

Abstract
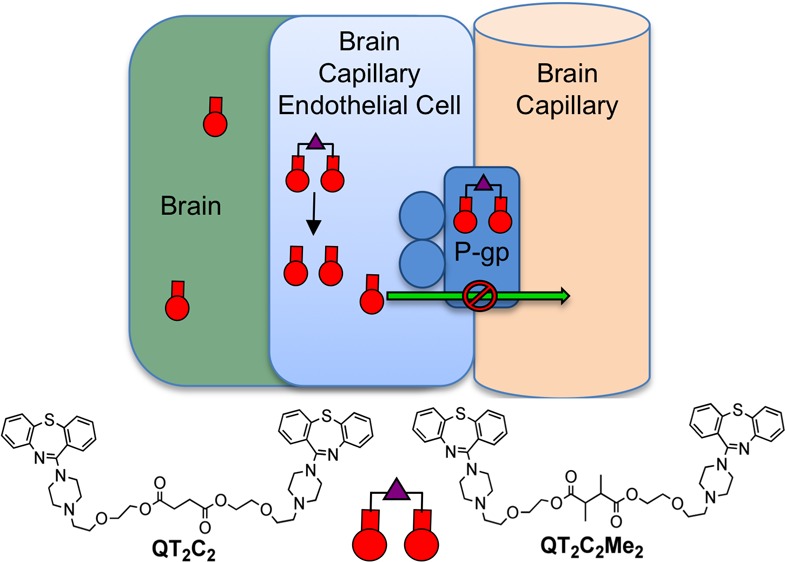
The multidrug resistance transporter P-glycoprotein (P-gp) is highly expressed in the capillary endothelial cells of the blood-brain barrier (BBB) where it functions to limit the brain penetration of many drugs, including antipsychotic agents used to treat schizophrenia. Therefore, in an effort to inhibit the transporter, we designed dimers of the antipsychotic drug and P-gp substrate quetiapine (QT), linked by variable length tethers. In P-gp overexpressing cells and in human brain capillary endothelial hCMEC/D3 cells, the dimer with the shortest tether length (QT2C2) (1) was the most potent inhibitor showing >80-fold better inhibition of P-gp-mediated transport than monomeric QT. The dimers, which are linked via ester moieties, are designed to revert to the therapeutic monomer once inside the target cells. We demonstrated that the addition of two sterically blocking methyl groups to the linker (QT2C2Me2, 8) increased the half-life of the molecule in plasma 10-fold as compared to the dimer lacking methyl groups (QT2C2, 1), while retaining inhibitory potency for P-gp transport and sensitivity to cellular esterases. Experiments with purified P-gp demonstrated that QT2C2 (1) and QT2C2Me2 (8) interacted with both the H- and R-binding sites of the transporter with binding affinities 20- to 30-fold higher than that of monomeric QT. Using isolated rat brain capillaries, QT2C2Me2 (8) was a more potent inhibitor of P-gp transport than QT. Lastly, we showed that QT2C2Me2 (8) increased the accumulation of the P-gp substrate verapamil in rat brain in situ three times more than QT. Together, these results indicate that the QT dimer QT2C2Me2 (8) strongly inhibited P-gp transport activity in human brain capillary endothelial cells, in rat brain capillaries, and at the BBB in an animal model.
Keywords: P-glycoprotein, blood-brain barrier, quetiapine, inhibitor, ABC transporter, antipsychotic agent
The first line treatment for patients with schizophrenia is the administration of atypical antipsychotic agents, such as quetiapine (QT), either alone or in combination.1,2 It is estimated that approximately 54 million annual prescriptions were written in 2011 for atypical antipsychotics for treatment of schizophrenia, bipolar disorder or as an adjuvant therapy for depression (IMS Health), with QT, aripiprazole and olanzapine being among the most common. The clinical efficacy of these drugs relies on their ability to enter the brain, an event that is controlled by the blood-brain barrier (BBB). The BBB is a system of capillary endothelial cells that protects the brain from damaging substances in the bloodstream, and comprises both tight junctions that prevent passage of molecules between cells, as well as transport proteins in the endothelial cells, which limit uptake.3−5 By these mechanisms, the BBB poses a major impediment to successful schizophrenia therapy by limiting the efficient delivery of antipsychotic agents to the brain. Therefore, to achieve efficacy, treatment of schizophrenia frequently requires increased doses of the therapeutic agents or an increase in the number of drugs administered. However, these clinical strategies can often result in toxic drug–drug interactions and unwanted physiological side effects, including movement disorders, agranulocytosis, and rapid major weight gain, the latter resulting in weight-related diseases, such as diabetes and hypertension.6−10
Of the numerous transporters expressed at the BBB, P-glycoprotein (P-gp), a member of the ATP-binding cassette (ABC) family of transporters, is the best characterized and arguably most important in limiting the brain accumulation of antipsychotic drugs used to treat schizophrenia.11 P-gp is an integral membrane transporter protein comprising two homologous halves each spanning the plasma membrane bilayer six times, and each containing an ATP binding site.12−16 P-gp is localized to the apical membrane of brain capillary endothelial cells and limits the accumulation of drugs in the brain by using the energy of ATP hydrolysis to actively transport compounds out of the capillary cell membrane and back into the bloodstream.17−19
Numerous antipsychotic drugs have been shown to be substrates of P-gp both in vitro and in vivo. QT, risperidone, olanzapine, and clozapine stimulated the ATPase activity of P-gp in crude membrane preparations, which is an indication that the compounds interact with the substrate binding site(s) of P-gp.20 In vivo, the accumulation of antipsychotic agents in the brain has been studied using P-gp null mice. The brain concentration of QT was determined to be approximately twice that found in wild-type mice, whereas the serum concentrations of QT were similar.21 These data suggest that QT is a substrate for P-gp at the BBB and that its entry into the brain is restricted. Similar results were also obtained for other atypical antipsychotic agents such as olanzapine,22 risperidone,23 and aripiprazole.24,25 Pharmacological inhibition of P-gp with cyclosporin A in rats treated with either fluphenazine or amisulpride resulted in significantly higher brain concentrations of the antipsychotic than animals treated with the antipsychotic alone.26,27 Furthermore, the behavioral effects of amisulpride27 and risperidone28 were increased significantly and lasted longer than in animals cotreated with a P-gp inhibitor. Together, these studies suggest that altering the biochemical properties of P-gp at the BBB may allow for better brain penetration of antipsychotic drugs and thus better clinical efficacy.
The drug binding region of P-gp has been shown biochemically to contain at least two, and perhaps more, distinct substrate binding sites that are localized to the transmembrane segments of the transporter.29−34 Furthermore, in the three-dimensional crystal structures of the inward-facing conformation of nucleotide-free P-gp from both mouse and C. elegans, the overall drug binding pocket is formed by the contacts between transmembrane helices, and consists of a large flexible internal cavity that is able to accommodate the binding of multiple molecules.35,36 For example, the structures of mouse P-gp cocrystallized with two stereoisomers of a cyclic P-gp peptide inhibitor demonstrated that three peptide molecules occupy different overlapping regions within the cavity, and each made a different set of interactions with the protein.35 Although these structures were obtained in the absence of nucleotide, it was recently determined that mouse P-gp is highly flexible and can adopt a wide range of conformations, even in the presence of nucleotide,37 further suggesting that the substrate binding pocket of P-gp can accommodate larger molecules. To exploit this multiplicity of binding sites, we have developed several potent bivalent P-gp inhibitors based on P-gp substrates. These inhibitors were designed to occupy the binding sites in P-gp and increase the molecular interactions with the transporter compared to the monomer, thereby increasing their binding affinity.38−44
Herein, we designed a set of dimeric analogues of QT, an antipsychotic drug and P-gp substrate,21 that are reversibly linked via ester bonds. The ester groups were introduced so the dimers have the capacity to revert back to the corresponding monomeric drugs upon cleavage by esterases after they have entered the cytoplasm of the cell. In this way, the QT dimers would act both as prodrugs of QT and inhibit P-gp transport at the BBB. Herein, we demonstrated that the dimer with the shortest tether linking the QT monomers inhibited P-gp-mediated drug transport most potently in cultured cells (QT2C2) (1). Furthermore, the introduction of two sterically blocking methyl groups to the tether linking the QT monomers (QT2C2Me2) (8) resulted in a more potent inhibitor that resisted hydrolysis in human plasma, but was susceptible to breakdown by purified pig liver esterase. Importantly, we also demonstrated that these dimeric QT compounds interacted with the H- and R-substrate binding sites of P-gp with binding affinities 20- to 30-fold greater than that of the QT monomer. Extending the work to models of the blood-brain barrier, we showed that QT2C2Me2 (8) was a potent P-gp inhibitor in isolated rat brain capillaries and increased the brain penetration of verapamil in an in situ rat brain perfusion assay.
