

Abstract
Estrogens play pivotal roles in development and function of many organ systems, including the reproductive system. We have generated estrogen receptor 1 (Esr1)-knockout rats using zinc finger nuclease (ZFN) genome targeting. mRNAs encoding ZFNs targeted to exon 3 of Esr1 were microinjected into single-cell rat embryos and transferred to pseudopregnant recipients. Of 17 live births, 5 had biallelic and 1 had monoallelic Esr1 mutations. A founder with monoallelic mutations was backcrossed to a wild-type rat. Offspring possessed only wild-type Esr1 alleles or wild-type alleles and Esr1 alleles containing either 482 bp (Δ482) or 223 bp (Δ223) deletions, indicating mosaicism in the founder. These heterozygous mutants were bred for colony expansion, generation of homozygous mutants, and phenotypic characterization. The Δ482 Esr1 allele yielded altered transcript processing, including the absence of exon 3, aberrant splicing of exon 2 and 4, and a frameshift that generated premature stop codons located immediately after the codon for Thr157. ESR1 protein was not detected in homozygous Δ482 mutant uteri. ESR1 disruption affected sexually dimorphic postnatal growth patterns and serum levels of gonadotropins and sex steroid hormones. Both male and female Esr1-null rats were infertile. Esr1-null males had small testes with distended and dysplastic seminiferous tubules, whereas Esr1-null females possessed large polycystic ovaries, thread-like uteri, and poorly developed mammary glands. In addition, uteri of Esr1-null rats did not effectively respond to 17β-estradiol treatment, further demonstrating that the Δ482 Esr1 mutation created a null allele. This rat model provides a new experimental tool for investigating the pathophysiology of estrogen action.
Estrogens are multifunctional hormones that exhibit essential regulatory roles in the development and function of many organ systems including the reproductive axis (1) and contribute to differences in female vs male physiology (2, 3). Estrogens are synthesized in the ovary and other tissues (4–6) and act on their cellular targets through physical interactions with specific ligand-activated transcription factors, termed estrogen receptor 1 (Esr1, also called estrogen receptor-α) and estrogen receptor 2 (Esr2, also called estrogen receptor-β), to regulate gene expression (1, 7). Consequently, dysregulation of signaling through estrogen receptors is implicated in the development and progression of a wide range of diseases, including infertility, endometriosis, cancer, neurodegeneration, osteoporosis, cardiovascular disease, lupus erythematosus, diabetes, and obesity (1, 8).
Our understanding of the physiology of estrogen action was significantly advanced following the generation of mutant mouse models possessing disruptions in Esr1 (9, 10) and Esr2 (10, 11) genes. However, there are limitations in using the mouse as a model in some research areas. In some cases, the rat is more similar to the human and is a superior model for investigating both physiological and pathological processes (12–14).
To meet the need for rat models of human disease, strategies have been developed to manipulate the rat genome, including genome editing of early embryos (15–17), transposon mutagenesis using spermatogonial stem cells (18), and gene targeting with rat embryonic stem cells (19). Over the past few years, genome editing using zinc finger nucleases (ZFNs) has proved to be an effective strategy for generating targeted mutations (15, 20–28).
ZFNs are engineered zinc finger proteins containing nucleotide sequence-specific DNA binding with the endonuclease activity of the restriction enzyme FokI (29). The endonuclease creates site-specific double-strand DNA breaks at the target locus, which activates DNA repair (29). Imperfect repair of double-strand DNA breaks by the nonhomologous end-joining pathway results in mutations, especially deletions, resulting in frameshifts, and functional knockouts of targeted genes at high efficiencies (15, 30).
In this study, we used ZFN-mediated genome editing to generate Esr1-knockout rats. Here we report the initial characterization of the Esr1-null rat.
Materials and Methods
Experimental animals
All animal experiments were performed with Holtzman Sprague-Dawley rats obtained from Harlan Sprague-Dawley. The University of Kansas Medical Center Animal Care and Use Committee approved the protocols for generation of targeted Esr1 mutations and phenotypic characterization.
Generation of targeted Esr1 mutation
ZFN constructs specific for the rat Esr1 gene were designed, assembled, and validated by Sigma-Aldrich. Selected ZFNs were targeted to exon 3 of the Esr1 gene, which encodes the DNA binding domain (target sequence: CTCTCCAGCAGCAGCgagaagGGAAACATGATCATG; nucleotides 717–752, NM_012689.1) (Figure 1A). ZFN mRNAs were microinjected into single-cell rat embryos. Injected embryos were transferred to the oviduct of day-0.5 pseudopregnant rats. Offspring were screened for ZFN-induced mutations at the target site of the Esr1 gene.
Figure 1.

ZFN generated targeted mutations at the Esr1 locus. A and B, Schematic representations of the ZFN target site within the rat Esr1 (NC_005100.3) locus (A) and PCR primer locations (B) used for mutation detection. C, PCR products from founder rat DNA samples were identified by agarose gel electrophoresis and ethidium bromide staining and mutations (mutant animal numbers are in bold) were resolved by DNA sequence analysis. D, Offspring of the monoallelic mutant founder (F871) possessed a wild-type allele and an allele containing either a 482-bp (**) or a 223-bp (*) deletion at the Esr1 locus, indicating mosaicism. E and F, DNA sequence analysis showing the 482-bp deletion (Δ482) (E) and the 223-bp deletion (Δ223) (F) at the Esr1 gene locus. Schematic diagrams beneath each DNA sequencing profile show the impact of the deletion on exon 3 of the Esr1 gene. E, The nucleotides in blue reflect the 9-bp remnant of exon 3 for Δ482. F, In contrast, Δ223 contains an exon 3 that is largely intact except for a 9-bp truncation at the 3′ end of the exon. The terminal 9 nucleotides of remaining exon 3 for the Δ223 deletion are highlighted in blue.
