

Abstract
The zebrafish has proven to be a valuable model system for exploring skeletal muscle function and for studying human muscle diseases. Despite the many advantages offered by in vivo analysis of skeletal muscle in the zebrafish, visualizing the complex and finely structured protein milieu responsible for muscle function, especially in whole embryos, can be problematic. This hindrance stems from the small size of zebrafish skeletal muscle (60 μm) and the even smaller size of the sarcomere. Here we describe and demonstrate a simple and rapid method for isolating skeletal myofibers from zebrafish embryos and larvae. We also include protocols that illustrate post preparation techniques useful for analyzing muscle structure and function. Specifically, we detail the subsequent immunocytochemical localization of skeletal muscle proteins and the qualitative analysis of stimulated calcium release via live cell calcium imaging. Overall, this video article provides a straight-forward and efficient method for the isolation and characterization of zebrafish skeletal myofibers, a technique which provides a conduit for myriad subsequent studies of muscle structure and function.
Keywords: Basic Protocol, Issue 81, Zebrafish, Neuromuscular Diseases, Muscular Diseases, Muscular Dystrophies, Primary Cell Culture, Immunohistochemistry (IHC), skeletal muscle, myofiber, live imaging
Introduction
Skeletal muscle is a highly specialized tissue responsible for generating the contractile forces necessary for motility. Contraction is initiated through a process known as excitation-contraction (EC) coupling that converts electric signals to calcium release from intracellular stores1,2 . Intracellular calcium release activates the sarcomere to shorten and generate force. The many specific components of the molecular machinery responsible for mediating neuromuscular junction transmission3, EC coupling4,5 , and actin-myosin dependent contractions6 continue to be the ongoing subject of intense research. In addition, proteins that stabilize the muscle membrane during contraction7,8 and that mediate signaling between the myofiber and the extracellular matrix7,9 have been identified and studied in great detail.
Mutations in a number of genes important for muscle structure and function have been identified as causes of human skeletal muscle diseases (http://www.musclegenetable.org/). These diseases, classified broadly as skeletal myopathies and muscular dystrophies based on clinical and histopathologic features, are associated with muscle weakness, lifelong disability, and early mortality10,11 . The zebrafish has proven to be an outstanding system for modeling and studying human skeletal muscle diseases8,12,13 . It has been employed to validate new gene mutations8, define new aspects of disease pathophysiology14,15 , and identify new therapeutic approaches15,16 . The power of the zebrafish for studying human muscle disease relates to the large number of offspring, the rapid development of muscle structure and function, the optical clarity of the zebrafish embryo, and the ease of genetic and pharmacologic manipulation of the developing zebrafish17.
We and others12,18,19 have recently developed a simple technique for the rapid and efficient isolation of myofibers from the developing zebrafish. This methodology has enabled the examination of myofibers in greater detail than can be provided by whole embryo analysis. The technique has been exploited for the characterization of protein subcellular localization20 as well as for the identification of important histopathologic characteristics as part of validation studies in newly developed disease models21. Furthermore, isolated myofibers can additionally be used for live imaging and for electrophysiological studies22, techniques that allow for the interrogation of key aspects of muscle function. The specific protocol for myofiber isolation, along with two examples of subsequent analytic experiments, is detailed in the remaining parts of this manuscript.
Protocol
1. Preparation of Poly-L-Lysine Coated Coverslips (Time: 1 hr)
Coating coverslips allows for rapid myofiber settling and adhesion. This may be performed during the dissociation step of the myofiber isolation (step 2 below).
Cut and place Parafilm on the bottom of a 60 mm Petri dish (any brand).
Place microscope cover glass slips (12 mm diameter) on Parafilm in a 60 mm tissue culture dish or place them individually in single wells of 24-well plates.
Note 1: These small round coverslips help to concentrate myofiber numbers and reduce excess antibody usage.
Note 2: Other size coverslips will work; however, the volumes of reagents and embryos used will need to be adjusted accordingly.