Results and Discussion
Design and Synthesis of QT Dimeric Prodrugs
P-gp binds a variety of structurally diverse compounds that are recognized by multiple binding sites within the transmembrane region of the protein.16−19,29−34 To take advantage of these multiple sites, we designed dimers of the antipsychotic agent and P-gp substrate QT. The primary hydroxyl group on QT was used to link monomers via bis-esters (Scheme 1) (1–7).40−42 These dimeric QT analogues have the potential to convert back to the monomeric drug QT by cleavage of the ester linkages by cellular and plasma esterases. In this way, the dimers could potentially act as both P-gp inhibitors and prodrugs of the therapy. One goal was for the dimers to have prolonged plasma stability, but to have a relatively facile reversion to monomer within the cell. We used a range of different tether lengths to evaluate the effect on P-gp inhibition and also employed tethers containing an additional methyl group α to carbonyl groups in the tether to evaluate how these groups affected the rate of breakdown by plasma and cellular esterases.
Scheme 1. Synthetic Scheme for QT Prodrug Dimers (n = 2–8) (1–7).

Dimers were named according to the number of methylene (−CH2−) groups located between the ester bonds, e.g., n = 2 is QT2C2 (1). All dimers were synthesized using dicarboxylic acid linkers and were purified to >95% by reverse phase HPLC. The pure compounds were characterized by MALDI-TOF mass spectrometry.
To synthesize the dimeric prodrugs, QT was treated with dicarboxylic acids of varied length, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and N,N-diisopropylethylamine to provide the desired diester compounds (Scheme 1). The dimers (QT2C2 through QT2C8) (1–7) were purified to >95% homogeneity by reverse phase HPLC and characterized by MALDI-TOF mass spectrometry. The compounds were named according to the number of methylene groups between the ester bonds. For example, QT2C2 (1) contained two methylene units between the ester linkages (Scheme 1).
QT and Its Dimers Inhibited P-gp-Mediated Transport in Cells Overexpressing Human P-gp
The set of QT dimeric prodrugs (QT2C2 through QT2C8) (1–7) were screened by flow cytometry for their ability to increase the cellular accumulation of the fluorescent P-gp substrate rhodamine 123 (R123) in human MCF-7/DX1 cells that overexpress P-gp. In this assay, an increase in cellular accumulation of R123 is indicative of inhibition of efflux by P-gp. The IC50 values for 1–7 for inhibition of P-gp activity presented in Table 1 were determined from assays performed in the presence and absence of increasing concentrations of 1–7 (Figure S1, Supporting Information). Dimers QT2C2 through QT2C4 were equipotent with respect to inhibition of R123 efflux and were approximately 80-fold more potent than monomeric QT. As the tether length was increased in compounds QT2C5–QT2C7, modest decreases in inhibitory potency were observed, followed by a further decrease for QT2C8 (17-fold less potent than QT2C2) (Table 1 and Figure S1). Our previous studies with dimers of P-gp substrates41−43 demonstrated that the tether length for optimal inhibition of P-gp transport varied from 6 to 10 carbon atoms. In the present study, however, the optimal number of carbon atoms in the tether ranged from two to four (Table 1). This difference may be due to the fact that QT was dimerized through the primary hydroxyl group at the end of a flexible ethylene glycol-like unit linked to the piperazine ring (Scheme 1), thus allowing shorter tether lengths to provide the optimal distance between the monomers.
Table 1. Inhibition of P-gp-Mediated R123 Transport by QT and QT Dimeric Prodrug Compounds in Human MCF-7/DX1 Cells That Overexpress P-gp.
| compd | Rhodamine 123 IC50 (μM)a |
|---|---|
| QT | 122 ± 8 |
| QT2C2 (1) | 1.5 ± 0.1 |
| QT2C3 (2) | 1.6 ± 0.1 |
| QT2C4 (3) | 1.8 ± 0.1 |
| QT2C5 (4) | 2.8 ± 0.2 |
| QT2C6 (5) | 4.0 ± 0.2 |
| QT2C7 (6) | 5.2 ± 0.3 |
| QT2C8 (7) | 26 ± 4 |
IC50 values are reported as the concentration of QT or QT dimer resulting in half-maximum fluorescence level ± SEM from a minimum of three independent experiments. For each experiment, the mean fluorescence values from each concentration point were used to generate a dose response curve and the IC50 was calculated using GraphPad Prism 4.0. The original flow cytometry data from which these data were determined are shown in Figure S1.
Based on these studies, and with a regard for maintaining the lowest molecular weight possible, the shortest dimer, QT2C2 (1), was selected for further analysis in P-gp substrate accumulation assays in human MCF-7/DX1 cells that overexpress P-gp. Substrates used include calcein-AM (calcein-acetoxymethylester), doxorubicin, 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY)-FL-verapamil, and [3H]daunorubicin (Figure S2). As summarized in Table 2, QT2C2 (1) was a potent inhibitor (IC50 = 1–3 μM) of the transport of all tested P-gp substrates, although it was 2-fold less potent for [3H]-daunorubicin as compared to the others. QT2C2 (1) was 8.5- to 45-fold more potent than monomeric QT for inhibition of transport of these substrates (Table 2).
Table 2. Inhibition of P-gp-Mediated Transport by QT and Its Dimers QT2C2 (1) and QT2C2Me2 (8) in Human MCF-7/DX1 Cells That Overexpress P-gp.
| IC50 (μM) |
|||||
|---|---|---|---|---|---|
| compd | Rhodamine 123b | Calcein-AMa | BVab | Doxorubicina | [3H]Daunorubicinc |
| QT | 122 ± 8b | 67 ± 3 | 31 ± 4 | 22 ± 1 | 23 ± 2 |
| QT2C2 (1) | 1.5 ± 0.1 | 1.5 ± 0.1 | 1.5 ± 0.1 | 1.2 ± 0.1 | 2.7 ± 0.2 |
| QT2C2Me2 (8) | 1.6 ± 0.1 | 1.1 ± 0.1 | 0.9 ± 0.1 | 1.1 ± 0.1 | 1.8 ± 0.1 |
BV = BODIPY-FL-verapamil.
IC50 values are reported as the concentration of QT or QT dimer resulting in half-maximum fluorescence level ± SEM from a minimum of three independent experiments. For each experiment, the mean fluorescence values from each concentration point were used to generate a dose response curve and the IC50 was calculated using GraphPad Prism 4.0. The original flow cytometry data from which these results were derived are shown in Figure S2.
IC50 values are reported as the concentration of QT or QT dimer resulting in half-maximum inhibition of radioactive substrate accumulation ± SD from a minimum of three independent experiments. For each experiment, the inhibition values from each concentration point were used to generate a dose response curve and the IC50 was calculated using GraphPad Prism 4.0.