Identification of founders and establishment of Esr1 mutant lines
For initial screening, genomic DNA was purified from tail-tip biopsies using the E.Z.N.A. tissue DNA kit (Omega Bio-Tek). PCR was performed on the purified DNA samples using multiple sets of primers (Figure 1B and Supplemental Table 1, published on The Endocrine Society's Journals Online website at http://endo.endojournals.org) directed to stepwise increases in the length of nucleotide sequences flanking the ZFN site (150 bp to ∼5 kbp on either side). PCR products were identified by agarose gel electrophoresis and ethidium bromide staining and the sites of mutations resolved by DNA sequence analysis. Attempts were made to mate all founders with wild-type rats (Supplemental Table 2). Genotyping of offspring was performed on genomic DNA extracted from tail-tip biopsies and multiplex PCR using the REDExtract-N-Amp tissue PCR kit (Sigma-Aldrich) and mutation-specific primers (Supplemental Tables 3 and 4). Esr1 mutant heterozygotes were intercrossed, and phenotypic characterization was performed on wild-type and homozygous mutant offspring.
Growth assessment and examination of the reproductive tract
Female rats were killed at 8 weeks of age, whereas male rats were killed at 10 weeks. Body weights and crown-rump lengths were measured at the time of killing. Rats were euthanized and blood samples were collected immediately by cardiac puncture. Testes, epididymides, and seminal vesicles were collected from male rats, and ovaries, uteri, and mammary glands were collected from female rats. Each organ weight was recorded before further processing. Tissue samples were preserved in fixative or snap-frozen in liquid nitrogen and stored at −80°C until processed. Detailed descriptions of the histological, immunohistochemistry, Western blotting, RT-PCR, and quantitative RT-PCR procedures are provided in the Supplemental Materials and Methods.
Detection of ESR1 expression
Mutant Esr1 mRNA was detected by RT-PCR using primers designed from exon sequences flanking the targeted exon 3 (Figure 1A and Supplemental Table 5), and changes in the open reading frame were determined by DNA sequencing. Expression of mutant ESR1 proteins was determined by Western blotting and immunofluorescence. A rabbit monoclonal antibody (E115; Epitomics) that recognizes a region flanking Ser106 of ESR1 was used in both of the immunodetection techniques.
Fertility tests
Fertility was assessed by cohabiting 8-week-old males with 8-week-old female rats for 12 weeks and recording pregnancies and litter sizes. The breeding combinations included wild-type males with wild-type or homozygous Esr1 mutant females and homozygous Esr1 mutant males with wild-type females.
Hormone measurements
Hormone levels were measured in male or female rats housed 2 per cage. Blood samples were collected from Esr1-null or age-matched wild-type female rats randomly irrespective of their estrous cycle. Serum LH and FSH concentrations were determined by the Ligand Assay and Analysis Core at the University of Virginia (Charlottesville, VA). Total serum 17β-estradiol (E2) and testosterone concentrations were measured by RIA as previously described (31).
Uterine responsiveness to E2
Adult wild-type and age-matched homozygous Esr1 mutant rats were ovariectomized at 8 weeks, rested for 2 weeks, and sc treated with E2 (40 μg/kg) or vehicle (sesame oil), and uterine responsiveness was assessed. Uterine tissues were collected 6 hours after a single injection or 24 hours after 3 daily injections (see Figure 6, A and G). Uterine weight, histology, and gene expression responses were examined. A list of quantitative RT-PCR primer sequences for E2-responsive genes (32) are shown in Supplemental Table 6.
