

Abstract
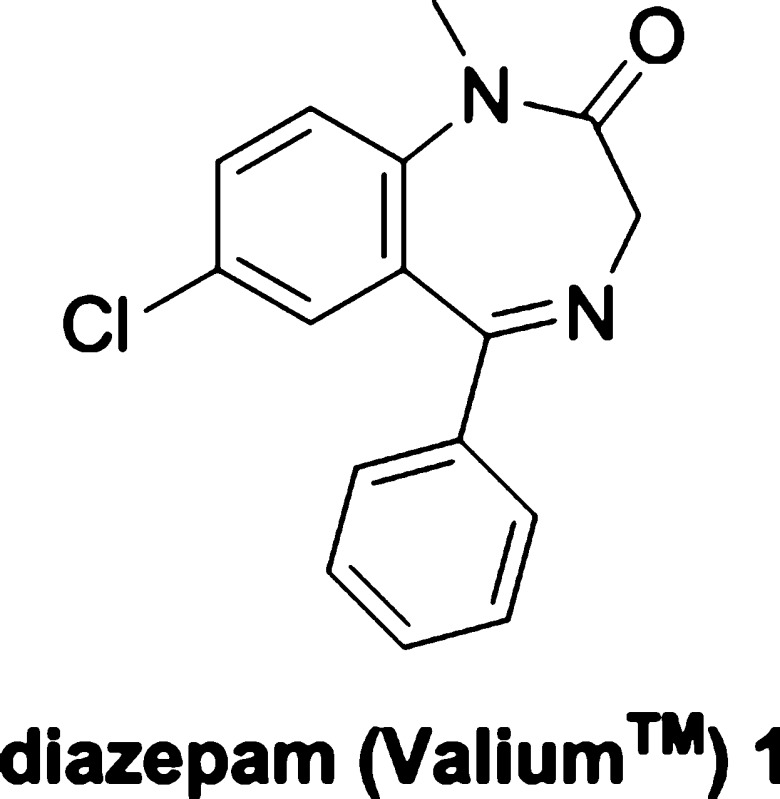
Diazepam (Valium) is among the most successful drugs from the onset of the psychopharmacological revolution that began during the 1950s. Efficacious in treating a wide-spectrum of CNS disorders, including anxiety and epilepsy, it set the standard for pharmacotherapy in terms of potency, onset of action, and safety. In this Review, the legacy of diazepam to chemical neuroscience will be considered along with its synthesis, pharmacology, drug metabolism, adverse events and dependence, clinical use, and regulatory issues.
Keywords: Diazepam, Valium, benzodiazepine, GABA, anxiety, hypnotic, sedative
Anxiety disorders have been the most prevalent of all psychiatric ailments. Along with generalized anxiety disorder (GAD), over 40 million people in the United States live with panic disorder, agoraphobia, post-traumatic stress disorder (PTSD), obsessive-compulsive disorder (OCD), or other forms of anxiety disorders. (1) While estimates for global prevalence vary, anxiety disorders and lack of treatment are common problems in both developed and developing countries.2
Historically, the first pharmacological treatments for anxiety consisted of using general depressants and sedatives such as alcohol, opiates, lithium bromide, and chloral hydrate. These were largely supplanted by carbamates (meprobamate 3) and barbiturates (phenobarbital 4) by the mid-20th century (Figure 1). However, it was not until 1960 when the pharmacotherapy for anxiety disorders became available with a greatly improved safety profile, in contrast to the narrow therapeutic range of sedatives and hypnotics. Beginning with chlordiazepoxide 2 (Librium), benzodiazepines proved to be a promising new class of potent anxiolytics without fatal adverse events. In 1963 an even more potent benzodiazepine, diazepam 1 (Valium), was released to the public, possessing “a greater dissociation between its sedative and anxiolytic properties.”2 Not only prescribed for anxiety, diazepam had therapeutic benefits for epilepsy, muscle spasms, and alcohol withdrawal.3 Diazepam quickly became one of the top selling drugs of all time, despite 15 years without significant knowledge of its mechanism of action.
Figure 1.

First marketed benzodiazepines and the classical sedative hypnotics.
Diazepam and other drugs in the benzodiazepine class are positive allosteric modulators (no functional response alone, but increase the response of the endogenous ligand) of the GABAA (γ-aminobutyric acid type A) receptor complex, binding to a unique site on the alpha-gamma subunit interface. Diazepam interaction with these sites increases neuronal chloride-ion influx upon GABA binding, resulting in hyperpolarized postsynaptic membranes, thus enhancing CNS depression response to endogenous GABA.4 These GABA potentiating actions are observed in the limbic system, thalamus, hypothalamus, and cerebral cortex and have calming effects on neuronal processes in these areas, resulting in anxiolytic and antiepileptic effects.5 Along with its targeted mechanism, diazepam’s potency, high bioavailability, and fast onset of action all account for its clinical efficacy and commercial success.
However, the efficacy and fast onset of diazepam brought about a higher risk of dependence and abuse. Benzodiazepine addiction and misuse arose from early overly optimistic expectations of the safety of diazepam in the 1970s, which led to inappropriate use in cases where pharmacotherapy was unnecessary. This resulted in a backlash against benzodiazepine prescriptions and ultimately engendered regulatory restriction. Chronic therapeutic diazepam usage was found to put patients at higher risks for dependence and withdrawal, although modern therapy seeks to monitor and address these concerns.2,6 In this Review, the synthesis, pharmacology, drug metabolism, and adverse events of diazepam are reviewed, as well as the legacy to chemical neuroscience, contemporary issues, and future role of diazepam.