Adding Sterically Blocking Methyl Groups to QT2C2 Increased Stability to Esterases while Preserving Potent P-gp Inhibition
The QT prodrug dimers were designed to inhibit P-gp while concurrently accumulating and subsequently breaking down to monomeric QT inside the cell by esterases. To be efficacious P-gp inhibitors at the BBB, however, the dimeric prodrugs should have limited breakdown in the bloodstream, but hydrolyze within target cells. To determine the stability of QT2C2 (1) to plasma esterases, reversion to monomer was monitored following incubation in human plasma (55%) at 37 °C. The half-life (t1/2) of QT2C2 (1) was relatively short (t1/2 = 3.0 ± 0.4 h), prompting the synthesis of a modified, more hindered QT2C2 (1) dimer containing two additional methyl groups in the tether (QT2C2Me2) (8) (Figure 1A). We hypothesized that this hindered dimer would have improved stability in plasma due to the increased steric hindrance surrounding both ester bonds, slowing tether release.45,46 QT2C2Me2 (8), demonstrated a significantly longer t1/2 of ∼38 h (Figure 1B). In vitro experiments with purified pig liver esterase, a model of cellular esterases, demonstrated that the dimeric prodrugs were susceptible to breakdown, with t1/2 values of 0.8 and 2.2 h for QT2C2 (1) and QT2C2Me2 (8), respectively (Figure 1B). Thus, it is possible to engineer the tether as to control the breakdown of dimeric agents in human plasma. In this way, the dimers should persist long enough to reach their cellular target and still generate monomeric drugs in the presence of cellular esterases.
Figure 1.

Structures of QT2C2 (1) and QT2C2Me2 (8) and stability analysis. (A) The hindered dimer prodrug, QT2C2Me2 (8), was synthesized as in Scheme 1 using a dicarboxylic acid linker (2,3-dimethylsuccinic acid). The prodrugs were purified to homogeneity by reverse phase HPLC, purity confirmed by analytical HPLC, and structures confirmed by MALDI-TOF mass spectrometry. (B) QT2C2 (1) and QT2C2Me2 (8) (40 μM each) were incubated in the presence of human plasma (55% in 10 mM phosphate buffer, pH 7.4) or 10 units of pig liver esterase at 37 °C. Reversion of the prodrugs to monomer was quantified via analytical HPLC and normalized to an internal standard. t1/2 values for stability in human plasma represent duplicate experiments ± SD.
To determine whether QT2C2Me2 (8) inhibited P-gp activity similarly to the unhindered QT2C2 (1) dimer, P-gp transport activity was evaluated via quantification of fluorescent and radiolabeled substrate accumulation using MCF-7/DX1 cells that overexpress P-gp. QT2C2Me2 (8) inhibited P-gp transport of R123, calcein-AM, doxorubicin, BODIPY-FL-verapamil, and [3H]-daunorubicin similarly to QT2C2 (1), with IC50 values in the low micromolar range (Table 2 and Figure S2). These IC50 values were 13- to 75-fold lower than those for the QT monomer. These results indicated that both dimers are effective P-gp inhibitors.
QT Dimers Stimulated P-gp ATPase Activity
A number of inhibitors of P-gp are also transport substrates.12,47 Furthermore, substrate transport is directly coupled to ATP hydrolysis. A commonly employed assay to determine if compounds are substrates of P-gp is to measure their ability to stimulate ATP hydrolysis. Therefore, we determined the effects of QT, QT2C2 (1) and QT2C2Me2 (8) on P-gp mediated ATP hydrolysis, crude membranes from Sf9 cells expressing human P-gp were analyzed for stimulation of ATP hydrolysis activity. For QT, stimulation of ATP hydrolysis peaked between 50 and 100 μM. For the dimers, peak stimulation was observed from 0.5 to 1.0 μM (Figure 2). For each of the compounds tested, a drop in ATPase activity was observed at higher concentrations, which has been observed with other P-gp modulators.12,47 As stimulation of ATPase activity is often an indication of transport activity, these data indicate that QT2C2 (1) and QT2C2Me2 (8) interact with P-gp and may be substrates for the transporter at low concentrations.
Figure 2.

Effect of QT and dimers QT2C2 (1) and QT2C2Me2 (8) on P-gp-mediated ATPase activity in crude membranes from Sf9 cells overexpressing human P-gp. (A) P-gp-mediated ATP hydrolysis was stimulated by QT with a peak at 50 to 100 μM. Verapamil (Ver., 30 μM) served as a positive control. (B) Stimulation of P-gp mediated ATP hydrolysis by QT2C2 (1) (white bars) and QT2C2Me2 (8) (gray bars). Verapamil (Ver., 30 μM) served as a positive control. Sf9 crude membrane vesicles (10 μg) were incubated with verapamil or various concentrations of QT or dimer for 20 min at 37 °C. ATPase activity was determined by quantifying the amount of inorganic phosphate release spectrophotometrically. Each data point represents the mean of two independent experiments performed in duplicate ± SEM.
QT Dimers Competed for Substrate Binding to P-gp
The QT prodrug dimers were designed to potentially interact with multiple binding sites in P-gp, and as such should compete for these sites more effectively than the QT monomer. To verify that the dimeric QT inhibitors were, in fact, interacting with the drug binding region of P-gp, photoaffinity cross-linking experiments were performed with a radiolabeled azido-analog of the P-gp substrate prazosin, [125I]iodoarylazidoprazosin ([125I]IAAP). [125I]IAAP is known to bind specifically to the drug binding sites of P-gp.29,48 Sf9 insect membranes expressing human P-gp were coincubated with [125I]IAAP (11 nM) and increasing concentrations of QT, QT2C2 (1), or QT2C2Me2 (8), followed by photo-cross-linking, SDS-PAGE, and autoradiographic analysis and quantification (Figure 3). QT2C2 (1) (Figure 3B) and QT2C2Me2 (8) (Figure 3C) potently inhibited the incorporation of [125I]IAAP into P-gp with IC50 values of 180 ± 40 nM and 150 ± 20 nM, respectively. These data demonstrate that QT2C2 (1) and QT2C2Me2 (8) effectively competed with [125I]IAAP for binding, suggesting that they specifically interact with the drug binding regions of the transporter. On the other hand, the QT monomer stimulated the cross-linking of [125I]IAAP to P-gp at low concentrations, reducing binding only at concentrations greater than 100 μM; even with 400 μM QT binding was reduced by only 50% (Figure 3A). These data indicate that QT competed less effectively for the substrate binding site region of P-gp than the dimers. Furthermore, at lower concentrations, QT may bind at an allosteric site that promotes an increase in the affinity of [125I]IAAP for P-gp. This type of stimulatory effect was previously reported for photoaffinity labeling of P-gp using [3H]tamoxifen aziridine.49
Figure 3.

P-gp drug binding competition photo-cross-linking assay. P-gp containing crude Sf9 membranes (25 μg) were incubated for 10 min at room temperature in the presence of the P-gp substrate [125I]IAAP (11 nM) and increasing concentrations of (A) QT, (B) QT2C2 (1), or (C) QT2C2Me2 (8). Following photo-cross-linking at 365 nm on ice for 20 min, proteins were separated by SDS-PAGE, visualized by autoradiography, and incorporation of [125I]IAAP into P-gp was quantified using Image J (NIH). Nonspecific binding, defined as labeling in the presence of the known inhibitor GF210918 (10 μM), was subtracted from each data point.
