

Abstract
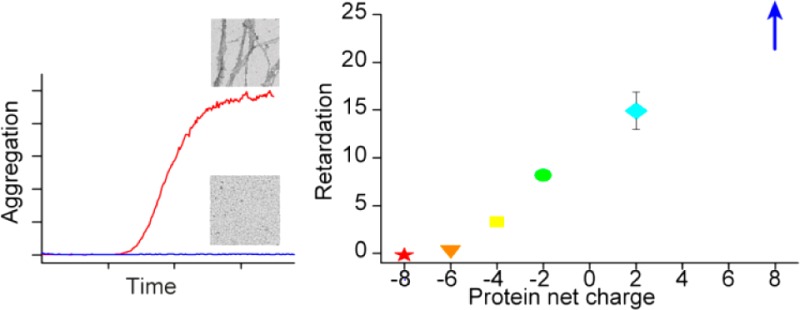
The aggregation of amyloid β peptides (Aβ) into amyloid fibrils is implicated in the pathology of Alzheimer’s disease. In light of the increasing number of proteins reported to retard Aβ fibril formation, we investigated the influence of small hydrophilic model proteins of different charge on Aβ aggregation kinetics and their interaction with Aβ. We followed the amyloid fibril formation of Aβ40 and Aβ42 using thioflavin T fluorescence in the presence of six charge variants of calbindin D9k and single-chain monellin. The formation of fibrils was verified with transmission electron microscopy. We observe retardation of the aggregation process from proteins with net charge +8, +2, −2, and −4, whereas no effect is observed for proteins with net charge of −6 and −8. The single-chain monellin mutant with the highest net charge, scMN+8, has the largest retarding effect on the amyloid fibril formation process, which is noticeably delayed at as low as a 0.01:1 scMN+8 to Aβ40 molar ratio. scMN+8 is also the mutant with the fastest association to Aβ40 as detected by surface plasmon resonance, although all retarding variants of calbindin D9k and single-chain monellin bind to Aβ40.
Keywords: Amyloid β, hydrophobicity, inhibitors, protein−protein interactions, kinetics, Alzheimer’s disease
Alzheimer’s disease (AD) is the most common neurodegenerative disease.1 The symptoms of memory deficits and gradual loss of brain functions were first described in 19072 and still there is no cure or consensus on the etiology of the disease. AD is characterized by neurofibrillary tangles and neuritic plaques.3 The main component of the plaques is the amyloid β peptide (Aβ), which is cleaved from the membrane spanning amyloid precursor protein. There is variability in the cleavage site, which generates Aβ peptides of different lengths with variation in fibril forming propensities. The ratio between the lengths can be used as a tool for characterization of pathological conditions as reviewed by Finder et al.4,5
According to the amyloid cascade hypothesis, Aβ plays a causative role in AD by forming oligomers and fibrils from the unstructured monomers.3,6,7 Fibrils of Aβ are thermodynamically stable and in equilibrium with a low concentration of soluble Aβ.6,7 The fibrils have cross-β structure, where each Aβ monomer contributes two β-strands perpendicular to the axis of the fibril.8−10
In vivo, the aggregation of Aβ occurs in a complex environment composed of a number of different proteins, other macromolecules, salts, and small molecules, as well as different surfaces including the phospholipid membranes with associated proteins. This may influence the aggregation pattern of Aβ through crowding effects, electrostatic interactions, and screening effects, as well as interactions involving hydrophobic patches on chaperones and other proteins.
One approach toward understanding the aggregation behavior of Aβ in vivo is to study the aggregation process of pure peptide in vitro and introduce various components one at a time. This makes it possible in a controlled manner to analyze which factors of the solution accelerate or retard aggregation. A number of substances and materials have been found to retard Aβ fibrillation kinetics,11 including N-methylated peptides,12,13 synthetic nanoparticles,14−17 and dendrimers.18 There are also many proteins that have been reported to retard aggregation, for instance: human serum albumin (HSA),19 lysozyme,20 αs1-casein,21 the designed affibody Zaβ3,22 and the chaperones αB-crystallin23,24 and the BRICHOS domain.25 Some of these macromolecules cause significant retardation at substoichiometric levels in relation to Aβ, whereas others require up to a 1:1 ratio. Some of the retarding proteins may be of direct biological relevance by colocalization with Aβ in vivo.26,27 The retarding effects are often rationalized in terms of interactions involving hydrophobic groups due to the amphiphatic properties of Aβ, and the presence of exposed hydrophobic areas on several of the proteins that bind to Aβ or retard the aggregation process.19,21−23
The current work investigated the effects of hydrophilic proteins on the aggregation of Aβ(M1–40), hereafter Aβ40, and Aβ(M1–42), hereafter Aβ42. Two proteins, single-chain monellin (scMN; Mw 11 kDa) and calbindin D9k (CB; Mw 9 kDa), were selected based on their high thermal stability, small hydrophobic surface area (Figure 1), and their tolerance to charge modulation without compromising folding.28−32 Here we ask the question whether there is a charge dependent inhibitory protein effect on Aβ fibrillation by interrogating the effects of charge variations in these two globular proteins. The protein surface charges were modified by charge substitution mutations to generate a charge series from −8 to +8. The hydrophilic nature of all proteins in this series was confirmed using 8-anilinonaphthalene-1-sulfonic acid (ANS) fluorescence spectroscopy, and the interaction with Aβ40 was studied using surface plasmon resonance (SPR) technology. The results show an inhibitory effect on the aggregation process that correlates with the protein net charge.
Figure 1.
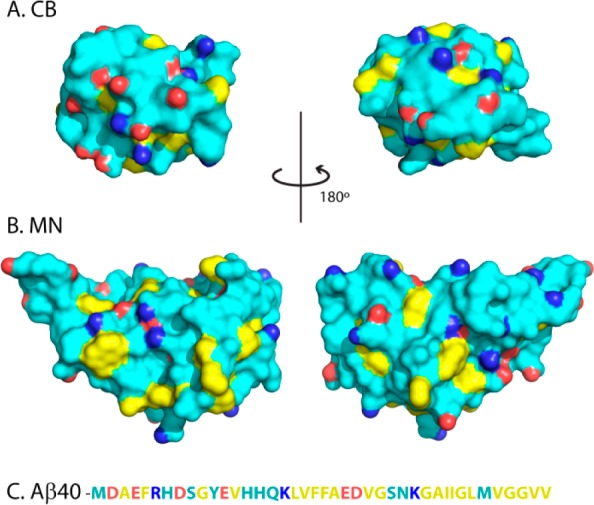
Surface representation of wild type form of CB (A), and monellin (B). Hydrophobic side chains are yellow, acidic side chains have one carboxylic oxygen marked in red, and basic side chains have one nitrogen marked in blue. Panel (C) displays the sequence of Aβ40 with the same color coding. The figure was prepared from the PDB files 1ICB and 4MON using Pymol.33
Results
In this work, we study the charge-dependent effects of hydrophilic proteins on the formation of amyloid fibrils from Aβ40 and Aβ42. To evaluate the role of protein net charge, we prepared in total six variants of the two model proteins scMN and CB. The proteins were modified with charge substitutions to cover a wide range of net charges; +8 (scMN+8), +2 (scMN+2), −2 (scMN-2), −4 (CB-4), −6 (CB-6), and −8 (CB-8). The net charge of Aβ40 is roughly −3 at physiological pH. The mutations do not affect the structure of any of the proteins based on previous NMR studies,28−32 and circular dichroism (CD) spectroscopy (Figure S1, Supporting Information).
