

Abstract
Inflammation and extracellular matrix (ECM) remodeling are important components regulating the response of the left ventricle (LV) to myocardial infarction (MI). Significant cellular and molecular level contributors can be identified by analyzing data acquired through high-throughput genomic and proteomic technologies that provide expression levels for thousands of genes and proteins. Large scale data provide both temporal and spatial information that need to be analyzed and interpreted using systems biology approaches in order to integrate this information into dynamic models that predict and explain mechanisms of cardiac healing post-MI. In this review, we summarize the systems biology approaches needed to computationally simulate post-MI remodeling, including data acquisition, data analysis for biomarker classification and identification, data integration to build dynamic models, and data interpretation for biological functions. An example for applying a systems biology approach to ECM remodeling is presented as a reference illustration.
Keywords: mathematical modeling, differential equations, biclustering, myocardial infarction, matrix metalloproteinase, LV remodeling, review
CARDIAC REPAIR POST-MYOCARDIAL INFARCTION: THE PROBLEM INTRODUCED
Following the onset of myocardial infarction (MI), the left ventricle (LV) undergoes a series of molecular, cellular, and extracellular matrix alterations that combined are known as LV remodeling. LV remodeling includes four overlapping phases: cardiomyocyte death, an acute inflammatory response, granulation tissue formation, and scar formation.1 Irreversible cardiomyocyte death begins after approximately 20 min of ischemia, and myocytes loss has been attributed to necrosis, apoptosis, and other cell death mechanisms. Cardiomyocyte death releases complement C5a, which stimulates the inflammatory response and begins the wound healing cascade.2 Figure 1 shows the time course of cardiac healing and the processes involved at each time period.
Figure 1.
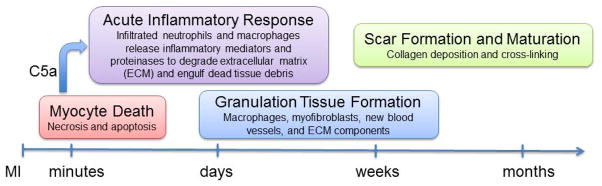
The time course of cardiac healing after myocardial infarction (MI), demonstrating the overlap of processes involved at each time period. About 20 min following MI, myocytes undergo irreversible necrosis and/or apoptosis to induce complement C5a release and initiates an acute inflammation. During the acute response period, infiltrated neutrophils and macrophages release inflammatory mediators and proteinases to degrade extracellular matrix (ECM) and engulf dead tissue debris. Thereafter, granulation tissue is formed, characterized by the presence of macrophages, myofibroblasts, new blood vessels, and ECM components. Eventually, a scar enriched in collagen forms and matures.
An early increase in cytokines and chemokines stimulates neutrophil infiltration into the ischemic area, making neutrophils the first responder leukocyte. Neutrophils release inflammatory mediators and proteinases to degrade ECM and provide chemotaxis signals for subsequent macrophage infiltration.3 During the initial macrophage response, classical M1 pro-inflammatory macrophages prevail and facilitate inflammation and ECM degradation. Subsequently, macrophages convert to an alternative M2 anti-inflammatory subtype that promote resolution of inflammation, ECM synthesis, and scar formation.4, 5 A timely switch and dynamic balance between M1 and M2 macrophages is believed to be required to promote appropriate cardiac repair.
Granulation tissue is formed in the infarct area between 2 and 14 days after MI and is characterized by the enhanced presence of macrophages, myofibroblasts, new blood vessels, and ECM components. At this stage, myofibroblasts in both the infarct and remote non-infarct regions produce abundant ECM, with the myofibroblasts in the infarct region being more active than those in the non-infarct region.6 In the final stages of wound healing, granulation tissue matures into a collagen-rich, cross-linked scar that spans the entire infarct region and infiltrates into the remote region. The time period for completion of each phase varies depending on the species, with rodents showing an accelerated wound repair and healing compared to larger animal models and humans.7
During the different phases post-MI, inflammation and ECM turnover play significant roles in LV remodeling. Inflammation is a bi-directional response, with both necessary and deleterious roles in the repair process. Appropriate inflammation fosters removal of tissue and cell debris to form a stable scar. Excessive and prolonged inflammation or insufficient inflammation both hinder normal cardiac repair by stimulating continual tissue destruction and infarct expansion.8
ECM provides the scaffolding structural support for granulation tissue formation. The balance between ECM degradation and synthesis is critical for stable scar formation. Excessive ECM accumulation can increase wall stiffness and stimulate the development of diastolic dysfunction. On the other hand, insufficient or improper ECM deposition can promote cardiac rupture and maladaptive cardiac remodeling.5, 9 The ECM environment plays a key role in scar formation by transducing mechanical, electrical, and chemical signaling pathways.10 The ECM coordinates responses between myocytes in the viable non-infarcted remote region and neutrophils, macrophages, endothelial cells, and fibroblasts in the infarct region. Therefore, investigating the mechanisms of inflammation and ECM remodeling post-MI will help us to better understand MI responses that can stimulate adverse LV remodeling and lead to the development of congestive heart failure.
As described above, one critical feature of post-MI LV remodeling is the importance of the time course of inflammation and matrix turnover. For example, early neutrophil recruitment provides a key signal for subsequent macrophage trafficking and polarization, which coordinate myofibroblast phenotype and function. Due to different roles of these cellular and molecular players in MI response, significance of the players varies with time and temporal profiles of these players carry such information for us to discover. With this consideration, time course data are necessary to identify key markers and their underlying regulatory mechanisms. To address this need, systems biology provides useful techniques including temporal clustering for identification of key markers and differential equation models to characterize and interpret regulatory mechanisms with time course data.
Challenges of systems biology approaches using –omics technologies
During the last three decades, advancements in molecular and cellular biology have yielded dramatic progress in biomarker discovery and functional characterization that improve our understanding of mechanisms of LV remodeling.11 There is a vast amount of experimental data deposited in public databases with respect to gene and protein expression, cellular responses, and tissue remodeling post-MI. These data cover temporal as well as the spatial response between infarct and remote non-infarcted myocardium.12 The recent emergence of genomics and proteomics datasets provides a chance for researchers to examine the whole system instead of focusing on only one or several molecules.13, 14 The generation of these very large datasets has also generated issues on how to best analyze and extract useful information from the data.
To address this need, systems biology approaches have been proposed and developed to elucidate the mechanisms of biological response that can be applied to our example of post-MI LV remodeling.15 Systems biology is the conceptual framework that analyzes and integrates complex and highly diverse information acquired largely from isolated high-throughput -omics datasets.16 This approach is a transition from the molecular cell biology method that studies each data set individually to a more complete systemic analysis and integration to interpret the dynamics of the entire biological system. According to the different levels of the organism being studied, systems biology has adopted different processing methods. It incorporates all -omics technologies, including genomics, proteomics (and its subspecialties, such as matridomics and degradomics), matabolomics, and interactomics with signal processing algorithms. It also allows characterization of the supramolecular networks and functional modules that represent the most essential aspects of cell organization and physiology with mathematical modeling and graphical networks.12
One problem encountered when applying systems biology approaches is how to handle very large datasets. The advanced high-throughput –omics technology can offer expression levels for >1,000 genes or proteins in one experiment. Additionally, such experiments might be quickly repeated many times for temporal progression information, in different organs from different species for comparative information, or with different dosages of interventions, which increases the dimensionality of the data compared to previous snap-shot data.17–19 In addition to this big data set problem, integrating datasets collected from gene, protein, and cellular components to depict whole spectra of the dynamic progression is another challenge for systems biology generally and the cardiac research field specifically.
In this review, we will summarize current methods of systems biology for data acquisition, analysis, integration, and interpretation to understand cardiac inflammation and ECM deposition in the setting of MI. A systemic flow chart of this approach is presented in Figure 2.
Figure 2.