QT Dimers Have Increased Binding Affinity for Purified P-gp
To further characterize the interaction of QT and its dimer prodrugs with P-gp, the binding affinities of monomeric QT, QT2C2 (1), and QT2C2Me2 (8) for purified P-gp from Chinese hamster ovary cells were determined by measuring the saturable quenching of the intrinsic Trp fluorescence of the protein.50 This method was used previously to evaluate binding of compounds to P-gp and to obtain quantitative estimates of the Kd values for binding.50−52 Values for the maximal % quenching (ΔFmax) and the dissociation constant (Kd) were obtained by fitting the experimental data to a hyperbolic equation for monophasic quenching (Table 3).50,51 The maximal quenching of Trp fluorescence in P-gp was ∼64% for the QT monomer, which bound with a Kd of ∼29 μM. The dimers did not quench the Trp fluorescence as well as the monomer; however, the Kd values obtained for the dimers were much lower than that of the monomer, 1–1.5 μM (Table 3), indicative of a substantially higher binding affinity. This type of effect is expected if the dimers have more discrete interactions than the monomer with the substrate-binding pocket of the protein. The QT dimers with the highest P-gp binding affinity have ∼20% lower ΔFmax values than those with lower affinities. These data suggest that the higher affinity dimers may orient in a slightly different way in the substrate binding pocket so that their ability to quench Trp residues is altered. These findings are consistent with the cellular accumulation data demonstrating that the dimers were more potent inhibitors of P-gp-mediated transport than monomeric QT. Taken together, the creation of a covalently linked QT dimer is an effective strategy for increasing the strength of interaction with P-gp.
Table 3. Binding Parameters for Quenching of P-gp Trp Fluorescence by Binding of QT and QT Dimersa.
| compd | ΔFmax (%) | Kd (μM) |
|---|---|---|
| QT | 65 ± 2.7 | 29 ± 2.9 |
| QT2C2 (1) | 45 ± 0.8 | 1.5 ± 0.1 |
| QT2C2Me2 (8) | 43 ± 0.9 | 1.0 ± 0.1 |
Kd values were estimated by fitting of the quenching data from 3 independent experiments, and represents the mean ± fitting error.
The QT Dimer QT2C2Me2 (8) bound to both the H- and R-sites of P-gp
P-gp is known to have at least two positively cooperative drug binding sites within a larger flexible binding pocket, named the H-site and the R-site.31,34,52−54 Interaction of substrates and/or inhibitors of P-gp with either or both of these sites can be distinguished by their behavior in transport assays using fluorescent P-gp substrates that preferentially bind to each site.47,55 Thus, to gain a better understanding as to where our dimeric compounds interact with P-gp, we performed real-time transport assays using the substrates Hoechst 33342 (H33342) as a reporter for the H-site, and tetramethylrosamine (TMR) as a reporter for the R-site, and 2-{4-[4-(dimethylamino)phenyl]-l,3-butadienyl}-3-ethylbenzothiazoliumperchlorate (LDS-751), which binds to both sites.55 The ability of QT and QT2C2Me2 (8) to compete for transport of these fluorescent substrates was examined using purified P-gp from Chinese hamster ovary cells reconstituted into proteoliposomes in the presence of increasing concentrations of QT and QT2C2Me2 (8) (Figure 4). The concentration of QT and QT2C2Me2 (8) that caused 50% inhibition of the initial transport rate for each substrate was defined as the IC50 value (Table 4), and was estimated from the plot of % initial rate of transport versus concentration (Figure 4). Pure R-site agents inhibit transport of TMR at low concentrations but typically require >10-fold higher concentrations to inhibit H33342 transport.55 On the other hand, drugs that bind preferentially to the H-site demonstrate a reverse pattern of inhibition.
Figure 4.

Concentration-dependent inhibition of transport of (A) TMR, (B), H33342, and (C) LDS-751. QT (●) and QT2C2Me2 (8) (○). Results are shown for representative experiments with each data point carried out in duplicate. Where error bars are not visible, they are contained within the symbols. Inhibition plots were derived from real-time fluorescence data as described in Methods.
Table 4. Inhibition of P-gp-Mediated Transport in Reconstituted Proteoliposomes by QT and the Dimer QT2C2Me2 (8).
| IC50 for inhibition of transport (μM)a |
|||
|---|---|---|---|
| compd | TMR | H33342 | LDS-751 |
| QT | 59 ± 9 | 42.5 ± 1.5 | 83.6 ± 1.6 |
| QT2C2Me2 (8) | 21 ± 1 | 18.1 ± 7.2 | 36.3 ± 0.1 |
IC50 values were determined from inhibition plots as shown in Figure 4 and represent the mean ± range for two independent experiments.
Both QT and QT2C2Me2 (8) inhibited transport of TMR and H33342 in a similar concentration range and both also inhibited the transport of LDS-751 (Figure 4). These data suggest that the compounds are not pure H-site or pure R-site compounds and interact, perhaps in an overlapping way, with both of the sites.55 QT2C2Me2 (8) inhibited the transport of all three fluorescent substrates 2.3–2.8-fold better than QT (Table 4), which is consistent with its higher affinity for the transporter (Table 3). Although the difference in inhibition of transport is less than what was observed for the differences between Kd values for the two molecules (Table 3), the transport assay system is, by its nature, an indirect measure of binding that may be further complicated by the presence of multiple substrates. It is also likely that the P-gp substrate binding pocket is more complex than simply two or three sites and is made up of many subsites within a larger space in which compounds can bind.56,57 We also observed that the IC50 values for transport were higher than the Kd values for binding. This phenomenon has been observed previously55 and is likely due to differences in the two assay systems. The transport assay contains a large amount of lipid compared to the binding assay, which likely acts as a “sink” for drug thus reducing its aqueous concentration.58
The Dimer QT2C2Me2 (8) Inhibited P-gp Mediated Transport in Cells Derived from the Human Blood-Brain Barrier
The blood-brain barrier resides within the brain capillary endothelium. The endothelial cells within these vessels are the functional unit of the barrier, protecting the brain from neurotoxicants and limiting entry of therapeutic drugs. P-gp is expressed on the apical membrane of brain capillary endothelial cells and actively excludes agents from the brain by pumping them back into the blood. To determine the efficacy of the QT dimers in a cell-based blood-brain barrier model, we measured the ability of QT2C2Me2 (8) to inhibit P-gp-mediated transport in immortalized human brain capillary endothelial (hCMEC/D3) cells. These cells express endogenous levels of P-gp and retain the major protein expression patterns and functional and morphological features of normal brain endothelial cells.59,60 Furthermore, these cells have been used as a model for studies of barrier function and disruption.
hCMEC/D3 cells were incubated with the fluorescent P-gp substrates R123 or calcein-AM in the presence or absence of increasing concentrations of QT monomer (Figure 5A and B) or QT2C2Me2 (8) (Figure 5C and 5D). An increase in intracellular fluorescence, which is indicative of inhibition of P-gp, was quantified by flow cytometry (Figure S3). Mean fluorescence values obtained from the dose response curve shown in Figure 5 were used to determine the IC50 values for each compound (Figure 5, insets). The data show that the dimer QT2C2Me2 (8) was 100- and 20-fold more potent than the QT monomer for increasing R123 and calcein-AM accumulation, respectively, with IC50 values of 0.6 ± 0.1 and 0.4 ± 0.1 μM. Thus, QT2C2Me2 (8) was an effective inhibitor of P-gp in vitro in cells derived from the BBB and thus has potential to inhibit P-gp in vivo.
Figure 5.

Inhibition of P-gp transport in immortalized human hCMEC/D3 cells derived from the blood-brain barrier. IC50 values ± SEM for inhibition of P-gp transport of R123 (○) (0.5 μg/mL) and calcein-AM (□) (0.5 μM) by QT (A and B) and QT2C2Me2 (8) (C and D) in immortalized human hCMEC/D3 cells. Each curve represents the mean of three independent experiments ± SEM. The original flow cytometry data from which these results were derived are shown in Figure S3.
QT2C2Me2 (8) Inhibited P-gp Transport in Isolated Rat Brain Capillaries
To more accurately mimic the in vivo state of the blood brain barrier, we performed transport experiments with the fluorescent P-gp substrate [N-ε(4-nitrobenzofurazan-7-yl)-d-Lys8]cyclosporin A (NBD-CsA) in isolated rat brain capillaries that express native levels of P-gp (Figure 6).61,62 In this assay, luminal accumulation of NBD-CsA is a measure of P-gp transport activity. Upon inhibition of P-gp, the lumen of the capillary will accumulate less NBD-CsA and appear darker. Representative confocal images of capillaries incubated to steady-state are shown in Figure 6A–D. The control capillaries show intense luminal fluorescence (Figure 6A) that is substantially reduced in the presence of the specific P-gp inhibitor, PCS833 (Figure 6B). Although these capillaries also express other ABC transporters, including ABCG2 and ABCC2, NBD-CsA is only a substrate for P-gp under the conditions of our assay.61,62
Figure 6.