Properties and Chemical Synthesis
Diazepam (CAS No: [439-14-5]) is a classical aryl 1,4-benzodiazepine with no hydrogen bond donors, three acceptors, a low molecular weight (MW = 284.7), and a ClogP of 3 with a TPSA of 32.7 (calculated in ref (7)). While it is now accepted that these properties are consistent with good CNS penetration and drug metabolism properties, diazepam and its numerous relatives can be credited with significantly influencing these criteria in a number of comparative databases used to define the guidelines.7
First synthesized by Leo Sternbach at Hoffman-La Roche in the late 1950s (Scheme 1), the aryl 1,4-benzodiazepine’s syntheses came from the observation that quinazoline-3-oxides such as 7(8) produced ring-expanded products on treatment with hydroxide, ammonia or primary amines via initial attack at the 2-position of the quinoxaline ring (rather than simple displacement of the chloride).9 The first clinically approved benzodiazepine, chlordiazepoxide (Librium 2), was prepared by reaction of methylamine with compound 7.10 Given the interesting pharmacological properties of 2,11 further exploration of the reactions of the benzodiazepine structure found that acidic hydrolysis of 2 produced N-oxide 8 which could be alkylated at the secondary amide nitrogen with methyl iodide, and the N-oxide reduced with either PCl3 or Raney nickel to provide diazepam.12 However, this synthesis was cumbersome and not permissive for the rapid development of analogues.13
Scheme 1. Several Routes to Diazepam Disclosed by Sternbach and Colleagues at Roche.

Another route was developed by Sternbach and colleagues using the same amino-benzophenone starting material 5 used to generate 7. The subsequent synthetic route consisted of cyclo-condensation with glycine ethyl ester hydrochloride to form the benzodiazepine core (10), followed by the alkylation of the 1 position nitrogen via dimethyl sulfate in sodium methoxide and methanol at reflux. Despite the one-step simplicity of converting 5 to 10, treatment of 5 with chloroacteyl chloride followed by ammonia and heat provided 10 in higher yields with easier purification.14 Additionally, the original patent also reported synthesis by initial alkylation. This was accomplished via tosylation of the 2-amino for the ensuing selective amino methylation, followed by deprotection and cyclo-condensation in heated pyridine with the glycine ethyl ester. (15) Since Sternbach’s preliminary routes, other notable methods have been reported. Gates described a method utilizing 7-chloro-1-methyl-3,4-dihydro-1H-1,4-benzodiazepine-2,5-dione, from 5-chloro-N-methylisatoic anhydride and glycine, in a series of efficient steps resulting in an overall yield of about 50% from starting material.16 Ishikura and colleagues were able to create the benzodiazepine core using palladium-catalyzed carbonylation to insert CO into aryl halides and cyclize into the seven membered ring through a concerted mechanism.17
Manufacturing Information
Diazepam is the generic name of compound 1, which was originally manufactured by Hoffman-La Roche as Valium. First synthesized in 1959, diazepam entered the market in 196318 and became the top selling pharmaceutical in the United States from 1968 until 1982 with a peak of 2.3 billion tablets sold in 1978.19 Roche sells Valium in 10, 5, and 2 mg tablets in the United States.20 After the patent expired in 1985, over 500 different brands of generic diazepam are now sold in a variety of tablets, suppositories, suspensions, gels, and other formulations.21 Over 14 million prescriptions in the United States were reported in 2011.22
Drug Metabolism
Diazepam is a bioavailable, widely distributed (Vd 0.95–2.0 l/kg) lipid soluble CNS penetrant compound.23 Bioavailability ranges from 93 to 100% orally, and 90% rectally with high levels of plasma protein binding (96–99%).24 Peak plasma levels are reached after 30–90 min via oral delivery, 30–60 min from intramuscular injection, and 10–45 min rectally. Steady state levels during chronic dosing are reached after 5–14 days. Elimination occurs via a biphasic process, redistributing into the muscle and adipose tissue after absorption, with a half-life of 24–48 h.23 Drug half-life and free fraction increases in aged populations due to reduced carrier albumin levels in serum, in correlation with the number of other medications taken by the patient.25
Diazepam is well metabolized by liver CYP450 enzymes and glucuronidated for elimination according to Scheme 2. Typically, diazepam is demethylated (by CYP 2C9, 2C19, 2B6, 3A4, and 3A5), yielding 10 (desmethyldiazepam, equipotent to diazepam64), and then is 3′ hydroxylated (CYP 3A4, 3A5, and 2C19), giving 12, although 11 (CYP 3A4 3A5) is also detected. Marginal amounts of unchanged diazepam are found in the urine, with nearly the entire original dose being excreted as metabolites.26
Scheme 2. Metabolic Fate of Diazepam through the Principally Acting CYP 450 Enzymes, Predominately via Demethylation to 10 Followed by Hydroxylation.

Notably, each major metabolite was developed and separately marketed.
These metabolites have been well characterized in the literature, with the primary active metabolite being 10, as 11 and 12 are eliminated nearly at the same rate of production. Due to its longer half-life (50–120 h), 10 accumulates as the majority of the circulating dose during daily administration and can take longer than 3 weeks to reach steady state.27
The principle CYPs affecting diazepam metabolism are CYP2C19 and the CYP 3A isoforms. In vitro data suggests total demethylation activity by either enzyme to be 33% and 44% respectively, although some reports suggest equal activity with diazepam being a better substrate for CYP2C19.26d Total hydroxylation toward 11 is 9% and 83% respectively. CYP2C19 is genetically polymorphic with 5% of Caucasians and 15–20% of Asian populations being poor metabolizers.28 It has been found that CYP2C19 inhibitors decrease the clearance of diazepam and 10. For example, coadministration of diazepam with fluvoxamine, a clinically relevant SSRI (selective serotonin reuptake inhibitor) and potent CYP2C19 inhibitor, increases the diazepam peak plasma concentrations, reduces its clearance as well as increases its half-life. Fluvoxamine also increases the exposure and time to reach steady state of principle metabolite 10. CYP2C19 inducers have been identified, including anticonvulsant and mood stabilizer carbamazepine and antiepileptic phenytoin, and these are implicated in inducing diazepam clearance.29
The three main diazepam metabolites 10 (nordiazepam), 11 (temazepam), and 12 (oxazepam) have also been developed and marketed as drugs. Oxazepam (12) has a slow onset of action and does not require hepatic oxidation for clearance, making it preferable for elderly or hepatically impaired patients.30 By bypassing the long-lived metabolite nordiazepam (10), the shorter overall half-life of temazepam 11 makes it preferred for treatment of insomnia.31
Pharmacology, Medicinal Chemistry, and Structure–Activity Relationship
Diazepam and its principle active metabolite 10 defined the historical “benzodiazepine receptor”, which was later identified as the ionotropic GABAA chloride channel via receptor binding studies.32 In addition to GABAA receptor, there is evidence of diazepam binding to voltage gated sodium channels which may contribute to some of its anticonvulsant properties.33 Diazepam shows activity as a neuronal voltage gated calcium channel blocker in rats.34 It also binds to a unique benzodiazepine receptor, initially deemed the “peripheral benzodiazepine receptor” and later characterized as the “translocator protein (18 kDa)” which appears to be localized on the mitochondrial membrane.35 Furthermore, diazepam has been shown to inhibit acetylcholine release in mouse hippocampus,36 decrease histamine recycling in mouse brain,37 and suppress prolactin release in rats.38