Figure 6.
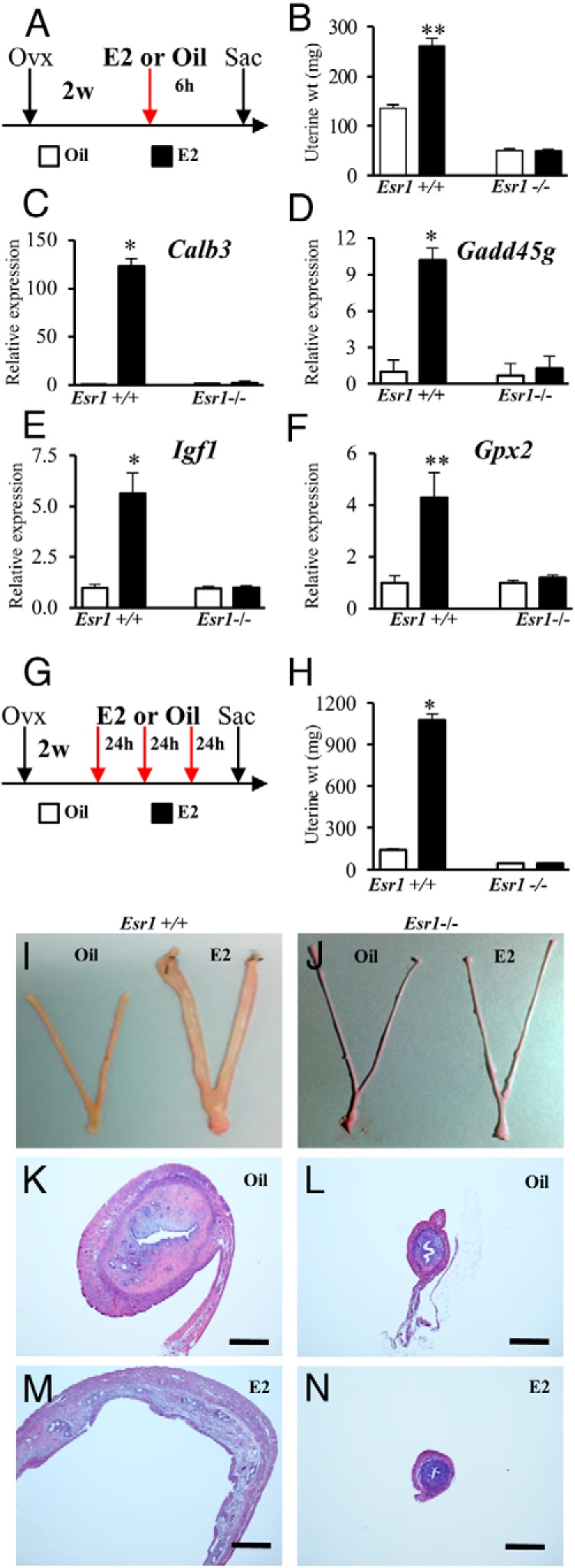
Effects of ESR1 disruption on uterine responses to E2. A, Wild-type and Esr1-null female rats were ovariectomized (Ovx), rested for 2 weeks (w), and treated (red arrow) with a single dose of vehicle (oil) or E2 (40 μg/kg) and killed (Sac) 6 hours after injection. B–F, Uteri were dissected, weighed (B), and processed for quantitative RT-PCR analysis of Calb3 (C), Gadd45g (D), Igf1 (E), and Gpx2 (F). G, In a second experiment, wild-type and Esr1-null female rats were similarly prepared and then treated once daily for 3 successive days (red arrows) with oil or E2 and killed 24 hours after the last injection. H–N, Uteri were dissected, grossly inspected (I and J), weighed (H), and prepared for histological analysis and hematoxylin and eosin staining (K–N). Scale bars, 0.5 mm. Sample sizes were ≥6 per treatment. Asterisks indicate a significant difference between oil and E2 treatments: *, P < .001; **, P < .002.
Statistical analysis
Each experimental group consisted of a minimum of 6 rats, and the procedures were repeated at least 3 times. Values reported graphically are expressed as mean ± SEM. Analyses were performed using the SPSS Statistical Package (IBM). Statistical comparisons between 2 means were performed using Student's t test. P values < .05 were considered significant.
Results
ZFN-generated targeted mutations at the rat Esr1 locus
ZFN-mediated genome editing was used to generate targeted mutations in exon 3 of rat Esr1. Transfer of the ZFN mRNA microinjected embryos to 5 recipients yielded 17 viable offspring. PCR-based screening of tail-biopsy DNA and sequencing identified 6 founders possessing either monoallelic or biallelic mutations at the targeted Esr1 locus (Figure 1C). Deletions ranged from 7 bp to >4 kbp (Supplemental Table 2). Biallelic deletions were detected in 5 of the 6 founders (2 males and 3 females), and monoallelic mosaic mutations were identified in the remaining founder female (Supplemental Table 2). Backcrossing to wild-type rats was attempted for all founders, but fertility was evident only in the monoallelic Esr1 mutant female. She gave birth to a litter consisting of 5 wild-type pups, 2 pups with a 482-bp deletion (Δ482) mutation and 4 pups with a 223-bp deletion (Δ223) mutation, demonstrating an efficient germline transmission (Figure 1D). Δ482 spans 300 bp of intron 2 including the branch and splice acceptor sites as well as 182 bp of exon 3 (Figure 1E and Supplemental Figure 1), whereas Δ223 is located immediately downstream of the Δ482 site and includes 9 bp of exon 3 and 214 bp of intron 3 (Figure 1F and Supplemental Figure 1).
ESR1 deficiency in rats possessing homozygous Esr1 mutations
Δ482 and Δ223 Esr1 mutations were further characterized at the mRNA and protein level in uterine tissue samples. RT-PCR analysis for Esr1 in uterine tissue from Δ482 mutants detected a short weakly expressed transcript lacking exon 3 (Figure 2B), whereas analysis of uterine tissues from Δ223 mutants resolved multiple bands (Figure 2C). Sequencing of PCR-amplified Δ482 cDNA indicated alternative splicing of the mutant Esr1 transcript between exon 2 and exon 4, which resulted in a frameshift creating 2 premature stop codons located immediately after the codon for Thr157 (Figure 2D). ESR1 protein was not detected by Western blot analysis or immunofluorescence in uterine tissue samples from rats harboring either homozygous Δ223 or Δ482 Esr1 mutations (Figure 2, E–H). Due to the detection of aberrant transcripts in Δ223 (Figure 2C), subsequent phenotypic analyses were focused on the Δ482 Esr1 mutation.
Figure 2.