P-gp-mediated transport in isolated rat brain capillaries. P-gp transport activity was determined using isolated rat brain capillaries as described in Methods. Representative images of capillaries preincubated with (A) DMSO, (B) 5 μM PSC833, (C) 2.5 μM QT, or (D) 2.5 μM QT2C2Me2 (8) followed by 60 min incubation with 2 μM NBD-CsA. Capillary fluorescence was visualized with a Zeiss 510 NLO confocal scanning microscope and quantitated from images using ImageJ software. (E, F) Capillaries were preincubated with the indicated concentrations of (D) QT and (E) QT2C2Me2 (8) followed by 60 min incubation with 2 μM NBD-CsA. Shown are mean ± SEM for 5–12 capillaries. Statistical comparisons: one-way ANOVA, ***P < 0.001.
Representative confocal images of capillaries treated with 2.5 μM QT and QT2C2Me2 (8) are shown in Figure 6C and D, respectively. Both agents reduced fluorescence, but the dimer QT2C2Me2 (8) appeared to be more effective. We quantified luminal fluorescence using ImageJ (NIH) from experiments containing increasing concentrations of QT and QT2C2Me2 (8) (Figure 6E and F). Clearly QT2C2Me2 (8) was a more effective inhibitor of P-gp-dependent transport than QT. These data indicate that dimeric prodrugs of antipsychotic agents can be effective inhibitors of P-gp-mediated transport at the BBB in vitro in rat brain capillaries.
QT2C2Me2 (8) Inhibited P-gp in an in Situ Brain Perfusion Rat Model
To determine directly whether QT2C2Me2 (8) also inhibits P-gp at the BBB in an animal model under near physiological conditions, we performed in situ rat brain perfusion experiments.63 Using this assay, we infused the P-gp substrate [3H]verapamil or the vascular marker [14C]sucrose, which does not permeate the intact BBB, into the carotid artery and measured radiolabel accumulation in the brain. In controls, accumulation of [3H]verapamil was time dependent and nearly at steady state by 20 min (Figure 7A). At 20 min, the accumulation of [3H]verapamil and [14C]sucrose (Rbr) in control animals was similar to values reported in previous studies, that is, about 800 and 20 μL g–1, respectively (Figure 7B and C).63,64
Figure 7.

Accumulation of [3H]-verapamil and [14C]-sucrose in rat brain following in situ brain perfusion. (A) Time course of [3H]-verapamil (0.1 μCi/mL) brain accumulation in rats perfused for 10, 15, or 20 min in the absence (●) or presence of QT (10 μM) or QT2C2Me2 (8) (10 μM) (▲). Brain samples were processed as described in Methods, and the accumulated radioactivity was quantified by scintillation counting. Data (Rbr, μL g–1) are expressed as the ratio of radioactivity in the brain (dpm g–1) to that of perfusate (dpm μL–1). (B, C) Brain accumulation of (B) [3H]verapamil or (C) [14C]sucrose in rats perfused for 20 min in the presence or absence of 10 μM QT or QT2C2Me2 (8). Cyclosporin A (CsA) was used as a positive control. Shown are mean ± SEM for 6–8 rats. Statistical comparisons: one-way ANOVA, ***P < 0.001, ****P < 0.0001.
To test the effects of monomeric QT or 10 μM QT2C2Me2 (8) on brain, [3H]verapamil or [14C]sucrose accumulation in brain, the agents were added to the perfusate at 10 μM. As demonstrated above, QT2C2Me2 (8) was stable to plasma esterases with a half-life ∼38 h, so breakdown of the dimer during the course of the perfusion experiments was not a concern. Addition of QT or QT2C2Me2 (8) to the perfusate significantly increased Rbr for [3H]verapamil without changing Rbr for [14C]sucrose (Figure 7B and C). Indeed, the accumulation of [3H]verapamil tripled in the presence of QT2C2Me2 (8) (10 μM). Of the two compounds, QT2C2Me2 (8) was clearly more potent than monomeric QT. QT2C2Me2 (8) demonstrated ∼50% of the inhibitory activity of the P-gp inhibitor CsA (8 μM), which essentially abolishes P-gp activity in intact rats without affecting sucrose uptake.63,64 This result indicates that the increase in [3H]verapamil was not due to increases in paracellular permeability caused by QT and the dimer (Figure 7C). Together, these data show that QT and QT2C2Me2 (8) increased brain uptake of the P-gp substrate [3H]verapamil through inhibition of P-gp, with the dimer being more effective.
Together, these data using human capillary endothelial cells, rat brain capillaries and an in situ rat brain model demonstrated that the prodrug P-gp inhibitor, QT2C2Me2 (8), effectively inhibits P-gp at the BBB more effectively than its monomeric counterpart, QT. Importantly, both QT2C2 (1) and QT2C2Me2 (8) demonstrated minimal cytotoxic effects under the conditions and durations of the experiments described herein. In a more extended cytotoxicity assay (72 h), MCF-7 cells treated with 10 μM QT2C2 (1) or QT2C2Me2 (8) displayed ∼80% and ∼50% cell viability, respectively. Previous studies have shown that inhibition of P-gp increases the brain penetration of antipsychotics;27,28,65 however, adverse interactions may occur between the P-gp inhibitor and substrate.27,66 The use of dimeric prodrugs of already approved therapeutics potentially reduces the risk of drug–drug interactions and should increase brain penetration, thus lowering the effective dosage. In this study, we provide the first evidence that prodrug dimers can inhibit P-gp within an intact BBB, thus increasing the brain penetration of P-gp substrates without affecting BBB tight junction permeability.
Conclusions
In the present study, we used the antipsychotic agent QT as a model for therapeutic agents with poor brain penetration. By dimerizing QT with reversible linkages, we converted the molecule from a P-gp substrate to a potent P-gp inhibitor that specifically interacts with the drug binding sites on P-gp with substantially higher affinity than the monomeric drug. These dimers were stable in human plasma but were hydrolyzed to their monomeric forms using pig liver esterase as a model of cellular esterases. Ultimately, dimers that gain entry into the brain endothelial cells could revert to their monomeric forms in the esterase-rich environment of the cytosol, thus delivering the original therapy. We further demonstrated that QT dimers were potent P-gp inhibitors in several biological models of the BBB. Our best inhibitor QT2C2Me2 (8) was significantly more effective at blocking P-gp function than monomeric QT in human cells derived from brain capillary endothelial cells in culture, in isolated rat brain capillaries in vitro and ultimately in an in situ rat brain perfusion animal model.
P-gp at the blood-brain barrier presents a major therapeutic hurdle in the treatment of numerous brain disorders, including schizophrenia. This ATP-driven efflux transporter limits the brain penetration of drugs by actively removing them from brain capillary endothelial cells back into the blood. Thus, the drugs are unable to reach their molecular targets in the brain parenchyma at a sufficient concentration to have a therapeutic effect. Recently, the International Transporter consortium published a paper positing a low probability of modulating P-gp at the BBB with currently marketed drugs.67 Our overall strategy to overcome P-gp-based drug resistance at the BBB can be distilled down to the concept that one agent can serve as both the P-gp inhibitor, in the latent dimeric form, and the therapeutic agent itself. By eliminating combination therapies and their risk of toxic drug–drug interactions,68 along with the facile synthesis of the dimers and the dramatic increase in the affinity of the dimers for P-gp over monomeric therapies, this dimerization drug delivery strategy could be useful for increasing the brain penetration and thus clinical efficacy of schizophrenia and other brain disorder therapies. Alternatively, uncleavable dimers of these therapies could be used solely as P-gp inhibitors to augment the brain delivery of a variety of monomeric drugs.