Despite these alternative binding sites for diazepam, most of the pharmacology appears to be mediated through the GABAA chloride channel, where it acts as a positive allosteric modulator. As such, it has no effect on GABA levels and binds to an allosteric modulating site (BZ site), distinct from other ligands (GABA, barbiturates, channel blockers). The GABAA receptor class consists of heteropentameric channels containing two α, two β, and one γ subunit. The BZ site is located at the interface of the γ and α subunits of the GABAA pentamer, although heterogeneity of the subunit composition has discrete effects on diazepam pharmacodynamics. Upon the binding of diazepam, the GABAA complex undergoes a conformational change, resulting in increased affinity for the endogenous GABA ligand. This potentiates GABA’s effect on CNS depression through increasing postsynaptic flux and accumulation of the chloride anion, resulting in hyperpolarization and subsequent inhibition of reaching firing threshold.4,39,40
There are six known α subunit isoforms composing various GABAA receptor subtypes. Two of these isoforms (α4 and α6) are labeled “benzodiazepine insensitive” due to their lack of benzodiazepine binding and action. The others can be bundled in two classes: those resulting in anxiolytic effects (α2/α3) and those responsible for sedation, ataxia, and amnesia (α1/α5). The most prevalent receptor subtype in the brain is α1,β2,γ2. Diazepam itself is essentially nonselective for binding to any single “benzodiazepine sensitive” α subunit isoform (Table 1),40 resulting in its broad efficacy, along with its other non GABAA interactions.41 The binding potency corresponds to similar potency as measured by electrophysiology (Table 1).42
Table 1. Recombinant Receptor Binding and Potentiation of Cl– Ion Current in Cells.
At therapeutic concentrations, only 2–3% of diazepam and metabolite nordiazepam 10 is found in the CSF compared to total plasma concentrations, correlating well with plasma free-fraction.43 At these concentrations, only about 20% of available GABAA receptors are occupied; although this has a significant effect on GABA mediated Cl– ion potentiation and clinical anxiolytic effects. Larger doses, typically when treating status epilepticus, result in increased receptor occupancy, sedative effects, and anterograde amnesia.44
Following the success of diazepam, thousands of variations have been reported, and over 20 have been approved for human use in various jurisdictions. Most of the medicinal chemistry leading to approved drugs was carried out before the detailed knowledge of the GABAA receptors. Nevertheless, structure–activity relationship studies reveal modest tolerance for substitution on the benzo-fused ring (electron-withdrawing groups at the 7-position are preferred) as well as the pendant phenyl where unsubstituted or ortho-halo phenyl is in most approved drugs. The best combination identified by the Sternbach group at Roche is flunitrazepam 13, and many other substituents are tolerated on the lactam nitrogen such as the solubilizing diethylamino group in flurazepam 14 (Figure 2).18 This area can also be modified into 5-membered rings as exemplified by triazolam 16 and midazolam 17. Removal of the pendant phenyl produces compounds that bind to the BZ site but do not potentiate GABA (neutral allosteric modulators) such as flumazenil 15.45 This compound has proved quite valuable as a radiotracer for GABAA receptors.46 In terms of subtype selectivity across the different subtypes of GABAA receptors, most close analogues of diazepam show similar activity across α1, α2, α3, and α5 containing receptors (Table 1).40 In order to separate the sedative, anxiolytic, and cognitive effects of the benzodiazepines, pharmacophore models of the GABAA receptor have been proposed to guide more recent work.47
Figure 2.

Notable diazepam analogues and second generation compounds.
Approved Indications
Diazepam is approved for the treatment of anxiety, acute alcohol withdrawal, skeletal muscle spasm, and convulsive disorders (e.g., status epilepticus).3 It is used off-label for numerous other conditions including insomnia, restless leg syndrome, and pre/post-operative sedation. It is a Schedule IV controlled substance.2
Adverse Effects and Dosage
Serious and fatal adverse events associated with diazepam are extremely rare and are most often a consequence of interaction with another drug (such as opiates or alcohol). The most common fatal events are respiratory arrest and prolonged seizures resulting from prolonged habitual use, rather than acute overdose. In fact, reported cases of overdoses up to 2000 mg diazepam have resulted in an induced temporary coma with speedy recovery. More moderate adverse effects from chronic diazepam use include amnesia, dizziness, ataxia, confusion, sedation, depression, and tachycardia. Also, worsening of seizures or anxiety can occur in some patients being treated for epilepsy or anxiety disorders.48
Chronic diazepam use is associated with tolerance, dependence, and withdrawal. Diazepam exhibits a lower risk profile for addiction and dependence than other benzodiazepines, and it is typically used for treatment of withdrawal symptoms from other benzodiazepines and alcohol. Nevertheless, caution must be taken with dosage and duration for longer-term anxiety symptoms. Benzodiazepine withdrawal symptoms can range from mild headaches and pains during gradual cessation, to psychosis, hallucinations, seizures, mania, and death from convulsions if high-dose chronic treatment is abruptly stopped.49
Studies suggest diazepam and other benzodiazepines build tolerance through desensitizing GABA-ergic neurons through receptor internalization and possibly transcriptional regulation. It is also possible the NMDA glutamate excitatory system becomes over sensitized, resulting in excitotoxicity. Additionally, benzodiazepines are known to decrease norepinephrine, serotonin, acetylcholine, and dopamine, further perturbing normal neuronal network equilibria. To prevent dependence and withdrawal effects from chronic prescription, dosages can be slowly tapered off before complete discontinuation.50
Initial dosages for anxiolytic and antiepileptic therapy in normal adults range from 2 to 10 mg orally, administered 2–4 times daily. Maximum recommended doses range up to 30 mg every 8 h. Hypnotic uses typically require no more than 30 mg orally before sleep. In treatment of status epilepticus, 5–10 mg IV/IM every 10–15 min, up to 30 mg with adjunct antiseizure medications then an option.20
History and Importance in Neuroscience
As pharmacological probes, diazepam and its derivatives are essential tools for translational neuroscience given their well-characterized effects in humans across a range of indications. Laboratory models in rodents for anxiety and sedation commonly use diazepam as a control for establishing validity of the assay due to its ease of dosing (usually 1–10 mg/kg) and reproducible effects in laboratory rodents.51
During its 50 year history on the market, diazepam has undoubtedly become one of the most influential and salient psychotropic compounds produced by the pharmaceutical industry. From what now seems as the cliché accidental discovery, to the conflicts between its booming success and controversy, the legacy of “Valium” is one of achievement and therapeutic wisdom.