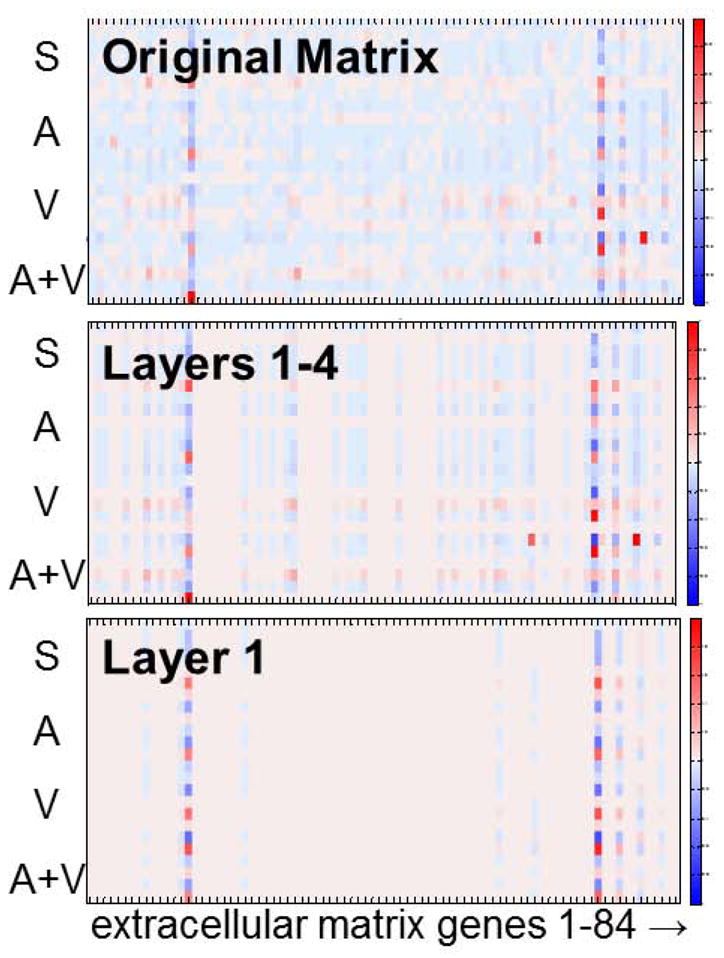
Comparison between the gene expression levels of 84 extracellular matrix (ECM) genes after myocardial infarction (MI) in the infarct region of saline treated mice and 3 treatment groups (aliskiren- A; valsartan- V; and combination-A+V) for the 4 most significant layers obtained by SSVD biclustering. The biclustering method was used to find potential ECM biomarkers of cardiac remodeling after MI. The original matrix is shown at the top, the reconstructed matrix containing the four most significant layers is shown in the middle, and the reconstructed matrix containing layer 1 is shown at the bottom. The highlighted columns represent the genes identified as potential ECM biomarkers. In the color legend, the red color means that a gene expression is up-regulated, while dark blue represents down-regulation of a gene expression with respect to the expression levels at day 0.
ACQUIRING LARGE DATASETS TO OBTAIN SUFFICIENT INFORMATION
Recent technologies such as next generation sequencing techniques can produce huge amounts of genomic data in less than a week, while assembling the first human genome data took about ten years and the cooperation of >200 research groups worldwide.17 As another example, proteomics studies frequently utilize multidimensional techniques for data acquisition, including different approaches for separation of proteins (1D vs. 2D polyacrylamide gel electrophoresis in buffers of differing solubility), several techniques for separating peptides (1D liquid chromatography (LC) vs. 2D LC vs. isoelectric focusing), and a variety of methods for mass spectrometry (MS) such as matrix-assisted laser desorption/ionization, electrospray ionization, and ion trap or time of flight analysis to generate MS and LC-tandem MS data.18 Measurement of a large number of biomarkers (e.g., covering the whole genome) of a single cell for several samples over a period of time is also an example that shows the high dimensionality possible in data acquisition.19
Using arrays to examine the molecular level
Gene and protein arrays provide a relatively high-throughput method for examining multiple molecules simultaneously, and it is useful to segregate molecules based on their potential involvement according to patterns of change in levels. Genes or proteins that do not increase or decrease post-MI are assumed to maintain a steady state during the response, assuming that the right time window was sampled. Both inflammatory and ECM gene and protein arrays are commercially available (e.g., from Qiagen, Biorad, and R&D). While it is well accepted that mRNA levels do not always reflect protein levels, measuring multiple gene levels is technically easier than measuring multiple protein levels. Additionally, measuring mRNA or protein levels does not give information on protein quality. This is an especially important consideration for ECM, as ECM proteins generally undergo at least one post-translational modification. Typical modifications include glycosylation and proteolytic processing. Therefore, proteomics data will be much more important for studying the dynamics of ECM remodeling for protein quantification. Additionally, MS-based proteomics has the capability of evaluating proteins and their complex interactions. Techniques to obtain proteomics data for ECM specifically are reviewed and summarized below.
Using matridomics to examine the extracellular matrix level
Matridomics is defined as the comprehensive study of all ECM protein components expressed in a tissue at the time of evaluation. This proteomic array technique describes the global, integrated changes of ECM in a given condition, which has several advantages over transcriptomics or traditional one target studies.20 Matridomics provides direct information on ECM protein quantity and quality. Barallobre-Barreiro and colleagues have used an innovative proteomic approach to analyze the cardiac ECM in a porcine model of ischemia/reperfusion.21 The detection and identification of the relatively low abundant cardiac ECM is a challenge that requires a sequential extraction procedure to enrich for ECM proteins. Using such an approach led to the identification of 139 cardiac ECM proteins by liquid chromatography/mass spectrometry, and 15 of these ECM proteins had not been previously shown to be involved in cardiac remodeling. A comparison between the ischemic region and border zone revealed much higher proteins in the infarct area but similar mRNA levels in both regions, which highlights the disparity between mRNA and protein levels. Further, the ECM proteins delineated a signature of early- and late-phase LV remodeling, with transforming growth factor β1 (TGFβ1) signaling at the core of the interaction network. Importantly, these novel cardiac ECM proteins identified by proteomics approach in the pig were also validated in human samples.
Our team has expanded on this approach by recently exploring a decellularization technique where the mouse LV tissue was extracted into fractions using three extraction buffers of increasing solubility potential, which we named the Texas-3 step protocol.22 The LV tissue was first incubated in a neutral low salt buffer with proteinase inhibitors to extract the most easily soluble components. The LV tissue was then decellularized with 1% sodium dodecyl sulfate with proteinase inhibitors to collect soluble cellular proteins, including the large amount of mitochondrial proteins present in LV tissue. As a final step, the decellularized tissue was subjected to acid extraction, deglycosylation, and solubilization using a 4 M GnHCl and 50 mM sodium acetate buffer with proteinase inhibitors to collect the insoluble ECM fraction. This protocol allows qualification and quantification of ECM components in tissue samples by removing highly abundant cellular and mitochondrial proteins.22
Using degradomics to examine the matrix metalloproteinase level
Degradomics is defined as a proteomic approach to catalogue all proteinases, inhibitors, and substrates in a tissue at the given time of evaluation.23 Multiple matrix metalloproteinases (MMPs) and the tissue inhibitors of metalloproteinases (TIMPs) have been shown to be involved in post-MI cardiac remodeling by regulating ECM turnover, inflammation, apoptosis, and angiogenesis.5, 24 Accumulating evidence suggests that ECM fragments cleaved by proteinases, the matricryptins, have biological effects and actively regulate angiogenesis, inflammation, and fibrosis.10, 25 Matricryptins are active ECM fragments that contain a cryptic domain that is only exposed after conformational changes of the parent ECM.26 The presence of cryptic sites may provide key signals to regulate cell migration, proliferation, differentiation, morphogenesis, survival, ECM turnover, angiogenesis, and tissue repair by binding to corresponding receptors (e.g., integrins).10, 27
Collagen fragments generated due to MMP-9 cleavage have been shown to inhibit angiogenesis and stimulate MMP-9 expression, which forms a positive feedback loop to maintain MMP-9 levels.28, 29 Collagen IV can be cleaved by MMPs to generate both proangiogenic and antiangiogenic fragments.30 In addition, matricryptins mediate the inflammatory response by modulating chemotactic activity for inflammatory cells, amplifying phagocytic function, activate immune responses, and altering the gene expression profiles of leukocytes.31
Our group has identified in vivo candidate substrates for MMP-7 and MMP-9 in 7 day post-MI infarct tissue using 2-dimensional gel electrophoresis analysis and mass spectrometry based approaches.32, 33 In the MMP-7 null mice, the infarct area showed a lower intensity of spots that were identified to include fibronectin (Fn1) and tenascin-C (TnC). In vitro cleavage assays verified that Fn1 and TnC fragments were generated by MMP-7. Further, infusion of exogenous recombinant MMP-7 restored the production of Fn1 and TnC fragments in MMP-7 null mice, confirming that Fn1 and TnC are in vivo MMP-7 substrates.32 This was the first report to identify Fn1 and TnC as in vivo MMP-7 substrates using a proteomics approach.