Methods
Materials
QT fumarate was obtained from AvaChem Scientific (San Antonio, TX). 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) and Eco-Lite (+) scintillation cocktail were obtained from MP Biomedicals (Solon, OH). GF120918 was synthesized in our laboratory using established methodologies.42 R123, (BODIPY)-FL-verapamil, calcein-AM, Sf9 cells, and antibiotic-antimycotic were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Lawrenceville, GA). l-Glutamine, penicillin-streptomycin, and Basal Medium Eagle were obtained from Mediatech (Herndon, VA). [3H]Daunorubicin and 125I]-IAAP were purchased from Perkin-Elmer (Waltham, MA). EDC was purchased from AK Scientific (Union City, CA). [3H]Verapamil and [14C]sucrose were purchased from American Radiolabeled Chemicals (St. Louis, MO). Cultrex rat collagen I was obtained from Trevigen (Gaithersburg, MD). 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) was purchased from AK Scientific (Union City, CA). [N-ε(4-Nitrobenzofurazan-7-yl)-d-Lys8]cyclosporin A (NBD-CsA) was custom synthesized by R. Wenger (Basel, Switzerland). PSC833 was a gift from Novartis. The immortalized human brain capillary endothelial cell line, hCMEC/D3, was obtained under license from Institut National de la Santé et de la Recherche Médicale (INSERM, Paris, France). All other reagents and chemicals were purchased from either Sigma-Aldrich (St. Louis, MO) or Invitrogen (Carlsbad, CA).
General Synthesis of QT Dimers
The free base of QT was obtained from QT fumarate as previously described.69 To a solution of commercially available dicarboxylic acid (0.04 mmol) in dry DMF (500 μL) at 0 °C was added EDC (0.17 mmol), 4-dimethylaminopyridine (0.02 mmol), and N,N-diisopropylethylamine (0.46 mmol). The solution stirred for 20 min 0 °C. QT (0.13 mmol) was added and the mixture was allowed to warm to room temperature and stirred for 12 h. The solvent was removed in vacuo, and the crude residue was separated by reverse phase HPLC using a C5 column (Phenomenex, Torrance, CA) and an eluent consisting of solvent A (water with 0.1% trifluoroacetic acid (TFA)) and solvent B (methanol with 0.1% TFA) with a gradient of 30–95% solvent B, a flow rate of 8 mL/min and UV detection at 214 and 254 nm. The compounds were purified to >95% homogeneity by HPLC and characterized by MALDI-TOF mass spectrometry. QT2C2 (1) [M + H]+: 849.1 (calculated), 849.3 (observed); QT2C3 (2) [M + H]+: 863.1 (calculated), 863.6 (observed); QT2C4 (3) [M + H]+: 877.1 (calculated), 877.6 (observed); QT2C5 (4) [M + H]+: 891.2 (calculated), 892.0 (observed); QT2C6 (5) [M + H]+: 905.2 (calculated), 905.1 (observed); QT2C7 (6) [M + H]+: 919.2 (calculated), 919.0 (observed); QT2C8 (7) [M + H]+: 933.2 (calculated), 934.0 (observed); QT2C2Me2 (8) [M + H]+: 877.1 (calculated), 877.6 (observed).
Cell Culture
MCF-7/DX1 cells were cultured at 37 °C with 5% carbon dioxide in RPMI 1640 medium supplemented with 10% FBS, 2 mM l-glutamine, 50 units/mL penicillin, 50 μg/mL streptomycin, and 1 μM doxorubicin. Sf9 cells were cultured at 27 °C in Sf-900 II SFM medium supplemented with 0.5× antibiotic–antimycotic.42 hCMEC/D3 cells were cultured as described previously.59,60,70 Cells were cultured in EBM-2 medium supplemented with 5% fetal bovine serum “gold”, 1% penicillin-streptomycin, 1.4 μM hydrocortisone, 5 μg/mL ascorbic acid, 1:100 dilution of chemically defined lipid concentrate, 10 mM HEPES, and 1 ng/mL basic fibroblast growth factor (FGF). The culture flasks were coated with 0.1 mg/mL rat tail collagen for at least 1 h at 37 °C before use and cells were incubated at 37 °C with 5% CO2.
Expression of P-gp in Insect Cells
Sf9 cells in 150 cm2 flasks (1.86 × 107 cells/flask) were infected with BV-MDR1 at a multiplicity of infection of five in 5 mL of culture medium as previously described.71 After a 2 h incubation at 27 °C, cells were fed with 15 mL of culture medium and incubated at 27 °C for another 72 h.
Preparation of Crude Insect Cell Membranes
BV-MDR1 infected Sf9 cells were collected, and crude membrane extracts prepared as described previously with minor modifications.71 Cells were harvested by centrifugation approximately 72 h after infection. The cell pellet was resuspended at a density of 1 × 107 cells/mL in homogenization buffer (50 mM Tris, pH 7.5, 50 mM mannitol, 2 mM EGTA, 1 mM 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF), 2 mM dithiothreitol (DTT), and 1% (v/v) aprotinin). After a 40 min incubation on ice, the cells were homogenized by 30 strokes with a Dounce homogenizer (pestle A) followed by 30 strokes with pestle B. The lysate was centrifuged at 500g for 10 min at 4 °C to remove nuclear debris. The supernatant was centrifuged at 100 000g for 60 min at 4 °C to isolate the crude cell membranes. The resulting pellet was resuspended using blunt-ended and bent 18-, 20-, 22-, and 25-gauge needles sequentially in buffer containing 50 mM Tris, pH 7.5, 300 mM mannitol, 1 mM EGTA, 1 mM AEBSF, 1 mM DTT, 1% (v/v) aprotinin, and 10% glycerol. The membranes were assayed for total protein concentration and then stored at −80 °C.71 P-gp expression was verified by immunoblot with C219 primary antibody (1:4000) and horseradish peroxidase (HRP)-conjugated goat anti-mouse secondary antibody (1:4000).
Flow Cytometry Assays
Flow cytometry assays were performed as described previously with minor modifications.72 MCF-7/DX1 or hCMEC/D3 cells (125 000 cells) were suspended in 1 mL Basal Medium Eagle (BME) medium supplemented with 5% FBS and were incubated with R123 (0.5 μg/mL), doxorubicin (3 μM), or BODIPY-FL-verapamil (0.5 μM) and increasing concentrations of the compounds of interest for 30 min at 37 °C. Cells were incubated at 37 °C for 10 min for calcein-AM (0.5 μM) before collection by centrifugation at 300g at 4 °C for 5 min. GF120918 (1 μM) served as a positive control. All compounds were dissolved in DMSO with a final DMSO concentration of 1% (v/v). For R123 and doxorubicin-containing samples, cells were collected by centrifugation at 300g, resuspended in 1 mL of BME medium containing 1 μM GF120918 or increasing concentrations of the compounds of interest, and incubated another 30 min at 37 °C. These cells and those treated with BODIPY-FL-verapamil were collected by centrifugation at 300g, resuspended in ice cold PBS, pH 7.4, and analyzed using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA) equipped with a 488 nm argon laser and a 530 nm band-pass filter (FL1) for R123 and BODIPY-FL-verapamil, and a 585/42 band-pass filter (FL2) for doxorubicin. Calcein accumulation studies were performed as above with the following modifications. Calcein-AM (0.5 μM) was incubated with MCF-7/DX1 cells in the presence of increasing concentrations of the compounds of interest and incubated at 37 °C for 10 min. For calcein-AM treated samples, the DMSO concentration was 5% (v/v). Cells were collected and analyzed as with R123. Ten thousand cells were counted for each data point, and the mean fluorescence was utilized to determine the IC50 values using GraphPad Prism 4.