Prior to the benzodiazepines, the world was not entirely without treatments to anxiety. Nevertheless, nearly all available options between chloral hydrate, barbiturates, and bromide salts were of low therapeutic index or long half-lives, and thus dosing was difficult to manage. Habitual administration of these earlier treatments commonly caused tolerance. Overdoses resulted in heavy sedation, inhibition of motor function, cognitive delays, comas, respiratory failure, and sometimes death. Compounding the detection of adverse events was that they were often confused with the expected side-effects. Naturally, these drugs focused on general sedation of behavior and agitation rather than addressing the underlying molecular causes. Without specific drugs or an identified molecular mechanism, the field of psychiatry focused on Freudian psychoanalysis rather than drugs associated with unwanted effects. Even at the advent of the psychopharmacological revolution in the 1950s, the first line of anxiolytics such as meprobamate still suffered from narrow therapeutic indexes and only meager improvements to standards of care.2,49b,52
The entrance of the benzodiazapine class and diazepam became a watershed moment for the standard of psychotropic drugs. Patients who were resistant to other treatments instead responded to diazepam, without the risk of potential overdose of conventional sedatives. Previously, sleeping pills commonly caused accidental death, a phenomenon largely curtailed upon widespread use of benzodiazepines. Psychoanalysis, while still strongly championed, became overshadowed by the convenience of doctors prescribing drugs rather than finding a trained psychiatrist for time-consuming analysis. From a toxicity and therapeutic standpoint, diazepam was touted as the worry free panacea; being able to treat anxiety, insomnia, epilepsy, alcohol and opiate withdrawal, panic attacks, and muscle spasms.2
Drawn to the potency and safety of the benzodiazepines, much effort was put into elucidating the still unknown mechanism of action. Early reports in 1974 by Roche and other groups showed that diazepam promoted the effect of GABA in a specific fashion,53 and it was later discovered in 1977 that diazepam bound specifically to receptors in the CNS. These receptors were revealed to be critical areas on the GABAA receptor complex. (32) Once advances were made in molecular biology, the GABAA complex was identified, sequenced, cloned,54 and analyzed with electron microscopy.55 Diazepam’s binding site and selectivity were structurally justified. It is this specificity to the GABAA receptors, and indirect activation through allosteric modulation, that results in diazepam’s safety and adjustable dosage for various applications.20
But diazepam’s fast onset of action, low toxicity, and high potency did more than prove itself as an efficacious drug, it became a pop-culture icon. Inevitably, this celebrity status shed light on the temporarily ignored danger with its blockbuster sales and production: over prescription and addiction. While initial inquiries into the addictive potential of benzodiazepines quickly dismissed any concerns, the safety profile was taken for granted, as some patients were irresponsibly given greater doses for longer durations than necessary. More thorough critiques of the clinical and preclinical literature suggested dependence was a substantial factor, although mostly in cases of higher dosage and duration. Normal dose dependence was suspected during the 1970s, and provocatively exclaimed by many United Kingdom groups, but not confirmed until the 1980s. By the 1990s, the scope of benzodiazepine addiction had been expanded to not only dosage escalation, but even chronic low-level administration.52a,56
Despite the vast majority of prescriptions validated for legitimate medical use, and abuse mostly occurring in individuals abusing other illicit drugs (especially heroin and alcohol), diazepam and other benzodiazepines fell out of favor with much of the medical community and were stigmatized by public regulatory institutions. While the restrictions in the United States, under the Controlled Substances Act as a schedule IV drug, have been relatively soft, action taken in European countries and in certain domestic areas has been more severe.57 Trialozam 16, one of the most frequently prescribed benzodiazepines in the United States and the United Kingdom, was suspended and then removed from the market in The Netherlands in 1979, the United Kingdom in 1991, and other western European nations quickly followed suit.58 Several countries placed hard restrictions on other benzodiazepines with high potentials for abuse (especially through intravenous routes), such as temazepam 11.59 The state of New York passed rigorous compliance regulations in 1989, most notably the requirement of state-issued triplicate copy prescription forms. This resulted in a 30–60% decrease in benzodiazepine prescriptions, and large increases in the use of inappropriate therapeutics (such as chloral hydrate and meprobamate) at rates over 100%.60 In 1994, the World Health Organization expert committee on drug dependence considered flunitrazepam (13) abuse as a substantial threat to public health.61 On top of self-abuse, flunitrazepam is used as a “date rape” drug, and therefore has historically received tighter regulations and scheduling than other benzodiazepines.48a While single actions against diazepam have not been taken, it certainly assumes the role as the face of general benzodiazepine class regulations, especially as Valium, “Mother’s Little Helper”. With the arrival of new tricyclic and SSRI compounds, in addition to the lingering backlash and fears from overuse in the 1970s and 1980s, diazepam prescriptions fell dramatically.62
However, there have been recent calls to dispel the “end of the benzodiazapine era”. While treatment for OCD has yielded to newer classes of drugs, benzodiazepines are still considered first line treatment for most other anxiety disorders and phobias. This is partly due to major side effects in SSRIs, lackluster efficacy versus specific conditions, and slower onset of action, despite many groups still championing SSRIs as first line treatment.63
The future of diazepam is still unfolding, but its addition to the World Health Organization’s list of essential medicines highlights its crucial role to modern medicine. Diazepam’s 50 year history of raising the standard of care and revolutionizing treatments in anxiety and epilepsy, among other illnesses, unequivocally earns the status as a classic in chemical neuroscience
The authors declare no competing financial interest.