Using a similar proteomics approach, we found Fn1 could also be cleaved by MMP-9 in the MI setting.33 In these studies, using only infarct tissue provided a way to naturally focus on ECM, because ECM proteins are enriched in the scar tissue at day 7 post-MI. Under normal conditions, mitochondria accounts for >30% of the myocyte volume, and myocyte comprise >90% of the LV volume. Compared to these levels, ECM components are in very low abundance and harder to analyze by mass spectrometry because of the noise contributed by mitochondria and other intracellular components. One goal of our ongoing projects is to investigate ways to further enhance ECM representation in our analyses.
Using genomics and proteomics deposition databases
Tremendous amounts of gene expression data have been deposited in public databases. Currently, a query of NCBI Gene Expression Omnibus (GEO) leads to public data repositories including 11,752 platforms, 61,727 samples, 39,713 series, and 3341 datasets- as of July 15, 2013. A total of over 1 million microarray results were available by the year of 2008, and the number of available microarrays doubles every 2–3 years. A summary of the data deposition resources available online is provided in Table 1.
Table 1.
Public resources of available data and tools for systems biological approaches
| Database | Specification | Website |
|---|---|---|
| Genomics | GEO | http://www.ncbi.nlm.nih.gov/geo/ |
| Proteomics | UniProt | http://www.uniprot.org |
| PRIDE | http://www.ebi.ac.uk/pride/ | |
| GPM | http://gpmdb.thegpm.org | |
| DIP | http://dip.doembi.ucla.edu/dip/Main.cgi | |
| MIPS | http://mips.helmholtzmuenchen.de/proj/ppi/ | |
| HPRD | http://www.hprd.org | |
| BioGRID | http://thebiogrid.org | |
| IntAct | http://www.ebi.ac.uk/intact/ | |
| HomoMINT | http://mint.bio.uniroma2.it/HomoMINT/Welcome.do | |
| Data integration | Mathematical models | http://bioeng.washington.edu/Models/). |
| Data interpretation | DAVID | http://david.abcc.ncifcrf.gov |
| iProClass | http://pir.georgetown.edu/pirwww/dbinfo/iproclass.shtml |
GEO- Gene Expression Omnibus; UniProt-Universal Protein Resource; PRIDE-Proteomics Identifications Database; GPM-The Global Proteome Machine; DIP-Database of Interacting Proteins; MIPS-Mammalian Protein-Protein Interaction Database; HPRD-Human Protein Reference Database; BioGRID-Biological General Repository for Interaction Datasets; IntAct-Mollecular Interaction Database; HomoMINT- Human Protein Interactions in the MINT Database); DAVID-The Database for Annotation, Visualization and Integrated Discovery; PIR-Protein Information Resource.
Similar to genomics databases, several worldwide organizations have provided online proteomics database repositories, including the PRoteomics IDEntifications (PRIDE) and Global Proteome Machine (GPM) databases. PRIDE is a public, user-populated proteomics data repository.34, 35 Users can upload, download, and view raw data generated by mass spectrometry proteomics experiments, including raw spectral data, peptides, protein identifications, and associated statistics through a free web interface as listed in Table 1. GPM gains the advantage of allowing researchers to use its proteomics data and tools to interrogate a number of proteomes.36 Currently, there is no specific data service focusing on cardiovascular research proteomics, and such a resource would significantly benefit the field.
It is worth mentioning that the protein-protein interaction databases such as Database of Interacting Proteins (DIP), mammalian protein-protein interaction database (MIPS), Human Protein Reference Database (HPRD), Biological General Repository for Interaction Datasets (BioGRID), IntAct database, and HomoMINT database, among others, provide information on a total of >70,000 proteins and 330,000 interactions.37–42 The reported data represent gene and protein expression profiles from different species under varying experimental conditions. Therefore, there is an urgent need to systemically integrate and analyze such data to elucidate the underlying regulatory mechanisms.
PERFORMING DATA ANALYSIS TO HARNESS THE INFORMATION
Before data analysis can be performed, the data must be cleaned to remove possible sources of noise, such as those coming from experimental design errors, measurement noise, and technical errors. These noises overlap with the inherited individual differences of biological processes, leading to a difficulty in determining true measurements. Thus, data cleaning is needed to filter noises and control the quality of data before any further investigation of the biological system can be undertaken. Several statistical methods have been used for detecting and correcting measurement outliers. These methods include finding the average or median of the data, computing the p-value, using boxplots for visualizing outliers, and using boot strapping methods to assign measures of accuracy to sample estimates.43
Data analysis or mining is defined as an automatic process of retrieving and discovering important and useful information in a large data set. Many computational methods have been proposed to find new significant biomarkers with relevant biological meaning. These techniques include unsupervised machine learning techniques such as clustering and principal component analysis (PCA), as well as supervised learning approaches that utilize the prior knowledge of the system under investigation such as support vector machine (SVM).
Clustering methods are generally used to classify objects of a large population into smaller groups called clusters. The similarity between members in each cluster is higher than that among members of different clusters. Clustering is often applied to biological studies to find gene or protein co-regulatory partners. The underlying assumption is that genes or proteins sharing similar behavior may regulate a biological process together and can be classified as one cluster. This approach has also been used to find sets of biomarkers associated with a specific function.44 Clustering methods are useful for predicting which group a biomarker belongs to and what functions that biomarker has. As more and more data are accumulated, researchers face the problem of comparing and integrating data collected under different attributes such as treatment dosages, species, and genotypes. This has led to biclustering methods that can identify not only significant markers but also the contribution of each attribution.
We have used a biclustering method to find the significant biomarkers of MI that could differentiate treatment strategies.45 This biclustering algorithm was performed on gene expression levels in the LV of mice before MI (control) and 28 days after MI with saline, aliskiren, valsartan, or a combination of both drugs treatment that was started at 3h post-MI (Figure 3). In another study, the differentially expressed genes were classified based on their functions, and >200 genes were identified as being related to MI.46
Figure 3.

Flow chart of data acquisition, analysis, integration, and interpretation used for systems biology approaches.
PCA is a mathematical method that converts a set of possibly related observed variables into a set of linearly uncorrelated variables by orthogonal transformation. PCA is usually used to reduce the dimensionality of expression data that will eventually identify groups of genes that have similar characteristics.47 Barallobre and colleagues used a PCA approach to identify the 20 most significantly expressed proteins in the ECM that were related to MI.21 These applications provide a promising direction to integrate systems biology approaches into experimental research to focus on hypothesis generation and data analysis.
In addition to clustering approaches, SVM is another popular classification method that was originally developed by Vapnik and Cortes in 1995.48 The SVM algorithm can predict which group the input belongs to by maximizing the margin between support points and using nonlinear mapping based on the model obtained from the training dataset. It has been widely applied to breast cancer diagnosis, protein sequencing, and ECG signal analysis for cardiovascular research on arrhythmias.49–52 To date, no application of SVM on inflammation and ECM remodeling has been conducted.
INTEGRATING DATA TO DEVELOP INTERPRETATIONS
Data analysis can elucidate the relation of data based on correlation analysis, an approach familiar to engineers, bioinformaticians, and biologists. More advanced integration, however, is required to interpret the spatiotemporal dependence embedded in the data. Furthermore, clear biological meaning of the data analysis should be presented to bridge the knowledge gap between biologists and bioinformaticians. Different integration schemes including differential equation and graphical network modeling have been proposed to integrate experimental data.
