

Abstract
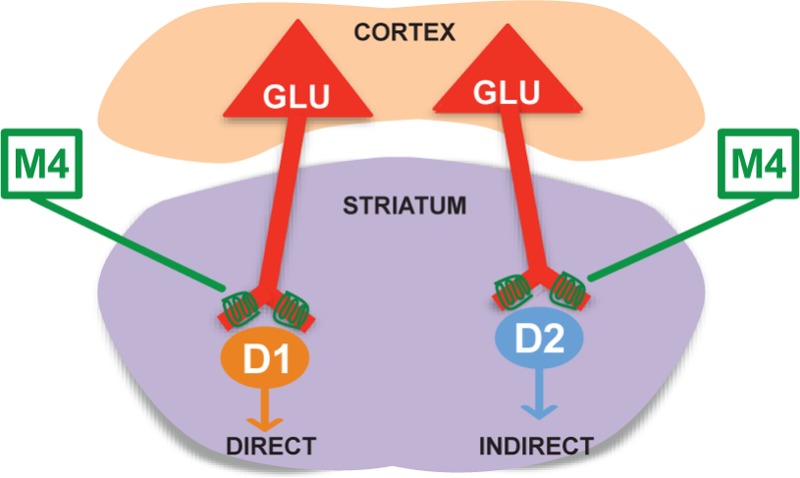
The striatum is the main input station of the basal ganglia and is extensively involved in the modulation of motivated behavior. The information conveyed to this subcortical structure through glutamatergic projections from the cerebral cortex and thalamus is processed by the activity of several striatal neuromodulatory systems including the cholinergic system. Acetylcholine potently modulates glutamate signaling in the striatum via activation of muscarinic receptors (mAChRs). It is, however, unclear which mAChR subtype is responsible for this modulatory effect. Here, by using electrophysiological, optogenetic, and immunoelectron microscopic approaches in conjunction with a novel, highly selective M4 positive allosteric modulator VU0152100 (ML108) and M4 knockout mice, we show that M4 is a major mAChR subtype mediating the cholinergic inhibition of corticostriatal glutamatergic input on both striatonigral and striatopallidal medium spiny neurons (MSNs). This effect is due to activation of presynaptic M4 receptors, which, in turn, leads to a decrease in glutamate release from corticostriatal terminals. The findings of the present study raise the interesting possibility that M4 mAChR could be a novel therapeutic target for the treatment of neurological and neuropsychiatric disorders involving hyper-glutamatergic transmission at corticostriatal synapses.
Keywords: Basal ganglia, glutamate, acetylcholine, excitotoxicity, muscarinic, dopamine
The striatum is the main entry to the basal ganglia circuitry. It is mostly comprised of medium spiny neurons (MSNs), accounting for more than 95% of all striatal neuronal population.1 MSNs are generally subdivided in two broad subpopulations, D1 MSNs (direct pathway) and D2 MSNs (indirect pathway). A balanced activation of these two pathways is necessary for normal motor and nonmotor functions. MSNs activity is driven by conspicuous glutamatergic afferents from the cerebral cortex and thalamus,2 while neuromodulators such as dopamine (DA) and acetylcholine (ACh) regulate neuronal excitability and responses to glutamate. Abnormalities in striatal neuromodulators signaling that affect glutamatergic transmission have been implicated in the etiology of neuropsychiatric and neurodegenerative disorders including Parkinson’s disease, Huntington’s disease, schizophrenia, and depression.3−5
ACh reduces corticostriatal glutamate release through the activation of presynaptic muscarinic receptors.6−8 Recent studies have implicated specific changes in transmission at corticostriatal synapses, including alterations of cholinergic modulation of corticostriatal transmission in multiple basal ganglia disorders such as dystonia,9 obsessive compulsive disorder (OCD),10 and addictive disorders.11 Based on this, it has been postulated that selectively targeting mAChR involved in regulating corticostriatal transmission could provide a novel approach for the treatment of these and other basal ganglia disorders. Previous anatomical and pharmacological studies suggested that the M2 and possibly M3 mAChR subtypes play a dominant role in regulating corticostriatal transmission.12−16 However, the lack of highly selective subtype-specific pharmacological tools has made it difficult to determine the exact role of individual mAChR subtypes in the regulation of striatal glutamatergic signaling. Interestingly, while M4 is expressed postsynaptically on striatal projection neurons and interneurons,17,18 it is also found together with M2, in presynaptic glutamatergic terminals in the rodent striatum.19
In the present study, we use the novel, highly selective, M4 positive allosteric modulator (M4-PAM), VU0152100 (ML108),20 in combination with electrophysiology, optogenetics, and mice bearing genetic deletion of M4 to demonstrate that M4 mAChRs play an essential role in the cholinergic inhibition of corticostriatal glutamatergic transmission.