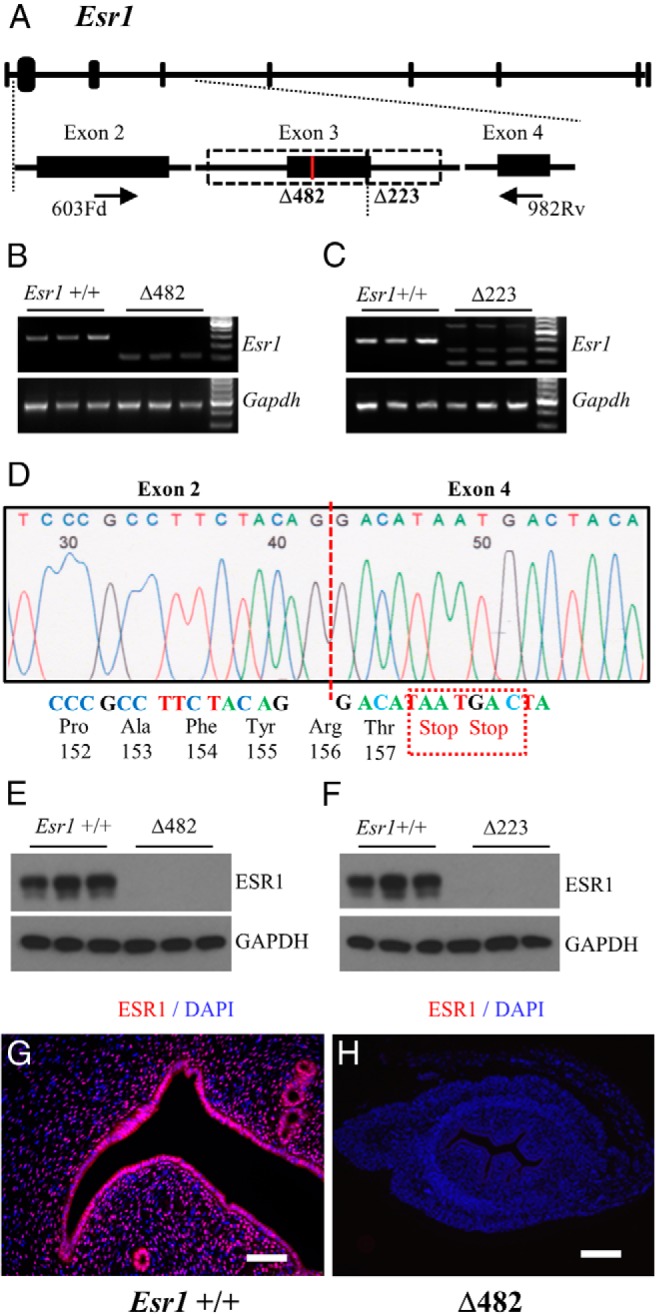
ESR1 deficiency in rats possessing homozygous Esr1 mutations. A, Schematic representation of the Esr1 gene (NC_005100.3) and the locations of primers used for RT-PCR. B and C, RT-PCR analyses for Esr1 detected a weakly expressed shorter transcript in uterine tissues from Δ482 mutants (B), whereas multiple transcripts of various lengths were detected in uterine tissues from Δ223 mutants (C). D, DNA sequencing of the Δ482 RT-PCR product demonstrated aberrant splicing between exons 2 and 4 and resulted in a frameshift creating two stop codons located immediately after the codon for Thr157. E–G, ESR1 protein was not detected in uterine tissues of the Esr1 mutant rats by western blotting (E and F) or immunofluorescence (G and H). Scale bars, 0.25 mm.
Postnatal growth and fertility
Esr1 mutant heterozygotes were intercrossed and phenotypic characterization performed on homozygous mutant and wild-type offspring. Heterozygous Esr1 mutant rats generated litters of expected size and Mendelian ratio (Supplemental Figure 2). At birth, rats of all Esr1 genotypes exhibited similar size and appearance. However, postnatal growth patterns differed. Esr1-null males were significantly smaller than their wild-type littermates, whereas Esr1-null females were significantly larger (Figure 3, A and B). Both males and females possessing homozygous null mutations at the Esr1 locus were infertile. Cohabiting Esr1-null rats with wild-type rats for 12 weeks did not generate any progeny.
Figure 3.

Effects of ESR1 disruption on postnatal growth and serum gonadotropin and sex steroid hormone levels. Adult wild-type and Esr1-null males (10 weeks of age) and adult wild-type and Esr1-null females (8 weeks of age) were weighed (A) and crown-rump length (B) and serum levels of LH (C), FSH (D), testosterone (E), and 17β-estradiol (F) measured. Sample size was ≥10 per genotype. Asterisks indicate a significant difference between the genotypes: *, P < .001; **, P < .002; ***, P < .02.
Serum gonadotropins and sex steroid hormones
Serum gonadotropin and sex steroid hormone levels were measured in adult homozygous Esr1 mutant rats and compared with levels in wild-type littermates. Serum LH and FSH levels were significantly higher in Esr1 mutant rats (Figure 3, C and D). Despite elevated gonadotropin levels, serum testosterone levels were lower in Esr1-null males. In contrast, Esr1 disruption in females resulted in elevated levels of serum testosterone and E2 (Figure 3, E and F). Serum E2 levels were below the detection limit (4 pg/mL) in males of both genotypes.
Male reproductive tract phenotype
Esr1-null male rats displayed normal-appearing external genitalia. However, testes, epididymides, and seminal vesicles were smaller in Esr1-null rats compared with wild-type littermates (Figure 4, A–F). Testes of Esr1-null rats exhibited a pronounced disorganization in tissue structure. Testicular pathology was due to the accumulation of fluid in the testes resulting in distension and atrophic changes in the seminiferous tubules (Figure 4, G and H). Only trace numbers of sperm were visible in histological sections of testes (Figure 4H) and epididymides (Figure 4, J and L) of 10-week-old Esr1-null rats.
Figure 4.