Mathematical modeling for temporal dynamics using differential equations
Ordinary differential equations (ODE) and partial differential equations (PDE) have been applied to model the temporal and spatial changes of biological and physical variables in a continuous format.53–55 Due to the complexity of PDE solving and modeling techniques, we use ODE to illustrate the modeling method here. ODE-based algorithms are able to infer the nonlinear dynamic nature of high dimensional regulatory mechanisms and predict the behavior of a system based on the constructed model.53, 54 There are two approaches in studying regulatory relationships characterized by physical and influence methodologies.56–59 In the physical approach, exact physical interactions between molecules such as transcription factors and their promoters are investigated and the impact of other factors in the regulatory mechanism is ignored. In the influence approach, regulatory interactions among molecules are studied such that every regulation is considered as an input and output. The regulatory process is not controlled by real interactive molecular bindings, although the model sheds light on hidden indirect associations among molecules. The general format of ODEs for this approach is written as follows:
| (1) |
where Xi denotes molecular concentration of the ith gene or protein at time t, with time-derivative term , the regulatory effect of other external stimuli such as chemical compounds or genetic perturbation are illustrated by Y, and the network dynamic model is characterized by fi(.), which reveals dynamic interactions among all molecule Xis. θ is represented as the model-parameter vector. To simplify the ODE equation (1) and reduce the computation load, the network dynamic model is approximated such that the changing rate of molecular concentration of the ith gene or protein is affected by weighted accumulation of all molecular expressions. The kernel of such ODE model is in linear format and represented in equation (2): 58, 59
| (2) |
The construction of the model described in equation (2) is formulated by determining associated weights, θijs, that describe the influence of molecule j on molecule i. In the presence of some prior biological knowledge about their kinetic rates, only a subset of parameters, θij, is required to be estimated. Least square and Bayesian approaches are wildly applied to estimate the unknown parameters.60, 61
The difficulties of parameter estimation lie in the fact that many parameters in the model need to be estimated, and there is a lack of data samples to estimate the parameters accurately. In this case, we need to limit the number of unknown parameters or take advantage of sparsity of such regulatory networks to reduce the dimension of the dataset.58, 62, 63 Different parameter estimation approaches with time series data have been proposed for both linearly and nonlinearly parameterized systems.54, 61, 64–68
While using such a linear format simplifies the modeling techniques, problems that arise with such a simplification approach include 1) how to accommodate the innate nonlinear dynamics of biological regulation due to multiple protein binding sites for protein interaction; and 2) how to approximate nonlinearly parameterized chemical reactions such as a hill equation with linear format.
ODEs and PDEs are deterministic mathematical model, i.e., the output of the system is determined given one set of parameters, initial condition, and input, while real biological processes demonstrate strong individual differences. With the same input, same biological processes may have different initial conditions and parameters. In most current research, variations of the system outputs are due to different initial conditions and noises. Sensitivity of parameters are examined to further simplify the differential equation model by focusing on key sensitive parameters and ignoring non-sensitive parameters in the model.69 Under such direction, bifurcation analysis has been applied to illustrate the effect of parameter variation on system outputs.70 Additionally, the effect of noises is confined within a stable system, i.e., a bounded input leads to bounded outputs. Currently, very few studies have been reported to use stochastic differential equation model to represent individual differences.71–73 There is a lack of research linking the distribution of key parameters to distribution of the output.
Many differential equation models of cardiovascular systems can be found in the website supported by the Bassingthwaighte group (http://bioeng.washington.edu/Models/). The complexity of ECM metabolism and inflammation signaling in the post-MI setting lends itself to ODE approaches. Our team has established an ODE model of cardiac remodeling post-MI to quantify the balance between the construction and destruction of ECM.74 To establish this ODE model, we first determined key variables representing ECM metabolism using our experimental results. We examined the expression of 84 ECM genes at 7 days post-MI and identified 17 differentially expressed genes (>2.5 fold and p<0.05). Among these genes, collagen I was selected as a key factor, because it is the major collagen in the myocardium. Osteopontin expression levels increased 206-fold in the infarct region compared to the control group, suggesting strong macrophage activation at day 7 post-MI. TIMP-1 expression increased 31-fold in the infarct compared to the control group, suggesting strong inhibition of proteolytic activity. Specifically, TIMP-1 inhibits MMP-9 and MMP-9 degrades collagen. Periostin gene expression level was increased 5.5-fold, and TGFβ1 gene expression level was increased 2.6-fold in the infarct sample at day 7, suggesting significant fibroblast functions post-MI. Based on these experimental results, the regulatory network of ECM metabolism includes the cellular contributions of macrophages and fibroblasts, and molecular contributions of TGFβ1, MMP-9, collagen I, and TIMP-1 as main components influencing LV remodeling.
With the determination of these variables, we further examined the regulatory relationship with in vitro experiments and published literature to determine the parameters in the ODE model. For example, we determined the fibroblast secretion rate of collagen stimulated by TGFβ1 and the macrophage infiltration rate stimulated by TGFβ1. After we determined the variables and parameters of the ODE model, we simulated the model and validated the model by comparing our computational results against temporal profiles of these variables from other research groups. Our computational predictions agree well with the observed experimental data.
Besides our group, Vovodotz and colleagues have developed several differential equation models for inflammatory responses by combining experimental and computational approaches.75 The Popel laboratory has developed theoretical model for collagen degradation by MMPs at the molecular level.76, 77 ODE models have also been widely adopted for other cardiovascular research topics in addition to ECM metabolism and inflammatory signaling. ODE models on cardiac electrophysiological from cellular level to organ level have been generated.78–81 Noble and colleagues developed a cardiac electrical activity model in 1985.82 Since then, Luo-Rudy models and Jafri-Rice-Winslow model have been reported and widely used to describe mechanisms of cardiac Ca2+ regulation in myocytes.83 All of these models illustrate the use of ODE as a promising way to interpret time course progression such as that seen in the MI response.
While differential equation models can represent biological measures, such as molecular concentration and cellular density, with respect to temporal and spatial dynamics, most models are built with influence methodologies without attempting to interpret the detailed binding information. In addition, the modeling procedure and analysis heavily depend on engineers and mathematicians due to the technical details, and this process is not very user unfriendly for typical biologists to understand or use the model. Graphical network models, including Bayesian network and protein-protein interaction (PPI) network, are new emerging representations that can improve model usage by biologists.
Using protein-protein interaction networks
A PPI network visualizes the interactions among proteins by representing each protein as a vertex and each interaction as an edge. Different measurements have been used to investigate structural properties of a network, including centrality analysis (shortest path, betweenness centrality, closeness centrality, and eccentricity) and clustering analysis (local/global clustering coefficient for community detection, average clustering coefficient with respect to degree). The shortest path is a pairwise concept that defines the minimum number of edges required to move from one vertex to another. Betweenness centrality measures the frequency of appearance of a vertex or link in the set of all shortest paths.84, 85 High betweenness centrality indicates that a vertex or edge is more frequently used to flow the information in the network. Closeness centrality measures how many steps are required to access every other vertex from a given vertex.84 The vertex with the smallest closeness centrality requires the least number of edges to spread information to other reachable vertices in the network. Eccentricity of a vertex measured the shortest path distance from the farthest vertex in the graph.86 Local clustering coefficient describes the connectivity of the neighbors of a vertex.87 A higher clustering coefficient means the neighbors are densely connected to each other. A tremendous amount of recent research has investigated the structural properties of PPI networks using these network concepts.88–90 However, studies are needed to identify key proteins of PPI network using only the structural properties.
Key proteins in a PPI network can be identified with enrichment analysis from DAVID with respect to Gene Ontology (GO) for functional linkage.91, 92 GO is a controlled vocabulary of terms that characterizes gene products in terms of their cellular components, biological processes, and molecular functions in different species. Significant effort has been contributed to such interpretation and available public resources such as Uniprot (http://www.uniprot.org/) and iProClass (http://proteininformationresource.org/).93–97
When the number of proteins studied in a disease is small, it is possible to predict and validate functionality of each important protein. When many proteins are involved in the process, finding the most useful proteins as biomarkers is tedious and hard if not impossible. LV remodeling is a highly complicated process that includes vast amounts of protein changes with respect to signaling pathways, inflammatory response, and angiogenesis, all of which are coordinated by ECM remodeling.