Results and Discussion
M4 mAChR Activation Decreases Glutamatergic Transmission at Corticostriatal Synapses
Although cholinergic muscarinic receptors (mAChRs) play an important role in regulating glutamatergic transmission at corticostriatal synapses, the specific mAChR subtype(s) responsible for this effect has not been determined. In this study, we used a highly selective M4-PAM, VU0152100, to define the role of M4 in regulating glutamatergic transmission at the corticostriatal synapse onto MSNs. Consistent with previous studies,6,7,14 mAChR activation with carbachol (CCh) caused a concentration-dependent inhibition of EPSCs evoked in MSNs by stimulating the corticostriatal afferent fibers (12.4 ± 4.7% inhibition at 1 μM, n = 5; 36.9 ± 5.9% inhibition at 3 μM, n = 10; Figure 1; p < 0.05, t test). When coapplied with VU0152100 (5 μM), both concentrations of CCh induced significantly greater depression of EPSC amplitude compared with CCh alone (Figure 1, p < 0.01). Application of 5 μM VU0152100 alone also caused a small, but significant, reduction of EPSC amplitude (n = 10, p < 0.01), likely due to potentiation of endogenous ACh action on M4. Furthermore, in M4-KO mice CCh (3 and 100 μM) failed to inhibit EPSCs (Figure 2; p > 0.05, paired t test), which were significantly different from the CCh effects in WT (Figure 2; p < 0.01 for 3 μM CCh and p < 0.001 for 100 μM CCh). Taken together, these results suggest that muscarinic depression of excitatory transmission at corticostriatal synapses is mediated mainly by M4.
Figure 1.

VU0152100 potentiates CCh-induced reduction of EPSC amplitude at corticostriatal synapses in MSNs. (A,B) Representative traces of averaged EPSCs showing the effects of 1 μM CCh (A1), 3 μM CCh (B1), 1 μM CCh with 5 μM VU0152100 (A2), and 1 μM CCh with 5 μM VU0152100 (B2) on EPSCs. (C,D) Bar graphs summarizing the potentiation effect of VU0152100 on the CCh-induced reduction of EPSC amplitude by 1 μM CCh (C) and 3 μM CCh (D). Data are presented as mean ± SEM (**p < 0.01, t test). Insets show the effects of VU0152100 + CCh on PPR (inset in C: 5 μM VU0152100 with 1 μM CCh; inset in D: 5 μM VU0152100 with 3 μM CCh, *p < 0.05, paired t test).
Figure 2.

Loss of CCh effect on EPSCs in M4 KO mice. (A,B) Representative traces of averaged EPSCs showing the effects of 3 μM CCh (A) and 100 μM CCh (B) on EPSCs in WT mice (A1 and B1) and M4 KO mice (A2 and B2). (C) Bar graph summarizing the effects of 3 μM and 100 μM CCh on EPSC amplitude in WT and M4-KO mice. Bars represent the percentage inhibition compared to baseline shown as mean ± SEM (**p < 0.01, ***p < 0.001, t test).
M4-Mediated Depression of EPSCs is Due to Decrease in Glutamate Release by Activation of Presynaptic M4 mAChRs
The potentiation of CCh-induced depression of EPSCs by VU0152100 is associated with an increase in paired pulse ratio (PPR) (Figure 1, n = 6, p < 0.05, paired t test), thereby indicating that M4-mediated depression of EPSCs in MSNs is likely due to decrease in presynaptic glutamate release.
Because basal ganglia function relies on balanced output from D1 and D2 MSNs (see above), we sought to determine if M4 activation has differential modulatory effects on corticostriatal synapses in D1 vs D2 MSNs. Using D1-EGFP mice, we recorded EPSCs from D1 MSNs and non-D1 MSNs (putative D2 MSNs). As in WT mice (Figure 1B, D), CCh (3 μM) reduced EPSC amplitude in both D1 and non-D1 MSNs to a similar extent (n = 4 and n = 6, respectively, p > 0.05; Figure 3C, D). In the presence of VU0152100 (5 μM), CCh (3 μM) caused a significantly greater depression of EPSC amplitude than CCh alone in both D1 (Figure 3C, n = 4) and non-D1 MSNs (Figure 3D, n = 5; p < 0.01 for both D1 and non-D1 MSNs). Also, the M4-dependent potentiation of CCh-induced depression of EPSCs was associated with increases in PPR for D1 and non-D1 MSNs (Figure 3C and D insets; p < 0.05 in a paired t test, for both D1 and non-D1 MSNs). These results indicate that M4 is critically involved in cholinergic modulation of glutamate release at corticostriatal synapses on both direct and indirect pathway MSNs.