References
- Kessler R. C.; Chiu W. T.; Demler O.; Walters E. E. (2005) Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiat. 62(6), 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baenninger A., Costa e Silva J. A., Hindmarch I., Moeller H. J., and Rickels K. (2004) Good Chemistry: The Life and Legacy of Valium Inventor Leo Sternbach, McGraw Hill, New York. [Google Scholar]
- Valium (Diazepam), Physician’s Desk Reference Online http://www.pdr.net/drug-summary/valium?druglabelid=2100. Accessed December 20, 2013.
- Nutt D. J.; Malizia A. L. (2001) New insights into the role of the GABA(A)-benzodiazepine receptor in psychiatric disorder. Br. J. Psychiatry 179(5), 390–396. [DOI] [PubMed] [Google Scholar]
- Zakusov V. V.; Ostrovskaya R. U.; Kozhechkin S. N.; Markovich V. V.; Molodavkin G. M.; Voronina T. A. (1977) Further Evidence for Gaba-Ergic Mechanisms in Action of Benzodiazepines. Arch. Int. Pharmacol. 229(2), 313–326. [PubMed] [Google Scholar]
- Greenblatt D. J.; Shader R. I. (1978) Dependence, Tolerance, and Addiction to Benzodiazepines - Clinical and Pharmacokinetic Considerations. Drug Metab. Rev. 8(1), 13–28. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Hou X. J.; Verhoest P. R.; Villalobos A. (2010) Moving beyond Rules: The Development of a Central Nervous System Multiparameter Optimization (CNS MPO) Approach To Enable Alignment of Druglike Properties. ACS Chem. Neurosci. 1(6), 435–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternbach L. H.; Kaiser S.; Reeder E. (1960) Quinazoline 3-Oxide Structure of Compounds Previously Described in the Literature as 3.1.4-Benzoxadiazepines. J. Am. Chem. Soc. 82(2), 475–479. [Google Scholar]
- Stempel A.; Reeder E.; Sternbach L. H. (1965) Quinazolines and 1,4-Benzodiazepines. XXVII. Mechanism of Ring Enlargement of Quinazoline 3-Oxides with Alkali to 1,4-Benzodiazepin-2-1 4-Oxides. J. Org. Chem. 30(12), 4267–4271. [DOI] [PubMed] [Google Scholar]
- Sternbach L.; Reeder E. (1961) Quinazolines and 1,4-Benzodiazepines. II. Rearrangement of 6-Chloro-2-Chlormethyl-4-Phenylquinazoline 3-Oxide into 2-Amino Derivatives of 7-Chloro-5-Phenyl-3H-1,4-Benzodiazepine 4-Oxide. J. Org. Chem. 26(4), 1111–1118. [Google Scholar]
- Randall L. O.; Schallek W.; Heise G. A.; Keith E. F.; Bagdon R. E. (1960) The Psychosedative Properties of Methaminodiazepoxide. J. Pharmacol. Exp. Ther. 129(2), 163–171. [PubMed] [Google Scholar]
- Sternbach L.; Reeder E. (1961) Quinazolines and 1,4-Benzodiazepines 0.4. Transformations of 7-Chloro-2-Methylamino-5-Phenyl-3h-1,4-Benzodiazepine 4-Oxide. J. Org. Chem. 26(12), 4936–4941. [Google Scholar]
- a Sternbach L.; Keller O.; Metlesics W.; Reeder E. (1961) Quinazolines and 1,4-Benzodiazepines. III. Substituted 2-Amino-5-Phenyl-3H-1,4-Benzodiazepine 4-Oxides. J. Org. Chem. 26(11), 4488–4497. [Google Scholar]; b Sternbach L. (1959) 1,4-Benzodiazepine 4-Oxides. U.S. Patent US2893992.
- Sternbach L. H. (1971) 1,4-Benzodiazepines - Chemistry and Some Aspects of Structure-Activity Relationship. Angew. Chem., Int. Ed. 10(1), 34–43. [DOI] [PubMed] [Google Scholar]
- Reeder E., and Sternbach L. (1968) 5-Aryl-3H-1,4-benzodiazepin-2(1h)-ones. U.S. Patent US3371085.
- Gates M. (1980) New Synthesis of Diazepam. J. Org. Chem. 45(9), 1675–1681. [Google Scholar]
- Ishikura M.; Mori M.; Ikeda T.; Terashima M.; Ban Y. (1982) New Synthesis of Diazepam and the Related 1,4-Benzodiazepines by Means of Palladium-Catalyzed Carbonylation. J. Org. Chem. 47(12), 2456–2461. [Google Scholar]
- Sternbach L. H. (1979) Benzodiazepine Story. J. Med. Chem. 22(1), 1–7. [DOI] [PubMed] [Google Scholar]
- Ainsworth S. (2013) Mother’s Little Helper Still Rocking. Nurse Prescribing 11(5), 255–256. [Google Scholar]
- Valium Prescribing Information: http://www.gene.com/download/pdf/valium_prescribing.pdf. Accessed December 20, 2013.
- International League Against Epilepsy, Worldwide AED Database. http://www.ilae.org/Visitors/Centre/AEDs/index.cfm. Accessed December 20, 2013.