Targeting the MI-specific proteins provides the opportunity to study MI responses in detail and propose preventive or treatment strategies. Our team has established a PPI network based on a set of known seed proteins and compared it with 100,000 randomly generated networks to demonstrate the specificity of the MI-specific network.98 Azuaje and colleagues have established a PPI network focusing on the inflammatory response post-MI to find the new prognostic biomarkers for LV dysfunction.28, 99 They identified inflammatory proteins, including TNF receptor-associated factor 2, SH3 domain-containing-binding protein 1, and Ubiquitin C protein family members. Their associations of these proteins with clinical outcomes post-MI provided the functional characterization of the network.
Ren and colleagues provided a NetCAD platform to construct a PPI network for coronary artery disease. Their PPI network provides physicians and scientists important information about the molecular mechanisms and potential targets for therapies of coronary artery disease.100 There have also been several groups that studied the effects of drugs on MI outcomes using PPI networks.101–103 In a recent study, we introduced a multiple-delayed linear regression model to estimate the time delay between the transcription factors and their targeting genes post-MI.104 A regulatory network was generated with different time delays where genes with high degree of connectivity were identified as potential biomarkers for MI, including specificity protein 1 (Sp1), histocompatibility 2-K1 (H2-K1), v-rel reticuloendotheliosis viral oncogene homolog A (Rela), nuclear factor NF-kappa-B p105 subunit (Nfkb1), and interleukin 18 (Il18).
BIOMARKER IDENTIFICATION: A CASE STUDY
We applied a sparse singular value decomposition (SSVD) biclustering algorithm to analyze gene expression data for significant biomarkers post-MI under different treatments methods (Figure 2).45
Data acquisition (Figure 3A and B): data includes expression of 84 ECM genes in the LV infarct region of C57BL/6J wild type (WT, n=30) mice. The mice were divided into 5 groups; control group at day 0 (number = 6) and 4 treatment groups at 28 days post-MI. The treatment groups include MI with saline treatment (S, number = 6), MI with Aliskiren treatment (A, 50 mg/kg/day, number = 6), MI with Valsartan treatment (V, 40 mg/kg/day, number = 6), and MI with Aliskiren and Valsartan treatment (A+V, number = 6).
Data analysis (Figure 3 C and D): We examined the p-value of gene expression to control the data quality. We then computed fold changes of genes expression at day 28 compared to its average value at day 0 and applied the SSVD biclustering algorithm. The most significant feature of the fold change data found by this algorithm is shown in Figure 3. Out of 84 ECM genes evaluated, 10 were differentially regulated in the infarct region.
Data integration (Figure 3 F): We further examined the biological functions involved with these significantly expressed genes to confirm the coherence between analytical results and biological meanings. For each gene set, we searched DAVID for enriched gene ontology (GO) terms for their associated biological processes.80 We observed 23 biological processes that are significantly involved with the 10 genes in the infarct region.
LIMITATIONS AND FUTURE DIRECTIONS
While the above approaches give promise for uncovering underlying mechanisms of LV remodeling post-MI using systems biology approaches, there are several areas where future work is needed (Table 2).
Table 2.
Future advances needed for better use of experimental and computational approaches to define post-MI LV remodeling.
| Experimental | Computational |
|---|---|
|
|
For cardiac ECM proteomic analysis, tissue solubility is a challenge that needs to be considered. Although the decellularization procedure greatly removes intracellular components and enriches ECM, not all of the ECM proteins are easily dissolved, especially high molecular weight glycoproteins, proteoglycans, and cross-linked ECM. This may mask potentially important ECM protein changes. Therefore, future studies are warranted to further improve proteomic protocols that focus on ECM.
Plasma or serum can be a valuable source of biomarkers for MI patients, and more research is needed to determine how well changes in the blood mirror changes in the LV tissue. Most studies using animal models analyze cardiac tissue to understand the molecular mechanisms of MI-induced remodeling. Indeed, tissue examination offers invaluable information on the pathogenesis. Because cardiac tissue samples cannot easily be collected in clinics to evaluate patient prognosis, however, having biomarkers that spill over into the blood to serve as surrogate indicators will be critical. The Human Proteome Organisation (HUPO) has an ongoing project to identify all proteins in the human plasma. Berhane and colleagues identified 3020 proteins in human plasma using mass spectrometry.105 Subsequently, a subset of these proteins has been annotated as cardiovascular related proteins based on the published literature. Several groups have analyzed the plasma proteomics after depletion of albumin and identified hundreds of proteins.106, 107 Given the magnitude of differences among the least and most highly expressed proteins, plasma evaluations remain challenging. Identifying which of these proteins best track with LV remodeling is the next step.
Applying systems biology approaches to achieve personalized medicine has been a hot topic over the past five years. In personalized medicine, there is a paradigm change from diagnosis and treatment of disease for an overall population to personalized monitoring and preventive medicine for individuals.108 This aim may not be achievable without gathering the whole dataset for each patient through –omics technologies, together with incorporating novel methods to find the most informative biomarkers of the disease and will likely include methods that are able to combine spatial and temporal data and integrating the relevant –omics data into more comprehensive models to predict the behavior of the biological system.109, 110 Whether we can reduce the number of variables that must be assessed for accurate model building remains to be determined.
Synthetic biology is another trend of systems biology, which builds up a biological system from its cells to the whole organ or living organism.111 In this approach, each cell is considered to be a computer that is involved in the flow of information by signaling interactions from and to their neighborhood environment. The living organism is the integration of all these modules (cells). This approach requires extensive data acquisition, big data management, and integration that is currently challenging on several levels.
CONCLUSIONS
In this review, we introduced the significance and challenges of applying systems biology approaches, using the inflammatory response and ECM remodeling following MI as an example. We summarized possible systems biology approaches to handle vast sums of data at multiple stages from acquisition, analysis, integration, and interpretation to elucidate the spectra for systemic studies. This review should provide a useful reference guide for researchers new to this area by summarizing the possible algorithms and resources available for computational modeling.
Acknowledgments
We acknowledge support from NIH/NHLBI HHSN 268201000036C (N01-HV-00244) for the San Antonio Cardiovascular Proteomics Center, R01 HL075360 and HL051971, and from the Biomedical Laboratory Research and Development Service of the Veterans Affairs Office of Research and Development Award 5I01BX000505 to MLL and from NIH/NHBLI SC2HL101430 to YFJ.