Pipette 50-200 μl of poly-L-lysine solution (0.01%) on each cover glass in the Petri dish.
Allow the coverslips to sit at least one hour at 37 °C. Longer incubations will not negatively influence results.
Remove poly-L-lysine solution from the cover glass and wash twice with a minimum of 100 μl 1x PBS.
Allow coverslips to dry.
Coverslips are now ready for myofibers.
Note 3: To ensure sterility, coating can be done in a hood and on autoclaved coverslips.
Note 4: Keep the 60 mm Petri dish with Parafilm on the bottom. This setup will be used during myofiber plating and immunolabeling (later steps).
Alternative 1: Instead of coating coverslips immediately prior to use, a coated coverslip stock can be used. A 60 mm Petri dish containing a minimum of 2 ml poly-L-lysine can be stored at 4 °C containing numerous coverslips. With this alternative start at step 1.5 to process the coverslips for myofibers.
Alternative 2: We also coat coverslips in poly-L-ornithine. This is more labor intensive, but is useful for longer term culturing because poly-L-ornithine coated coverslips can be UV treated. With UV treatment and careful sterile technique live myofibers can typically be maintained in culture from 4-7 days.
2. Dissociation of Zebrafish Embryos and Plating of Myofibers (Time: 1 to 3 hr)
Note: the standard protocol applies best to 3 dpf (days post fertilization) embryos.
- Transfer zebrafish embryos into a standard 1.5 ml centrifuge tube and remove as much excess fish water as possible. Typically, 10-20 embryos per tube, though less can be used. Using more than 20-25 embryos often results in a preparation of excessive density.
- Remove chorions prior to dissociation, manually using #5 forceps. Alternatively, chemical chorion removal is achieved with a 10-15 min Pronase treatment. Typically, the previous removal of the chorion is only needed when prior confirmation of a mutant phenotype or morphology is required, or when using stages where embryos are naturally hatched from their chorion.
Add 900 μl of CO2 independent media to the tube containing the embryos.
Add 100 μl of collagenase type II, final concentration 3.125 mg/ml (Stock collagenase solution at 31.25 mg/ml diluted in 1x PBS) to begin dissociation. Collagenase IV can also be used for the dissociation.
Rotate embryos on an orbital shaker, and triturate every 30 min at RT using a P1000 Pipetman for the trituration.
Note 2: Carefully monitor embryo dissociation; over or under dissociation (especially over) are the most common reasons for protocol failure. Times for digestion will vary depending on intensity of trituration, number of embryos per tube, and age of embryos. It is also often less (in comparison to wild types) for embryos from skeletal muscle mutants.
Note 3: Average digestion time per embryo age: 1 dpf = 1 hr, 2 dpf = 1.5 hr, 3 dpf = 2 hr, and 4 dpf = 2-2.5 hr.
Stop digestion when no whole embryos are visible, yet solid pieces are still visible - this is essential to prevent overdigestion.
Centrifuge tubes with dissociated embryos at 0.8-2.3 x g for 3-5 min to pellet cells.
Remove supernatant from pelleted cells and wash 2x with CO2 independent media.
Add fresh CO2 independent media to resuspend cells. 1 ml is typically used for preps of 10-20 embryos. The volume can be scaled based on embryo number.
Using a P1000 pipette, pass embryo suspension through a 70 μm filter . This helps remove unwanted debris from the prep.
Note 4: Embryo suspension can be filtered a second time through a 40 μm filter to further remove unwanted debris. From a double filtration, recovery is approximately 800 μl from a starting volume of 1 ml.
Add approximately 50-100 μl of myofiber suspension to each poly-L-lysine coated coverslip.
Note 5: Perform step 2.9 in the Petri dishes with Parafilm on the bottom, previously prepared. The Parafilm keeps the myofiber suspension from running off the coated coverslips. Keep Petri dishes covered to prevent evaporation.
Allow myofibers to settle approximately 1 hr onto the coated coverslips at room temperature.