On a macroscopic level, the time course of fibril formation follows a sigmoidal curve; beginning with a lag phase, continuing with a steep growth phase, and then an equilibrium phase at the plateau, where nearly all of the monomers have formed fibrils.34 This kinetic process can be followed using thioflavin T (ThT) fluorescence. The quantum yield of ThT increases upon binding to extended β-sheet structures such as in amyloid fibrils.5,35
Effect of Protein Charge Mutants on Aβ40 Fibril Formation Kinetics
We followed the amyloid fibril formation of 10 μM Aβ40 with ThT fluorescence in the absence and presence of the six charge mutants. The presence of the proteins results in either no measurable effect (CB-8 and CB-6) or a concentration-dependent retardation of the fibril formation (Figure 2). Samples without or with the proteins at 0.01:1 up to 3:1 protein to Aβ40 molar ratio were studied. CB-4 retards Aβ40 aggregation only above equimolar ratio, whereas all scMNs retard aggregation also at substoichiometric levels. scMN+8 has the strongest effect on Aβ40 aggregation, and no fibrils are formed above 300 nM scMN+8 during the time course of the experiment (66 h). The effect seen in the presence of added proteins is mainly a prolonged lag phase, whereas the intensity values at the equilibrium plateau are roughly the same as for Aβ40 aggregated without additives (Figure S2). Charge-dependent retardation by scMN and CB mutants is observed also for Aβ42 aggregation (Figure S3). Again, the strongest retarding effect is observed in the presence of scMN+8. Aβ42 has higher intrinsic aggregation propensity than Aβ40 and the proteins need to be added at higher concentration to elicit the same relative effect on Aβ42 compared to Aβ40 aggregation kinetics.
Figure 2.
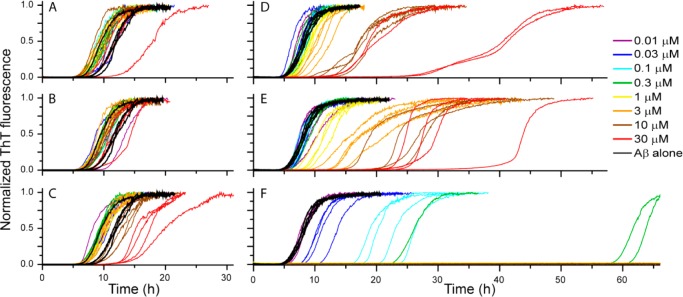
Normalized ThT fluorescence for 10 μM Aβ40 aggregating in the absence and presence of CB-8 (A), CB-6 (B), CB-4 (C), scMN-2 (D), scMN+2 (E), and scMN+8 (F). Each protein was added at seven or eight concentrations ranging from 0.01 to 30 μM. There are four technical replicates of each concentration, and the whole set of experiments has been repeated with similar results.
To compare the retardation of the aggregation in the presence of the different proteins, we derived from the experimental data t1/2, which is defined as the time of half completion of the process where half the maximum ThT fluorescence intensity is reached (eq 1). This is correspondingly also the time point where approximately half of the monomer is consumed.36 In Figure 3A, t1/2 is plotted against the concentration of added protein, showing the concentration-dependent retardation of the aggregation. The charge dependence is shown in Figure 3B where the retardation of Aβ40 shows a monotonic trend with net charge of the added proteins. For each protein, the variant with the most positive (or least negative) net charge (scMN+8 and CB-4) has the largest effect on t1/2. This suggests a contribution to the overall retardation based on charge rather than protein identity in the context of hydrophilic proteins.
Figure 3.
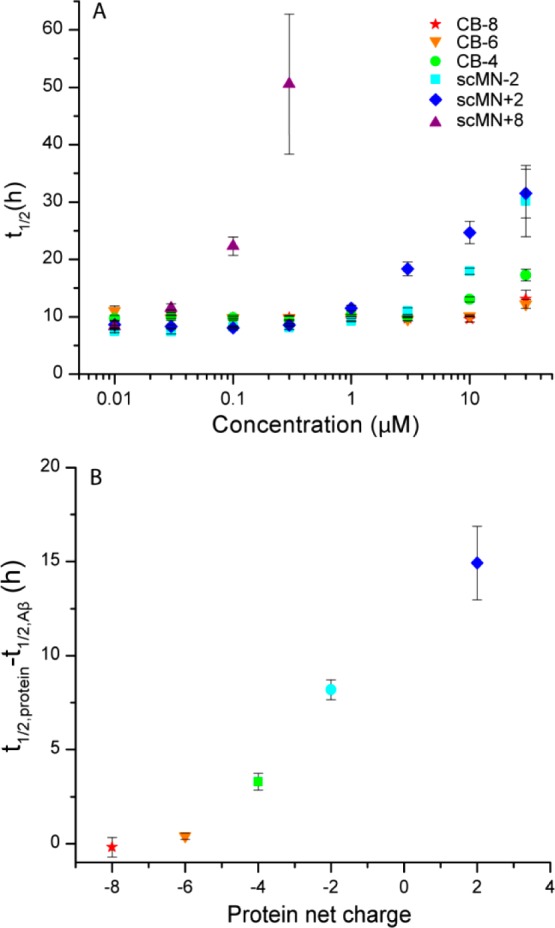
Half time, t1/2, of Aβ40 aggregation based on ThT fluorescence. The error bars indicate standard error of the mean from three or four replicates. (A) t1/2 vs concentration (log scale) of added protein (CB or scMN). t1/2 could not be calculated for the samples with high concentrations of scMN+8, which did not form fibrils within the time course of the experiment. The t1/2 for aggregation of Aβ40 without added proteins was 9.8 ± 0.6 h. (B) Increase in t1/2 of Aβ40 upon protein addition relative to t1/2 of Aβ40 alone plotted against the net charge of the added proteins. The concentration of both the proteins and Aβ40 was 10 μM. scMN+8 is omitted from the graph because it did not form fibrils under these conditions.
Morphology of Aβ40 Fibrils
The morphology of Aβ40 fibrils formed in the absence and presence of the added protein variants was studied using transmission electron microscopy (TEM). TEM grids were prepared with samples from the kinetic experiments which had reached the plateau in the ThT fluorescence, except in the case of Aβ40 with scMN+8 (see below). The samples contained equimolar amounts of Aβ40 and scMN or CB variants (Figure 4A–G). All ThT positive samples contained fibrils when imaged by TEM. The fibrils were 10–20 nm wide and up to several micrometers long, in agreement with previous reports on Aβ40 fibril morphology.8,37
Figure 4.
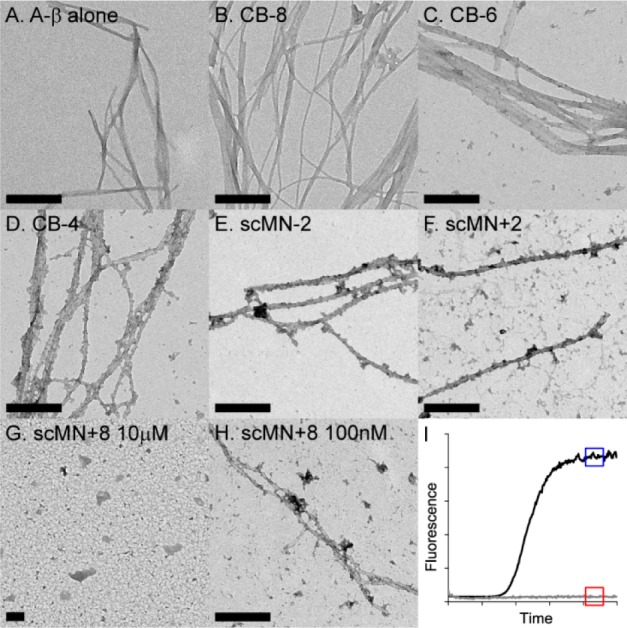
TEM images of 10 μM Aβ40 in the absence (A) and presence of 10 μM CB-8 (B), CB-6 (C), CB-4 (D), scMN-2 (E), scMN+2 (F), scMN+8 (G), and 100nM scMN+8 (H). The fibrillar samples (A–F, H) were taken when the ThT fluorescence had reached the plateau value (blue box in (I)). The red box indicates the time point for the nonfibrillar sample with 10 μM scMN+8 (G). The scale bar is 200 nm.