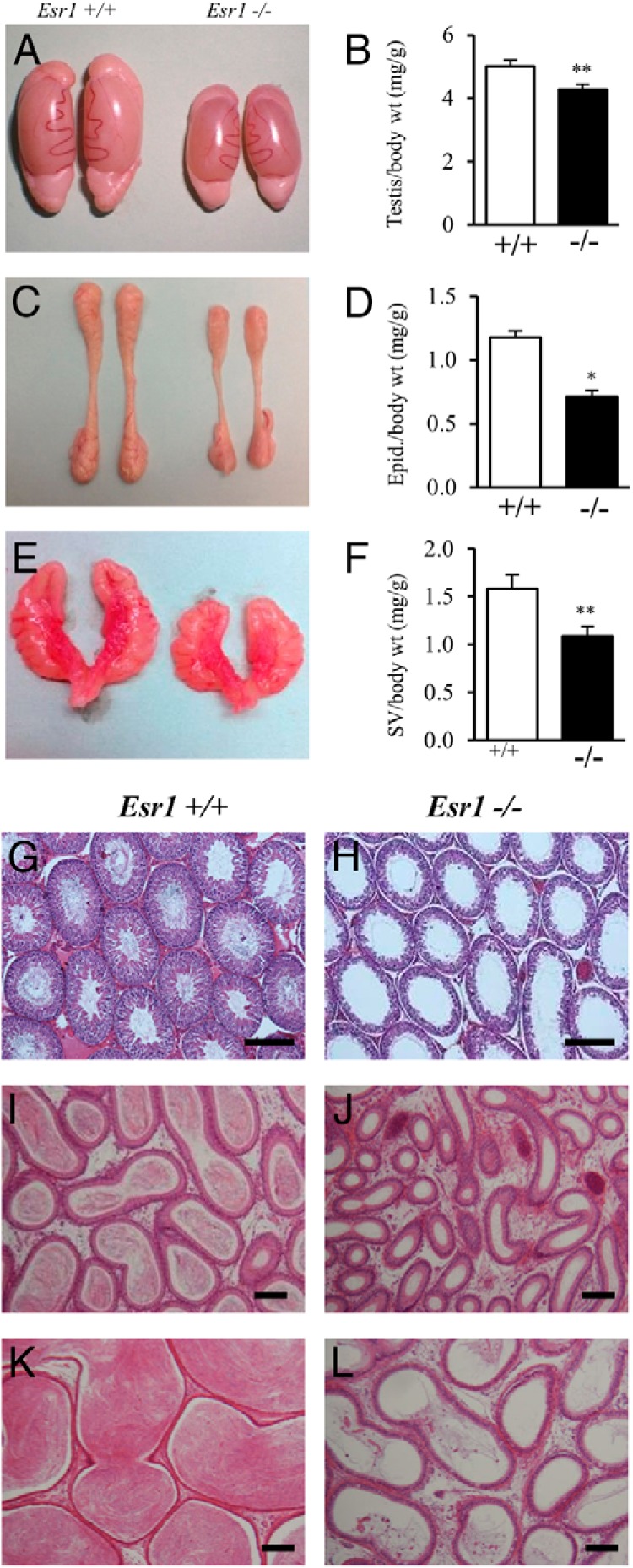
Effects of ESR1 disruption on the male reproductive tract. A–F, The reproductive tracts of adult wild-type and Esr1-null males (10 weeks of age) were examined, including gross appearance and weights for testes (A and B), epididymides (C and D), and seminal vesicles (E and F). Sample sizes for the organ weight measurements were ≥10 per genotype. Asterisks indicate a significant difference between the genotypes: *, P < .001. Abbreviations: Epid, epididymis; SV, seminal vesicle; wt, weight. G–L, Representative hematoxylin- and eosin-stained tissue sections of testis (G and H), caput epididymis (I and J), and cauda epididymis (K and L) from wild-type and Esr1-null males are presented. Please note the distention and atrophic changes in the seminiferous tubules and the trace numbers of sperm in the testis and epididymis of the Esr1-null rats (H, J, and L). Scale bars, 0.25 mm.
Female reproductive tract phenotype
Esr1-null female rats exhibited vaginal opening between postnatal days 33 and 35, which was similar to wild-type rats. Inspection of vaginal cytology revealed that unlike the cyclic changes observed in wild-type rats, Esr1-null females showed persistent leukocytic vaginal cytology and a failure to cycle. The Esr1-null mutation disrupted adult ovarian, uterine, and mammary gland morphologies. In contrast to the ovaries of wild-type females, Esr1-null females contained large polycystic ovaries, which were devoid of corpora lutea (Figure 5, A, B, E, and F). Esr1-null females possessed narrow, thread-like uteri (Figure 5, C and D). Each definitive layer of the uterus, including the myometrium, stroma, and epithelium was present in Esr1-null uteri; however, they were each hypoplastic compared with wild-type controls (Figure 5, G and H). Whole-mount staining of inguinal mammary glands (Figure 5, I and J) showed marked impairments of growth and differentiation in Esr1-null females. Only rudimentary ductal and alveolar structures were present in the mutant mammary gland (Figure 5J).
Figure 5.