Note 6: Myofibers will begin to settle within 5-10 min. However, for good myofiber attachment, a minimum of 1 hr (1-2 hr) is recommended. Allowing myofibers to settle for longer will not harm the prep. For longer incubations (including overnight), antibiotics can be added to the media. With antibiotics and sterile technique live cultures can typically be maintained for 1-2 days.
Live myofibers can be observed at this point. Myofibers from embryos 2 dpf and later will be seen as striated and elongated cells (Figure 1). At this point, myofibers are now ready for live analysis or for immunolabeling.
Note 7: Skeletal muscle from 1 dpf embryos does not plate as elongated and clearly striated fibers. Instead, large myoballs are visible. In addition, during the pelleting phase post dissociation (step 2.6), 1 dpf myoballs need to be centrifuged for a minimum of 8 min at 5,000 x g to achieve a pellet. For analysis of embryos at this stage, it is recommended to use the transgenic line expressing EGFP specifically in skeletal muscle23. This will allow identification of cells from muscle origin versus other sources.
3. Fluorescence Immunostaining of Dissociated Zebrafish Myofibers (Time: approximately 1 day)
3.1. Immunolabeling
Remove a portion of media from myofibers adhered to the coated coverslip
Fix cells using 4% paraformaldehyde or methanol. For PFA, remove ½ the volume of media and replace with the same amount of PFA. Fix for 10 min at RT, then remove total volume, replace with 50-100 μl of PFA, and fix for an additional 10 min. For alcohol fixation, ice cold methanol or acetone can be applied at 4 °C for 10 min or 5 min, respectively.
Wash myofibers at least 3-5x with 1x PBS or 1x PBS plus 0.1% TWEEN. Average wash volume is 50-100 μl per coverslip.
Add blocking solution to myofibers (working stock) and incubate 20-60 min at room temperature. Blocking solution will vary depending on the primary and secondary antibodies. The two most common used in the lab are (1) 1x PBS, 2 mg/ml BSA, 1% sheep serum, 0.25% Triton X-100 final and (2) 0.2% Triton X-100, 2 mg/ml (0.2% BSA) and 5% sheep/goat serum.
Remove blocking solution and add primary antibody diluted in either blocking solution or PBS. Incubate myofibers overnight at 4 °C. Make sure to either Parafilm or wrap in saran wrap the Petri dish containing the myofiber-loaded coverslips coated with antibody. Small pieces of moistened paper towel can be added to create a humidified chamber.
Remove primary antibody from coverslips and wash coverslips 3-5x for 5 min with PBS or blocking solution.
Add secondary antibody at appropriate concentration diluted in 1x PBS or blocking solution for 1-2 hr at RT.
Remove secondary antibody and wash myofibers 3-5x in PBS for 5 min each at RT.
3.2. Mounting coverslips
Apply 1-2 drops of antifade reagent to a microscope slide.
Carefully pick-up the coated and immunolabled coverslip using forceps and place (myofibers down) the coverslip on the antifade reagent on the microscope slide.
Note 1: Attention to the orientation of the coated coverslip is critical.
Lay 1-2 Kimwipes on a hard solid surface. Quickly invert the slide with the coated coverslips resting on the antifade reagent and place it (coverslip down) on the Kimwipes.
Apply light pressure to the corners of the microscope slide to remove excess antifade and form a tight seal between the slide and coverslip
Allow the antifade remaining to dry for 5-10 min at RT, then the myofibers are ready to image (Figures 2A and B) or store at 4 °C.
4. Live Cell Calcium Imaging Using GCaMP3
Live cell experiments can be performed on myofibers prior to fixation (following step 2.10). The following protocol describes live imaging in myofibers expressing GCaMP324, a genetically encoded calcium indicator, expressed by the skeletal muscle specific zebrafish α-actin promoter (pSKM)23. Alternatively, this technique can be readily adapted to use calcium indicator dyes such as Fura-2.
