |||||
| 2009-Beaumont, TX | RM254–RM224 | 66.1–78.1 | 77.2 | 11 | 14.4 | −0.4 | 3.8 | 5.6 | 5.6 | 124 | qkwp-8 | Unnamed* | |
| 1000 grain wt. | |||||||||||||
| 2009-Stuttgart, AR | RM413–RM169 | 13.3–45.4 | 14.5 | 5 | 9.2 | 1.3 | 3.7 | 7.8 | 6.5 | 279 | qtgw-2 # | gw5a, gw5 | |
| 2009-Beaumont, TX | RM437–RM169 | 23–45.4 | 28.6 | 5 | 17.7 | 1.5 | 3.6 | 4.8 | |||||
| 2011-Stuttgart, AR | RM437–RM169 | 23–45.4 | 29.6 | 5 | 13.5 | 1.4 | 3.4 | 5.6 | |||||
| 2012-Stuttgart, AR | RM437–RM169 | 23–45.4 | 28.6 | 5 | 15.3 | 1.3 | 3.7 | 7.8 | |||||
| 2009-Stuttgart, AR | RM5371–RM340 | 63.8–82.6 | 80.7 | 6 | 8.0 | −1.1 | 3.7 | 6.3 | 6.3 | 190 | qtgw-5 | qGW-6, gw6c, gw6b | |
| Grain yield per plant | |||||||||||||
| 2009-Beaumont, TX | RM6863–RM310 | 2.8–16.5 | 11.6 | 8 | 14.1 | −10.4 | 3.5 | 5.4 | 5.7 | 442 | qtyp-4 # | qYI-8, yld8.3 | |
| 2011-Stuttgart, AR | RM310–MJIndel1 | 16.5–31.5 | 23 | 8 | 14.2 | −9.5 | 3.8 | 6.1 |
Number of expressed genes identified in leaves, roots and meristem tissues based on their location in the mapped yield and yield related QTLs. LOD score ≥5 was used to identify the genes located in the mapped yield and yield related QTLs. Average LOD score was calculated for each QTL detected based on 6 experiments (more details are given in the supplemental table 1 and supplemental table 5).
Note: *- indicate the QTL is unnamed in Gramene rice QTL database-http://gramene.org/qtl/ or Japanese Rice QTL database - http://qtaro.abr.affrc.go.jp/.
#-Qtl identified in multiple locations.
∧- A positive additive effect indicates the Nipponbare allele increases the phenotype.
Of the seven QTLs for DTH, five showed average LOD score ≥5. The QTL qdth5 located at 21–33.5 cM on chromosome 6 [close to the flanking markers Con673 and RM527] had the highest mean LOD score of 25. Nine QTL were identified for PHT, of which three showed an average LOD score of ≥5. The QTL qpht1 located at 124.7–131.4 cM on chromosome 1 [close to the flanking markers RM486 and RM315] showed a mean LOD score of 16.8. Both parents Nipponbare and 93-11 have intermediate tiller angle [about 45°], but the F1 hybrid has erect tillers and an angle less than 30° from the perpendicular. Three QTLs were detected for tiller angle, of which qpty1 and qpty2 were not reported in gramene [ftp://ftp.gramene.org/pub/gramene/CURRENT_RELEASE/data/qtl/] and Japanese database [http://qtaro.abr.affrc.go.jp/qtab/table]. The qpty3 allele located on chromosome 9 contained 477 differentially expressed genes between marker intervals [RM434-LJSSR1] and was detected in all six field experiments with a mean LOD score of 31.8. Seven QTL have been identified for NOT, of which five showed mean LOD scores of ≥5. The QTLs qnot5 [chromosome 4; 74.4–104 cM] and qnot7 [chromosome 9; 48.3–77.1 cM] showed a mean LOD score of 6.1 and 6.3, respectively.
We identified seven QTLs related for PLE, of which three showed average LOD scores of ≥5. We identified six QTLs related to NOB, two of which showed an average LOD score of ≥5. A total of ten QTLs have been identified for NOK. Of these QTL only qnok3 showed a significant LOD score of 5 or higher (LOD 6.5). Of the nine QTLs detected for Total kernel weight per panicle, qkwp7 was a novel QTL. The allele qkwp8 was detected in the public rice QTL database, but was unnamed. QTLs for KWP were found on chromosomes 1, 2, 6, 7, 8 and 11 and four of these had a LOD ≥5. A total of seven QTLs were detected for TGW. The qtgw1, qtgw3, qtgw5 and qtgw7 alleles were only detected at the Stuttgart location. Of these, two QTLs had a LOD ≥5. Total grain yield per plant is the most important trait to be mapped on chromosomes for improving rice yield. The QTLs related to TYP were detected in the Beaumont 2009 and Stuttgart 2011. Six QTLs were found to be associated with TYP, of which four QTLs [qtyp1, qtyp2, qtyp4 and qtyp5] were detected at the Beaumont 2009 location. The QTL qtyp4 located on chromosome 8 at 2.8–31.5 cM showed a LOD score of 5.72, and contained 442 differentially expressed genes between the closely linked markers RM6863 and MJIndel1.
Expressed TF genes located in the mapped yield QTL were identified using homology search in the rice transcription factor database [http://ricetfdb.bio.uni-potsdam.de/v2.1/]. A total of 336 expressed TF genes belonging to 50 TF families, were identified in combined leaves, roots and meristem tissues, located in yield related QTL intervals (Table S7). Six TF genes were expressed at AHPL in all three tissues. This included three genes encoding helix-loop-helix [HLH] DNA binding domain containing proteins Os03g53020 [qdth2, qnot4], Os01g57580 [qple1] and Os02g47660 [qple2]. In addition, a ZIM motif family protein [Os03g08330], gibberellins response modulator gene belonging to GRAS TF family [Os03g49990], and homeobox associated leucine zipper family protein belonging to HB TF family [Os09g29460] located at mapped yield related QTL regions, showed the expression pattern AHPL in all three tissues-leaves, roots and meristems. Three TF genes showing expression pattern BLPL in all three tissues included a gene encoding homeobox domain containing protein belonging to HB TF family [Os02g49700; qple2], a transcriptional adaptor gene belonging to MYB TF family [Os03g53960; qnot4, qdth2], and a zinc-finger protein 1 gene belong to C2H2 TF family [Os03g55540; qnot4, qdth2] (Table S7).