References
- 1.Matsui Y, Morimoto J, Uede T. Role of matricellular proteins in cardiac tissue remodeling after myocardial infarction. World J Biol Chem. 2010;1:69–80. doi: 10.4331/wjbc.v1.i5.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Birdsall HH, Green DM, Trial J, Youker KA, Burns AR, MacKay CR, et al. Complement C5a, TGF-β1, and MCP-1, in Sequence, Induce Migration of Monocytes Into Ischemic Canine Myocardium Within the First One to Five Hours After Reperfusion. Circulation. 1997;95:684–92. doi: 10.1161/01.cir.95.3.684. [DOI] [PubMed] [Google Scholar]
- 3.Ma Y, Yabluchanskiy A, Lindsey ML. Neutrophil roles in left ventricular remodeling following myocardial infarction. Fibrogenesis & tissue repair. 2013;6:11. doi: 10.1186/1755-1536-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Troidl C, Mollmann H, Nef H, Masseli F, Voss S, Szardien S, et al. Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. Journal of cellular and molecular medicine. 2009;13:3485–96. doi: 10.1111/j.1582-4934.2009.00707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ma Y, Halade GV, Zhang J, Ramirez TA, Levin D, Voorhees A, et al. Matrix metalloproteinase-28 deletion exacerbates cardiac dysfunction and rupture after myocardial infarction in mice by inhibiting M2 macrophage activation. Circulation research. 2013;112:675–88. doi: 10.1161/CIRCRESAHA.111.300502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Squires CE, Escobar GP, Payne JF, Leonardi RA, Goshorn DK, Sheats NJ, et al. Altered fibroblast function following myocardial infarction. Journal of Molecular and Cellular Cardiology. 2005;39:699–707. doi: 10.1016/j.yjmcc.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 7.Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, et al. Of Mice and Dogs: Species-Specific Differences in the Inflammatory Response Following Myocardial Infarction. The American journal of pathology. 2004;164:665–77. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circulation research. 2012;110:159–73. doi: 10.1161/CIRCRESAHA.111.243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsui Y, Ikesue M, Danzaki K, Morimoto J, Sato M, Tanaka S, et al. Syndecan-4 prevents cardiac rupture and dysfunction after myocardial infarction. Circulation research. 2011;108:1328–39. doi: 10.1161/CIRCRESAHA.110.235689. [DOI] [PubMed] [Google Scholar]
- 10.Ma Y, Halade GV, Lindsey ML. Extracellular matrix and fibroblast communication following myocardial infarction. Journal of cardiovascular translational research. 2012;5:848–57. doi: 10.1007/s12265-012-9398-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Benjamin IJ, Schneider MD. Learning from failure: congestive heart failure in the postgenomic age. J Clin Invest. 2005;115:495–9. doi: 10.1172/JCI200524477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Albeck JG, MacBeath G, White FM, Sorger PK, Lauffenburger DA, Gaudet S. Collecting and organizing systematic sets of protein data. Nature reviews Molecular cell biology. 2006;7:803–12. doi: 10.1038/nrm2042. [DOI] [PubMed] [Google Scholar]
- 13.Kitano H. Computational systems biology. Nature. 2002;420:206–10. doi: 10.1038/nature01254. [DOI] [PubMed] [Google Scholar]
- 14.Bruggeman FJ, Westerhoff HV. The nature of systems biology. Trends Microbiol. 2007;15:45–50. doi: 10.1016/j.tim.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 15.Adams KF. Systems biology and heart failure: concepts, methods, and potential research applications. Heart Fail Rev. 2010;15:371–98. doi: 10.1007/s10741-009-9138-x. [DOI] [PubMed] [Google Scholar]
- 16.Chipman A. All systems go. Nature. 2006;439:136–7. doi: 10.1038/439136a. [DOI] [PubMed] [Google Scholar]
- 17.Wandelt S, Rheinlander A, Bux M, Thalheim L, Haldemann B, Leser U. Data management challenges in Next Generation Sequencing. Databank Spektrum [serial on the Internet] 2012;12(3) [Google Scholar]
- 18.Thiele H, Glandorf J, Hufnagel P, Korting G, Bluggel M. Managing Proteomics Data: From Generation and Data Warehouse to Central Data Repository. Journal of Proteomics & Bioinformatics [serial on the Internet] 2008;1(9) [Google Scholar]
- 19.Swedlow JR, Lewis SE, Goldberg IG. Modelling data across labs, genomes, space and time. Nat Cell Biol. 2006;8:1190–4. doi: 10.1038/ncb1496. [DOI] [PubMed] [Google Scholar]
- 20.Patterson NL, Iyer RP, Bras LD, Li Y, Andrews TG, Aune GJ, et al. Using proteomics to uncover extracellular matrix interactions during cardiac remodeling. Proteomics Clinical applications. 2013 doi: 10.1002/prca.201200100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barallobre-Barreiro J, Didangelos A, Schoendube FA, Drozdov I, Yin X, Fernandez-Caggiano M, et al. Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation. 2012;125:789–802. doi: 10.1161/CIRCULATIONAHA.111.056952. [DOI] [PubMed] [Google Scholar]
- 22.de Castro Bras LE, Ramirez TA, Deleon-Pennell KY, Chiao YA, Ma Y, Dai Q, et al. Texas 3-Step decellularization protocol: Looking at the cardiac extracellular matrix. Journal of Proteomics [serial on the Internet] 2013;86 doi: 10.1016/j.jprot.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butler GS, Overall CM. Updated biological roles for matrix metalloproteinases and new “intracellular” substrates revealed by degradomics. Biochemistry. 2009;48:10830–45. doi: 10.1021/bi901656f. [DOI] [PubMed] [Google Scholar]
- 24.Iyer RP, Patterson NL, Fields GB, Lindsey ML. The history of matrix metalloproteinases: milestones, myths, and misperceptions. American journal of physiology. 2012;303:H919–30. doi: 10.1152/ajpheart.00577.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang J-H, Javier JAD, Chang G-Y, Oliveira HB, Azar DT. Functional characterization of neostatins, the MMP-derived, enzymatic cleavage products of type XVIII collagen. FEBS Letters. 2005;579:3601–6. doi: 10.1016/j.febslet.2005.05.043. [DOI] [PubMed] [Google Scholar]
- 26.Davis GE, Bayless KJ, Davis MJ, Meininger GA. Regulation of tissue injury responses by the exposure of matricryptic sites within extracellular matrix molecules. Am J Pathol. 2000;156:1489–98. doi: 10.1016/S0002-9440(10)65020-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ricard-Blum S, Ballut L. Matricryptins derived from collagens and proteoglycans. Frontiers in bioscience: a journal and virtual library. 2011;16:674–97. doi: 10.2741/3712. [DOI] [PubMed] [Google Scholar]
- 28.Fichter M, Korner U, Schomburg J, Jennings L, Cole AA, Mollenhauer J. Collagen degradation products modulate matrix metalloproteinase expression in cultured articular chondrocytes. Journal of orthopaedic research: official publication of the Orthopaedic Research Society. 2006;24:63–70. doi: 10.1002/jor.20001. [DOI] [PubMed] [Google Scholar]
- 29.Hamano Y, Zeisberg M, Sugimoto H, Lively JC, Maeshima Y, Yang C, et al. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3:589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang C, Werb Z. The many faces of metalloproteases: cell growth, invasion, angiogenesis and metastasis. Trends in cell biology. 2001;11:S37–43. doi: 10.1016/s0962-8924(01)02122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adair-Kirk TL, Senior RM. Fragments of extracellular matrix as mediators of inflammation. The international journal of biochemistry & cell biology. 2008;40:1101–10. doi: 10.1016/j.biocel.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiao YA, Zamilpa R, Lopez EF, Dai Q, Escobar GP, Hakala K, et al. In vivo matrix metalloproteinase-7 substrates identified in the left ventricle post-myocardial infarction using proteomics. Journal of proteome research. 2010;9:2649–57. doi: 10.1021/pr100147r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zamilpa R, Lopez EF, Chiao YA, Dai Q, Escobar GP, Hakala K, et al. Proteomic analysis identifies in vivo candidate matrix metalloproteinase-9 substrates in the left ventricle post-myocardial infarction. Proteomics. 2010;10:2214–23. doi: 10.1002/pmic.200900587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martens L, Hermjakob H, Jones P, Adamski M, Taylor C, States D, et al. PRIDE: The proteomics identifications database. Proteomics. 2005;5:3537–45. doi: 10.1002/pmic.200401303. [DOI] [PubMed] [Google Scholar]
- 35.Jones P, Côté RG, Martens L, Quinn AF, Taylor CF, Derache W, et al. PRIDE: a public repository of protein and peptide identifications for the proteomics community. Nucleic Acids Research. 2006;34:D659–D63. doi: 10.1093/nar/gkj138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beavis R. New and Emerging Proteomic Techniques. Humana Press; 2006. Using the Global Proteome Machine for Protein Identification; pp. 217–28. [DOI] [PubMed] [Google Scholar]
- 37.Xenarios I, Rice DW, Salwinski L, Baron MK, Marcotte EM, Eisenberg D. DIP: the Database of Interacting Proteins. Nucleic Acids Research. 2000;28:289–91. doi: 10.1093/nar/28.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pagel P, Kovac S, Oesterheld M, Brauner B, Dunger-Kaltenbach I, Frishman G, et al. The MIPS mammalian protein-protein interaction database. Bioinformatics. 2005;21:832–4. doi: 10.1093/bioinformatics/bti115. [DOI] [PubMed] [Google Scholar]
- 39.Prasad TS, Kandasamy K, Pandey A. Human Protein Reference Database and Human Proteinpedia as discovery tools for systems biology. Methods in molecular biology. 2009;577:67–79. doi: 10.1007/978-1-60761-232-2_6. [DOI] [PubMed] [Google Scholar]
- 40.Breitkreutz BJ, Stark C, Reguly T, Boucher L, Breitkreutz A, Livstone M, et al. The BioGRID Interaction Database: 2008 update. Nucleic Acids Research. 2008;36:D637–40. doi: 10.1093/nar/gkm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, Chen C, et al. The IntAct molecular interaction database in 2012. Nucleic Acids Research. 2012;40:D841–6. doi: 10.1093/nar/gkr1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Persico M, Ceol A, Gavrila C, Hoffmann R, Florio A, Cesareni G. HomoMINT: an inferred human network based on orthology mapping of protein interactions discovered in model organisms. BMC bioinformatics. 2005;6 (Suppl 4):S21. doi: 10.1186/1471-2105-6-S4-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hellerstein JM. Quantitative Data Cleaning for Large Databases. 2008 Available from: http://db.cs.berkeley.edu/jmh/papers/cleaning-unece.pdf.