Inject embryos with pSKM: GCaMP3 construct at the 1-2 cell stage
At 3 dpf, collect larvae and prepare myofibers as described in sections 1 and 2. Allow myofibers to adhere for a minimum of 1 hr. For this technique, preparing coverslips in 24-well dishes is required.
Carefully remove any excess media, and add 300 μl of fresh CO2 independent media at RT.
Observe cells under an inverted microscope. GCaMP3 positive myofibers should be visible under green fluorescence.
Set up camera for recording, if desired.
Prepare a 30 mM caffeine solution in CO2 independent media. To stimulate cells, gently pipette 300 μl of caffeine solution into the well under magnification. Within 5-10 sec, GCaMP positive myofibers should respond to caffeine with a rapid increase in fluorescence (Figure 3). Myofibers may also contract. Of note, caffeine induces calcium release from the sarcoplasmic reticulum stores25. Other agents, such as KCl26 and ryanodine4, can be used to study inducible calcium release.
Representative Results
Fluorescent immunolabeling of myofibers (Figure 2)
Images showing expected fluorescent labeling pattern from myofibers immunostained after successful isolation and plating. The myofibers have been labeled with either anti-ryanodine receptor (1:100) (Figure 2A) or anti-α-actinin (1:100) (Figure 2B) antibodies, and reveal immunostaining of the triad and the Z-band respectively. Secondary antibodies used were Alexa Fluor 555 (1:500). Images captured using confocal microscopy.
Induced calcium release from a myofiber (Figure 3)
Panel series showing the representative calcium release from an isolated myofiber treated with caffeine. In brief, zebrafish embryos were injected at the one cell stage with the pSKM: GCaMP3 construct. At 3 dpf, embryos were dissociated and plated as described in the protocol. For analysis, GCaMP3 expressing myofibers were selected, as indicated by the presence of green fluorescence at baseline. Using a micropipette for drug administration, the selected fiber was bathed in a 30 mM caffeine solution to induce calcium release. Video recording was started prior to caffeine application and continued until peak fluorescence was reached. This figure demonstrates normal caffeine induced calcium release, and the technique can be used to interrogate the excitation-contraction coupling apparatus via live imaging.
 Figure 1. Schematic Timeline of Embryo Dissociation. Successive panels (top to bottom) illustrate individual steps, timing, and equipment required for embryo dissociation. A) Selection of embryos for dissociation. Scale bar 500 μm. B) Image of dissociation setup. Embryos are in media and collagenase while being rotated until sufficiently dissociated (Protocol step 2). C) Image of both myofiber plating techniques; round coverslips or 24-well plates (Protocol steps 1 and 4, respectively). D) A bright-field image of live dissociated zebrafish myofibers plated on poly-L Lysine coated coverslips. Arrows denote elongated myofibers from 48 hpf zebrafish. Scale bar 30 μm.
Figure 1. Schematic Timeline of Embryo Dissociation. Successive panels (top to bottom) illustrate individual steps, timing, and equipment required for embryo dissociation. A) Selection of embryos for dissociation. Scale bar 500 μm. B) Image of dissociation setup. Embryos are in media and collagenase while being rotated until sufficiently dissociated (Protocol step 2). C) Image of both myofiber plating techniques; round coverslips or 24-well plates (Protocol steps 1 and 4, respectively). D) A bright-field image of live dissociated zebrafish myofibers plated on poly-L Lysine coated coverslips. Arrows denote elongated myofibers from 48 hpf zebrafish. Scale bar 30 μm.