Figure 3.

M4-mediated depression of EPSCs is due to decrease in glutamate release by activation of presynaptic M4 mAChRs. (A,B) Representative traces of averaged EPSCs showing the effects of 3 μM CCh in the presence of 5 μM VU1052100 in D1 (A) and non-D1 (B) MSNs. (C,D) Bar graphs summarizing the potentiation effect of VU0152100 on the CCh-induced reduction of EPSC amplitude in D1 (C) and non-D1 (D) MSNs. Insets show the effect of 3 μM CCh + 5 μM VU0152100 on PPR in D1 (C) and non-D1 (D) MSNs. Bars represent the percentage inhibition compared to baseline shown as mean ± SEM (*p < 0.05, paired t test; **p < 0.01, t test). (E) Representative traces of averaged EPSCs (left) and summary bar graph (right) showing the effect of 3 μM CCh on EPSC amplitude in MSNs recorded with patch pipets with and without GDP-β-S (*p < 0.05, t test). (F) Electron micrographs showing M4-immunoreactive terminals (Te) forming asymmetric axo-spinous synapses with unlabeled and labeled spines (an unlabeled terminal also shown, u.Te). Scale bars: 0.5 μm. (G) Representative traces of averaged EPSCs (left) and summary bar graph (right) showing the effect of 3 μM CCh on EPSC amplitude in the presence of the cocktail of D1/D2 antagonists (*p < 0.05, paired t test).
To determine if postsynaptic M4 receptors played any roles in this effect, we substituted GTP in the intracellular solution with GDP-β-S (100 μM), a nonhydrolyzable analogue of GDP blocking G-protein-coupled-receptor signaling, and found that CCh (3 μM) caused a similar inhibition of EPSC amplitude under this condition (GDP-β-S n = 5; GTP, n = 10; p > 0.05 t test; Figure 3E). Collectively, these data suggest that cholinergic inhibition of corticostriatal synaptic transmission is primarily mediated by presynaptic M4 mAChRs. The anatomical substrate for these physiological effects was further confirmed at the electron microscopic level showing substantial presynaptic M4 expression in putative glutamatergic terminals forming axo-spinous asymmetric synapses in the rodent striatum (Figure 3F).
M4 activation modulates dopamine release in the striatum, while dopamine receptor activation regulates corticostriatal synaptic transmission.21,22 To determine if M4-mediated reduction of corticostriatal glutamatergic transmission is indirectly mediated through muscarinic modulation of the nigrostriatal dopaminergic system, we performed an experiment analogous to that in Figure 2, in the presence of a cocktail of D1 and D2 receptor antagonists (20 μM SCH23390/10 μM sulpiride). This cocktail had no significant effect on EPSC amplitude (p > 0.05, paired t test). Subsequent coapplication of 3 μM CCh with the D1/D2 antagonists, indeed, reduced EPSC amplitude compared to baseline (p < 0.05, paired t test), with a magnitude similar to that observed with 3 μM CCh alone (p > 0.05, Figure 3G). These data indicate that the M4-mediated modulation of glutamatergic transmission at corticostriatal synapses does not rely upon M4-mediated effects on the nigrostriatal dopaminergic system.