The ability to form amyloid fibrils is implied as an intrinsic property of any polypeptide backbone, and the fibrillar state is thus available for all proteins under appropriate conditions.38 Native monellin, but not scMN or CB, has been reported to form amyloid fibrils, under denaturing conditions at low pH.39,40 None of the CB or scMN charge variants formed fibrils on their own within 66 h under the conditions of this study (data not shown), which implies that all fibrils detected here originated from Aβ40. No significant differences between the fibrils from the different samples were detected within the resolution of our images. This indicates that the final structure of the amyloid fibrils is not affected by the presence of added protein.
No increase in ThT fluorescence intensity was observed for the Aβ40 sample with 10 μM scMN+8, at the time when all other samples had reached the equilibrium plateau (Figure 4I) and accordingly, no fibrils were detected by TEM (Figure 4G). No fibrils were detected in an identical sample with a ten times longer incubation time (150 h, data not shown), which supports a very strong inhibition of Aβ40 aggregation by scMN+8. To investigate whether fibrils could be formed in the presence of a lower concentration of scMN+8, a sample with 100 times lower concentration (100 nM) of scMN+8 was prepared. In this sample, with a moderate retardation of the Aβ40 fibril formation, we observe fibrils of the same general morphology as in the other TEM samples. This finding is supported by CD spectroscopy. No significant difference is detected between the CD spectra of Aβ40 fibrils formed in the absence and presence of 0.01 mol equiv of scMN+8 (Figure S4).
Low Surface Hydrophobicity of scMN and CB Mutants
Aβ is an amphiphatic peptide known to interact with hydrophobic surfaces.41,42 It is therefore necessary for the current study to verify that the differential effects on Aβ40 aggregation from the scMN and CB variants are not due to variations in hydrophobicity. A facile method to study surface hydrophobicity of proteins is to analyze modifications of the fluorescence spectra of ANS in the presence of proteins. The fluorescence intensity of ANS is increased and its emission spectrum is shifted to shorter wavelengths in the presence of hydrophobic protein surfaces.43 None of the scMN and CB variants show significant ANS binding (Figure 5), in contrast to the hydrophobic control protein calmodulin.44 The ANS fluorescence intensity is only slightly increased for the two proteins with the highest charges (scMN+2 and scMN+8), indicating that all charge variants in the present study are proteins with hydrophilic surface character.
Figure 5.
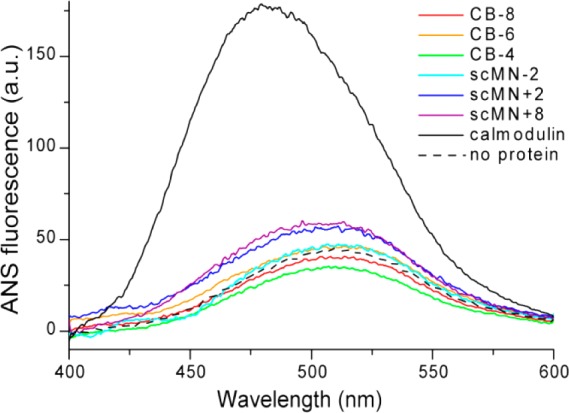
Emission spectra of 20 μM ANS in the absence and presence of the six scMN and CB variants, and the control protein calmodulin (see legend). The result was similar for all tested concentrations of ANS.
Direct Binding between Aβ40 and Retarding scMN and CB Mutants
The observation of a prolonged lag phase in the presence of added proteins (Figure 2) suggests that the early stages of amyloid fibril formation are affected. To study the molecular origin of the retarding effect, we investigated if there is any direct interaction between the added proteins and Aβ40. SPR experiments were performed with immobilized Aβ40 monomer and the protein variants flowed over the chip to study association kinetics, followed by buffer flow to study dissociation kinetics.
We observed binding between Aβ40 and most of the hydrophilic proteins; and the affinities correlate with the net charge and the induced retardation of Aβ40 fibril formation. No binding was detected for CB-6 and CB-8 at concentrations up to 30 μM, but all the other variants bind to Aβ40 at two or more of the injected concentrations (Figure S5). None of the mutants dissociated completely during the time course of the experiment (3 h) but could be removed by chip regeneration with GuHCl indicating an extremely slow dissociation process. scMN+8 binds to Aβ40 at a lower concentration than the other protein variants in agreement with its larger retarding effect on fibril formation kinetics. scMN+2 binds to the surface with Aβ40, as well as the reference surface without immobilized peptide (Figure S5B), and was therefore excluded from further analysis.
To gain more information on the interaction between Aβ40 and the binding protein variants, scMN+8, scMN-2, and CB-4, binding curves were fitted to the association and dissociation phases (see Methods and eqs 2–5), generating the rate constants kon and koff, and the equilibrium dissociation constant KD (Table 1). Inspection of the data revealed a second weaker binding to the chip for higher concentrations of scMN+8 and scMN-2, seen as a second exponential step in the association phase and an initial fast step in the dissociation phase. This has been observed in other SPR experiments on Aβ40 interactions.45 For each protein, one low concentration with significant binding was selected for curve fitting (Figure 6) to avoid the effects of a second weaker binding process (see Figure S5). The dissociation, apart from the initial step, is very slow for all three proteins. This means that only an upper limit of the dissociation rate (koff < 10–5 s–1) could be estimated (Figure S6) and that no significant difference between the proteins could be seen.
Table 1. Rate Constants for Aβ40 Interaction with CB and scMN Mutants Determined by SPR.
| kon (M–1 s–1) | koff (s–1) | KD (M) | |
|---|---|---|---|
| CB-4 | 200 | <1 × 10–5 | <5 × 10–8 |
| scMN-2 | 2000 | <1 × 10–5 | <5 × 10–9 |
| scMN+8 | 4000 | <1 × 10–5 | <2.5 × 10–9 |
Figure 6.
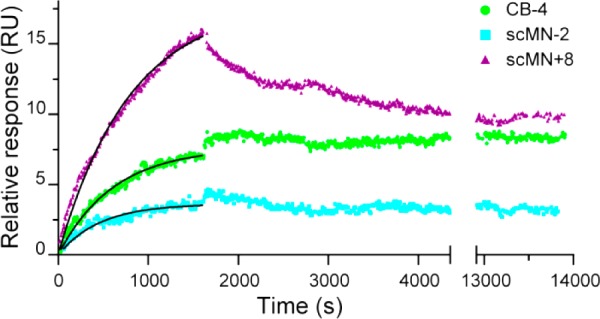
Binding between immobilized Aβ40 and 300 nM scMN+8, 1 μM scMN-2, and 10 μM CB-4 monitored by SPR technology. The proteins were injected at 3–7 concentrations in the range of 0.01–30 μM, but only one low concentration of each sample with significant binding is shown. Data from a reference cell without immobilized Aβ40 was subtracted from all curves. Black lines are fits of the association phase with koff set to 10–5 s–1.