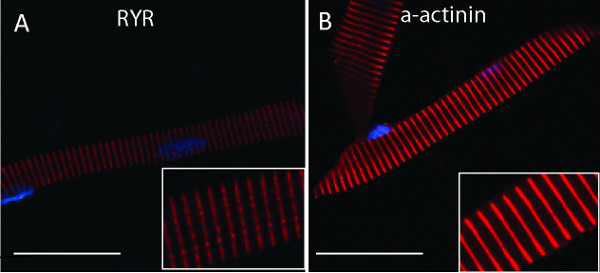 Figure 2. Immunolabeling of individual myofibers isolated from 48 hpf zebrafish embryos. A) Myofiber labeled with anti-ryanodine receptor (RyR1), revealing a banded pattern along the fiber consistent with localization to the triad/EC coupling apparatus. B) Myofiber labeled with anti-α-actinin, visualizing striations (red) along the myofiber localizing to the sarcomere and highlighting the Z bands. In both images, nuclei are labeled with DAPI, seen as blue ovals. Inserts show higher magnification images of the myofiber in the large image. A) and B) scale bar 30 μm.
Figure 2. Immunolabeling of individual myofibers isolated from 48 hpf zebrafish embryos. A) Myofiber labeled with anti-ryanodine receptor (RyR1), revealing a banded pattern along the fiber consistent with localization to the triad/EC coupling apparatus. B) Myofiber labeled with anti-α-actinin, visualizing striations (red) along the myofiber localizing to the sarcomere and highlighting the Z bands. In both images, nuclei are labeled with DAPI, seen as blue ovals. Inserts show higher magnification images of the myofiber in the large image. A) and B) scale bar 30 μm.
 Figure 3. Representative induced calcium release response from a single isolated zebrafish myofiber. All panels show a pseudocolored zebrafish myofiber expressing GCaMP3. The time course shows the increase of GCaMP3 fluorescence (red color) in response to application of 30 mM caffeine. Recording started prior to caffeine application (0 msec) and continued to maximum fluorescence response (992 msec). Scale bar 30 μm. Click here to view larger figure.
Figure 3. Representative induced calcium release response from a single isolated zebrafish myofiber. All panels show a pseudocolored zebrafish myofiber expressing GCaMP3. The time course shows the increase of GCaMP3 fluorescence (red color) in response to application of 30 mM caffeine. Recording started prior to caffeine application (0 msec) and continued to maximum fluorescence response (992 msec). Scale bar 30 μm. Click here to view larger figure.
Discussion
Zebrafish are a powerful vertebrate model system for studying muscle development and function in vivo25,27,28 . They have also emerged as a valuable asset for modeling human muscle diseases14,15,20,29 . While great strides have been taken to advance the use and application of zebrafish for the study of muscle function and muscle disease, there is a constant critical need to develop tools that will allow more in depth analysis that compliments the genetic, morphologic, behavioral and functional advantages zebrafish already offer17. We have therefore adapted and developed a simple and robust technique for the characterization of zebrafish myofibers from dissociated whole embryos.
The use of individual myofibers is commonplace in studies using the murine model system. In mice this has allowed for investigations and analyses that are impractical or impossible in whole animals, including subcellular protein localization studies30, live cell imaging31, and electrophysiologic measurements32. In the zebrafish, similar dissociation techniques have been previously developed to specifically identify and examine motorneurons33 as well as Rohon-Beard sensory neurons34, and these techniques have enabled detailed analysis of these neuron subtypes.
The broad applicability of isolated myofibers, combined with the many unique advantages the zebrafish offers as a vertebrate model17, are what motivated us to develop a technique for isolating and characterizing embryonic and larval zebrafish myofibers. We have utilized isolated myofibers in previous studies to determine subcellular localization of muscle proteins20,21 , to study localization of chimerically expressed transgenes35 and to define specific myofiber parameters including especially size20,21 . In addition, we have used the fibers as a source of validation for fish models of human muscle diseases20,21 . For example, we have identified nemaline bodies by immunofluorescent analysis in a model of nemaline myopathy20. We have also used myofibers as a means of interrogating specific intracellular pathways, including apoptosis and oxidative stress15, and for the application and testing of specific chemical modulators15.