M4-PAM Enhances EPSC Depression Induced by Synchronized Activation of Cholinergic Interneurons (ChIs)
To further characterize the impact of endogenous ACh release on M4-mediated regulation of corticostriatal glutamatergic transmission in MSNs, we used optogenetic techniques to induce a brief increase in ChIs activity and determine if M4 activation under such physiological conditions could modulate corticostriatal glutamatergic transmission in MSNs. This was achieved by using transgenic mice that express an improved channelrhodoposin-2/EYFP fusion protein (mhChR2::YFP) in ChIs under the control of mouse choline acetyltransferase promoter (31). Illuminating ChR2-expressing ChIs with blue light resulted in a rapid and reversible increase in action potential firing of these neurons that allowed for precise control of the timing of ACh release in a more physiologically relevant manner. As illustrated in Figure 4A and B, blue light stimulation (473 nm, 22 mW, 500 ms) caused a significant increase in ChIs firing rate in brain slices from ChAT-ChR2-EYFP mice (n = 6, p < 0.05, paired t test). Blue light stimulation (473 nm, 22 mW, 500 ms) caused a significant increase in ChIs firing rate in brain slices from ChAT-ChR2-EYFP mice (n = 6, p < 0.05, paired t test, Figure 4A and B). When the brief light pulse was delivered 25 ms prior to the electrical stimulation of the corpus-callosum, the amplitude of evoked EPSCs decreased by 9.7 ± 4.0% (n = 6, p > 0.05, paired t test; Figure 4C and D), similar to that reported in the previous study using electrical activation of ChIs in ChI-MSN pair recordings.8 In the presence of VU0152100 (5 μM), the light stimulation caused a significantly greater depression of EPSC amplitude (32.2 ± 4.2% inhibition, n = 5, vs 9.7 ± 4.0% inhibition without VU0152100, n = 6, p < 0.01, t test, Figure 4C and D). Scopolamine (20 μM) pretreatment completely abolished the effects of VU0152100 and light stimulation on EPSCs (n = 5, p < 0.01, t test, Figure 4C and D).
Figure 4.

M4-PAM enhances EPSC depression induced by synchronized activation of cholinergic interneurons (ChIs). (A) A representative trace of ChI firing in a slice taken from a ChAT-ChR2-EYFP mouse during control and light stimulation (473 nm, 22 mW, 500 ms, *p < 0.05, paired t test). (B) Bar graph summarizing the effect of light stimulation on firing rate of ChIs. (C) Representative traces of averaged EPSCs showing the effect of light (C1), light + VU0152100 (5 μM, C2) and light + VU0152100 (5 μM) + scopolamine (20 μM, C3) on EPSC amplitude. (D) Bar graph summarizing the effect on EPSC amplitude in the following conditions, light stimulation, light with VU0152100, and light with VU0152100 and scopolamine. Data are shown as mean ± SEM (**p < 0.01, t test).
The present results provide several lines of evidence that M4 is a key mAChR subtype mediating the muscarinic modulation of glutamatergic transmission at corticostriatal synapses on both striatonigral and striatopallidal MSNs in rodents. First, the highly selective M4-PAM VU0152100 potentiated the CCh-induced reduction of EPSC amplitude through a mechanism involving a decrease in glutamate release. This inhibitory effect of CCh on glutamatergic transmission was completely absent in M4-KO mice. In addition, our electrophysiological data, combined with electron microscopic observations from this and previous studies,16,17 indicate that the M4-PAM potentiation of CCh-induced effects is largely mediated through presynaptic mechanisms that affect corticostriatal synapses on both D1 and D2 striatal MSNs. Moreover, the fact that the blockade of postsynaptic GPCRs with GDP-β-S loaded intracellularly had no significant effect on CCh-induced depression of EPSCs argues against the role of postsynaptic M4 in regulating glutamate release at this synapse. Because M4 receptors are also expressed postsynaptically mostly in D1 MSNs,16 future studies are warranted to determine the role of postsynaptic M4 mAChRs in regulating MSNs activity.
Activation of mAChRs is known to strongly modulate dopamine release in the striatum, and previous studies have postulated that this effect is mediated by M2/M4 mAChR subtypes.20,23−26 The nigrostriatal dopamine system also plays an important role in modulating glutamatergic signaling and neuronal excitability in the striatum.21,27 Thus, the M4-mediated inhibitory effect on corticostriatal synapses could be confounded by the muscarinic-dependent modulation of striatal dopamine signaling that, in turn, can influence glutamate signaling. However, our results show that in the presence of D1/D2 receptor antagonists the CCh-induced depression of EPSCs is comparable to that seen in control condition, which argues against a significant role of the dopamine system in contributing to the M4-mediated presynaptic effects on glutamatergic transmission.
Interestingly, studies performed in mice lacking the M4 mAChR only in D1 dopamine receptor-expressing cells show that M4 plays a critical role in regulating dopamine signaling and dopamine dependent behavior.28 We show here that M4 plays an important part in modulation of glutamatergic transmission both in D1 and D2 MSNs. Although our data suggest that this effect is presynaptically mediated, we cannot entirely exclude the possibility that M4 activity at the postsynaptic membrane could counteract the facilitating role played by D1 on neuronal excitability21 particularly of the direct pathway MSNs where M4 is highly expressed.