Some of the highly expressed TF genes showing the AHPL expression pattern in both leaves and roots included: gene encoding for protein PHD finger protein gene [Os02g35600; qkwp3], gene encoding for ULTRAPETALA2 protein [Os01g57240; qple1], gene encoding auxin response factor 1 [Os02g35140; qkwp3] and gene encoding bZIP transcription factor [Os02g52780; qple2], gene encoding expressed protein [Os04g50120; qnot5] and, gene encoding WRKY DNA binding domain containing protein [Os04g51560; qnot5]. Some TF genes showed AHPL expression pattern in both leaves and meristems include gene encoding homeobox protein knotted-1-like 3 gene [Os06g43860; qtgw5], gene encoding AP2 domain containing protein [Os04g55520; qnot5], gene encoding RNA recognition motif family protein [Os07g48410; qdth7] and gene encoding myb like DNA binding domain protein [Os03g55590; qnot4]. TF genes showed AHPL expression pattern in both roots and meristems include gene encoding zinc finger protein [Os03g60570; qnot4], gene encoding DREB1A protein [Os09g35030; qpht7] and gene encoding GRAS family protein [Os04g50060; qnot5] (Table S7). In leaves, roots and meristems the highly expressed TF genes belonging to AHPL expression pattern encoding SET domain containing protein [Os02g50100, qple2], TF TGA4 protein [Os08g07970; qtyp4; bZIP family] and Myb-like DNA binding domain protein [Os09g31454; qpty3 or qnot7] respectively were identified.
Expression of genes involved in epigenetics
A total of 99 epigenetic/chromatin-related genes were expressed in leaves, roots and meristem tissues (Table S8). Several genes belonging to flowering, histone demethylases, histone deacetylases, genes encoding argonaute like proteins and polycomb group genes were highly represented. In leaves, genes belonging to the AHPL expression pattern were expressed including genes encoding flowering control-associated proteins [Os03g58070- qnot4; Os02g49230-qple2; Os06g15330-qdth5; Os06g44450-qtgw5; Os02g49840-qple2; Os06g16370-qdth5; Os03g50310-qdth2], genes encoding methyl binding domain proteins [Os04g52380-qnot5; Os09g29750-qpty3,qnot7], polycomb group gene [Os08g04270-qpty4]; jumonji domain group gene [Os05g10770-qtgw2], a gene encoding an argonaute-like protein [Os07g28850-qnot6, qple5, qkwp5, qdth6], piwi domain containing gene [Os03g57560-qnot4], a gene with a double stranded RNA-binding motif [Os05g05790-qtgw2], histone deacetylases [Os07g06980-qnot6, qple5, qkwp5], histone demethylases [Os08g04780 -qtyp4], histone H1 linker protein [Os03g58470 -qnot4], RNA helicases [Os03g06440-qdth1] and genes encoding bromodomain containing proteins [Os08g09340-qtyp4, Os09g33980-qpty3, qnot7, qpht7]. In roots, some of the genes belonging to the AHPL expression pattern were expressed including: genes encoding a histone H1 linker protein [Os03g58470-qnot4], bromodomain-containing proteins [Os08g39980-qnob4, Os08g09340-qtyp4], double-stranded RNA binding motif family protein [Os05g05790-qtgw2], histone ubiquitination proteins [group B Rad6 homolog] [Os03g57790-qnot4] and flowering control-associated protein [Os02g52340 -qple2] (Table S8).
Functional classification of genes showing AHPL expression pattern in F1 hybrids
The genes in the mapped yield related QTL showing AHPL expression patterns were classified into different groups based on KEGG's functional classification of genes (Figure S3). Biochemical pathways including carbohydrate metabolism, energy metabolism and metabolism of cofactors and vitamins were highly represented in leaves, roots and meristem tissues. Leaf and meristem tissues showed expression of more energy and nucleotide metabolism genes compared to roots (Figure S3). Because in leaves, seven biochemical pathways including carbohydrate metabolism, energy metabolism, nucleotide metabolism, metabolism of cofactors and vitamins, amino acid metabolism, translation, and sorting and degradation were highly represented, we further classified these genes based on mapped QTLs. More carbohydrate metabolism related genes were present in QTLs qdth2, qnot4, qnot6, qple5 and qkwp5. The QTLs qnot4 and qnot5 were highly represented by genes belonging to all seven biochemical pathways (Figure S3). Some of the genes belonging to the photosynthesis and carbon fixation [dark cycle/Calvin cycle] pathways showed AHPL expression patterns in leaves included the genes encoding ribulose-phosphate 3-epimerase [Os09g32810- qpty3 or qnot7], uridine/cytidine kinase-like 1 [Os09g32820- qpty3 or qnot7], ribose-5-phosphate isomerase [Os03g56869- qnot4], an expressed protein [Os03g56860- qnot4], vacuolar ATP synthase subunit D 1 [Os04g55040-qnot5], vacuolar ATP synthase catalytic subunit A [Os06g45120-tgw5], ferredoxin [Os09g33950-qpht7, qpty3, qnot7] and ferredoxin-NADP reductase [Os03g57120-qnot4]. The genes encoding phosphoglucomutase [Os03g50480-qdth2] and sucrose-phosphate synthase 1 [Os08g20660- qtyp4] (Table S6), belonging to sucrose biosynthesis also showed an AHPL expression pattern in leaves.
Mapped QTL regions covering previously cloned yield related genes
The gene for grain number Gn1a [Os01g56810; cytokinin dehydrogenase 5 precursor] was located in the QTL for PLE [qple1]. The gene for panicle number, panicle branching and high grain productivity was encoded by OsSPL14 [squamosa promoter-binding-like protein 9] and located in this study at the QTL for NOB [qnob4]. The gene for DTH, Hd1 [zinc finger protein CONSTANS] was located at the QTL qdth5. The gene for PTY TAC1, an expressed protein gene, was located at the QTL region for PTY/NOT/PHT [qpty3/qnot7/qpht7]. The gene for grain width and weight GW2, which showed homology to gene Os02g53140 [ubiquitin ligase protein COP1] was located in the QTL qple2. Similarly, another homologous gene Os02g19140 [ubiquitin ligase SINAT4] was located in the QTL qnot2/qnob2. The gene for dense panicle, high grain number per panicle and erect panicle, DEP1 [Os09g26999-keratin-associated protein 5-4] was located in the QTL qpty3/qnot7. Many of these genes were expressed in either of the parents and/or the F1 (Table S9).
Discussion
For the first time, genome-wide gene expression from the leaves, roots and meristems of rice were mapped onto 71 QTLs of yield related traits. Among them, sixty eight QTLs had have been previously reported by others; while, three QTLs [qpty1, qpty2, qkwp7] were novel and could be specific for 93-11 and Nipponbare. Three QTLs [qkwp8, qnok8 and qpht3] were reported in the Gramene/Q-TARO database without gene designations (Table S5). Of the seven QTLs detected for DTH, the alleles from Nipponbare decreased DTH at four loci while three alleles increased DTH. All seven QTLs for DTH had been reported previously (Table S5). The similarity of the regions associated with QTLs in this study for DTH compared to other studies involving indica and japonica cultivars suggests that the same alleles are responsible for DTH across different genetic and environmental backgrounds. Of the nine QTLs detected for PHT, Nipponbare alleles increased PHT at three QTLs. All nine QTLs for PHT had been previously reported (Table S5). All the QTLs for NOT had been reported earlier (Table S5). In addition, seven QTLs for NOT, TGW and PLE, and six QTLs for grain yield per plant were reported in multiple rice germplasm lines in rice production areas worldwide. These findings suggest that these QTLs may be intensively selected during the domestication and breeding process.