Grabner and colleagues have also used the myofiber system to great success19,22,36 . They are the only group to report prolonged culturing of isolated fibers. In addition, they have been able to use the fibers to measure the electrophysiological properties of the muscle. Specifically, they have tested the impact of the loss of the beta subunit of the dihydropyridine receptor on calcium release and measured the ability of a set of transgenes to rescue defects in this release. These studies illuminate some of the very powerful potential applications of isolated myofibers.
Another group to report use of the myofiber prep for studying muscle disease is Nixon et al. who used myofibers to study the impact of morpholino mediated caveolin 3 knockdown on muscle development and muscle structure18. These authors not only examined parameters by light microscopy, but also studied the fibers using electron microscopy. Their work identifies yet another advantage of the isolated myofiber system: the ability to study the ultrastructure of the myofiber in an isolated and controlled setting.
These studies, along with our work, point to the potential wide-ranging utility of this technique. Additional applications are certain to be developed, such as measuring rapid calcium spikes using indicators like Fura-2. Additionally, this preparation offers the opportunity to observe electrical activity, such as action potentials via voltage sensitive dyes (Di-4-ANEPPS) or direct electrophysiological recording. It seems clear that all current techniques used on murine myofibers should be applicable to zebrafish myofibers. In addition, the ability to express different transgenes in the developing zebrafish, as well as the availability of a large and growing number of transgenic zebrafish expressing various marker proteins (www.zfin.org), adds an additional layer of complexity and applicability to the system. We demonstrate this fact with the myofibers cultured from GCaMP3 expressing zebrafish, where we take advantage of the ability to express GCaMP3 in muscle to study induced calcium imaging in real time in isolated fibers (Figure 3).
As with most techniques, there are also some clear limitations to the isolated myofiber system. These issues are in addition to the standard shortcomings of studying a cell type in vitro that functions as a three dimensional syncytia in vivo and of performing electrophysiologic experiments with nonphysiologic stimuli. Using our methodology, one is not able to obtain a pure preparation of myofibers. This is not a problem for immunostaining or for electrophysiological measurements, but it does preclude certain experimental approaches. For example, we have yet to determine a suitable methodology for identifying a pure population of myofibers for transcriptomic analysis. We have attempted FACS sorting of GFP expressing myofibers followed by myofiber preparation, but this has not resulted in a pure culture with a suitable number of myofibers for either RNA or protein analysis.
Another challenge is the long term culturing of myofibers. We have been able to keep cultures for approximately 24 hr, and Grabner and colleagues report a preparation that has been suitable for multiple days of experimentation19. The challenge related to this aspect of the technique is preventing contamination, as these cultures (much like murine myofiber preps) are often difficult to keep sterile. Great care is needed if one is to attempt prolonged cultures. We also recommend the use of antimicrobials including antifungals.
In all, we demonstrate a practical method for the isolation of zebrafish myofibers as well as outline how to perform fluorescent immunolabeling and real time calcium imaging on the isolated fiber preparations. Continuing modification and development of this technique will offer an ever-expanding repertoire of tools to experiment on specific, isolated cell types in zebrafish. By dissociating zebrafish embryos into isolated individual myofibers, we can further enhance our understanding of muscle development, function and disease.
Disclosures
The authors declare no conflicting interests.
Acknowledgments
The authors wish to thank the members of the Dowling lab (Aaron Reifler, Trent Waugh, Angela Busta, and William Telfer) that contributed to the development of the technique and to the production of the manuscript. This work was funded by the Taubman Institute, the Department of Pediatrics at the University of Michigan, and in part from grants from the Muscular Dystrophy Association (JJD MDA186999) and the National Institutes of Health (JJD 1K08AR054835).