- Lindsley C. W. (2012) The Top Prescription Drugs of 2011 in the United States: Antipsychotics and Antidepressants Once Again Lead CNS Therapeutics. ACS Chem. Neurosci. 3(8), 630–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelli M.; Tognoni G.; Garattini S. (1978) Clinical Pharmacokinetics of Diazepam. Clin. Pharmacokinet. 3(1), 72–91. [DOI] [PubMed] [Google Scholar]
- Klotz U.; Antonin K. H.; Bieck P. R. (1976) Pharmacokinetics And Plasma Binding Of Diazepam In Man, Dog, Rabbit, Guinea-Pig And Rat. J. Pharmacol. Exp. Ther. 199(1), 67–73. [PubMed] [Google Scholar]
- Vozeh S. (1981) Pharmacokinetics of the Benzodiazepines in the Elderly Patient. Schweiz. Med. Wochenschr. 111(47), 1789–1793. [PubMed] [Google Scholar]
- a Zingales I. A. (1973) Diazepam Metabolism during Chronic Medication - Unbound Fraction in Plasma, Erythrocytes and Urine. J. Chromatogr. 75(1), 55–78. [DOI] [PubMed] [Google Scholar]; b Ono S.; Hatanaka T.; Miyazawa S.; Tsutsui M.; Aoyama T.; Gonzalez F. J.; Satoh T. (1996) Human liver microsomal diazepam metabolism using cDNA-expressed cytochrome P450s: Role of CYP2B6, 2C19 and the 3A subfamily. Xenobiotica 26(11), 1155–1166. [DOI] [PubMed] [Google Scholar]; c Greenblatt D. J.; Divoll M. K.; Soong M. H.; Boxenbaum H. G.; Harmatz J. S.; Shader R. I. (1988) Desmethyldiazepam Pharmacokinetics - Studies Following Intravenous and Oral Desmethyldiazepam, Oral Clorazepate, and Intravenous Diazepam. J. Clin. Pharmacol. 28(9), 853–859. [DOI] [PubMed] [Google Scholar]; d Andersson T.; Miners J. O.; Veronese M. E.; Birkett D. J. (1994) Diazepam Metabolism by Human Liver-Microsomes Is Mediated by Both S-Mephenytoin Hydroxylase and Cyp3A Isoforms. Br. J. Clin. Pharmacol. 38(2), 131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Hooper W. D.; Watt J. A.; Mckinnon G. E.; Reilly P. E. B. (1992) Metabolism of Diazepam and Related Benzodiazepines by Human Liver-Microsomes. Eur. J. Drug Metab. Pharmacokinet. 17(1), 51–59. [DOI] [PubMed] [Google Scholar]
- a Hillesta L.; Hansen T.; Melsom H.; Drivenes A. (1974) Diazepam Metabolism in Normal Man 0.1. Serum Concentrations and Clinical Effects after Intravenous, Intramuscular, and Oral-Administration. Clin. Pharmacol. Ther. 16(3), 479–484. [PubMed] [Google Scholar]; b Hillesta L.; Hansen T.; Melsom H. (1974) Diazepam Metabolism in Normal Man 0.2. Serum Concentration and Clinical Effect after Oral-Administration and Cumulation. Clin. Pharmacol. Ther. 16(3), 485–489. [PubMed] [Google Scholar]
- a Desta Z.; Zhao X. J.; Shin J. G.; Flockhart D. A. (2002) Clinical significance of the cytochrome P4502C19 genetic polymorphism. Clin. Pharmacokinet. 41(12), 913–958. [DOI] [PubMed] [Google Scholar]; b Bertilsson L. (1995) Geographical Interracial Differences in Polymorphic Drug Oxidation - Current State of Knowledge of Cytochromes P450 (Cyp) 2d6 and 2c19. Clin. Pharmacokinet. 29(3), 192–209. [DOI] [PubMed] [Google Scholar]; c Fukasawa T.; Suzuki A.; Otani K. (2007) Effects of genetic polymorphism of cytochrome P450 enzymes on the pharmacokinetics of benzodiazepines. J. Clin. Pharm. Ther. 32(4), 333–341. [DOI] [PubMed] [Google Scholar]
- a Perucca E.; Gatti G.; Cipolla G.; Spina E.; Barel S.; Soback S.; Gips M.; Bialer M. (1994) Inhibition of Diazepam Metabolism by Fluvoxamine - a Pharmacokinetic Study in Normal Volunteers. Clin. Pharmacol. Ther. 56(5), 471–476. [DOI] [PubMed] [Google Scholar]; b Clark W. G., Brater D. C., and Johnson A. R. (1992) Goth’s Medical Pharmacology, CV Mosby, St. Louis, MO. [Google Scholar]
- Greenblatt D. J.; Shader R. I.; Kochweser J. (1975) Pharmacokinetics In Clinical Medicine - Oxazepam Versus Other Benzodiazepines. Dis. Nerv. Syst. 36(5), 6–13. [PubMed] [Google Scholar]
- Heel R. C.; Brogden R. N.; Speight T. M.; Avery G. S. (1981) Temazepam - A Review Of Its Pharmacological Properties And Therapeutic Efficacy As An Hypnotic. Drugs 21(5), 321–340. [DOI] [PubMed] [Google Scholar]
- a Mohler H.; Okada T. (1977) Gaba Receptor-Binding with H-3(+) Bicuculline-Methiodide in Rat CNS. Nature 267, 65–67. [DOI] [PubMed] [Google Scholar]; b Squires R. F.; Braestrup C. (1977) Benzodiazepine Receptors in Rat-Brain. Nature 266, 732–734. [DOI] [PubMed] [Google Scholar]; c Braestrup C.; Squires R. F. (1977) Specific Benzodiazepine Receptors in Rat-Brain Characterized by High-Affinity [Diazepam-H-3] Binding (Affinity Binding Diazepam Anxiolytic Activity Brain Membranes Regional Distribution). Proc. Natl. Acad. Sci. U.S.A. 74(9), 3805–3809. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Mohler H.; Okada T. (1977) Benzodiazepine Receptor - Demonstration in Central Nervous-System. Science 198(4319), 849–851. [DOI] [PubMed] [Google Scholar]