Discussion
The aggregation process of Aβ40 and Aβ42 is affected by the presence of hydrophilic proteins. In this study we have used recombinant peptide with the high purity and sequence homogeneity (as opposed to synthetic peptide) required for reproducible kinetics. An initial methionine, is added to the sequence to allow expression in E. coli.46 We believe, based on previous studies,14,46 that the initial methionine does not compromise the effects on Aβ aggregation from the hydrophilic proteins. In the analysis of the data below, we focus on Aβ40, but we would like to point out that the effect of the hydrophilic proteins on the aggregation process is qualitatively similar for Aβ40 and Aβ42 (Figures 2 and S3).
The current data show that the aggregation of Aβ40 occurs at a much lower rate in the presence of the small hydrophilic proteins with net charge +8, +2, −2, and −4. This is a remarkable finding as most proteins previously reported to retard Aβ40 aggregation have some hydrophobic character or are naturally colocalized with Aβ40. No effect was observed for proteins with net charge of −6 and −8. The variant with the highest positive net charge in our series, scMN+8, has the largest retarding effect and causes a noticeable retardation of Aβ40 fibril formation at substoichiometric concentrations (0.01:1 scMN+8:Aβ40 and up). Moreover, the same CB and scMN charge variants which retard amyloid formation show binding to Aβ40 as observed in SPR analysis.
Whereas we observe here a clear retarding effect from positively charged scMN variants, positively charged polyamines and nanoparticles have been found to accelerate Aβ aggregation.14,47 Aggregation of Aβ40 is also accelerated in the presence of cationic lipid vesicles,48 whereas vesicles composed of anionic and zwitterionic lipids retard amyloid formation in a noncharge dependent manner.49 The lipids and nanoparticles are not directly comparable with the proteins used in this study due to differences in size and surface properties; but they serve as examples on how sensitive the Aβ aggregation process is to most forms of additives.
Aβ has been reported to interact with many different endogenous proteins both in pathogenic processes and as a way to reduce Aβ toxicity by complex formation.50−54 In contrast to the proteins in blood and cerebrospinal fluid, the model proteins in this study are not natural binding partners for Aβ40 and should not have any evolved interaction surfaces for Aβ40. scMN is a single chain variant of the plant protein monellin originating from Dioscoreophyllum cumminsii,55 without any evolved interaction with Aβ40, and CB is a calcium binding protein present in several mammalian tissues without any confirmed protein interaction partners.56 The variants of CB and scMN all have a hydrophilic surface according to the low fluorescence intensity in the ANS binding assay. This implies that surface hydrophobicity is not likely the major driving force for the interaction that we observe with Aβ40. This is in contrast to several of the other retarding proteins such as HSA, αB-crystallin, and Zaβ3. These have high binding capacity to ANS and where hydrophobicity is proposed to play a significant role in the interaction with Aβ40.19,57−60 Our results imply that Aβ40 interacts with proteins of very different surface character.
The affinity between the hydrophilic proteins and Aβ40 correlates with their retarding effect. The association rate constant, kon, is the lowest for CB-4, intermediate for scMN-2 and the highest for scMN+8 binding to Aβ40, implying that the net charge is an important factor that governs binding to Aβ40. The low value of kon for CB-4 implies that saturation is reached more slowly and that the rate of exchange between free and bound species is decreased compared to the other cases. The affinity for the protein variants binding to Aβ40 is similar to that observed for Aβ40 binding to the engineered protein Zaβ3, with a KD around 17 nM as determined by ITC,22 but higher than the affinity for HSA (KD = 5 μM)61 and αB-crystallin (KD = 2 μM).24
The saturation level of protein binding to Aβ40 on the SPR chips is quite low in all cases in this study, seen as small changes in the responses. The signal is proportional to the mass at the chip surface. From the amount of immobilized Aβ40 (ca. 470 pg/mm2), the largest amount of bound scMN+8 (ca. 32 pg/mm2) and the molecular weights of Aβ40 and scMN, the saturation level on the chip can be calculated to 1:36 scMN+8:Aβ40. An explanation for this low stoichiometry of binding could be that only a fraction of the Aβ40 on the chip has a significant affinity for the protein variants, because of interference from the coupling to the chip. Another possibility is that even though we have immobilized monomeric Aβ40, small oligomers might have formed on the chip. With a likely range of different species of Aβ40 on the chip, only some may bind to the CB and scMN protein variants. A third possibility is that several Aβ40 peptides bind to each scMN+8.
The prolonged lag phase of Aβ40 aggregation in the presence of hydrophilic proteins suggests retardation of the initial processes in the aggregation. The length of the lag phase depends on the rates of both nucleation and growth processes, as reviewed by Cohen et al.34 The nucleation processes can be divided into processes dependent on monomer only (primary nucleation) and processes dependent on preformed aggregates (secondary nucleation). The dominating nucleation process for Aβ40 and Aβ42 is monomer-dependent secondary nucleation, where new aggregates are formed by monomers interacting with preexisting fibrils.36,62 Monomers and fibrils are the two dominating populations at all time points with monomers dominating during the lag time and fibrils at the final plateau. Based on the rate equations of amyloid formation,34,63 it is possible to predict how individual aggregation curves will change in terms of t1/2 and slope based on which combinations of the underlying rate constants are altered. For example, if only the primary nucleation rate is reduced, the effect is increased t1/2 but no change in slope. If both the rate of fibril elongation and the rates of the nucleation processes are reduced, the equations predict a positive correlation between increased lag phase and flatter slopes. An example of this case would be decreased monomer concentration through depletion, as discussed below.
The main effect of the presence of the studied proteins on Aβ40 aggregation is an increased t1/2, but also the slopes of the individual aggregation curves are affected (Figure 2). For Aβ40 in the presence of scMN-2, the apparent elongation rate, that is, the slope, is reduced. This suggests that both the rates of primary nucleation and elongation or secondary nucleation are affected by this protein. The slopes of Aβ40 with scMN+2 seem to be somewhere in between scMN-2 and scMN+8, with variation between the protein concentrations, and the variation of the slope for Aβ40 with CB-4 is inconclusive. For Aβ40 in the presence of scMN+8, the consistent steep slopes for all aggregating conditions indicate that once the amount of fibrils reaches above a threshold, the rate of the reaction is the same independent of the lag time. A likely explanation for this behavior is that the main effect of this protein is on primary nucleation, while the effect on secondary nucleation and elongation are minor. This means that the monomers or small aggregates are affected by scMN+8. The SPR data indicates binding to monomers of Aβ40 or binding to small oligomers that might have formed on the chip, as discussed above. Binding of Aβ40 monomer to scMN+8 in equimolar ratio is not enough to explain the retarding effect observed at low concentrations of scMN+8. The retardation is significant even at a 0.01:1 molar ratio where the t1/2 is doubled (Figure 3). Sequestering 1% of the Aβ40 from the solution would not have any significant effect on the lag time.7,63 An example of retardation due to monomer binding in 1:1 ratio is reported for the engineered affibody Zaβ3 (as a dimer), which captures Aβ monomer in a beta hairpin structure, and accordingly equimolar ratios are required for complete inhibition of the aggregation.22
Another possibility is that Aβ40 binds evenly to the surface of scMN+8 in a similar manner as it may interact with nanoparticles.14−17 Up to 22 Aβ40 monomers would fit on the surface of scMN+8, in the limit of both molecules having spherical shape. At 33 Aβ40 peptides per scMN+8 (10 μM Aβ40 with 0.3 μM scMN+8), Aβ40 aggregation is significantly retarded with a 5-fold increase in lag time, and at a lower Aβ40:scMn+8 ratio the aggregation is retarded beyond the time course of the experiment. Depletion of Aβ40 monomers in a high affinity complex would reduce both the nucleation and elongation rates in the same way as having a lower concentration of Aβ40 monomers available for the aggregation process. This would lead to a reduction in the total amount of fibrils at equilibrium and also a less steep growth phase in the sigmoidal curve. In our experiments there is no systematic variation in fluorescence plateau for Aβ40 with and without the different proteins (Figure S2). ThT fluorescence measurements are not always quantitative but under optimized settings it varies linearly with the amount of fibrils in the sample.36 The polydispersity of the fibril sample makes it hard to quantify the amount of fibrils for instance by dynamic light scattering (DLS), but both the ThT plateau fluorescence and the CD data indicate that fibrils of the same amount and general secondary structure are formed in the absence and presence of scMN+8. This and the low stoichiometric ratio required for retardation makes it likely that scMN+8 acts on small oligomers of Aβ40 in addition to the monomers. The low amount (at most 1.5% for Aβ42)36 and the transient nature of on-pathway oligomers makes it very hard to detect an oligomer–protein complex directly. A detailed mechanistic analysis of the aggregation process might give more information on the microscopic events.