References
- Dulhunty AF. Excitation-contraction coupling from the 1950s into the new millennium. Clin. Exp. Pharmacol. Physiol. 2006;33:763–772. doi: 10.1111/j.1440-1681.2006.04441.x. [DOI] [PubMed] [Google Scholar]
- Bannister RA. Bridging the myoplasmic gap: recent developments in skeletal muscle excitation-contraction coupling. J. Muscle Res. Cell Motil. 2007;28:275–283. doi: 10.1007/s10974-007-9118-5. [DOI] [PubMed] [Google Scholar]
- Ohno K. Genetic defects and disorders at the neuromuscular junction. Brain Nerve. 2011;63:669–678. [PubMed] [Google Scholar]
- Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb. Perspect. Biol. 2010;2 doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Calcium channel diversity: multiple roles of calcium channel subunits. Curr. Opin. Neurobiol. 2009;19:237–244. doi: 10.1016/j.conb.2009.06.006. [DOI] [PubMed] [Google Scholar]
- Sparrow JC, Schock F. The initial steps of myofibril assembly: integrins pave the way. Nat. Rev. Mol. Cell Biol. 2009;10:293–298. doi: 10.1038/nrm2634. [DOI] [PubMed] [Google Scholar]
- Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ. Res. 2004;94:1023–1031. doi: 10.1161/01.RES.0000126574.61061.25. [DOI] [PubMed] [Google Scholar]
- Bassett D, Currie PD. Identification of a zebrafish model of muscular dystrophy. Clin. Exp. Pharmacol. Physiol. 2004;31:537–540. doi: 10.1111/j.1440-1681.2004.04030.x. [DOI] [PubMed] [Google Scholar]
- Barresi R, Campbell KP. Dystroglycan: from biosynthesis to pathogenesis of human disease. J. Cell Sci. 2006;119:199–207. doi: 10.1242/jcs.02814. [DOI] [PubMed] [Google Scholar]
- Emery AE. Population frequencies of inherited neuromuscular diseases-a world survey. Neuromuscul. Disord. 1991;1:19–29. doi: 10.1016/0960-8966(91)90039-u. [DOI] [PubMed] [Google Scholar]
- Nance JR, Dowling JJ, Gibbs EM, Bonnemann CG. Congenital myopathies: an update. Curr. Neurol. Neurosci. Rep. 2012;12:165–174. doi: 10.1007/s11910-012-0255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett DI, Currie PD. The zebrafish as a model for muscular dystrophy and congenital myopathy. Hum. Mol. Genet. 2003;12:265–270. doi: 10.1093/hmg/ddg279. [DOI] [PubMed] [Google Scholar]
- Dooley K, Zon LI. Zebrafish: a model system for the study of human disease. Curr. Opin. Genet. Dev. 2000;10:252–256. doi: 10.1016/s0959-437x(00)00074-5. [DOI] [PubMed] [Google Scholar]
- Dowling JJ, et al. Loss of myotubularin function results in T-tubule disorganization in zebrafish and human myotubular myopathy. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling JJ, et al. Oxidative stress and successful antioxidant treatment in models of RYR1-related myopathy. Brain. 2012;135:1115–1127. doi: 10.1093/brain/aws036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara G, et al. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. U.S.A. 2011;108:5331–5336. doi: 10.1073/pnas.1102116108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detrich HW, Westerfield M, 3rd, M , Zon LI. Overview of the Zebrafish system. Methods Cell Biol. 1999;59:3–10. doi: 10.1016/s0091-679x(08)61816-6. [DOI] [PubMed] [Google Scholar]
- Nixon SJ, et al. Zebrafish as a model for caveolin-associated muscle disease; caveolin-3 is required for myofibril organization and muscle cell patterning. Hum. Mol. Genet. 2005;14:1727–1743. doi: 10.1093/hmg/ddi179. [DOI] [PubMed] [Google Scholar]