In comparison with parents, changes including increased DTH, increased PHT, narrow PTY, increased NOT, slightly decreased PLE, moderate NOB, decreased number of NOK, decreased KWP, moderate TGW and decreased TYP were observed in the F1 hybrid (Table S1). The OsSUT1 gene at QTL [qdth1] was located on chromosome 3 for DTH and PHT [64]. Another QTL, Ghd7 on chromosome 7 was predicted to encode a CCT-domain protein controlling grain yield, PHT and DTH in rice [15]. For PTY, the TAC1gene on chromosome 9 was identified at qTA-9 in a 93-11xNipponbare cross [5]. The cytokinin dehydrogenase 5 precursor gene [Gn1a, Os01g56810] for grain number was located at qple1 for PLE [1]. The OsSPL14 gene encoding a squamosa promoter-binding-like protein 9 for panicle number, panicle branching and high grain productivity was located at qnob4 [64]. The Hd1 gene [zinc finger protein CONSTANS] for DTH was located at qdth5 [12]. The TAC1gene for PTY was located at qpty3 [65] (Table S9).
Consistently, In F1 hybrid, HPL and LPL may explain dominance and AHPL and BLPL may explain over-dominance (Table S4). For example, genes for the AHPL in F1 are involved in plant growth, development and signal transduction including granule-bound starch synthase I, growth regulator and phosphatidylinositol 3- and 4-kinase family protein. The genes involved in carbon fixation and photosynthesis pathways were the AHPL expression pattern in F1 leaf including vacuolar ATP synthase subunit D 1, vacuolar ATP synthase catalytic subunit A, ribulose-phosphate 3-epimerase, uridine/cytidine kinase-like 1, ribose-5-phosphate isomerase, ferredoxin and ferredoxin-NADP reductase. Genes involved in sucrose biosynthesis phosphoglucomutase and sucrose-phosphate synthase 1 belonged to AHPL (Table S6). Similarly, genes for photosynthesis, carbon fixation, starch and sucrose metabolism were mapped at yield related QTL, and their enhanced expressions were found in the super rice hybrid [4]. Besides photosynthesis, and sucrose and starch pathways, the oxidative phosphorylation, citrate cycle [TCA cycle], and stress-resistant pathway, etc., may also contribute to heterosis [46]. In our study, the gene for sucrose phosphate synthase [SPS], the major limiting enzyme for sucrose synthesis, mapped at ph1 responsible for plant height, and was highly expressed [AHPL] in F1 hybrid leaves compared to that of the parents (Table S6). The higher SPS activity was proposed to be responsible for increasing panicle length [9].
Heterosis may also be a combination of genetic and epigenetic regulation [26], [33], [43]. Altered gene expression caused by interactions between transcription factors and the allelic promoter region in the hybrids was one plausible mechanism for heterosis in rice [50]. Many differentially expressed TF genes in super hybrid rice were located in grain yield related QTLs [43]. In this study, the TF genes belonging to helix-loop-helix DNA binding domain containing protein genes of TF family HLH including Os03g53020 [qdth2, qnot4], Os01g57580 [qple1] and Os02g47660 [qple2] showed the AHPL expression pattern in leaves, roots and meristems, and located in the mapped yield related QTL. The LAX1 gene encoding a bHLH transcription factor was involved in the formation of all types of axillary meristems throughout the ontogeny of rice [66] and a mutant of LAX1 [lax 1–2] was shown to reduce tiller number [67] suggesting that LAX1 function may be required for the generation of axillary meristems of both tillers and panicles [67].
The TF family AP2-EREBP members were potential targets of miRNA [68]. Noncoding RNAs were involved in epigenetic regulations, and other epigenetic mechanisms including DNA methylation, acetylation and deacetylation of histones, and chromatin remodeling [69]–[71]. Three genes encoding AP2 domain containing protein belonging to TF family AP2-EREBP showed an AHPL expression pattern in F1 hybrid leaves (Table S6). In Arabidopsis epigenetic regulation of a few regulatory genes for growth and development were observed in hybrids [55]. In this study, a total of 99 chromatin-related genes were expressed in leaves, roots and meristem tissues. Specifically, several epigenetic related genes belonging to flowering, histone demethylases, histone deacetylases, argonaute like protein genes, and polycomb group genes were highly expressed F1 hybrids suggesting their potential roles heterosis.
In the past decade, oligoarrays, SAGE, MPSS, and SBS have been used for transcriptome profiling. Illumina's MPSS technology has been used to generate expression data for many organisms [58], [59], [77]–[79]. Thus far, MPSS was the most popular tag based technology for sequencing of the transcriptomes of various organisms [72]–[75], [76]–[78]. More genes were identified using MPSS technology than SAGE or oligoarrays [76]. In this study, MPSS technology was used to analyze the transcriptomes of the leaves, roots and meristem tissues obtained from Nipponbare, 93-11 and their F1 hybrid. A total of 1 to 3 million signatures were obtained from each library. The number of redundant and non-redundant signatures generated in this study was similar to those in previous reports in rice and Arabidopsis [58], [59], [72]–[76]. It is important to note that significant proportion of MPSS signatures failed to match Nipponbare genome. One of the most plausible explanations is alternate splicing. Published reports demonstrated that alternative splicing in rice ranged from 13 to 21% [79]. Other possibilities also include sequencing errors in the MPSS signatures and in the Nipponbare genome or un-sequenced regions of the Nipponbare genome and also unknown mechanisms.
In summary, MPSS technology was used to obtain genome wide expression profiles in leaves, roots and meristem from ‘Nipponbare’ and ‘93-11’, and their F1 hybrid. Commonly and specifically expressed rice genes were identified, and mapped to 71 yield related QTL regions for days to heading, plant height, plant type, number of tillers, main panicle length, number of primary branches per main panicle, number of kernels per main panicle, total kernel weight per main panicle, 1000 grain weight and total grain yield per plant. Differentially expressed genes at yield related QTLs are the important candidate genes for further functional validation to unravel their role in positive and negative heterosis in F1 hybrids. This study provides the starting genomic materials to elucidate the molecular basis of yield related traits and heterosis in rice.
Materials and Methods
The cross between japonica cultivar Nipponbare and indica cultivar 93-11 was made at Ohio State University. The RIL population an F5-7 was developed using an F2 and F3 single seed decent method under greenhouse conditions at the USDA Agricultural Research Service Dale Bumpers National Rice Research Center [DB NRRC].