- 44.Sotiriou C, Neo SY, McShane LM, Korn EL, Long PM, Jazaeri A, et al. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:10393–8. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghasemi O, Nguyen N, Ramirez TA, Zhang J, Lindsey ML, Jin Y, editors. A biclustering approach to analyze drug effects on extracellular matrix remodeling post-myocardial infarction; 2012 IEEE International Conference on Bioinformatics and Biomedicine Workshops (BIBMW 2012); Philadelphia, USA: IEEE; 2012. [Google Scholar]
- 46.Stanton LW, Garrard LJ, Damm D, Garrick BL, Lam A, Kapoun AM, et al. Altered patterns of gene expression in response to myocardial infarction. Circulation research. 2000;86:939–45. doi: 10.1161/01.res.86.9.939. [DOI] [PubMed] [Google Scholar]
- 47.Ringner M. What is principal component analysis? Nature biotechnology. 2008;26:303–4. doi: 10.1038/nbt0308-303. [DOI] [PubMed] [Google Scholar]
- 48.Cortes C, Vapnik V. Support-vector networks. Mach Learn. 1995;20:273–97. [Google Scholar]
- 49.Dhawan A, Wenzel B, George S, Gussak I, Bojovic B, Panescu D, editors. Detection of Acute Myocardial Infarction from serial ECG using multilayer support vector machine; Engineering in Medicine and Biology Society (EMBC), 2012 Annual International Conference of the IEEE; Aug. 28 2012–Sept. 1 2012; 2012. [DOI] [PubMed] [Google Scholar]
- 50.Asl BM, Setarehdan SK, Mohebbi M. Support vector machine-based arrhythmia classification using reduced features of heart rate variability signal. Artif Intell Med. 2008;44:51–64. doi: 10.1016/j.artmed.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 51.Iorio MV, Ferracin M, Liu C-G, Veronese A, Spizzo R, Sabbioni S, et al. MicroRNA Gene Expression Deregulation in Human Breast Cancer. Cancer Research. 2005;65:7065–70. doi: 10.1158/0008-5472.CAN-05-1783. [DOI] [PubMed] [Google Scholar]
- 52.Hua S, Sun Z. A novel method of protein secondary structure prediction with high segment overlap measure: support vector machine approach. Journal of Molecular Biology. 2001;308:397–407. doi: 10.1006/jmbi.2001.4580. [DOI] [PubMed] [Google Scholar]
- 53.Penfold CA, Wild DL. How to infer gene networks from expression profiles, revisited. Interface Focus. 2011;1:857–70. doi: 10.1098/rsfs.2011.0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin Y, Lindsey ML. Stability analysis of genetic regulatory network with additive noises. BMC genomics. 2008;9 (Suppl 1):S21. doi: 10.1186/1471-2164-9-S1-S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stelling J, Gilles ED. Mathematical modeling of complex regulatory networks. IEEE Trans Nanobioscience. 2004;3:172–9. doi: 10.1109/tnb.2004.833688. [DOI] [PubMed] [Google Scholar]
- 56.Yeang CH, Ideker T, Jaakkola T. Physical network models. J Comput Biol. 2004;11:243–62. doi: 10.1089/1066527041410382. [DOI] [PubMed] [Google Scholar]
- 57.Tavazoie S, Hughes JD, Campbell MJ, Cho RJ, Church GM. Systematic determination of genetic network architecture. Nature genetics. 1999;22:281–5. doi: 10.1038/10343. [DOI] [PubMed] [Google Scholar]
- 58.Gardner TS, di Bernardo D, Lorenz D, Collins JJ. Inferring Genetic Networks and Identifying Compound Mode of Action via Expression Profiling. Science. 2003;301:102–5. doi: 10.1126/science.1081900. [DOI] [PubMed] [Google Scholar]
- 59.Chen T, He HL, Church GM. Modeling gene expression with differential equations. Pacific Symposium on Biocomputing Pacific Symposium on Biocomputing; 1999. pp. 29–40. [PubMed] [Google Scholar]
- 60.Huang Y, Liu D, Wu H. Hierarchical Bayesian Methods for Estimation of Parameters in a Longitudinal HIV Dynamic System. Biometrics. 2006;62:413–23. doi: 10.1111/j.1541-0420.2005.00447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ghasemi O, Lindsey M, Yang T, Nguyen N, Huang Y, Jin Y-F. Bayesian parameter estimation for nonlinear modelling of biological pathways. BMC Systems Biology. 2011;5:S9. doi: 10.1186/1752-0509-5-S3-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yeung MKS, Tegnér J, Collins JJ. Reverse engineering gene networks using singular value decomposition and robust regression. Proceedings of the National Academy of Sciences. 2002;99:6163–8. doi: 10.1073/pnas.092576199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.van Someren EP, Wessels LF, Reinders MJ. Linear modeling of genetic networks from experimental data. Proceedings/International Conference on Intelligent Systems for Molecular Biology; ISMB International Conference on Intelligent Systems for Molecular Biology. 2000;8:355–66. [PubMed] [Google Scholar]
- 64.Escobar M, West M. Bayesian density estimation and inference using mixtures. Journal of the american statistical association. 1995:577–88. [Google Scholar]
- 65.Verstraete F, Doherty AC, Mabuchi H. Sensitivity optimization in quantum parameter estimation. Physical Review A. 2001;64:032111. [Google Scholar]
- 66.Jin Y, Qu Z, editors. Synchronization of Lorenz systems by adaptive observation; American Control Conference, 2003 Proceedings of the 2003; 2003. [Google Scholar]
- 67.Tang Y, Ghosal S, Roy A. Nonparametric Bayesian estimation of positive false discovery rates. Biometrics. 2007;63:1126–34. doi: 10.1111/j.1541-0420.2007.00819.x. [DOI] [PubMed] [Google Scholar]
- 68.Qu Z. Adaptive and robust controls of uncertain systems with nonlinear parameterization. IEEE Transactions on Automatic Control. 2003;48:1817–24. [Google Scholar]
- 69.Saltelli A, Chan K, Scott M. Sensitivity Analysis. New York: John Wiley and Sons; 2000. [Google Scholar]
- 70.Khalil HK. Nonlinear systems. 3. Prentice Hall; 2001. [Google Scholar]
- 71.Samoilov M, Plyasunov S, Arkin AP. Stochastic amplification and signaling in enzymatic futile cycles through noise-induced bistability with oscillations. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:2310–5. doi: 10.1073/pnas.0406841102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Manninen T, Linne M-L, Ruohonen K. Developing Itô stochastic differential equation models for neuronal signal transduction pathways. Computational Biology and Chemistry. 2006;30:280–91. doi: 10.1016/j.compbiolchem.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 73.Arkin A, Ross J, McAdams HH. Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia coli cells. Genetics. 1998;149:1633–48. doi: 10.1093/genetics/149.4.1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jin Y, Han HC, Berger J, Dai Q, Lindsey ML. Combining experimental and mathematical modeling to reveal mechanisms of macrophage-dependent left ventricle remodeling. BMC Systems Biology [serial on the Internet] 2011;5(60) doi: 10.1186/1752-0509-5-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chow CC, Clermont G, Kumar R, Lagoa C, Tawadrous Z, Gallo D, et al. The Acute Inflammatory Response in Diverse Shock States. Shock. 2005;24:74–84. doi: 10.1097/01.shk.0000168526.97716.f3. [DOI] [PubMed] [Google Scholar]
- 76.Karagiannis ED, Popel AS. A Theoretical Model of Type I Collagen Proteolysis by Matrix Metalloproteinase (MMP) 2 and Membrane Type 1 MMP in the Presence of Tissue Inhibitor of Metalloproteinase 2. J Biol Chem. 2004;279:39105–14. doi: 10.1074/jbc.M403627200. [DOI] [PubMed] [Google Scholar]