Concluding Remarks
Here, we show that charge variants of two hydrophilic proteins retard amyloid formation of Aβ40 in a manner highly dependent on their net charge. The hydrophilic proteins with positive or low negative net charge retard amyloid fibril formation and cause an increase in lag time. The proteins display interactions with Aβ40 monomers, small aggregates, or both, and the mechanism behind the retardation seems to involve decreased rates of the nucleation processes. The electrostatic component of the interaction is important. As both proteins with positive net charge and low negative net charge retard amyloid formation to some degree, we conclude that opposite net charge is not necessary for retardation of aggregation or interaction with Aβ40 peptides. However, in the presence of highly negatively charged proteins, offering substantial electrostatic repulsion of Aβ, the effect seems to be abolished. The absence of an evolutionary connection between Aβ40 and the model proteins suggests a general protein effect leading to retardation of Aβ40 fibril formation. The high surface activity of Aβ40 gives the peptide a multitude of interaction partners, which poses a challenge in the search for Aβ targeting molecules for treatment of AD.
Methods
Materials
All chemicals were of analytical grade.
The Aβ40 and Aβ42 peptides were expressed in Escherichia coli from a synthetic gene. The initial methionine has no significant effect on the aggregation rate.46 In short, the purification procedure involved sonication of E. coli cells, dissolution of inclusion bodies in 8 M urea, anion-exchange in batch mode on DEAE cellulose resin, centrifugation through a 30 kDa molecular weight cutoff (MWCO) filter, and finally concentration using a 3 kDa MWCO filter, as previously described.46 The purified peptide was frozen in identical 1 mL aliquots.
scMN charge substitution mutants (C41S+Q13E+N14D+Q28E+N50E for scMN-2, C41S+M42L for scMN+2, and C41S+E2Q+E4Q+D21N+E22Q+E48Q+E59Q for scMN+8) were expressed in E. coli from synthetic genes and purified using cation and/or anion exchange and gel filtration chromatography as previously described.28 Native monellin consists of two peptide chains, but here we use a single chain variant with a glycine (scMN-2, scMN+8) or a methionine (scMN+2) as a linker between the two chains. The C41S mutation is added to eliminate disulfide bond formation. The noncharge substitution mutation M42L has no effect on the Aβ40 aggregation (data not shown).
CB (bovine minor A) charge substitution mutants (N21D for CB-8, E26Q for CB-6, and E17Q+D19N+E26Q for CB-4) were expressed in E. coli from synthetic genes and purified using ion anion-exchange and gel filtration chromatography as described.30,64,65
Calmodulin was expressed in E. coli and purified as described.66
Preparation of Proteins
The freeze-dried CB mutants, scMN+2, scMN-2, and calmodulin were dissolved in the appropriate buffer and centrifuged 7 min at 13 000 g to remove any nondissolved particles. Frozen aliquots of scMN+8 were reconditioned to the correct buffer with a NAP-10 column (GE Healthcare, Buckinghamshire, U.K.). The protein concentrations were spectrophotometrically determined at 280 nm with the extinction coefficients 1490 M–1 cm–1 for CB,67 14600 M–1 cm–1 for scMN,68 and 3200 M–1 cm–1 for calmodulin.69 To retain CB in the apo form, 200 μM EDTA was added to all buffers.
Preparation of Multiwell Plates for Kinetic Experiments
For kinetic experiments with Aβ40, aliquots of purified peptide were freeze-dried, dissolved in 6 M GuHCl, and subjected to gel filtration on a Superdex 75 column in 20 mM sodium phosphate buffer, pH 7.4, with 200 μM EDTA. The monomer fraction was collected and kept on ice. The monomer concentration was calculated from integration of the chromatogram absorbance at 280 nm using an extinction coefficient of 1440 M–1 cm–1.46,49 The collected monomer was diluted to prepare a solution of 20 μM Aβ in 20 mM sodium phosphate buffer, 200 μM EDTA, pH 7.4 supplemented with 14 μM ThT (Calbiochem) from a 1.4 mM stock solution. The Aβ40 solution was then added, 50 μL per well, to a 96-well half area plate of black polystyrene with clear bottom and nonbinding surface (Corning 3881) on ice. Before adding Aβ40, each well had been provided with 50 μL degassed buffer or 50 μL of the protein to be tested in the same working buffer with pH set to 7.4. The buffer concentration of 20 mM sodium phosphate is enough to keep the pH stable throughout the aggregation process. All concentrations of Aβ and the other proteins given in the Results section and figure legends are the final concentrations after mixing the samples in the wells. Before incubation in the plate reader, the plate was sealed with a plastic film (Corning 3095).
Aβ42 was prepared for kinetic experiments in the same manner as Aβ40, with the following exceptions: the pH of the buffer for the gel filtration and kinetic experiment was 8.0, the final peptide concentration in the well was 3 μM, and the ThT concentration was 6 μM.
Kinetic Aggregation Experiment
The experiment was initiated by placing the 96-well plate at 37 °C without shaking in a plate reader (Fluostar Omega or Fluostar Optima, BMG Labtech, Offenburg, Germany). The ThT fluorescence was measured through the bottom of the plate every 5 min (with excitation filter 440 nm, and emission filter 480 nm) for 66 h.
The sigmoidal aggregation curves were normalized in OriginPro (OriginLab corporation, Northampton, MA) and fitted to
| 1 |
where A1 and A2 are the minimum and maximum fluorescence, t1/2 is the halftime of the reaction, and dx is the inverse apparent elongation rate.
Transmission Electron Microscopy
Samples from the kinetics studies with 10 μM Aβ40 and 10 μM of the scMN and CB mutants were spotted on 300 mesh Formvar carbon film grids (Electron Microscopy Sciences, Hatfield, PA) after the aggregation had reached equilibrium. An aliquot of 5 μL of the sample was placed on the grid for 3 min. Then the grid was blotted and placed on a drop of 1.5% uranyl acetate (Merck) for another 3 min, blotted, and rinsed with two drops of water (Milli-Q). The samples were analyzed with a Philips CM120 BioTWIN Cryo apparatus at 6200× and 31 000× magnification.
Circular Dichroism Spectroscopy
CD spectra were recorded at 37 °C in a 10 mm quartz cuvette using a JASCO J-810 spectropolarimeter. Far-UV spectra were recorded at 0.5 nm intervals between 185 and 260 nm using a scan rate of 20 nm/min, with response time of 8 s and a band-pass of 1 nm. Aβ40 monomer was isolated by gel filtration in 5 mM sodium phosphate buffer, pH 7.4, with 40 mM NaF and diluted to 4 μM in the same buffer with or without 0.04 μM scMN+8. Spectra of agitated samples were recorded during the lag phase and in the final stage reached after 24 h incubation.