Phenotype evaluation
Phenotypes of RILs and their parents were evaluated in two locations in a randomized complete block design with three replications in the fields at the DBNRRC, Stuttgart, Arkansas and Rice Research Unit [RRU], Beaumont, Texas in 2009, 2010, 2011 and 2012. Population of 257, 254, 205 and 231 RILs were planted in the field at the DBNRRC, Stuttgart, Arkansas in 2009 [Stuttgart-2009, AR], 2010 [Stuttgart-2010, AR], 2011 [Stuttgart-2011, AR] and 2012 [Stuttgart-2012, AR], respectively. A planting represented by 36 plants with 20 cm spacing was performed at Stuttgart for the years 2009 and 2010. In 2011 and 2012 at Stuttgart, a planting was represented by 3 plants in a row [2 meter single rows], 61 cm alleys, and 61 cm row spacing was maintained. Similarly, a subset of 82 and 252 RILs was planted in the field of RRU, Beaumont, Texas in 2009 [Beaumont-2009, TX] and 2010 [Beaumont-2010, TX], respectively, represented by 12 plants with 20 cm spacing. For the Stuttgart and Beaumont locations in 2009 and 2010, three central plants per RIL were marked and the main panicle of each marked plant was tagged at the heading stage. These marked plants were representative of the corresponding RIL. The panicles of each marked plant were harvested individually for characterization of yield components. For the Stuttgart location in 2011 and 2012, all three plants representing each RIL were tagged at the heading stage and subsequently harvested to measure yield components (Table 1). The trait data collected per location varied depending on the weather conditions during that growing season [ex: hot summer]. The traits DTH, PHT and PTY were obtained in six field experiments. The KWP, LOG, NOT, NOK, PLE, TGW and TYP were obtained from four field experiments. NOB was obtained in three field experiments (Table 1).
Evaluation of yield related traits
Yield related traits were measured in RILs using a modified procedure from Moncada et al. [80] and a source book from the International Rice Research Institute [IRRI] [November 2002] entitled ‘Standard Evaluation System for Rice’. Number of days to heading was recorded when 50% of plants had flowers on at least one panicle. Lodging was measured using 0–9 scale, where 0 stands for no lodging, 1 stands for up to 10% lodged, 2–11 to 20% lodged, 3–21 to 30% lodged, 4–31 to 40% lodged, 5–41 to 50% lodged, 6–51 to 60% lodged, 7–61 to 70% lodged, 8–71 to 80% lodged, 9–81 to 100% lodged. Tiller angle [plant type] was measured using 1–9 scale, where 1-tillers were erect with an angle less than 30° from the perpendicular; 3-tillers were intermediate -the angle was about 45°; 5-tillers were open- the angle was about 60°; 7-tillers were spreading- angle was more than 60° but the culms do not rest on the ground; 9-procumbent the culm or its lower part rests on ground surface. Plant height [cm] was measured for each plot. Averages were calculated for each RIL and each trait in all 6 experiments [mentioned above]. Number of tillers per marked plant was counted. One ‘main panicle’ from the marked plant was harvested to record panicle length [length of the panicle from the base to tip of the panicle], number of primary branches per main panicle, number of kernels per main panicle, and 1000 grain weight per main panicle. Main panicle data was collected for each marked RIL. The total grain yield per plant was calculated collecting all the kernels from the entire plant [for each marked RIL]. Phenotypic data were analyzed using Microsoft access and JMP Genomics [version 5.1] software (Table 1).
Genotyping and data analysis
A total of 266 F5-F7 RILs were used for SSR analysis. DNA extraction and quantification was performed as previously described [81], [82]. Except for two indels and two SSR primers designed in house the primer sequences and map position of the SSR markers were obtained from the Gramene database [http://www.gramene.org/qtl/index.html] [83]–[85]. LJSSR1 and Con673 sequences were described by Li et al. [85]. MJIndel1 and MJIndel 2 were designed using the annotated Nipponbare and 93-11 genomes. The sequences for MJIndel 1 were F: attggatcaacacaccacac R: cagtcgaactccatcttcct and MJIndel 2 were F: aacttcaacaccaccctttga R: tttccaggtccagctcctaa. Marker amplification and allele calling were determined as described by Liu et al. [81]. A linkage map was constructed using JoinMap 4 based on the Kosambi function. Composite interval mapping [CIM] was used for phenotypic data obtained from 6 field experiments using Windows QTL Cartographer version 2.5 to identify QTLs affecting each yield related trait. The threshold was estimated by 1000 permutations at P <0.01 by QTL cartographer for each trait (Table S5). This LOD threshold was used to declare the presence of a putative QTL in order to compare with the previously identified yield related QTLs on rice chromosomes in the Gramene QTL database. -Known QTLs were identified in the public gramene rice QTL database [November 19th 2012 release] [ftp://ftp.gramene.org/pub/gramene/CURRENT_RELEASE/data/qtl/] [82] and Japanese rice QTL database - http://qtaro.abr.affrc.go.jp/qtab/table [86].
Plant materials for gene expression analysis and total RNA extraction
Nipponbare, 93-11 and their F1 hybrid were grown in a Conviron growth chamber at 70% relative humidity with 12 h of light [500 µmol photons m-2 sec-1] at 26°C followed by 12 h of dark at 20°C temperature. Plants from eight week old parents and their F1 hybrid were used to collect leaves, roots and meristem tissues. Total RNA was extracted separately from leaves, roots and meristem tissues from each plant using Trizol reagent [Invitrogen] according to the manufacturer's instructions.
A total of 21 MPSS libraries were constructed from leaf, root and meristem tissues obtained from Nipponbare, 93-11 and their F1 hybrid. The libraries included 4 replications from Nipponbare leaves [NLA, NLB, NLC, NLD], 2 replications from Nipponbare roots [NRA, NRB], 1 replication from Nipponbare meristems [NME], 4 replications from 93-11 leaves [I9LA, I9LB, I9LC, I9LD], 2 replications from 93-11 roots [I9RO, I9RR], 1 replication from 93-11 meristems [I9ME], 4 replications from F1 hybrid leaves [FLA, FLB, FLC, FLD], 2 replications from F1 hybrid roots [FRO, FRR], and 1 replication from F1 hybrid meristems [FME]. Each replication represented 4 individual plants. Since MPSS technology was very expensive, we restricted the number of replications to 2 and 1 in root and meristem tissues, respectively.
Construction of the MPSS libraries, sequencing, and bioinformatics
MPSS library construction, sequencing and annotation were performed essentially as previously described [58], [59], [72]–[76]. Briefly, to get high quality data, sequences obtained from MPSS technology were passed through two filters – reliability and significance. The ‘reliability’ filter determines if the given signature is found in more than one library (reliable signatures) or present in only one library (unreliable signature). The ‘significance’ filter determines if a given signature is found in any library at ≥4TPM (transcripts per million) (significant signature) or <4TPM (non-significant signature) in a normalized library. The significant and reliable distinct signatures were identified in 21 MPSS libraries as previously described. For leaf tissue, the signature frequencies data obtained from 4 replications were used to calculate the mean value. This included the transcripts expressed either in only one, two, three or all four replications. Similarly, mean values were calculated for 2 replications of root tissues. The expression levels of Nipponbare and 93-11 were compared with that of their F1 hybrid and the differentially expressed signatures were classified into 8 expression patterns [AHPL, HPL, MPL, LPL, BLPL, SEF1, EOPF1 and EPAF1] with some modifications [26], [44]. To avoid sequence bias we performed cluster analysis on the signatures matching to both Nipponbare and 93-11 genomes to identify commonly and specifically expressed genes among Nipponbare, 93-11 and F1 hybrid. Clustering analysis was carried out using Microsoft Access and JMP Genomics [version 5.1] software to identify the genes specifically and commonly expressed in Nipponbare, 93-11 and their F1 hybrid. Bioinformatic analyses including identification of antisense transcripts, alternate transcripts, TFs and functional classification of genes using KEGG database were conducted as previously described [58], [59], [72]–[76]. The entire dataset is available at the NCBI's Gene Expression Omnibus database.