- a Willow M.; Kuenzel E. A.; Catterall W. A. (1984) Inhibition of Voltage-Sensitive Sodium-Channels in Neuro-Blastoma Cells and Synaptosomes by the Anticonvulsant Drugs Diphenylhydantoin and Carbamazepine. Mol. Pharmacol. 25(2), 228–234. [PubMed] [Google Scholar]; b Mclean M. J.; Macdonald R. L. (1988) Benzodiazepines, but Not Beta-Carbolines, Limit High-Frequency Repetitive Firing of Action-Potentials of Spinal-Cord Neurons in Cell-Culture. J. Pharmacol. Exp. Ther. 244(2), 789–795. [PubMed] [Google Scholar]
- Taft W. C.; Delorenzo R. J. (1984) Micromolar-Affinity Benzodiazepine Receptors Regulate Voltage-Sensitive Calcium Channels In Nerve-Terminal Preparations. Proc. Natl. Acad. Sci. U.S.A. 81(10), 3118–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Papadopoulos V.; Baraldi M.; Guilarte T. R.; Knudsen T. B.; Lacapere J. J.; Lindemann P.; Norenberg M. D.; Nutt D.; Weizman A.; Zhang M. R.; Gavish M. (2006) Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol. Sci. 27(8), 402–409. [DOI] [PubMed] [Google Scholar]; b Venneti S.; Lopresti B. J.; Wiley C. A. (2006) The peripheral benzodiazepine receptor (Translocator protein 18 kDa) in microglia: From pathology to imaging. Prog. Neurobiol. 80(6), 308–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J. A.; Richter J. A. (1985) Effects of Anticonvulsants In vivo on High-Affinity Choline Uptake In vitro in Mouse Hippocampal Synaptosomes. Br. J. Pharmacol. 84(1), 19–25. [PMC free article] [PubMed] [Google Scholar]
- Oishi R.; Nishibori M.; Itoh Y.; Saeki K. (1986) Diazepam-Induced Decrease in Histamine Turnover in Mouse-Brain. Eur. J. Pharmacol. 124(3), 337–342. [DOI] [PubMed] [Google Scholar]
- Grandison L. (1982) Suppression of Prolactin Secretion by Benzodiazepines In vivo. Neuroendocrinology 34(5), 369–373. [DOI] [PubMed] [Google Scholar]
- Battistin L.; Varotto M.; Berlese G.; Roman G. (1984) Effects of Some Anticonvulsant Drugs on Brain Gaba Level and Gad and Gaba-T Activities. Neurochem. Res. 9(2), 225–231. [DOI] [PubMed] [Google Scholar]
- Sieghart W. (1995) Structure and Pharmacology of Gamma-Aminobutyric Acid(a) Receptor Subtypes. Pharmacol. Rev. 47(2), 181–234. [PubMed] [Google Scholar]
- a Atack J. R. (2005) The benzodiazepine binding site of GABA(A) receptors as a target for the development of novel anxiolytics. Expert Opin. Invest. Drugs 14(5), 601–618. [DOI] [PubMed] [Google Scholar]; b Mamalaki C.; Stephenson F. A.; Barnard E. A. (1987) The Gabaa/Benzodiazepine Receptor Is a Heterotetramer of Homologous Alpha-Subunit and Beta-Subunit. EMBO J. 6(3), 561–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puia G.; Vicini S.; Seeburg P. H.; Costa E. (1991) Influence of Recombinant Gamma-Aminobutyric Acid-a Receptor Subunit Composition on the Action of Allosteric Modulators of Gamma-Aminobutyric Acid-Gated Cl- Currents. Mol. Pharmacol. 39(6), 691–696. [PubMed] [Google Scholar]
- Kanto J.; Kangas L.; Siirtola T. (1975) Cerebrospinal-Fluid Concentrations of Diazepam And Its Metabolites In Man. Acta Pharmacol. Toxicol. 36(4), 328–334. [DOI] [PubMed] [Google Scholar]
- Facklam M.; Schoch P.; Bonetti E. P.; Jenck F.; Martin J. R.; Moreau J. L.; Haefely W. E. (1992) Relationship between Benzodiazepine Receptor Occupancy and Functional-Effects In vivo of 4 Ligands of Differing Intrinsic Efficacies. J. Pharmacol. Exp. Ther. 261(3), 1113–1121. [PubMed] [Google Scholar]
- Brogden R. N.; Goa K. L. (1988) Flumazenil - A Preliminary Review Of Its Benzodiazepine Antagonist Properties, Intrinsic Activity And Therapeutic Use. Drugs 35(4), 448–467. [DOI] [PubMed] [Google Scholar]
- Lassen N. A.; Bartenstein P. A.; Lammertsma A. A.; Prevett M. C.; Turton D. R.; Luthra S. K.; Osman S.; Bloomfield P. M.; Jones T.; Patsalos P. N.; Oconnell M. T.; Duncan J. S.; Andersen J. V. (1995) Benzodiazepine Receptor Quantification In-Vivo In Humans Using C-11 Flumazenil And Pet - Application Of The Steady-State Principle. J. Cereb. Blood Flow Metab. 15(1), 152–165. [DOI] [PubMed] [Google Scholar]
- Huang Q.; He X. H.; Ma C. R.; Liu R. Y.; Yu S.; Dayer C. A.; Wenger G. R.; McKernan R.; Cook J. M. (2000) Pharmacophore/receptor models for GABA(A)/BzR subtypes (alpha 1 beta 3 gamma 2, alpha 5 beta 3 gamma 2, and alpha 6 beta 3 gamma 2) via a comprehensive ligand-mapping approach. J. Med. Chem. 43(1), 71–95. [DOI] [PubMed] [Google Scholar]
- a Mullins M. E. (1999) Laboratory confirmation of flunitrazepam in alleged cases of date rape. Acad. Emerg. Med. 6(9), 966–968. [DOI] [PubMed] [Google Scholar]; b Greenblatt D. J.; Woo E.; Allen M. D.; Orsulak P. J.; Shader R. I. (1978) Rapid Recovery from Massive Diazepam Overdose. J. Am. Med. Assoc. 240(17), 1872–1874. [PubMed] [Google Scholar]; c Finkle B. S.; McCloskey K. L.; Goodman L. S. (1979) Diazepam and Drug-Associated Deaths - Survey in the United-States and Canada. J. Am. Med. Assoc. 242(5), 429–434. [PubMed] [Google Scholar]
- a Breier A.; Charney D. S.; Nelson J. C. (1984) Seizures Induced by Abrupt Discontinuation of Alprazolam. Am. J. Psychiat. 141(12), 1606–1607. [DOI] [PubMed] [Google Scholar]; b Brunton L., Chabner B., and Knollman B. (2010) Goodman and Gilman’s The Pharmacological Basis of Therapeutics, Twelfth ed., The McGraw-Hill Companies, New York. [Google Scholar]