ANS Fluorescence Measurement
The ANS fluorescence was measured using a Perkin-Elmer luminescence spectrometer LS 50 B connected to a Julabo thermostatic water bath at 37 °C. The emission spectra were recorded between 400 and 600 nm with excitation at 385 nm. In addition to the six scMN and CB mutants in phosphate buffer (20 mM sodium phosphate buffer, 200 μM EDTA, pH 7.4) we measured the ANS binding to calmodulin (in 5 mM Tris-HCl, pH 7.4, 20 mM NaCl and 100 μM CaCl2) as positive control and phosphate buffer alone as negative control. The proteins were tested at 10 μM together with five concentrations of ANS between 5 and 30 μM. Spectra from the proteins without ANS were subtracted from the result to compensate for background fluorescence. The data have been smoothened with a sliding average over three measuring points.
Surface Plasmon Resonance
Monomeric Aβ40, prepared as for the kinetic experiment, was immobilized on a CM5 chip with standard amine coupling in a Biacore 3000 (GE Healthcare, Uppsala, Sweden) as described by the manufacturer and Brännström et al.70 Briefly, 10 μM Aβ40 was titrated with 100 mM NaAc pH 2.75, to a final pH of 5, and immobilized on the chip surface, activated with N-ethyl-N-(dimethylaminopropyl) carbodiimide (EDC) and N-hydroxysuccinimide (NHS), to a level of about 470 RU. Both the flow cell with immobilized Aβ40 and a reference cell without any peptide bound were inactivated with ethanolamine to block any free amine binding sites. The interaction between Aβ40 and the scMN and CB mutants was studied in filtered and degassed PBS (pH 7.4) with 200 μM EDTA and 0.005% Tween 20. The flow rate was generally 10 μL/min, and the temperature 25 °C. Each protein injection was 270 μL, which gives an association time of 27 min. The dissociation time was typically 3 h and then the surface was regenerated twice with 5 μL 6 M GuHCl in running buffer to reset the signal to the initial level.
Analysis of SPR Data
The response signal (R) from the reference cell without immobilized Aβ40 was subtracted from the R of the sample cell before analyzing the data. The signal at the start of the injection was subtracted to facilitate comparison between the samples. Graphpad Prism (GraphPad Software, San Diego, CA) was used for all curve fitting.
To minimize undesired effects from secondary binding events, we selected one of the lower concentrations with significant binding for each protein. This corresponds to 300 nM for scMN+8, 1 μM for scMN-2, and 10 μM for CB-4. Assuming a 1:1 binding model, the dissociation phase was fitted to the following equation:
| 2 |
t1 is the time at the start of the dissociation phase, and R0 is the response at t1. The dissociation rate, as opposed to the association rate, is not dependent on protein concentration due to the buffer flowing over the chip during the dissociation phase. The initial fast decrease in the dissociation is considered an artifact based on the second binding event and the latter part of the curve is so flat that only a maximal koff can be determined. The koff cannot be determined more specific than <10–5 s–1 (see Figure S6) for any of the protein variants due to noise levels and instrumental drift. The upper limit in koff translates to an upper limit of KD through
| 3 |
The association phase of the data was fitted to a one phase association
| 4 |
where A is a constant. The observed association rate constant, kobs, reflects a combination of association and dissociation events through
| 5 |
c is the analyte concentration, and koff is 10–5 s–1 as the maximal value determined above. kobs is 2 orders of magnitude larger than koff, which means that the uncertainty in determination of koff does not affect kon.
Acknowledgments
We thank Birgitta From, Eva Thulin and Hanna Nilsson at the divisions of Biochemistry and Structural Biology and Biophysical Chemistry, Lund university, for help with preparation of the proteins used in this study. We also thank Gunnel Karlsson at the Biomicroscopy Unit, Polymer and Materials Chemistry, Chemical Centre, Lund University, for the assistance with the TEM work.
Supporting Information Available
Figures S1–S6 with raw data and complementary analyses. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
S.L., A.A., and E.H. designed the study. A.A. and S.L. performed the experiments. A.A., E.H., C.C.-L, and S.L. analyzed the data and prepared the manuscript.
This study was funded by the Swedish Research Council (VR) and its Linneaus Centre Organizing Molecular Matter (OMM) (S.L.), nanometer structure consortium at Lund University (S.L., C.C.-L.), the Crafoord Foundation (S.L., C.C.-L.), the Royal Physiographic Society (C.C.-L., E.H.).
The authors declare no competing financial interest.
Supplementary Material
References
- Brookmeyer R.; Johnson E.; Ziegler-Graham K.; Arrighi H. M. (2007) Forecasting the global burden of Alzheimer’s disease. Alzheimer’s Dementia 3, 186–191. [DOI] [PubMed] [Google Scholar]
- Alzheimer A. (1907) Über eine eigenartige Erkrankung der Hirnrinde. Allg. Z. Psychiatr. 64, 146–148. [Google Scholar]
- Selkoe D. J. (1991) The molecular pathology of Alzheimer’s disease. Neuron 6, 487–498. [DOI] [PubMed] [Google Scholar]
- Finder V. H. (2010) Alzheimer’s disease: a general introduction and pathomechanism. J. Alzheimer’s Dis. 22, 5–19. [DOI] [PubMed] [Google Scholar]
- Finder V. H.; Glockshuber R. (2007) Amyloid-beta aggregation. Neurodegener. Dis. 4, 13–27. [DOI] [PubMed] [Google Scholar]
- Baldwin A. J.; Knowles T. P. J.; Tartaglia G. G.; Fitzpatrick A. W.; Devlin G. L.; Shammas S. L.; Waudby C. A.; Mossuto M. F.; Meehan S.; Gras S. L.; Christodoulou J.; Anthony-Cahill S. J.; Barker P. D.; Vendruscolo M.; Dobson C. M. (2011) Metastability of Native Proteins and the Phenomenon of Amyloid Formation. J. Am. Chem. Soc. 133, 14160–14163. [DOI] [PubMed] [Google Scholar]