Supporting Information
Transgressive variation among the F6–8 generation RILs of Nipponbare X 93–11 cross in the field at Stuttgart, Arkansas. Also, phenotypic variation (plant height, number of tillers and maturity) of F1 hybrids and their parents in both field and growth chamber conditions.
(TIF)
Filter results for 21 MPSS libraries. A total of 179,151 distinct 17-base expressed signatures from 21 MPSS libraries were processed according to three filters- significance, reliability, and genomic match as described by Meyers et al. [58] – [59] .
(TIF)
‘Kyoto Encyclopedia of Genes and Genomes’ based functional classification of genes induced in F1 hybrid (leaves, roots and meristem tissues) compared to their parents Nipponbare and 93–11. Genes showing expression level pattern AHPL were only used for this analysis.
(TIF)
Phenotypic variation for yield related traits among the RILs in six field studies (Stuttgart-2009, AR; Stuttgart-2010, AR; Stuttgart-2011, AR; Stuttgart-2012, AR; Beaumont-2009, TX and Beaumont-2010, TX). Phenotypic variation was observed for days to heading, plant height, tiller angle, tiller number, panicle length, number of primary branches per panicle, number of kernels per panicle, total kernel weight per panicle, 1000 grain weight and total grain yield per plant.
(XLSX)
Characteristics of MPSS libraries. Library statistics of Nipponbare, 93-11 and F1 hybrid from leaves, roots and meristem tissues.
(DOCX)
Classification of the MPSS signatures based on their location on the annotated gene (hits = 1) (See Meyers et al. [58]–[59] for details). Reliable and significant (≥4TPM) MPSS signatures obtained from leaf, root and meristem libraries (mean of replications in each tissue) of Nipponbare, 93–11 and their F1 hybrid are summarized.
(DOCX)
Gene expression levels in leaf, root and meristem tissues of F1 hybrids and their parents Nipponbare and 93–11. Mean signature frequencies (copy number) were calculated from four leaf replications and two root replications separately. The mean signature value of F1 hybrids was compared with their parents to classify the signature into one of the 8 expression patterns (AHPL, HPL, MPL, LPL, BLPL, SEF1, EOPF1 and EPAF1). Detailed annotation of each signature is presented.
(XLSX)
List of known and novel yield related QTL identified in this study based on Gramene QTL database ( ftp://ftp.gramene.org/pub/gramene/CURRENT_RELEASE/data/qtl/ ) and Japanese rice QTL database (Q-TARO database) - http://qtaro.abr.affrc.go.jp/qtab/table . SSR marker intervals, percent variation, additive effect and LOD score for each QTL identified in each location is presented. Average LOD score was calculated for QTL identified in more than one location. Similar QTL locations identified in other studies (indicated in RED font color) are presented for the known QTL (in our study). Yellow highlighted QTL are reported in Japanese Q-TARO database.
(XLSX)
List of all expressed genes identified in mapped yield related QTL regions. The expressed genes located between the two flanking SSR markers of yield related QTL are listed. Also, the expressed genes belonging to SEF1 and AHPL expression pattern categories located in mapped yield related QTL regions are also listed. The QTL identified by mean LOD score ≥5 are used for this analysis.
(XLSX)
List of TF genes identified in mapped yield related QTL regions. The TF genes located between the two flanking SSR markers of yield related QTL are listed. The QTL identified by mean LOD score ≥5 are used for this analysis. TF family names and the expression level patterns of TF genes in different tissues are presented here.
(XLSX)
List of chromatin related genes identified in mapped yield related QTL regions. The chromatin related genes located between the two flanking SSR markers of yield related QTL are listed. The QTL identified by mean LOD score ≥5 are used for this analysis.
(XLSX)
Cloned yield related genes located in mapped QTL regions in our study. The genes responsible for yield related traits have been isolated in different studies and the information is deposited in Japanese Q-TARO database (http://qtaro.abr.affrc.go.jp/ogro/table).
(XLSX)
Acknowledgments
We thank the staff members of DB NRRC, Stuttgart, AR; Rice Research Unit, Beaumont, TX and, Rice Research and Extension Center, University of Arkansas Division of Agriculture, Stuttgart, AR, Jane Khatenje, Pooja Ghai and Saritha Kontham of Arkansas State University for their help in phenotyping RILs. This work was supported in part by the Plant Genome Research Program of the National Science Foundation [Award numbers: 0701745 and 0321437]. USDA is an equal opportunity provider and employer.