Early studies suggest that the effect of mAChRs activation on glutamatergic transmission might be mediated by M2-like and/or M3.11−15 Nonetheless, the muscarinic receptor ligands used in those early reports lacked proper specificity for individual mAChRs subtypes. Here, using a highly selective M4-PAM VU0152100 in combination with electrophysiology and M4-KO mice, we show that M4 is the essential mAChR subtype that mediates the cholinergic depression of corticostriatal glutamatergic transmission. It is also worth noting that this effect is not unique for the corticostriatal synapse. In fact, our previous studies showed that M4 is also involved in regulating the Shaffer collateral (SC)-CA1 synapse in the hippocampus.29 Interestingly, in contrast to the corticostriatal synapse where M4 seems to be the main mAChR subtype mediating the cholinergic inhibition as demonstrated by the entire lack of the CCh-mediated effects in M4-KO mice, other mAChR subtypes are also involved in regulating transmission at the SC-CA1 synapse because CCh is still able to depress the glutamatergic transmission, albeit to a lesser extent, in M4-KO mice.11 The striking difference between the corticostriatal and SC-CA1 synapses in terms of CCh effects in M4-KO mice, especially the entire lack of action of CCh on glutamatergic transmission at this synapse in M4-KO mice, led us to conclude that M4 is the key, if not the sole, mAChR subtype mediating inhibition of glutamatergic transmission at corticostriatal synapses in rodents. It is noteworthy that this presynaptic M4-mediated effect on glutamatergic transmission is brain region and possibly synapse specific. Recent work in our laboratory, indeed, revealed that M4 does not play any significant role in the cholinergic inhibition of glutamatergic transmission in other basal ganglia nuclei, such as the subthalamic nucleus (Xiang et al., unpublished data). As M2 and M3 mAChRs are also present in putative glutamatergic axon terminals that form asymmetric synapses in the rodent striatum,17 their roles in muscarinic modulation of striatal glutamatergic transmission remain to be determined.
Concluding Remarks
Changes in striatal cholinergic and glutamatergic functions have been implicated in a number of neuropsychiatric and neurodegenerative disorders including Parkinson’s disease, Huntington’s disease, Tourette syndrome, obsessive-compulsive disorder, schizophrenia, addictive disorders, and major depression. These diseases are commonly associated with abnormal glutamatergic transmission at corticostriatal synapses. The data shown here indicate that M4 plays a pivotal role in the regulation of corticostriatal glutamatergic transmission. Therefore, selectively targeting the striatal M4 mAChR at these synapses might provide a significant therapeutic benefit for the treatment of these neurological and neuropsychiatric disorders. In particular, highly selective M4-PAMs may be of great therapeutic interest for various brain diseases involving hyperactivity of the corticostriatal glutamatergic system.
Methods
Animals
C57BL/J6 mice (male, 23 ± 3 days of age) were used in electrophysiologic studies. Male, adult rats were used in EM studies. All experimental procedures were approved by the Institutional Animal Care and Use committee at Vanderbilt University or Emory University. Bacterial artificial chromosome (BAC) D1-EGFP mice expressing EGFP under the control of the D1 receptor promoter (donated by Dr. Gregg Stanwood) were used to discriminate striatal D1 from non-D1 (putatively D2) MSNs in brain slices. M4 receptor knockout (M4-KO) mice were obtained from Taconic.30 ChAT-ChR2-EYFP line-5 mice (B6.CgTg(Chat-COP4*H134R/EYFP,Slc18a3)5Gfng/J,ChAT-ChR2 mice) expressing the mhChR2::YFP fusion protein in the cholinergic neurons via the mouse choline acetyltransferase promoter31 were from Jackson Laboratory.
Ex Vivo Electrophysiology
Coronal brain slices (300–350 μm) containing the striatum and cortex were prepared as previously described in our laboratory.32
Whole-cell or cell-attached recordings were made from MSNs or cholinergic interneurons (ChIs) in the dorsal striatum using an Axon Multiclamp 700B amplifier (Molecular Devices). All neurons recorded were visually identified using an Olympus BX50WI microscope equipped with oblique illumination optics and epifluorescence (Olympus, Japan). Patch pipets were prepared from borosilicate glass (Sutter Instrument Company) using a P-97 Flaming/Brown micropipet puller (Sutter Instruments), and had resistance of 3–6 MΩ when filled with the following intracellular solution (mM): 120 K-MeSO3, 1 MgCl2, 0.1 CaCl2, 10 mM HEPES, 1 EGTA, 12 phosphocreatine, 0.4 GTP, and 2 ATP. The pH of the pipet solution was adjusted to 7.3, and osmolarity was adjusted to 283–292 mOsm. In some experiments GDP-β-S (0.1 mM) replaced the intracellular GTP. EPSCs were recorded from MSNs at −70 mV and evoked by stimulating the corpus callosum every 10 s with a concentric bipolar electrode (FHC). To isolate glutamatergic excitatory postsynaptic currents, GABAA receptor antagonist bicuculline (20 μM) was included in artificial cerebrospinal fluid (ACSF) containing (in mM): NaCl 126, KCl 2.5, NaHCO3 26.2, NaH2PO4 1.25, CaCl2 2, MgSO4 1.5, d-glucose 10, supplemented with l-ascorbic acid 0.6 mM. In all the experiments monitoring the effects of VU0152100 on Carbachol (CCh)-induced decrease in amplitude, the sequence of drug application was as follows: after 5 min stable baseline recordings, VU0152100 was applied for 5 min followed by 5 min coapplication of VU0152100 and CCh.