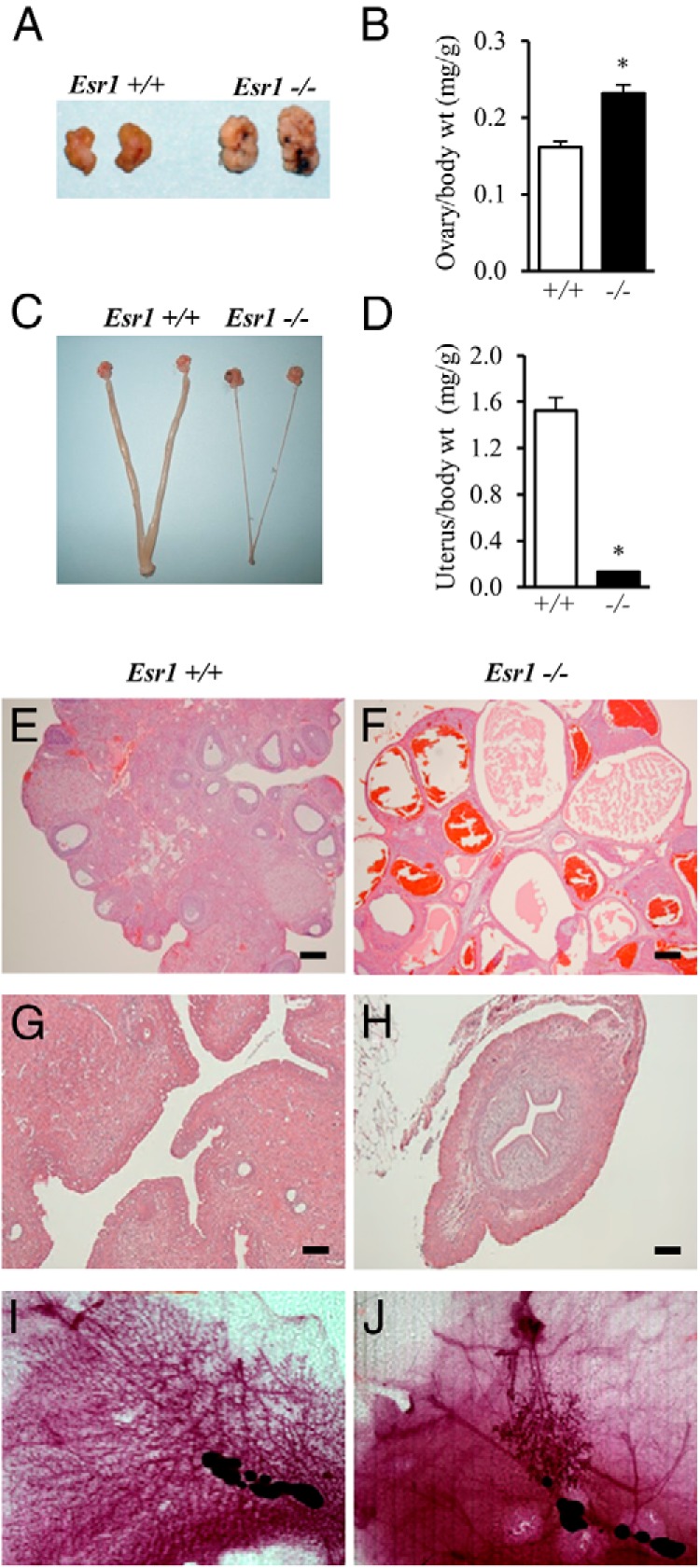
Effects of ESR1 disruption on the female reproductive system. A–D, Reproductive tracts of adult wild-type and Esr1-null females (8 weeks of age) were examined, including gross appearance and weights for ovaries (A and B) and uteri (C and D). Sample sizes for the organ weight measurements were ≥10 per genotype. Asterisks indicate a significant difference between the genotypes: *, P < .001. E–H, Representative hematoxylin- and eosin-stained tissue sections from ovaries (E and F) and uteri (G and H) from wild-type and Esr1-null females. I and J, Whole-mount staining of mammary glands from wild-type (I) and Esr1-null (J) females. Please note the cystic and hypoplastic features of ovaries and uteri, respectively, and the rudimentary ductal and alveolar structures in the mammary glands of Esr1-null females (F, H, and J). Scale bars, 0.25 mm.
Uterine responsiveness to E2
Estrogen responsiveness was evaluated in ovariectomized adult wild-type and Esr1-null rats treated with either a single dose of E2 and examined 6 hours later (Figure 6A) or after 3 daily doses of E2 and examined 24 hours after the last injection (Figure 6G). Uteri from wild-type rats responded to E2 within 6 hours with hyperemia, increased weight gain, and robust induction in the expression of a subset of estrogen-responsive genes (Calb3, Gadd45g, Igf1, and Gpx2), whereas uteri from Esr1-null rats did not exhibit any significant responses to E2 (Figure 6, B–F). Three daily injections of E2 resulted in uterine hyperemia, water imbibition, and marked weight gain in wild-type but not in Esr1 null rats (Figure 6, H–N). These findings further demonstrated the absence of any residual ESR1 protein or ESR1 activity in homozygous Esr1 mutant uteri.
Collectively, our characterization indicates that the Δ482 Esr1 mutation yields a rat with a null phenotype. These animals fail to express a functional ESR1 protein and exhibit prominent abnormalities in postnatal growth, fertility, development of male and female reproductive tracts, and the capacity to respond to E2.
Discussion
The purpose of this research was to expand the available model systems for studying estrogen action. More specifically, efforts were directed toward addressing a need to develop genetic models for estrogen action in species other than the mouse. Such tools are needed because of the breadth of physiological and pathological systems affected by estrogen (1, 2) and key differences in the physiology of the mouse vs other species, including the human (12, 14). The size of the mouse is also a potential limitation for some types of physiological monitoring, behavioral testing, brain mapping, repeated blood sampling, and toxicological analyses (12, 14). Furthermore, concepts are strengthened when investigated in multiple species. We responded to the need and successfully generated a rat model possessing an Esr1-null mutation.