- 77.Vempati P, Karagiannis ED, Popel AS. A Biochemical Model of Matrix Metalloproteinase 9 Activation and Inhibition. J Biol Chem. 2007;282:37585–96. doi: 10.1074/jbc.M611500200. [DOI] [PubMed] [Google Scholar]
- 78.Noble D. Modelling the heart: insights, failures and progress. Bioessays. 2002;24:1155–63. doi: 10.1002/bies.10186. [DOI] [PubMed] [Google Scholar]
- 79.Cassman MC, Arkin A, Doyle F, Katagiri F, Lauffenburger D, Stokes C. International Research and Development in Systems Biology. 2005. [Google Scholar]
- 80.Winslow RL, Scollan DF, Holmes A, Yung CK, Zhang J, Jafri MS. Electrophysiological modeling of cardiac ventricular function: from cell to organ. Annu Rev Biomed Eng. 2000;2:119–55. doi: 10.1146/annurev.bioeng.2.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Luo C, Rudy Y. A dynamic model of the cardiac ventricular action potential. II. Afterdepolarizations, triggered activity, and potentiation. Circ Res. 1994;74:1097–113. doi: 10.1161/01.res.74.6.1097. [DOI] [PubMed] [Google Scholar]
- 82.DiFrancesco D, Noble D. A Model of Cardiac Electrical Activity Incorporating Ionic Pumps and Concentration Changes. Philosophical Transactions of the Royal Society of London B, Biological Sciences. 1985;307:353–98. doi: 10.1098/rstb.1985.0001. [DOI] [PubMed] [Google Scholar]
- 83.Jafri MS, Rice JJ, Winslow RL. Cardiac Ca2+ Dynamics: The Roles of Ryanodine Receptor Adaptation and Sarcoplasmic Reticulum Load. Biophysical Journal. 1998;74:1149–68. doi: 10.1016/S0006-3495(98)77832-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Freeman LC. Centrality in Social Networks Conceptual Clarification. Soc Networks. 1979;1:215–39. [Google Scholar]
- 85.Junker BH, Schreiber F. Analysis of biological networks. Hoboken, N.J: Wiley-Interscience; 2008. [Google Scholar]
- 86.Harary F. Graph theory. Reading, Mass: Addison-Wesley Pub. Co; 1969. [Google Scholar]
- 87.Wasserman S, Faust K. Social network analysis: methods and applications. Cambridge; New York: Cambridge University Press; 1994. [Google Scholar]
- 88.Raman K. Construction and analysis of protein-protein interaction networks. Automated Experimentation. 2010;2:2. doi: 10.1186/1759-4499-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen L, Xuan J, Riggins RB, Wang Y, Clarke R. Identifying protein interaction subnetworks by a bagging Markov random field-based method. Nucleic Acids Research. 2013;41:e42. doi: 10.1093/nar/gks951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Jahid MJ, Ruan J. A Steiner tree-based method for biomarker discovery and classification in breast cancer metastasis. BMC genomics. 2012;13:S8. doi: 10.1186/1471-2164-13-S6-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 92.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chi A, Valencia JC, Hu ZZ, Watabe H, Yamaguchi H, Mangini NJ, et al. Proteomic and bioinformatic characterization of the biogenesis and function of melanosomes. Journal of proteome research. 2006;5:3135–44. doi: 10.1021/pr060363j. [DOI] [PubMed] [Google Scholar]
- 94.Hu ZZ, Valencia JC, Huang H, Chi A, Shabanowitz J, Hearing VJ, et al. Comparative Bioinformatics Analyses and Profiling of Lysosome-Related Organelle Proteomes. Int J Mass Spectrom. 2007;259:147–60. doi: 10.1016/j.ijms.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hu ZZ, Kagan BL, Ariazi EA, Rosenthal DS, Zhang L, Li JV, et al. Proteomic analysis of pathways involved in estrogen-induced growth and apoptosis of breast cancer cells. PLoS One. 2011;6:e20410. doi: 10.1371/journal.pone.0020410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hu ZZ, Huang H, Cheema A, Jung M, Dritschilo A, Wu CH. Integrated Bioinformatics for Radiation-Induced Pathway Analysis from Proteomics and Microarray Data. J Proteomics Bioinform. 2008;1:47–60. doi: 10.4172/jpb.1000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Raman K. Construction and analysis of protein-protein interaction networks. Automated experimentation. 2010;2:2. doi: 10.1186/1759-4499-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nguyen N, Zhang X, Wang Y, Han HC, Jin Y, Schmidt G, et al. Targeting Myocardial Infarction-Specific Protein Interaction Network Using Computational Analyses. 2011 IEEE International Workshop on Genomic Signal Processing and Statistics [serial on the Internet]; 2011. [Google Scholar]
- 99.Azuaje FJ, Rodius S, Zhang L, Devaux Y, Wagner DR. Information encoded in a network of inflammation proteins predicts clinical outcome after myocardial infarction. BMC medical genomics. 2011;4:59. doi: 10.1186/1755-8794-4-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ren G, Liu Z. NetCAD: a network analysis tool for coronary artery disease-associated PPI network. Bioinformatics. 2013;29:279–80. doi: 10.1093/bioinformatics/bts666. [DOI] [PubMed] [Google Scholar]
- 101.Wu LH, Wang Y, Nie J, Fan XH, Cheng YY. A Network Pharmacology Approach to Evaluating the Efficacy of Chinese Medicine Using Genome-Wide Transcriptional Expression Data. Evid-Based Compl Alt. 2013 doi: 10.1155/2013/915343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhu WL, Yang L, Shan HL, Zhang Y, Zhou R, Su Z, et al. MicroRNA Expression Analysis: Clinical Advantage of Propranolol Reveals Key MicroRNAs in Myocardial Infarction. Plos One. 2011:6. doi: 10.1371/journal.pone.0014736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Daskalopoulou SS, Delaney JAC, Filion KB, Brophy JM, Mayo NE, Suissa S. Discontinuation of statin therapy following an acute myocardial infarction: a population-based study. Eur Heart J. 2008;29:2083–91. doi: 10.1093/eurheartj/ehn346. [DOI] [PubMed] [Google Scholar]
- 104.Bonab M, Ghasemi O, Jamshidi M, Jin Y. Time Delay Estimation in Gene Regulatory Networks. Proceeding of IEEE SoSE Conference; Maui, HI. 2013; 2013. serial on the Internet. [Google Scholar]
- 105.Berhane BT, Zong C, Liem DA, Huang A, Le S, Edmondson RD, et al. Cardiovascular-related proteins identified in human plasma by the HUPO Plasma Proteome Project pilot phase. Proteomics. 2005;5:3520–30. doi: 10.1002/pmic.200401308. [DOI] [PubMed] [Google Scholar]
- 106.Rothemund DL, Locke VL, Liew A, Thomas TM, Wasinger V, Rylatt DB. Depletion of the highly abundant protein albumin from human plasma using the Gradiflow. Proteomics. 2003;3:279–87. doi: 10.1002/pmic.200390041. [DOI] [PubMed] [Google Scholar]
- 107.Tam SW, Pirro J, Hinerfeld D. Depletion and fractionation technologies in plasma proteomic analysis. Expert Rev Proteomics. 2004;1:411–20. doi: 10.1586/14789450.1.4.411. [DOI] [PubMed] [Google Scholar]
- 108.Chen R, Snyder M. Systems biology: personalized medicine for the future? Current Opinion in Pharmacology. 2012;12:623–8. doi: 10.1016/j.coph.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mayr M. From data gathering to systems medicine. Cardiovascular Research. 2013;97:599–600. doi: 10.1093/cvr/cvt017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Loscalzo J. Personalized Cardiovascular Medicine and Drug Development: Time for a New Paradigm. Circulation. 2012:638–45. doi: 10.1161/CIRCULATIONAHA.111.089243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Roukos HD. Biotechnological, genomics and systems–synthetic biology revolution: redesigning genetic code for a pragmatic systems medicine. Expert Review of Medical Devices. 2012;9:97–101. doi: 10.1586/erd.11.68. [DOI] [PubMed] [Google Scholar]