- Hellstrand E.; Boland B.; Walsh D. M.; Linse S. (2010) Amyloid beta-Protein Aggregation Produces Highly Reproducible Kinetic Data and Occurs by a Two-Phase Process. ACS Chem. Neurosci. 1, 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Gonnelli L.; Luchinat C.; Mao J.; Nesi A. (2011) A new structural model of Abeta40 fibrils. J. Am. Chem. Soc. 133, 16013–16022. [DOI] [PubMed] [Google Scholar]
- Luhrs T.; Ritter C.; Adrian M.; Riek-Loher D.; Bohrmann B.; Dobeli H.; Schubert D.; Riek R. (2005) 3D structure of Alzheimer’s amyloid-beta(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes A. J.; Parker K. D.; Atkins E. D.; Beighton E. (1968) “Cross-beta” conformation in proteins. J. Mol. Biol. 32, 343–358. [DOI] [PubMed] [Google Scholar]
- Härd T.; Lendel C. (2012) Inhibition of amyloid formation. J. Mol. Biol. 421, 441–465. [DOI] [PubMed] [Google Scholar]
- Hughes E.; Burke R. M.; Doig A. J. (2000) Inhibition of toxicity in the beta-amyloid peptide fragment beta-(25–35) using N-methylated derivatives: a general strategy to prevent amyloid formation. J. Biol. Chem. 275, 25109–25115. [DOI] [PubMed] [Google Scholar]
- Kokkoni N.; Stott K.; Amijee H.; Mason J. M.; Doig A. J. (2006) N-Methylated Peptide Inhibitors of β-Amyloid Aggregation and Toxicity. Optimization of the Inhibitor Structure. Biochemistry 45, 9906–9918. [DOI] [PubMed] [Google Scholar]
- Cabaleiro-Lago C.; Quinlan-Pluck F.; Lynch I.; Dawson K. A.; Linse S. (2010) Dual Effect of Amino Modified Polystyrene Nanoparticles on Amyloid beta Protein Fibrillation. ACS Chem. Neurosci. 1, 279–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabaleiro-Lago C.; Quinlan-Pluck F.; Lynch I.; Lindman S.; Minogue A. M.; Thulin E.; Walsh D. M.; Dawson K. A.; Linse S. (2008) Inhibition of amyloid beta protein fibrillation by polymeric nanoparticles. J. Am. Chem. Soc. 130, 15437–15443. [DOI] [PubMed] [Google Scholar]
- Rocha S.; Thunemann A. F.; Pereira Mdo C.; Coelho M.; Mohwald H.; Brezesinski G. (2008) Influence of fluorinated and hydrogenated nanoparticles on the structure and fibrillogenesis of amyloid beta-peptide. Biophys. Chem. 137, 35–42. [DOI] [PubMed] [Google Scholar]
- Liao Y. H.; Chang Y. J.; Yoshiike Y.; Chang Y. C.; Chen Y. R. (2012) Negatively charged gold nanoparticles inhibit Alzheimer’s amyloid-beta fibrillization, induce fibril dissociation, and mitigate neurotoxicity. Small 8, 3631–3639. [DOI] [PubMed] [Google Scholar]
- Heegaard P. M.; Boas U.; Otzen D. E. (2007) Dendrimer effects on peptide and protein fibrillation. Macromol. Biosci. 7, 1047–1059. [DOI] [PubMed] [Google Scholar]
- Stanyon H. F.; Viles J. H. (2012) Human serum albumin can regulate amyloid-beta peptide fiber growth in the brain interstitium: implications for Alzheimer disease. J. Biol. Chem. 287, 28163–28168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.; Warmlander S. K.; Graslund A.; Abrahams J. P. (2013) Human lysozyme inhibits the in vitro aggregation of Abeta peptides, which in vivo are associated with Alzheimer’s disease. Chem Commun (Cambridge, U.K.) 49, 6507–6509. [DOI] [PubMed] [Google Scholar]
- Carrotta R.; Canale C.; Diaspro A.; Trapani A.; Biagio P. L.; Bulone D. (2012) Inhibiting effect of alpha(s1)-casein on Abeta(1–40) fibrillogenesis. Biochim. Biophys. Acta 1820, 124–132. [DOI] [PubMed] [Google Scholar]
- Hoyer W.; Grönwall C.; Jonsson A.; Ståhl S.; Härd T. (2008) Stabilization of a beta-hairpin in monomeric Alzheimer’s amyloid-beta peptide inhibits amyloid formation. Proc. Natl. Acad. Sci. U.S.A. 105, 5099–5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecroyd H.; Carver J. A. (2009) Crystallin proteins and amyloid fibrils. Cell. Mol. Life Sci. 66, 62–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shammas S. L.; Waudby C. A.; Wang S.; Buell A. K.; Knowles T. P.; Ecroyd H.; Welland M. E.; Carver J. A.; Dobson C. M.; Meehan S. (2011) Binding of the molecular chaperone alphaB-Crystallin to Abeta amyloid fibrils inhibits fibril elongation. Biophys. J. 101, 1681–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willander H.; Presto J.; Askarieh G.; Biverstål H.; Frohm B.; Knight S. D.; Johansson J.; Linse S. (2012) BRICHOS domains efficiently delay fibrillation of amyloid beta-peptide. J. Biol. Chem. 287, 31608–31617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara H.; Inaguma Y.; Goto S.; Inagaki T.; Kato K. (1993) Alpha B Crystallin and HSP28 are enhanced in the cerebral cortex of patients with Alzheimer’s disease. J. Neurol. Sci. 119, 203–208. [DOI] [PubMed] [Google Scholar]
- Kuo Y. M.; Kokjohn T. A.; Kalback W.; Luehrs D.; Galasko D. R.; Chevallier N.; Koo E. H.; Emmerling M. R.; Roher A. E. (2000) Amyloid-beta peptides interact with plasma proteins and erythrocytes: implications for their quantitation in plasma. Biochem. Biophys. Res. Commun. 268, 750–756. [DOI] [PubMed] [Google Scholar]
- Xue W. F.; Szczepankiewicz O.; Bauer M. C.; Thulin E.; Linse S. (2006) Intra- versus intermolecular interactions in monellin: contribution of surface charges to protein assembly. J. Mol. Biol. 358, 1244–1255. [DOI] [PubMed] [Google Scholar]
- Akke M.; Forsen S. (1990) Protein stability and electrostatic interactions between solvent exposed charged side chains. Proteins 8, 23–29. [DOI] [PubMed] [Google Scholar]
- Fast J.; Hakansson M.; Muranyi A.; Gippert G. P.; Thulin E.; Evenas J.; Svensson L. A.; Linse S. (2001) Symmetrical stabilization of bound Ca2+ ions in a cooperative pair of EF-hands through hydrogen bonding of coordinating water molecules in calbindin D(9k). Biochemistry 40, 9887–9895. [DOI] [PubMed] [Google Scholar]
- Xue W. F.; Szczepankiewicz O.; Thulin E.; Linse S.; Carey J. (2009) Role of protein surface charge in monellin sweetness. Biochim. Biophys. Acta 1794, 410–420. [DOI] [PubMed] [Google Scholar]
- Linse S.; Johansson C.; Brodin P.; Grundstrom T.; Drakenberg T.; Forsen S. (1991) Electrostatic contributions to the binding of Ca2+ in calbindin D9k. Biochemistry 30, 154–162. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.4.1, Schrödinger, LLC.