Funding Statement
This project was supported in part by the Plant Genome Research Program of the National Science Foundation [Award numbers: 0701745 and 0321437]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ashikari M, Sakakibara H, Lin S, Yamamoto T, Takashi T, et al. (2005) Cytokinin oxidase regulates rice grain production. Science 309: 741–745. [DOI] [PubMed] [Google Scholar]
- 2. Song XJ, Huang W, Shi M, Zhu MZ, Lin HX (2007) A QTL for rice grain width and weight encodes a previously unknown RING type E3 ubiquitin ligase. Nat Genet 39: 623–630. [DOI] [PubMed] [Google Scholar]
- 3. Tanksley SD (1993) Mapping polygenes. Annu Rev Genet 27: 205–233. [DOI] [PubMed] [Google Scholar]
- 4. Xing Y, Zhang Q (2010) Genetic and molecular bases of rice yield. Annu Rev Plant Biol 61: 421–42. [DOI] [PubMed] [Google Scholar]
- 5. Wang L, Wang A, Huang X, Zhao Q, Dong G, et al. (2011) Mapping 49 quantitative trait loci at high resolution through sequencing-based genotyping of rice recombinant inbred lines. Theor Appl Genet 122: 327–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cai HY, Diao S, He YG, Zhang LP, Liu SJ, et al. (2012) Genetic and physical mapping of qHY-8, a pleiotropic QTL for heading date and yield-related traits in rice. Euphytica 184: 109–118. [Google Scholar]
- 7. Yano M, Kojima S, Takahashi Y, Lin H, Sasaki T (2001) Genetic control of flowering time in rice, a short-day plant. Plant Physiol 127: 1425–1429. [PMC free article] [PubMed] [Google Scholar]
- 8. Yano M, Harushima Y, Nagamura Y, Kurata N, Minobe Y, et al. (1997) Identification of quantitative trait loci controlling heading date in rice using a high-density linkage map. Theor Appl Genet 95: 1025–1032. [Google Scholar]
- 9. Lin SY, Sasaki T, Yano M (1998) Mapping quantitative trait loci controlling seed dormancy and heading date in rice Oryza sativa L., using backcross inbred lines. Theor Appl Genet 96: 997–1003. [Google Scholar]
- 10. Yamamoto T, Lin H, Sasaki T, Yano M (2000) Identification of heading date quantitative trait locus hd6 and characterization of its epistatic interactions with Hd2 in rice using advanced backcross progeny. Genetics 154: 885–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin H, Ashikari M, Yamanouchi U, Sasaki T, Yano M (2002) Identification and characterization of a quantitative trait locus, Hd9, controlling heading date in rice. Breed Sci 52: 35–41. [Google Scholar]
- 12. Yano M, Katayose Y, Ashikari M, Yamanouchi U, Monna L, et al. (2000) Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12: 2473–2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kojima S, Takahashi Y, Kobayashi Y, Monna L, Sasaki T, et al. (2003) Hd3a, a Rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol 43: 1096–1105. [DOI] [PubMed] [Google Scholar]
- 14. Doi K, Izawa T, Fuse T, Yamanouchi U, Kubo T, et al. (2004) Ehd1, a B-type response regulator in rice confers short-day promotion of flowering and controls FT-like gene expression independently of Hd1. Genes Dev 18: 926–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xue W, Xing Y, Weng X, Zhao Y, Tang W, et al. (2008) Natural variation in Ghd7 is an important regulator of heading date and yield potential in rice. Nat Genet 40: 761–767. [DOI] [PubMed] [Google Scholar]
- 16. Yan WH, Wang P, Chen HX, Zhou HJ, Li QP, et al. (2011) A major QTL, Ghd8, plays pleiotrophic roles in regulating grain productivity, plant height, and heading date in rice. Mol Plant 4: 319–330. [DOI] [PubMed] [Google Scholar]
- 17. Spielmeyer W, Ellis MH, Chandler PM (2002) Semidwarf (sd-1), ‘green revolution’ rice, contains a defective gibberellin 20-oxidase gene. Proc Natl Acad Sci USA 99: 9043–9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shomura A, Izawa T, Ebana K, Ebitani T, Kanegae H, et al. (2008) Deletion in a gene associated with grain size increased yields during rice domestication. Nat Genet 40: 1023–1028. [DOI] [PubMed] [Google Scholar]
- 19. Weng J, Gu S, Wan X, Gao H, Guo T, et al. (2008) Isolation and initial characterization of GW5, a major QTL associated with rice grain width and weight. Cell Res 18: 1199–1209. [DOI] [PubMed] [Google Scholar]
- 20. Xie XB, Song MH, Jin FX, Ahn SN, Suh JP, et al. (2006) Fine mapping of a grain weight quantitative trait locus on rice chromosome 8 using near-isogenic lines derived from a cross between Oryza sativa and Oryza rufipogon . Theor Appl Genet 113: 885–894. [DOI] [PubMed] [Google Scholar]
- 21. Xie X, Jin F, Song MH, Suh JP, Hwang HG, et al. (2008) Fine mapping of a yield-enhancing QTL cluster associated with transgressive variation in an Oryza sativa × O. rufipogon cross. Theor Appl Genet 116: 613–622. [DOI] [PubMed] [Google Scholar]
- 22. Tian F, Zhu ZF, Zhang BS, Tan LB, Fu YC, et al. (2006) Fine mapping of a quantitative trait locus for grain number per panicle from wild rice (Oryza rufipogon Griff.). Theor Appl Genet 113: 619–629. [DOI] [PubMed] [Google Scholar]
- 23. Liu TM, Mao DH, Zhang SP, Xing YZ (2009) Fine mapping SPP1, a QTL controlling the number of spikelets per panicle, to a BAC clone in rice (Oryza sativa). Theor Appl Genet 118: 1509–1517. [DOI] [PubMed] [Google Scholar]
- 24. Liu TM, Shao D, Kovi MR, Xing YZ (2010) Mapping and validation of quantitative trait loci for spikelets per panicle and 1,000-grain weight in rice (Oryza sativa L.). Theor Appl Genet 120: 933–942. [DOI] [PubMed] [Google Scholar]
- 25. Meyer RC, Kusterer B, Lisec J, Steinfath M, Becher M, et al. (2010) QTL analysis of early stage heterosis for biomass in Arabidopsis . Theor Appl Genet 120: 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He G, Zhu X, Elling AA, Chen L, Wang X, et al. (2010) Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. Plant Cell 22: 17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Garcia AA, Wang S, Melchinger AE, Zeng ZB (2008) Quantitative trait loci mapping and the genetic basis of heterosis in maize and rice. Genetics 180: 1707–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lippman ZB, Zamir D (2007) Heterosis: revisiting the magic. Trends Genet 23: 60–66. [DOI] [PubMed] [Google Scholar]
- 29. Bruce AB (1910) The Mendelian theory of heredity and the augmentation of vigor. Science 32: 627–628. [DOI] [PubMed] [Google Scholar]
- 30. Zhou G, Chen Y, Yao W, Zhang C, Xie W, et al. (2012) Genetic composition of yield heterosis in an elite rice hybrid. Proc Natl Acad Sci USA 109: 15847–15852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. East EM (1936) Heterosis. Genetics 21: 375–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yu SB, Li JX, Xu CG, Tan YF, Gao YJ, et al. (1997) Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc Natl Acad Sci USA 94: 9226–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chodavarapu RK, Feng S, Ding B, Simon SA, Lopez D, et al. (2012) Transcriptome and methylome interactions in rice hybrids. Proc Natl Acad Sci USA 109: 12040–12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Virmani SS, Sun ZX, Mou TM, Jauhar Ali A, Mao CX (2003) Two-line hybrid rice breeding manual. Los Banos (Philippines): International Rice Research Institute, 88p