For optogenetic stimulation, we used a CoolLED pE-100 illumination system (CoolLED, U.K.) connected to the Olympus microscope. The 473 nm blue light beam was applied to the slice through the 40× water immersion objective. To determine the effect of ChIs activation on EPSCs in MSNs, brief light stimulation (500 ms, 22mW) was delivered 25 ms prior to the electrical stimulation of the corpus callosum.
Data Acquisition and Analysis
Data was acquired using a Digidata 1440A and pClamp 9.2, and analyzed with Clampfit 9.2 (Molecular Devices). Statistical analysis was performed using Prism 5.0 (GraphPad Software). Data are reported as mean ± SEM.
Immunoelectron Microscopy
Rats were deeply anesthetized with pentobarbital (100 mg/kg, i.v.) and transcardially perfusion-fixed with a cold oxygenated Ringer’s solution and a fixative containing 0.1% glutaraldehyde and 4% paraformaldehyde in phosphate buffer (PB; 0.1 M, pH 7.4). Brains were then removed, postfixed for 12 h in 4% paraformaldehyde solution, cut into serial 60-μm-thick coronal sections that were collected in an antifreeze solution (1.4% NaH2PO4·H2O, 2.6% Na2HPO4·7H2O, 30% ethylene glycol, 30% glycerol dissolved in distilled water) and stored in a −20 °C freezer. Prior to the immunocytochemical processing, the sections were placed into a 1% sodium borohydride/PBS solution for 20 min, followed by washes with PBS.
Sections were incubated with specific antibodies raised against M4 as described in previous publications17 and then processed according to the avidin–biotin immunoperoxidase method (ABC) using 3,3′ diaminobenzidine (DAB) as chromogen. Finally, the tissue was processed for electron microscopy as previously described in detail in our laboratory.33−35
Reagents
All reagents were purchased from Sigma except for bicuculline (Abcam) and GDP-β-S 3Li (Calbiochem). VU0152100 was synthetized in our chemistry facility.
Acknowledgments
The authors thank Dr. Allen Levey for the generous gift of the M4 antibodies; Dr. Gregg Stanwood for generous gift of BAC D1-EGFP mouse breeders; Jean-Francois Pare and Susan Jenkins for their technical assistance; and Weimin Peng for maintaining colonies of transgenic mice.
This work was supported in part by National Institute of Health Grants (P50NS071669-01 to Y.S., U54MH084659 to C.W.L., P50NS071669 to P.J.C., and RO1 NS065867 to Z.X.), and a grant from Tourette Syndrome Associtation (Z.X. and P.J.C.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Kreitzer A. C. (2009) Physiology and pharmacology of striatal neurons. Annu. Rev. Neurosci. 32, 127–147. [DOI] [PubMed] [Google Scholar]
- Kemp J. M.; Powell T. P. (1971) The termination of fibres from the cerebral cortex and thalamus upon dendritic spines in the caudate nucleus: a study with the Golgi method. Philos. Trans. R. Soc., B 262, 429–439. [DOI] [PubMed] [Google Scholar]
- Do J.; Kim J. I.; Bakes J.; Lee K.; Kaang B. K. (2012) Functional roles of neurotransmitters and neuromodulators in the dorsal striatum. Learn. Mem. 20, 21–28. [DOI] [PubMed] [Google Scholar]
- Berton O.; Nestler E. J. (2006) New approaches to antidepressant drug discovery: beyond monoamines. Nat. Rev. Neurosci. 7, 137–151. [DOI] [PubMed] [Google Scholar]
- Foster D. J.; Jones C. K.; Conn P. J. (2012) Emerging approaches for treatment of schizophrenia: modulation of cholinergic signaling. Discovery Med. 14, 413–420. [PMC free article] [PubMed] [Google Scholar]