Radioactive Substrate Accumulation Assay
Radiolabeled substrate accumulation assays were performed as described previously with some modifications.72 MCF-7/DX1 cells (500 000 cells) were seeded in 6-well plates the night before assaying. The next day, the cells were washed with PBS, pH 7.4 (2 mL). Two milliliters of BME medium supplemented with 5% FBS containing [3H]daunorubicin (0.25 μCi/mL) and increasing concentrations of the compounds of interest was added to the cells. The known P-gp inhibitor GF12091873 (1 μM) served as a positive control. All compounds were dissolved in DMSO with a final DMSO concentration of 1%. The cells were incubated at 37 °C for 40 min and then washed twice with ice cold PBS, pH 7.4 (2 mL). Prewarmed trypsin-EDTA (1 mL) was added to the cells, and the cells were incubated at 37 °C for 1 h. The contents of the wells along with two washes with ice cold PBS (0.5 mL each) was then added to scintillation vials containing 18 mL Eco-Lite (+) scintillation cocktail. Each vial was counted using a Packard Tri-Carb 1600 CA liquid scintillation analyzer and data normalized to counts per 10 000 cells. IC50 values were determined using GraphPad Prism 4.
ATP Hydrolysis Assay
Vanadate-sensitive ATP consumption in the absence and presence of increasing concentrations of either QT or the QT dimer was analyzed as previously described.72 Crude membranes (10 μg) derived from Sf9 cells expressing human P-gp were incubated with either DMSO or the test compounds in a total volume of 100 μL assay buffer (45 mM Tris-HCl, pH 7.5, 5 mM sodium azide, 2 mM EGTA, 1 mM ouabain, 2 mM DTT, and 10 mM MgCl2) and 5 mM ATP, with or without sodium orthovanadate (300 μM) at 37 °C for 20 min. The reaction was terminated by the addition of 100 μL of 5% (w/v) SDS, and ATPase activity was measured by the colorimetric detection of inorganic phosphate released.72 All assays were performed in duplicate with two replicates per assay.
[125I]-IAAP Photoaffinity Cross-Linking Assay
Photoaffinity cross-linking of P-gp with ([125I]IAAP) (specific activity 2200 Ci/mmol) was performed as described previously with some modifications.72 Crude Sf9 membranes containing P-gp (25 μg) were incubated in the dark for 10 min at room temperature in assay buffer (50 mM Tris-HCl, pH 7.5, 1% aprotinin, 1 mM DTT, and 2 mM AEBSF) with either DMSO, GF120918 (10 μM; a positive control), or increasing concentrations of the compounds of interest and [125I]IAAP (11 nM). All compounds were dissolved in DMSO with a final DMSO concentration of 2.5% (v/v). The samples were exposed to UV light (365 nm) for 20 min on ice and separated by SDS-PAGE using a 7.5% Tris-glycine gel. The gel was fixed and allowed to dry overnight. Following exposure to X-ray film at −80 °C for 3 h, the band corresponding to P-gp was quantified using ImageJ (NIH, Bethesda, MD) to determine the amount of [125I]IAAP photo-cross-linked to P-gp. Values are represented as a percent of the DMSO control sample, and the IC50 values were determined by GraphPad Prism 4.
Plasma Stability Assay
Plasma stability studies were performed as described previously, with some modifications.74 QT dimers (40 μM) were incubated at 37 °C in 55% human plasma (diluted in PBS, pH 7.4) or in the presence of 10 units of purified pig liver esterase. At various time points, acetonitrile containing an internal standard (quinine, 30 μM) was added to precipitate proteins. The solution was vortexed for 20 s, and centrifuged at 6000 × g for 10 min. The supernatant was stored at −80 °C until analysis by analytical HPLC using a C5 column (Phenomenex). The conversion of QT dimers to QT monomer was monitored by HPLC over time. The formation of monomeric QT was verified by coelution with a QT standard. Peak areas of QT monomer were quantified and normalized to the internal standard, and the half-life of the dimers was determined using GraphPad Prism 4.
Purification of P-gp
Purified P-gp was isolated by two-step membrane extraction and purification from the colchicine-resistant Chinese hamster ovary cell line CHRB30 as previously described,50 with some modifications. Briefly, plasma membrane vesicles were treated with 15 mM CHAPS in HEPES buffer (20 mM HEPES, 5 mM MgCl2, 100 mM NaCl, 2 mM dithioerythritol, pH 7.4), and after centrifugation the resulting pellet was solubilized in 45 mM CHAPS in HEPES buffer. The supernatant obtained after centrifugation was diluted to 15 mM CHAPS and purified on a concanavalin A-Sepharose 4B column equilibrated with 2 mM CHAPS in HEPES buffer. Protein in the column eluent was quantitated by Bradford assay,75 and ATPase activity was measured using a colorimetric assay for inorganic phosphate with 2 mM ATP and 20 min incubation at 37 °C.76 The final P-gp preparation was 90–95% pure, with a concentration of 0.2–0.3 mg/mL protein and ATPase activity of 1.8–2.8 μmol Pi/min/mg. The purified protein was stored at −70 °C.
Measurement of Binding Affinity by Tryptophan Fluorescence Quenching of Purified P-gp
All fluorescence measurements were made on a PTI QM-8 steady-state fluorimeter at 22 °C, with excitation and emission bandwidths of 2 and 4 nm, respectively. Intrinsic Trp fluorescence was measured by exciting 250 μL of 50 μg/mL purified P-gp in 2 mM CHAPS/20 mM HEPES in a quartz microcuvette (0.5 cm path length) at 290 nm and monitoring emission at 330 nm.50 Titrations of P-gp with various QT compounds was carried out by successive addition of 0.5 μL aliquots in DMSO. Fluorescence measurements were corrected for dilution, scattering, and the inner filter effect as previously described.51,77 All experiments were carried out in triplicate and the data were fitted to a hyperbolic equation for monophasic quenching.50,51 The maximum percent quenching, ΔFmax, and the dissociation constant for binding to P-gp, Kd, were extracted from fitting of the data using SigmaPlot. Three independent quenching experiments were conducted for each compound.
Inhibition of Drug Transport by Reconstituted P-gp
The ability of QT and QT2C2Me2 (8) to compete for transport of three fluorescent substrates was examined using purified reconstituted Pgp as described previously,58,78,79 with some modifications. Purified protein was reconstituted into proteoliposomes of egg phosphatidylcholine (PC) at a lipid/protein ratio of 10:1 (w/w). The initial rate of transport was measured at 37 °C with TMR (1 μM), H33342 (1 μM), and LDS-751 (4 μM), in the presence of increasing concentrations of QT and QT2C2Me2 (8). The concentration of QT and QT2C2Me2 (8) that resulted in 50% inhibition of the transport rate for each substrate was defined as the IC50 value, and was estimated from a plot of % initial rate of transport vs concentration.
Rat Brain Capillary Isolation
Male Sprague–Dawley rats (Taconic, Hudson, NY) were euthanized by CO2, decapitated, and the brains removed for capillary isolation as described previously.62,80 Brains from 5–10 rats were placed in ice cold PBS complete (2.7 mM KCl, 1.46 mM KH2PO4, 139.9 mM NaCl, 8.1 mM Na2HPO4, 0.9 mM CaCl2, 1.05 mM MgCl2), pH 7.4, supplemented with 5 mM glucose and 1 mM sodium pyruvate. After removal of the cerebellum, choroid plexuses, meninges, and white matter, the gray matter was homogenized in a 4-fold volume of PBS complete using ∼30 strokes of a Teflon-tipped drill mounted homogenizer (200 μm clearance) followed by ∼5 strokes of a Dounce homogenizer (150 μm clearance). Ficoll solution (30% w/v) was added to the homogenate with a ∼1.2:1 ratio of Ficoll to homogenate, and the samples were centrifuged for 20 min at 5800g, 4 °C. The pellet containing capillaries was resuspended in PBS complete containing 1% BSA (w/v) and applied to a column containing 40 mL of glass beads (Sartorius, Goettingen, Germany) pre-equilibrated with PBS complete containing 1% BSA. Following several washes with PBS complete with 1% BSA, capillaries were collected from the beads by gentle agitation and with several washes with ice cold PBS complete. The capillaries were then collected by centrifugation at 500g for 5 min at 4 °C and washed three times with PBS complete. The final capillary-containing pellet was resuspended in 300–500 μL ice cold PBS complete and stored on ice.