- a Allison C.; Pratt J. A. (2003) Neuroadaptive processes in GABAergic and glutamatergic systems in benzodiazepine dependence. Pharmacol. Ther. 98(2), 171–195. [DOI] [PubMed] [Google Scholar]; b Longone P.; Impagnatiello F.; Guidotti A.; Costa E. (1996) Reversible modification of GABA(A) receptor subunit mRNA expression during tolerance to diazepam-induced cognition dysfunction. Neuropharmacology 35(9–10), 1465–1473. [DOI] [PubMed] [Google Scholar]; c Marley R. J.; Gallager D. W. (1989) Chronic Diazepam Treatment Produces Regionally Specific Changes in Gaba-Stimulated Chloride Influx. Eur. J. Pharmacol. 159(3), 217–223. [DOI] [PubMed] [Google Scholar]; d Uusi-Oukari M.; Korpi E. R. (2010) Regulation of GABA(A) Receptor Subunit Expression by Pharmacological Agents. Pharmacol. Rev. 62(1), 97–135. [DOI] [PubMed] [Google Scholar]
- Crawley J. N. (2007) What’s Wrong with My Mouse, 2nd ed., John Wiley and Sons, Hoboken, NJ. [Google Scholar]
- a Ninan P. T.; Cole J. O., and Yonkers K. A. (1998) Nonbenzodiazepine Anxiolytics. In Textbook of Psychopharmacology (Schatzberg A. F., and Nemeroff C. B., Eds.), Second ed., American Psychiatric Publishing, Washington, D.C. [Google Scholar]; b Lader M. (1991) History of Benzodiazepine Dependence. J. Subst. Abuse Treat. 8(1–2), 53–59. [DOI] [PubMed] [Google Scholar]
- Polc P.; Mohler H.; Haefely W. (1974) Effect Of Diazepam On Spinal-Cord Activities - Possible Sites And Mechanisms Of Action. Arch. Pharmacol. 284(4), 319–337. [DOI] [PubMed] [Google Scholar]
- a Richards J. G.; Schoch P.; Haring P.; Takacs B.; Mohler H. (1987) Resolving Gaba-a Benzodiazepine Receptors - Cellular and Subcellular-Localization in the CNS with Monoclonal-Antibodies. J. Neurosci. 7(6), 1866–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sieghart W. (2006) Structure, Pharmacology, and Function of GABA(A) Receptor Subtypes. Adv. Pharmacol. 54, 231–263. [DOI] [PubMed] [Google Scholar]
- Nayeem N.; Green T. P.; Martin I. L.; Barnard E. A. (1994) Quaternary Structure of the Native Gaba(a) Receptor Determined by Electron-Microscopic Image-Analysis. J. Neurochem. 62(2), 815–818. [DOI] [PubMed] [Google Scholar]
- a File S. E. (1990) The History of Benzodiazepine Dependence - a Review of Animal Studies. Neurosci. Biobehav. Rev. 14(2), 135–146. [DOI] [PubMed] [Google Scholar]; b Hallstrom C.; Lader M. (1981) Benzodiazepine Withdrawal Phenomena. Int. Pharmacopsychiatry 16(4), 235–244. [DOI] [PubMed] [Google Scholar]
- Abraham J. (2002) Transnational industrial power, the medical profession and the regulatory state: adverse drug reactions and the crisis over the safety of Halcion in the Netherlands and the UK. Soc. Sci. Med. 55(9), 1671–1690. [DOI] [PubMed] [Google Scholar]
- Wysowski D. K.; Barash D. (1991) Adverse Behavioral Reactions Attributed to Triazolam in the Food-and-Drug-Administrations Spontaneous Reporting System. Arch. Intern. Med. 151(10), 2003–2008. [PubMed] [Google Scholar]
- Degenhardt L.; Roxburgh A.; Van Beek I.; Hall W. D.; Robinson M. K. F.; Ross J.; Mant A. (2008) The effects of the market withdrawal of temazepam gel capsules on benzodiazepine injecting in Sydney, Australia. Drug Alcohol Rev. 27(2), 145–151. [DOI] [PubMed] [Google Scholar]
- Weintraub M.; Singh S.; Byrne L.; Maharaj K.; Guttmacher L. (1991) Consequences of the 1989 New-York-State Triplicate Benzodiazepine Prescription Regulations. J. Am. Med. Assoc. 266(17), 2392–2397. [PubMed] [Google Scholar]
- Druid H.; Holmgren P.; Ahlner J. (2001) Flunitrazepam: an evaluation of use, abuse and toxicity. Forensic Sci. Int. 122(2–3), 136–141. [DOI] [PubMed] [Google Scholar]
- Woods J. H. (1998) Problems and opportunities in regulation of benzodiazepines. J. Clin. Pharmacol. 38(9), 773–782. [PubMed] [Google Scholar]
- a Stahl S. M. (2002) Don’t ask, don’t tell, but benzodiazepines are still the leading treatments for anxiety disorder. J. Clin. Psychiatry 63(9), 756–757. [DOI] [PubMed] [Google Scholar]; b Uhlenhuth E. H.; Balter M. B.; Ban T. A.; Yang K. (1999) Trends in recommendations for the pharmacotherapy of anxiety disorders by an international expert panel, 1992–1997. Eur. Neuropsychopharmacol. 9, 393–398. [DOI] [PubMed] [Google Scholar]; c Baldwin D. S.; Anderson I. M.; Nutt D. J.; Bandelow B.; Bond A.; Davidson J. R. T.; den Boer J. A.; Fineberg N. A.; Knapp M.; Scott J.; Wittchen H. U.; (2005) Evidence-based guidelines for the pharmacological treatment of anxiety disorders: recommendations from the British Association for Psychopharmacology. J. Psychopharmacol. 19, 567–596. [DOI] [PubMed] [Google Scholar]
- Braestrup C.; Squires R. F. (1978) Pharmacological Characterization Of Benzodiazepine Receptors In The Brain. Eur. J. Pharmacol. 48, 263–270. [DOI] [PubMed] [Google Scholar]