- Higley M. J.; Soler-Llavina G. J.; Sabatini B. L. (2009) Cholinergic modulation of multivesicular release regulates striatal synaptic potency and integration. Nat. Neurosci. 12, 1121–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka R. C.; Kocsis J. D. (1988) Presynaptic actions of carbachol and adenosine on corticostriatal synaptic transmission studied in vitro. J. Neurosci. 8, 3750–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakhotin P.; Bracci E. (2007) Cholinergic interneurons control the excitatory input to the striatum. J. Neurosci. 27, 391–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciamanna G.; Tassone A.; Mandolesi G.; Puglisi F.; Ponterio G.; Martella G.; Madeo G.; Bernardi G.; Standaert D. G.; Bonsi P.; Pisani A. (2012) Cholinergic dysfunction alters synaptic integration between thalamostriatal and corticostriatal inputs in DYT1 dystonia. J. Neurosci. 32, 11991–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguiere E.; Monteiro P.; Feng G.; Graybiel A. M. (2013) Optogenetic stimulation of lateral orbitofronto-striatal pathway suppresses compulsive behaviors. Science 340, 1243–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Darvas M.; Storey G. P.; Bamford I. J.; Gibbs J. T.; Palmiter R. D.; Bamford N. S. (2013) Acetylcholine encodes long-lasting presynaptic plasticity at glutamatergic synapses in the dorsal striatum after repeated amphetamine exposure. J. Neurosci. 33, 10405–10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolders I.; Bogaert L.; Ebinger G.; Michotte Y. (1997) Muscarinic modulation of striatal dopamine, glutamate, and GABA release, as measured with in vivo microdialysis. J. Neurochem. 68, 1942–1948. [DOI] [PubMed] [Google Scholar]
- Calabresi P.; Centonze D.; Gubellini P.; Pisani A.; Bernardi G. (1998) Blockade of M2-like muscarinic receptors enhances long-term potentiation at corticostriatal synapses. Eur. J. Neurosci. 10, 3020–3023. [DOI] [PubMed] [Google Scholar]
- Hernandez-Echeagaray E.; Galarraga E.; Bargas J. (1998) 3-α-Chloro-imperialine, a potent blocker of cholinergic presynaptic modulation of glutamatergic afferents in the rat neostriatum. Neuropharmacology 37, 1493–1502. [DOI] [PubMed] [Google Scholar]
- Hsu K. S.; Huang C. C.; Gean P. W. (1995) Muscarinic depression of excitatory synaptic transmission mediated by the presynaptic M3 receptors in the rat neostriatum. Neurosci. Lett. 197, 141–144. [DOI] [PubMed] [Google Scholar]
- Sugita S.; Uchimura N.; Jiang Z. G.; North R. A. (1991) Distinct muscarinic receptors inhibit release of gamma-aminobutyric acid and excitatory amino acids in mammalian brain. Proc. Natl. Acad. Sci. U.S.A. 88, 2608–2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince E.; Ciliax B. J.; Levey A. I. (1997) Differential expression of D1 and D2 dopamine and m4 muscarinic acetylcholine receptor proteins in identified striatonigral neurons. Synapse 27, 357–366. [DOI] [PubMed] [Google Scholar]
- Hersch S. M.; Gutekunst C. A.; Rees H. D.; Heilman C. J.; Levey A. I. (1994) Distribution of m1-m4 muscarinic receptor proteins in the rat striatum: light and electron microscopic immunocytochemistry using subtype-specific antibodies. J. Neurosci. 14, 3351–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hersch S. M.; Levey A. I. (1995) Diverse pre- and post-synaptic expression of m1-m4 muscarinic receptor proteins in neurons and afferents in the rat neostriatum. Life Sci. 56, 931–938. [DOI] [PubMed] [Google Scholar]
- Brady A. E.; Jones C. K.; Bridges T. M.; Kennedy J. P.; Thompson A. D.; Heiman J. U.; Breininger M. L.; Gentry P. R.; Yin H.; Jadhav S. B.; Shirey J. K.; Conn P. J.; Lindsley C. W. (2008) Centrally active allosteric potentiators of the M4 muscarinic acetylcholine receptor reverse amphetamine-induced hyperlocomotor activity in rats. J. Pharmacol. Exp. Ther. 327, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umemiya M.; Raymond L. A. (1997) Dopaminergic modulation of excitatory postsynaptic currents in rat neostriatal neurons. J. Neurophysiol. 78, 1248–1255. [DOI] [PubMed] [Google Scholar]