- Cohen S. I.; Vendruscolo M.; Dobson C. M.; Knowles T. P. (2012) From macroscopic measurements to microscopic mechanisms of protein aggregation. J. Mol. Biol. 421, 160–171. [DOI] [PubMed] [Google Scholar]
- Levine H. (1993) Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 2, 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S. I.; Linse S.; Luheshi L. M.; Hellstrand E.; White D. A.; Rajah L.; Otzen D. E.; Vendruscolo M.; Dobson C. M.; Knowles T. P. (2013) Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. U.S.A. 110, 9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fandrich M.; Meinhardt J.; Grigorieff N. (2009) Structural polymorphism of Alzheimer Abeta and other amyloid fibrils. Prion 3, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson C. M. (1999) Protein misfolding, evolution and disease. Trends Biochem. Sci. 24, 329–332. [DOI] [PubMed] [Google Scholar]
- Konno T. (2001) Multistep nucleus formation and a separate subunit contribution of the amyloidgenesis of heat-denatured monellin. Protein Sci. 10, 2093–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno T.; Murata K.; Nagayama K. (1999) Amyloid-like aggregates of a plant protein: a case of a sweet-tasting protein, monellin. FEBS Lett. 454, 122–126. [DOI] [PubMed] [Google Scholar]
- Shen L.; Adachi T.; Vanden Bout D.; Zhu X. Y. (2012) A Mobile Precursor Determines Amyloid-β Peptide Fibril Formation at Interfaces. J. Am. Chem. Soc. 134, 14172–14178. [DOI] [PubMed] [Google Scholar]
- Rocha S.; Krastev R.; Thunemann A. F.; Pereira M. C.; Mohwald H.; Brezesinski G. (2005) Adsorption of amyloid beta-peptide at polymer surfaces: a neutron reflectivity study. ChemPhysChem 6, 2527–2534. [DOI] [PubMed] [Google Scholar]
- Stryer L. (1965) The interaction of a naphthalene dye with apomyoglobin and apohemoglobin. A fluorescent probe of non-polar binding sites. J. Mol. Biol. 13, 482–495. [DOI] [PubMed] [Google Scholar]
- Berggård T.; Silow M.; Thulin E.; Linse S. (2000) Ca2+- and H+-dependent conformational changes of calbindin D-28k. Biochemistry 39, 6864–6873. [DOI] [PubMed] [Google Scholar]
- Grönwall C.; Jonsson A.; Lindström S.; Gunneriusson E.; Ståhl S.; Herne N. (2007) Selection and characterization of Affibody ligands binding to Alzheimer amyloid beta peptides. J. Biotechnol. 128, 162–183. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Thulin E.; Minogue A. M.; Gustavsson N.; Pang E.; Teplow D. B.; Linse S. (2009) A facile method for expression and purification of the Alzheimer’s disease-associated amyloid beta-peptide. FEBS J. 276, 1266–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J.; Yu C. H.; Yu H.; Borstnar R.; Kamerlin S. C.; Graslund A.; Abrahams J. P.; Warmlander S. K. (2013) Cellular polyamines promote amyloid-beta (abeta) peptide fibrillation and modulate the aggregation pathways. ACS Chem. Neurosci. 4, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrand E. (2012) Protein-Lipid Association and Aggregation From Neurodegenerative Disease to Nanosafety. Ph.D. Thesis, Lund University, Lund. ISBN 978-91-7422-313-2. [Google Scholar]
- Hellstrand E.; Sparr E.; Linse S. (2010) Retardation of Abeta fibril formation by phospholipid vesicles depends on membrane phase behavior. Biophys. J. 98, 2206–2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Zimbron L. F.; Luna-Munoz J.; Mena R.; Vazquez-Ramirez R.; Kubli-Garfias C.; Cribbs D. H.; Manoutcharian K.; Gevorkian G. (2012) Amyloid-beta peptide binds to cytochrome C oxidase subunit 1. PLoS One 7, e42344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustbader J. W.; Cirilli M.; Lin C.; Xu H. W.; Takuma K.; Wang N.; Caspersen C.; Chen X.; Pollak S.; Chaney M.; Trinchese F.; Liu S.; Gunn-Moore F.; Lue L. F.; Walker D. G.; Kuppusamy P.; Zewier Z. L.; Arancio O.; Stern D.; Yan S. S.; Wu H. (2004) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304, 448–452. [DOI] [PubMed] [Google Scholar]
- Bohrmann B.; Tjernberg L.; Kuner P.; Poli S.; Levet-Trafit B.; Näslund J.; Richards G.; Huber W.; Dobeli H.; Nordstedt C. (1999) Endogenous proteins controlling amyloid beta-peptide polymerization. Possible implications for beta-amyloid formation in the central nervous system and in peripheral tissues. J. Biol. Chem. 274, 15990–15995. [DOI] [PubMed] [Google Scholar]
- Chauhan V. P. S.; Ray I.; Chauhan A.; Wisniewski H. M. (1999) Binding of gelsolin, a secretory protein, to amyloid beta-protein. Biochem. Biophys. Res. Commun. 258, 241–246. [DOI] [PubMed] [Google Scholar]
- Goldgaber D.; Schwarzman A. I.; Bhasin R.; Gregori L.; Schmechel D.; Saunders A. M.; Roses A. D.; Strittmatter W. J. (1993) Sequestration of amyloid beta-peptide. Ann. N.Y. Acad. Sci. 695, 139–143. [DOI] [PubMed] [Google Scholar]
- Morris J. A.; Martenson R.; Deibler G.; Cagan R. H. (1973) Characterization of monellin, a protein that tastes sweet. J. Biol. Chem. 248, 534–539. [PubMed] [Google Scholar]
- O’Connell D. J.; Bauer M.; Linse S.; Cahill D. J. (2011) Probing calmodulin protein-protein interactions using high-content protein arrays. Methods Mol. Biol. 785, 289–303. [DOI] [PubMed] [Google Scholar]
- Weber G.; Laurence D. J. R. (1954) Fluorescent indicators of adsorption in aqueous solution and on the solid phase. Biochem. J. 56, R31–R31. [PubMed] [Google Scholar]
- Narayanan S.; Kamps B.; Boelens W. C.; Reif B. (2006) αB-Crystallin competes with Alzheimer’s disease β-amyloid peptide for peptide–peptide interactions and induces oxidation of Abeta-Met35. FEBS Lett. 580, 5941–5946. [DOI] [PubMed] [Google Scholar]
- Hoyer W.; Härd T. (2008) Interaction of Alzheimer’s A beta peptide with an engineered binding protein—thermodynamics and kinetics of coupled folding-binding. J. Mol. Biol. 378, 398–411. [DOI] [PubMed] [Google Scholar]
- Asomugha C. O.; Gupta R.; Srivastava O. P. (2011) Structural and functional roles of deamidation of N146 and/or truncation of NH2- or COOH-termini in human alphaB-Crystallin. Mol. Vision 17, 2407–2420. [PMC free article] [PubMed] [Google Scholar]
- Rozga M.; Kloniecki M.; Jablonowska A.; Dadlez M.; Bal W. (2007) The binding constant for amyloid Abeta40 peptide interaction with human serum albumin. Biochem. Biophys. Res. Commun. 364, 714–718. [DOI] [PubMed] [Google Scholar]
- Meisl G., Yang X., Hellstrand E., Frohm B., Cohen S., Dobson C., Linse S., and Knowles T. (2013) Differences in nucleation behaviour underlie the contrasting aggregation pathways of the Aβ40 and Aβ42 peptide. Manuscript in preparation. [DOI] [PMC free article] [PubMed]
- Knowles T. P.; Waudby C. A.; Devlin G. L.; Cohen S. I.; Aguzzi A.; Vendruscolo M.; Terentjev E. M.; Welland M. E.; Dobson C. M. (2009) An analytical solution to the kinetics of breakable filament assembly. Science 326, 1533–1537. [DOI] [PubMed] [Google Scholar]
- Johansson C.; Brodin P.; Grundström T.; Thulin E.; Forsen S.; Drakenberg T. (1990) Biophysical studies of engineered mutant proteins based on calbindin D9k modified in the pseudo EF-hand. Eur. J. Biochem. 187, 455–460. [DOI] [PubMed] [Google Scholar]
- Linse S.; Brodin P.; Johansson C.; Thulin E.; Grundström T.; Forsen S. (1988) The role of protein surface charges in ion binding. Nature 335, 651–652. [DOI] [PubMed] [Google Scholar]
- Waltersson Y.; Linse S.; Brodin P.; Grundström T. (1993) Mutational effects on the cooperativity of Ca2+ binding in calmodulin. Biochemistry 32, 7866–7871. [DOI] [PubMed] [Google Scholar]
- Pace C. N.; Vajdos F.; Fee L.; Grimsley G.; Gray T. (1995) How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 4, 2411–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J. A.; Cagan R. H. (1980) Formation of oligomeric monellin in protein denaturants. Proc. Soc. Exp. Biol. Med. 164, 351–354. [DOI] [PubMed] [Google Scholar]
- Bauer M. C.; O’Connell D.; Cahill D. J.; Linse S. (2008) Calmodulin binding to the polybasic C-termini of STIM proteins involved in store-operated calcium entry. Biochemistry 47, 6089–6091. [DOI] [PubMed] [Google Scholar]
- Brännström K.; Öhman A.; Olofsson A. (2011) Abeta peptide fibrillar architectures controlled by conformational constraints of the monomer. PLoS One 6, e25157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.