In our rat mutagenesis strategy, we targeted exon 3 of the Esr1 gene. Exon 3 encodes a portion of the ESR1 DNA binding domain and has also been targeted in the mouse for the purpose of generating conditional Esr1-knockout models (10, 33–38). These rat and mouse exon 3 mutations yield a similar alteration in transcript processing. Exon 3 deletions result in aberrant splicing of exons 2 and 4, leading to a nucleotide frameshift and the generation of premature stop codons, which compromise expression of the ESR1 protein and uterine responsiveness to E2 (35, 37, 38) (present study). Furthermore, disruption of ESR1 signaling caused infertility in both male and female rats. Mutant males exhibited gonadal pathologies characterized by distension and atrophic changes in seminiferous tubules that disrupted spermatogenesis, whereas mutant females possessed polycystic ovaries and hypoplastic uteri. The phenotype of the Esr1-null rat model described in this report is similar to mutant mouse models that targeted the third exon of the Esr1 gene (10, 35, 37–39). These phenotypic similarities between rat and mouse Esr1-null models contribute to the validation of the rat Esr1-null model. Potential species differences in postnatal growth and serum gonadotropin responses to ESR1 deficiency in the rat (present study) vs those previously reported in the mouse were noted (37, 39). At this juncture, comparisons of phenotypes between rat and mouse Esr1-knockout models are premature and potentially problematic. Much of the existing literature on ESR1 action using mouse models was generated with a hypomorphic Esr1 mutation (9, 40), which precludes comparison with our null rat model. Additionally, phenotypic analysis for mouse Esr1-null models was performed on inbred C57BL/6 mice (10, 35, 37–39), whereas our characterization of the Esr1-null rat used the Holtzman Sprague-Dawley outbred rat. Thus, we cannot effectively determine whether apparent differences between phenotypes of our rat Esr1-null model and the previously characterized mouse Esr1-null models are attributed to strain differences within a species or whether they represent true species differences. Resolution of these issues will require additional experimentation.
The mutant rat model described in this report was generated using ZFN-mediated genome editing. Several studies have used ZFNs to efficiently create mutant rat models (15, 20–28). Recently, TALEN and CRISPR/Cas9 genome editing systems have also been shown to be effective in rat mutagenesis (16, 17). Based on these observations, it is evident that genome editing is the preferred method for mutagenesis of the rat. These technologies have been extended to other species, including the rabbit, sheep, and pig (41–43). The impact of these achievements is profound and represents a paradigm shift. Thus, it is no longer necessary to adapt research to the mouse for the purpose of creating genetic models, and instead, the most appropriate species can be genetically manipulated and selected as a model system based on its intrinsic scientific merits.
In conclusion, the Esr1-null rat model described in this report represents a valuable new experimental tool for a wide range of research efforts directed at investigating the physiology and pathophysiology of estrogen action.
Acknowledgments
We thank Shondra Miller (Washington University, St Louis, MO); Howard Jacob, Aron Guerts, Melinda Dwinnell, and Hartmut Weiler (Medical College of Wisconsin, Milwaukee, WI) for their expert advice in using ZFNs to generate mutant rat models; and Fariba Behbod (University of Kansas Medical Center) for guidance in the preparation and staining of rat mammary gland whole mounts.
This work was supported by a pilot grant from the University of Kansas School of Medicine and from National Institutes of Health grants (HD066406, OD01478, and P20GM104936). P.D. was supported by a Lalor Foundation Fellowship. K.K. was supported by fellowships from the American Heart Association and the Japan Society for the Promotion of Science. D.C. was supported by a fellowship from the American Heart Association.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- E2
- 17β-estradiol
- Esr
- estrogen receptor
- ZFN
- zinc finger nuclease.
References
- 1. Deroo BJ, Korach KS. Estrogen receptors and human disease. J Clin Invest. 2006;116:561–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Simpson ER, Misso M, Hewitt KN, et al. Estrogen–the good, the bad, and the unexpected. Endocr Rev. 2005;26:322–330 [DOI] [PubMed] [Google Scholar]
- 3. Simpson ER, Jones ME. 2006 Of mice and men: the many guises of estrogens. Ernst Schering Found Symp Proc. 2006;1:45–67 [DOI] [PubMed] [Google Scholar]
- 4. Adashi EY. Endocrinology of the ovary. Hum Reprod. 1994;9:815–827 [DOI] [PubMed] [Google Scholar]
- 5. Simpson ER, Clyne C, Speed C, Rubin G, Bulun S. Tissue-specific estrogen biosynthesis and metabolism. Ann N Y Acad Sci. 2001;949:58–67 [DOI] [PubMed] [Google Scholar]
- 6. Simpson ER. Sources of estrogen and their importance. J Steroid Biochem Mol Biol. 2003;86:225–230 [DOI] [PubMed] [Google Scholar]
- 7. Zhao C, Dahlman-Wright K, Gustafsson JA. Estrogen signaling via estrogen receptor β. J Biol Chem. 2010;285:39575–39579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Burns KA, Korach KS. Estrogen receptors and human disease: an update. Arch Toxicol. 2012;86:1491–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci U S A. 1993;90:11162–11166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. 2000 Effect of single and compound knockouts of estrogen receptors α (ERα) and β (ERβ) on mouse reproductive phenotypes. Development. 2000;127:4277–4291 [DOI] [PubMed] [Google Scholar]
- 11. Krege JH, Hodgin JB, Couse JF, et al. Generation and reproductive phenotypes of mice lacking estrogen receptor β. Proc Natl Acad Sci U S A. 1998;95:15677–15682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jacob HJ, Kwitek AE. Rat genetics: attaching physiology and pharmacology to the genome. Nat Rev Genet. 2002;3:33–42 [DOI] [PubMed] [Google Scholar]