- Tzavara E. T.; Bymaster F. P.; Davis R. J.; Wade M. R.; Perry K. W.; Wess J.; McKinzie D. L.; Felder C.; Nomikos G. G. (2004) M4 muscarinic receptors regulate the dynamics of cholinergic and dopaminergic neurotransmission: relevance to the pathophysiology and treatment of related CNS pathologies. FASEB J. 18, 1410–1412. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Yamada M.; Gomeza J.; Basile A. S.; Wess J. (2002) Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1-M5 muscarinic receptor knock-out mice. J. Neurosci. 22, 6347–6352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudernatsch M.; Sutor B. (1994) Cholinergic modulation of dopamine overflow in the rat neostriatum: a fast cyclic voltammetric study in vitro. Neurosci. Lett. 181, 107–112. [DOI] [PubMed] [Google Scholar]
- Ding J.; Guzman J. N.; Tkatch T.; Chen S.; Goldberg J. A.; Ebert P. J.; Levitt P.; Wilson C. J.; Hamm H. E.; Surmeier D. J. (2006) RGS4-dependent attenuation of M4 autoreceptor function in striatal cholinergic interneurons following dopamine depletion. Nat. Neurosci. 9, 832–842. [DOI] [PubMed] [Google Scholar]
- Threlfell S.; Clements M. A.; Khodai T.; Pienaar I. S.; Exley R.; Wess J.; Cragg S. J. (2010) Striatal muscarinic receptors promote activity dependence of dopamine transmission via distinct receptor subtypes on cholinergic interneurons in ventral versus dorsal striatum. J. Neurosci. 30, 3398–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch N. X.; Sabatini B. L. (2012) Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 76, 33–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dencker D.; Wortwein G.; Weikop P.; Jeon J.; Thomsen M.; Sager T. N.; Mork A.; Woldbye D. P.; Wess J.; Fink-Jensen A. (2011) Involvement of a subpopulation of neuronal M4 muscarinic acetylcholine receptors in the antipsychotic-like effects of the M1/M4 preferring muscarinic receptor agonist xanomeline. J. Neurosci. 31, 5905–5908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirey J. K.; Xiang Z.; Orton D.; Brady A. E.; Johnson K. A.; Williams R.; Ayala J. E.; Rodriguez A. L.; Wess J.; Weaver D.; Niswender C. M.; Conn P. J. (2008) An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat. Chem. Biol. 4, 42–50. [DOI] [PubMed] [Google Scholar]
- Gomeza J.; Zhang L.; Kostenis E.; Felder C.; Bymaster F.; Brodkin J.; Shannon H.; Xia B.; Deng C.; Wess J. (1999) Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M(4) muscarinic acetylcholine receptor knockout mice. Proc. Natl. Acad. Sci. U.S.A. 96, 10483–10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S.; Ting J. T.; Atallah H. E.; Qiu L.; Tan J.; Gloss B.; Augustine G. J.; Deisseroth K.; Luo M.; Graybiel A. M.; Feng G. (2011) Cell type-specific channelrhodopsin-2 transgenic mice for optogenetic dissection of neural circuitry function. Nat. Methods 8, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr J. C.; Turlington M. L.; Reid P. R.; Utley T. J.; Sheffler D. J.; Cho H. P.; Klar R.; Pancani T.; Klein M. T.; Bridges T. M.; Morrison R. D.; Blobaum A. L.; Xiang Z.; Daniels J. S.; Niswender C. M.; Conn P. J.; Wood M. R.; Lindsley C. W. (2012) Targeting selective activation of M(1) for the treatment of Alzheimer’s disease: further chemical optimization and pharmacological characterization of the M(1) positive allosteric modulator ML169. ACS Chem. Neurosci. 3, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogenpohl J. W.; Ritter S. L.; Hall R. A.; Smith Y. (2012) Adenosine A2A receptor in the monkey basal ganglia: ultrastructural localization and colocalization with the metabotropic glutamate receptor 5 in the striatum. J. Comp. Neurol. 520, 570–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan A.; Hu X.; Smith Y.; Wichmann T. (2011) Localization and pharmacological modulation of GABA-B receptors in the globus pallidus of parkinsonian monkeys. Exp. Neurol. 229, 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales K. K.; Pare J. F.; Wichmann T.; Smith Y. (2013) GABAergic inputs from direct and indirect striatal projection neurons onto cholinergic interneurons in the primate putamen. J. Comp. Neurol. 521, 2502–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]