- 35. Hitzemann R, Malmanger B, Reed C, Lawler M, Hitzemann B, et al. (2003) A strategy for the integration of QTL, gene expression, and sequence analyses. Mamm Genome 14: 733–747. [DOI] [PubMed] [Google Scholar]
- 36. Fan C, Xing Y, Mao H, Lu T, Han B, et al. (2006) GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theor Appl Genet 112: 1164–1171. [DOI] [PubMed] [Google Scholar]
- 37. Xu X, Liu X, Ge S, Jensen JD, Hu F, et al. (2011) Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotechnol 30: 105–111. [DOI] [PubMed] [Google Scholar]
- 38. Zhao K, Tung CW, Eizenga GC, Wright MH, Ali ML, et al. (2011) Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa . Nat Commun 2: 467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ishimaru K, Ono K, Kashiwagi T (2004) Identification of a new gene controlling plant height in rice using the candidate-gene strategy. Planta 218: 388–395. [DOI] [PubMed] [Google Scholar]
- 40. Ashikari M, Wu J, Yano M, Sasaki T, Yoshimura A (1999) Rice gibberellin insensitive dwarf mutant gene Dwarf 1 encodes the alpha-subunit of GTP-binding protein. Proc Natl Acad Sci USA 96: 10284–10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sasaki A, Ashikari M, Ueguchi-Tanaka M, Itoh H, Nishimura A, et al. (2002) Green revolution: a mutant gibberellin-synthesis gene in rice. Nature 416: 701–702. [DOI] [PubMed] [Google Scholar]
- 42. Huang Y, Zhang L, Zhang J, Yuan D, Xu C, et al. (2006) Heterosis and polymorphisms of gene expression in an elite rice hybrid as revealed by a microarray analysis of 9198 unique ESTs. Plant Mol Biol 62: 579–591. [DOI] [PubMed] [Google Scholar]
- 43. Wei G, Tao Y, Liu G, Chen C, Luo R, et al. (2009) A transcriptomic analysis of super-hybrid rice LYP9 and its parents. Proc Natl Acad Sci USA 106: 7695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Song GS, Zhai HL, Peng YG, Zhang L, Wei G, et al. (2010) Comparative transcriptional profiling and preliminary study on heterosis mechanism of super-hybrid rice. Mol Plant 3: 1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Swanson-Wagner RA, Jia Y, DeCook R, Borsuk LA, Nettleton D, et al. (2006) All possible modes of gene action are observed in a global comparison of gene expression in a maize F1 hybrid and its inbred parents. Proc Natl Acad Sci USA 103: 6805–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yao Y, Ni Z, Zhang Y, Chen Y, Ding Y, et al. (2005) Identification of differentially expressed genes in leaf and root between wheat hybrid and its parental inbreds using PCR-based cDNA subtraction. Plant Mol Biol 58: 367–384. [DOI] [PubMed] [Google Scholar]
- 47. Zhai R, Feng Y, Wang H, Zhan X, Shen X, et al. (2013) Transcriptome analysis of rice root heterosis by RNA-Seq. BMC Genomics 14: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nuruzzaman M, Alam MF, Ahmed MG, Shohael AM, Biswas MK, et al. (2002) Studies on parental variability and heterosis in rice. Pakistan J Biol Sci 5: 1006–1009. [Google Scholar]
- 49. Rahimi M, Rabiei B, Samizadeh H, Kafi Ghasemi A (2010) Combining Ability and Heterosis in Rice (Oryza sativa L.) Cultivars. J Agr Sci Tech 12: 223–231. [Google Scholar]
- 50. Zhang HY, He H, Chen LB, Li L, Liang MZ, et al. (2008) A genome-wide transcription analysis reveals a close correlation of promoter INDEL polymorphism and heterotic gene expression in rice hybrids. Mol Plant 1: 720–731. [DOI] [PubMed] [Google Scholar]
- 51. Manning K, Tor M, Poole M, Hong Y, Thompson AJ, et al. (2006) A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat Genet 38: 948–952. [DOI] [PubMed] [Google Scholar]
- 52. Shindo C, Lister C, Crevillen P, Nordborg M, Dean C (2006) Variation in the epigenetic silencing of FLC contributes to natural variation in Arabidopsis vernalization response. Genes Dev 20: 3079–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Henderson IR, Jacobsen SE (2007) Epigenetic inheritance in plants. Nature 447: 418–424. [DOI] [PubMed] [Google Scholar]
- 54. Birchler JA, Auger DL, Riddle NC (2003) In search of the molecular basis of heterosis. Plant Cell 15: 2236–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ni Z, Kim ED, Ha M, Lackey E, Liu J, et al. (2009) Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 457: 327–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang J, Tian L, Lee HS, Wei NE, Jiang H, et al. (2006) Genome wide non-additive gene regulation in Arabidopsis allotetraploids. Genetics 172: 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang Z, Gerstein M, Snyder M (2009) RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10: 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Meyers BC, Tej SS, Vu TH, Haudenschild CD, Agrawal V, et al. (2004a) The use of MPSS for whole-genome transcriptional analysis in Arabidopsis . Genome Res 14: 1641–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Meyers BC, Vu TH, Tej SS, Ghazal H, Matvienko M, et al. (2004b) Analysis of the transcriptional complexity of Arabidopsis thaliana by massively parallel signature sequencing. Nat Biotechnol 22: 1006–11. [DOI] [PubMed] [Google Scholar]
- 60. Goff SA, Ricke D, Lan TH, Presting G, Wang R, et al. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science 296: 92–100. [DOI] [PubMed] [Google Scholar]
- 61. Yu J, Hu S, Wang J, Wong GK, Li S, et al. (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296: 79–92. [DOI] [PubMed] [Google Scholar]
- 62. International Rice Genome Sequencing Project: The map-based sequence of the rice genome (2005) Nature 436: 793–800. [DOI] [PubMed] [Google Scholar]
- 63. Ishimaru K (2003) Identification of a locus increasing rice yield and physiological analysis of its function. Plant Physiol. 133: 1083–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jiao Y, Wang Y, Xue D, Wang J, Yan M, et al. (2010) Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat Genet 42: 541–544. [DOI] [PubMed] [Google Scholar]
- 65. Yu B, Lin Z, Li H, Li X, Li J, et al. (2007) TAC1, a major quantitative trait locus controlling tiller angle in rice. Plant J 52: 891–898. [DOI] [PubMed] [Google Scholar]
- 66. Komatsu K, Maekawa M, Ujiie S, Satake Y, Furutani I, et al. (2003) LAX and SPA: major regulators of shoot branching in rice. Proc Natl Acad Sci USA 100: 11765–11770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oikawa T, Kyozuka J (2009) Two-step regulation of LAX PANICLE1 protein accumulation in axillary meristem formation in rice. Plant Cell 21: 1095–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shigyo M, Hasebe M, Ito M (2006) Molecular evolution of the AP2 subfamily. Gene 366: 256–265. [DOI] [PubMed] [Google Scholar]
- 69. Feng S, Jacobsen SE, Reik W (2010) Epigenetic reprogramming in plant and animal development. Science 330: 622–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Feng S, Jacobsen SE (2011) Epigenetic modifications in plants: an evolutionary perspective. Curr Opin Plant Biol 14: 179–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Simon SA, Meyers BC (2011) Small RNA-mediated epigenetic modifications in plants. Curr Opin Plant Biol 14: 148–155. [DOI] [PubMed] [Google Scholar]
- 72. Venu RC, Sheshu Madhav M, Sreerekha MV, Nobuta K, Zhang Y, et al. (2010) Deep and comparative transcriptome analysis of rice plants infested by the beet armyworm (Spodoptera exigua) and water weevil (Lissorhoptrus oryzophilus). Rice 3: 22–35. [Google Scholar]