- Schredelseker J, et al. The beta 1a subunit is essential for the assembly of dihydropyridine-receptor arrays in skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 2005;102:17219–17224. doi: 10.1073/pnas.0508710102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telfer WR, Nelson DD, Waugh T, Brooks SV, Dowling JJ. neb: a zebrafish model of nemaline myopathy due to nebulin mutation. Dis. Model Mech. 2012;5:389–396. doi: 10.1242/dmm.008631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling JJ, Low SE, Busta AS, Feldman EL. Zebrafish MTMR14 is required for excitation-contraction coupling, developmental motor function and the regulation of autophagy. Hum. Mol. Genet. 2010;19:2668–2681. doi: 10.1093/hmg/ddq153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schredelseker J, Dayal A, Schwerte T, Franzini-Armstrong C, Grabner M. Proper restoration of excitation-contraction coupling in the dihydropyridine receptor beta1-null zebrafish relaxed is an exclusive function of the beta1a subunit. J. Biol. Chem. 2009;284:1242–1251. doi: 10.1074/jbc.M807767200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashijima S, Okamoto H, Ueno N, Hotta Y, Eguchi G. High-frequency generation of transgenic zebrafish which reliably express GFP in whole muscles or the whole body by using promoters of zebrafish origin. Dev. Biol. 1997;192:289–299. doi: 10.1006/dbio.1997.8779. [DOI] [PubMed] [Google Scholar]
- Tian L, et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W, et al. Non-sense mutations in the dihydropyridine receptor beta1 gene, CACNB1, paralyze zebrafish relaxed mutants. Cell Calcium. 2006;39:227–236. doi: 10.1016/j.ceca.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Dora KA, Doyle MP, Duling BR. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc. Natl. Acad. Sci. U.S.A. 1997;94:6529–6534. doi: 10.1073/pnas.94.12.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata H, et al. accordion, a zebrafish behavioral mutant, has a muscle relaxation defect due to a mutation in the ATPase Ca2+ pump SERCA1. Development. 2004;131:5457–5468. doi: 10.1242/dev.01410. [DOI] [PubMed] [Google Scholar]
- van Eeden FJ, et al. Mutations affecting somite formation and patterning in the zebrafish, Danio rerio. Development. 1996;123:153–164. doi: 10.1242/dev.123.1.153. [DOI] [PubMed] [Google Scholar]
- Gupta V, et al. The zebrafish dag1 mutant: a novel genetic model for dystroglycanopathies. Hum. Mol. Genet. 2011;20:1712–1725. doi: 10.1093/hmg/ddr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leuranguer V, Papadopoulos S, Beam KG. Organization of calcium channel beta1a subunits in triad junctions in skeletal muscle. J. Biol. Chem. 2006;281:3521–3527. doi: 10.1074/jbc.M509566200. [DOI] [PubMed] [Google Scholar]
- Capote J, Bolanos P, Schuhmeier RP, Melzer W, Caputo C. Calcium transients in developing mouse skeletal muscle fibres. J. Physiol. 2005;564:451–464. doi: 10.1113/jphysiol.2004.081034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams BA, Tanabe T, Mikami A, Numa S, Beam KG. Intramembrane charge movement restored in dysgenic skeletal muscle by injection of dihydropyridine receptor cDNAs. Nature. 1990;346 doi: 10.1038/346569a0. [DOI] [PubMed] [Google Scholar]
- Sakowski SA, et al. A novel approach to study motor neurons from zebrafish embryos and larvae in culture. J. Neurosci. Methods. 2012;205:277–282. doi: 10.1016/j.jneumeth.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y, et al. Biogenesis of GPI-anchored proteins is essential for surface expression of sodium channels in zebrafish Rohon-Beard neurons to respond to mechanosensory stimulation. Development. 2010;137:1689–1698. doi: 10.1242/dev.047464. [DOI] [PubMed] [Google Scholar]
- Davidson AE, et al. Novel deletion of lysine 7 expands the clinical, histopathological and genetic spectrum of TPM2-related myopathies. Brain. 2013;136:508–521. doi: 10.1093/brain/aws344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schredelseker J, Shrivastav M, Dayal A, Grabner M. Non-Ca2+-conducting Ca2+ channels in fish skeletal muscle excitation-contraction coupling. Proc. Natl. Acad. Sci. U.S.A. 2010;107:5658–5663. doi: 10.1073/pnas.0912153107. [DOI] [PMC free article] [PubMed] [Google Scholar]