- 13. Abbott A. Return of the rat. Nature. 2009;460:788. [DOI] [PubMed] [Google Scholar]
- 14. Dow LE, Lowe SW. Life in the fast lane: mammalian disease models in the genomics era. Cell. 2012;148:1099–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Geurts AM, Cost GJ, Freyvert Y, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325:433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tesson L, Usal C, Ménoret S, et al. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol. 2011;29:695–696 [DOI] [PubMed] [Google Scholar]
- 17. Li D, Qiu Z, Shao Y, et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol. 2013;31:681–683 [DOI] [PubMed] [Google Scholar]
- 18. Izsvák Z, Fröhlich J, Grabundzija I, et al. Generating knockout rats by transposon mutagenesis in spermatogonial stem cells. Nat Methods. 2010;7:443–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tong C, Li P, Wu NL, Yan Y, Ying QL. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature. 2010;467:211–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mashimo T, Takizawa A, Voigt B, et al. Generation of knockout rats with X-linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One. 2010;5:e8870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moreno C, Hoffman M, Stodola TJ, et al. Creation and characterization of a renin knockout rat. Hypertension. 2011;57:614–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chu X, Zhang Z, Yabut J, et al. Characterization of multidrug resistance 1a/P-glycoprotein knockout rats generated by zinc finger nucleases. Mol Pharmacol. 2012;81:220–227 [DOI] [PubMed] [Google Scholar]
- 23. Feng D, Yang C, Geurts AM, et al. Increased expression of NAD(P)H oxidase subunit p67(phox) in the renal medulla contributes to excess oxidative stress and salt-sensitive hypertension. Cell Metab. 2012;15:201–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gopalakrishnan K, Kumarasamy S, Abdul-Majeed S, et al. Targeted disruption of Adamts16 gene in a rat genetic model of hypertension. Proc Natl Acad Sci U S A. 2012;109:20555–20559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kawamata M, Ochiya T. Two distinct knockout approaches highlight a critical role for p53 in rat development. Sci Rep. 2012;2:945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McCoy A, Besch-Williford CL, Franklin CL, Weinstein EJ, Cui X. Creation and preliminary characterization of a Tp53 knockout rat. Dis Model Mech. 2013;6:269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vaira S, Yang C, McCoy A, et al. Creation and preliminary characterization of a leptin knockout rat. Endocrinology. 2012;153:5622–5628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brown AJ, Fisher DA, Kouranova E, et al. Whole-rat conditional gene knockout via genome editing. Nat Methods. 2013;10:638–640 [DOI] [PubMed] [Google Scholar]
- 29. Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93:1156–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Urnov FD, Miller JC, Lee YL, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651 [DOI] [PubMed] [Google Scholar]
- 31. Terranova PF, Garza F. Relationship between the preovulatory luteinizing hormone (LH) surge and androstenedione synthesis of preantral follicles in the cyclic hamster: detection by in vitro responses to LH. Biol Reprod. 1983;29:630–636 [DOI] [PubMed] [Google Scholar]
- 32. Kwekel JC, Burgoon LD, Burt JW, Harkema JR, Zacharewski TR. A cross-species analysis of the rodent uterotrophic program: elucidation of conserved responses and targets of estrogen signaling. Physiol Genomics. 2005;23:327–342 [DOI] [PubMed] [Google Scholar]
- 33. Wintermantel TM, Campbell RE, Porteous R, et al. Definition of estrogen receptor pathway critical for estrogen positive feedback to gonadotropin-releasing hormone neurons and fertility. Neuron. 2006;52:271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Feng Y, Manka D, Wagner KU, Khan SA. Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc Natl Acad Sci U S A. 2007;104:14718–14723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen M, Wolfe A, Wang X, Chang C, Yeh S, Radovick S. Generation and characterization of a complete null estrogen receptor alpha mouse using Cre/LoxP technology. Mol Cell Biochem. 2009;321:145–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martin-Millan M, Almeida M, Ambrogini E, et al. The estrogen receptor-alpha in osteoclasts mediates the protective effects of estrogens on cancellous but not cortical bone. Mol Endocrinol. 2010;24:323–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hewitt SC, Kissling GE, Fieselman KE, Jayes FL, Gerrish KE, Korach KS. Biological and biochemical consequences of global deletion of exon 3 from the ER alpha gene. FASEB J. 2010;24:4660–4667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Antonson P, Omoto Y, Humire P, Gustafsson JÅ. Generation of ERalpha-floxed and knockout mice using the Cre/LoxP system. Biochem Biophys Res Commun. 2012;424:710–716 [DOI] [PubMed] [Google Scholar]
- 39. Goulding EH, Hewitt SC, Nakamura N, Hamilton K, Korach KS, Eddy EM. Ex3αERKO male infertility phenotype recapitulates the αERKO male phenotype. J Endocrinol. 2010;207:281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Couse JF, Curtis SW, Washburn TF, et al. Analysis of transcription and estrogen insensitivity in the female mouse after targeted disruption of the estrogen receptor gene. Mol Endocrinol. 1995;9:1441–1454 [DOI] [PubMed] [Google Scholar]
- 41. Duranthon V, Beaujean N, Brunner M, et al. On the emerging role of rabbit as human disease model and the instrumental role of novel transgenic tools. Transgenic Res. 2012;21:699–713 [DOI] [PubMed] [Google Scholar]
- 42. Lillico SG, Proudfoot C, Carlson DF, et al. Live pigs produced from genome edited zygotes. Sci Rep. 2013;3:2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang C, Wang L, Ren G, et al. Targeted disruption of the sheep MSTN gene by engineered zinc-finger nucleases. Mol Biol Rep. 2014;41:209–215 [DOI] [PubMed] [Google Scholar]