- 73. Venu RC, Sreerekha MV, Nobuta K, Beló A, Ning Y, et al. (2011a) Deep sequencing reveals the complex and coordinated transcriptional regulation of genes related to grain quality in rice cultivars. BMC Genomics 12: 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Venu RC, Zhang Y, Weaver B, Carswell P, Mitchell TK, et al. (2011b) Large scale identification of genes involved in plant-fungal interactions using Illumina's sequencing-by-synthesis technology. Methods Mol Biol 722: 167–178. [DOI] [PubMed] [Google Scholar]
- 75. Nobuta K, Venu RC, Lu C, Beló A, Vemaraju K, Kulkarni K, et al. (2007) An expression atlas of rice mRNAs and small RNAs. Nature Biotechnol 25: 473–477. [DOI] [PubMed] [Google Scholar]
- 76. Gowda M, Venu RC, Raghupathy MB, Nobuta K, Li H, et al. (2006) Deep and comparative analysis of the mycelium and appressorium transcriptomes of Magnaporthe grisea using MPSS, RL-SAGE, and oligoarray methods. BMC Genomics 8: 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Vega-Sanchez ME, Gowda M, Wang GL (2007) Tag-based approaches for deep transcriptome analysis in plants. Plant Science 173: 371–380. [Google Scholar]
- 78. Simon SA, Zhai J, Nandety RS, McCormick KP, Zeng J, et al. (2009) Short-read sequencing technologies for transcriptional analyses. Annu Rev Plant Biol 60: 305–333. [DOI] [PubMed] [Google Scholar]
- 79. Wang BB, Brendel V (2006) Genomewide comparative analysis of alternative splicing in plants. Proc Natl Acad Sci USA 103: 7175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Moncada P, Martinez CP, Borrero J, Chatel M, Gauch H, et al. (2001) Quantitative trait loci for yield and yield components in an Oryza sativa x Oryza rufipogon BC2F2 population evaluated in an upland environment. Theor Appl Genet 102: 41–52. [Google Scholar]
- 81. Liu G, Bernhardt JL, Jia M, Wamishe YA, Jia Y (2008) Molecular characterization of the recombinant inbred line population derived from a japonica-indica rice cross. Euphytica 159: 73–82. [Google Scholar]
- 82. Ware DH, Jaiswal P, Ni J, Yap IV, Pan X, et al. (2002) Gramene, a tool for grass genomics. Plant Physiol 130: 1606–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. McCouch S, Kochert G, Yu Z, Wang Z, Khush GS, et al. (1988) Molecular mapping of rice chromosomes. Theor Appl Genet 76: 815–29. [DOI] [PubMed] [Google Scholar]
- 84. McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, et al. (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9: 199–207. [DOI] [PubMed] [Google Scholar]
- 85. Li X, Yan W, Agrama H, Hu B, Jia L, et al. (2010) Genotypic and phenotypic characterization of genetic differentiation and diversity in the USDA rice mini-core collection. Genetica 138: 1221–1230. [DOI] [PubMed] [Google Scholar]
- 86. Yonemaru J, Yamamoto T, Fukuoka S, Uga Y, Hori K, et al. (2010) Q-TARO:QTL Annotation Rice Online Database. Rice 3: 194. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Transgressive variation among the F6–8 generation RILs of Nipponbare X 93–11 cross in the field at Stuttgart, Arkansas. Also, phenotypic variation (plant height, number of tillers and maturity) of F1 hybrids and their parents in both field and growth chamber conditions.
(TIF)
Filter results for 21 MPSS libraries. A total of 179,151 distinct 17-base expressed signatures from 21 MPSS libraries were processed according to three filters- significance, reliability, and genomic match as described by Meyers et al. [58] – [59] .
(TIF)
‘Kyoto Encyclopedia of Genes and Genomes’ based functional classification of genes induced in F1 hybrid (leaves, roots and meristem tissues) compared to their parents Nipponbare and 93–11. Genes showing expression level pattern AHPL were only used for this analysis.
(TIF)
Phenotypic variation for yield related traits among the RILs in six field studies (Stuttgart-2009, AR; Stuttgart-2010, AR; Stuttgart-2011, AR; Stuttgart-2012, AR; Beaumont-2009, TX and Beaumont-2010, TX). Phenotypic variation was observed for days to heading, plant height, tiller angle, tiller number, panicle length, number of primary branches per panicle, number of kernels per panicle, total kernel weight per panicle, 1000 grain weight and total grain yield per plant.
(XLSX)
Characteristics of MPSS libraries. Library statistics of Nipponbare, 93-11 and F1 hybrid from leaves, roots and meristem tissues.
(DOCX)
Classification of the MPSS signatures based on their location on the annotated gene (hits = 1) (See Meyers et al. [58]–[59] for details). Reliable and significant (≥4TPM) MPSS signatures obtained from leaf, root and meristem libraries (mean of replications in each tissue) of Nipponbare, 93–11 and their F1 hybrid are summarized.
(DOCX)
Gene expression levels in leaf, root and meristem tissues of F1 hybrids and their parents Nipponbare and 93–11. Mean signature frequencies (copy number) were calculated from four leaf replications and two root replications separately. The mean signature value of F1 hybrids was compared with their parents to classify the signature into one of the 8 expression patterns (AHPL, HPL, MPL, LPL, BLPL, SEF1, EOPF1 and EPAF1). Detailed annotation of each signature is presented.
(XLSX)
List of known and novel yield related QTL identified in this study based on Gramene QTL database ( ftp://ftp.gramene.org/pub/gramene/CURRENT_RELEASE/data/qtl/ ) and Japanese rice QTL database (Q-TARO database) - http://qtaro.abr.affrc.go.jp/qtab/table . SSR marker intervals, percent variation, additive effect and LOD score for each QTL identified in each location is presented. Average LOD score was calculated for QTL identified in more than one location. Similar QTL locations identified in other studies (indicated in RED font color) are presented for the known QTL (in our study). Yellow highlighted QTL are reported in Japanese Q-TARO database.
(XLSX)
List of all expressed genes identified in mapped yield related QTL regions. The expressed genes located between the two flanking SSR markers of yield related QTL are listed. Also, the expressed genes belonging to SEF1 and AHPL expression pattern categories located in mapped yield related QTL regions are also listed. The QTL identified by mean LOD score ≥5 are used for this analysis.
(XLSX)
List of TF genes identified in mapped yield related QTL regions. The TF genes located between the two flanking SSR markers of yield related QTL are listed. The QTL identified by mean LOD score ≥5 are used for this analysis. TF family names and the expression level patterns of TF genes in different tissues are presented here.
(XLSX)
List of chromatin related genes identified in mapped yield related QTL regions. The chromatin related genes located between the two flanking SSR markers of yield related QTL are listed. The QTL identified by mean LOD score ≥5 are used for this analysis.
(XLSX)
Cloned yield related genes located in mapped QTL regions in our study. The genes responsible for yield related traits have been isolated in different studies and the information is deposited in Japanese Q-TARO database (http://qtaro.abr.affrc.go.jp/ogro/table).
(XLSX)