P-Glycoprotein Transport Assay with Rat Brain Capillaries
Capillary transport assays performed as described previously.62,63 Rat brain capillaries (30 μL) were incubated at room temperature in PBS complete for 5 min before the addition of PBS complete (470 μL) containing QT, QT2C2Me2 or PSC833 as a control. Following incubation at room temperature for 45 min, an aliquot of PBS complete (500 μL) containing NBD-CsA (2 μM) was added and the capillaries incubated for an additional 30 min at room temperature. The capillaries were then imaged with a Zeiss 510 NLO confocal scanning microscope using an argon laser (488 nm excitation) and 40× water-immersion objective (NA = 1.2). For each capillary imaged, mean luminal fluorescence was quantified using ImageJ (NIH).
In Situ Rat Brain Perfusion Assay
In situ brain perfusion assays were performed as described previously with minor modifications.63 Briefly, male rats were anaesthetized by i.p. injection with 1 mL/kg ketamine cocktail (79 mg/mL ketamine, 3 mg/mL xylazine, 0.6 mg/mL acepromazine) and administered heparin (10 kU/kg). The common carotid arteries were exposed and cannulated using silicone tubing connected to a perfusion circuit system, and perfused with oxygenated Ringer’s solution (37 °C) (in mM: NaCl 117, KCl 4.7, MgSO4 0.8, NaHCO3 24.8, KH2PO4 1.2, CaCl2 2.5, d-glucose 10, plus 39 g/L 170 kDa dextran, 1 g/L BSA, and 0.055 g/L Evans Blue) at 3 mL/min. [14C]Sucrose (0.5 μCi/mL) or [3H]verapamil (0.1 μCi/mL) and either QT (10 μM) or QT2C2Me2 (10 μM) were infused into the circuit via syringe pump at 0.5 mL/min for 20 min. Following perfusion, a sample of perfusate was collected from each cannula as a reference. Cerebral hemisphere sections were separated and stripped of meninges, midbrain and choroid plexuses, and minced. Tissue and 100 μL perfusate samples were digested for 2 days with tissue solubilization solution (Hyamine Hydroxide, MP Biomedicals, Santa Ana, CA). Samples were prepared for scintillation counting by adding 100 μL of 30% acetic acid and 4 mL of liquid scintillation cocktail (CytoScint ES, MP Biomedicals), and incubated overnight in the dark. Results were expressed as the ratio of radioactivity in the brain to that of perfusate (Rbr μL/g): (Rbr = Cbrain/Cperfusate) where Cbrain is the dpm/g in the brain tissue and Cperfusate is the dpm/μL in the perfusate.
Glossary
Abbreviations
- ABC
ATP-binding cassette
- AEBSF
4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride
- BBB
blood-brain barrier
- BME
Basal Medium Eagle
- BODIPY
4,4-difluoro-4-bora-3a,4a-diaza-s-indacene
- calcein-AM
calcein-acetoxymethylester
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- DTT
dithiothreitol
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide
- FBS
fetal bovine serum
- H33342
Hoechst 33342
- [125I]IAAP
[125I]iodoarylazidoprazosin
- LDS-751
2-{4-[4-(dimethylamino)phenyl]-l,3-butadienyl}-3-ethylbenzothiazoliumperchlorate
- NBD-CsA
[N-ε(4-nitrobenzofurazan-7-yl)-d-Lys8]cyclosporin A
- PBS
phosphate buffered saline
- P-gp
P-glycoprotein
- QT
quetiapine
- R123
rhodamine 123
- TFA
trifluoroacetic acid
- TMR
tetramethylrosamine
Supporting Information Available
Supplementary Figure S1 shows the histograms used to determine the IC50 values for the QT dimers presented in Table 1. Supplementary Figure S2 shows the concentration curves used to generate the IC50 values for QT, QT2C2 (1), and QT2C2Me2 (8) presented in Table 2. Supplementary Figure S3 shows the histograms used to generate the concentration curves and IC50 values for QT, QT2C2 (1), and QT2C2Me2 (8) presented in Figure 5. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ D.E. and C.R.C. contributed equally to this work. D.E. synthesized the dimers and conducted the transport and photo-cross-linking experiments with the assistance of K.B. H.A.N. assisted with the synthesis of the dimers and conducted the stability and breakdown experiments. D.W. purified P-gp and carried out the Trp quenching experiments. P.L. performed the transport inhibition assays with purified P-gp. C.R.C. and D.E. performed the rat brain capillary assays, and C.R.C. performed the rat brain perfusion assays. J.C., F.J.S., D.S.M., and C.A.H. interpreted the data with the other authors. C.A.H. prepared the manuscript with contributions from all authors. All authors have given approval to the final version of the manuscript.
This work was supported by a grant from NIH/NEI 1R21EY018481-01(C.A.H. and J.C.) and a research grant (#770248) from the Canadian Cancer Society (F.J.S.).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Meltzer H. Y. (2002) Mechanism of action of atypical antipsychotic drugs. In Neuropsychopharmacology: The fifth generation of progress(Davis K. L., Charney D., Coyle J. T., and Nemeroff C., EdS.), American College of Neuropsychopharmacology, Reven Press, New York. [Google Scholar]
- Meltzer H. Y.; Matsubara S.; Lee J. C. (1989) Classification of typical and atypical antipsychotic drugs on the basis of dopamine D-1, D-2 and serotonin2 pKi values. J. Pharmacol. Exp. Ther. 251, 238–246. [PubMed] [Google Scholar]
- Hitchcock S. A. (2008) Blood-brain barrier permeability considerations for CNS-targeted compound library design. Curr. Opin. Chem. Biol. 12, 318–323. [DOI] [PubMed] [Google Scholar]
- Pardridge W. M. (2007) Blood-brain barrier delivery. Drug Discovery Today 12, 54–61. [DOI] [PubMed] [Google Scholar]
- Weiss N.; Miller F.; Cazaubon S.; Couraud P. O. (2009) The blood-brain barrier in brain homeostasis and neurological diseases. Biochim. Biophys. Acta 1788, 842–857. [DOI] [PubMed] [Google Scholar]
- Raggi M. A.; Mandrioli R.; Sabbioni C.; Pucci V. (2004) Atypical antipsychotics: Pharmacokinetics, therapeutic drug monitoring and pharmacological interactions. Curr. Med. Chem. 11, 279–296. [DOI] [PubMed] [Google Scholar]
- Bebawy M.; Chetty M. (2008) Differential pharmacological regulation of drug efflux and pharmacoresistant schizophrenia. Bioessays 30, 183–188. [DOI] [PubMed] [Google Scholar]
- Glaeser H. (2011) Importance of P-glycoprotein for drug-drug interactions. Handb. Exp. Pharmacol. 201, 285–297. [DOI] [PubMed] [Google Scholar]
- Meyer J. (2007) Drug-drug interactions with antipsychotics. CNS Spectrums 12, 6–9. [DOI] [PubMed] [Google Scholar]
- Sandson N. B.; Armstrong S. C.; Cozza K. L. (2005) An overview of psychotropic drug-drug interactions. Psychosomatics 46, 464–94. [DOI] [PubMed] [Google Scholar]
- Moons T.; de Roo M.; Claes S.; Dom G. (2011) Relationship between P-glycoprotein and second-generation antipsychotics. Pharmacogenomics 12, 1193–211. [DOI] [PubMed] [Google Scholar]
- Ambudkar S. V.; Dey S.; Hrycyna C. A.; Ramachandra M.; Pastan I.; Gottesman M. M. (1999) Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol. 39, 361–398. [DOI] [PubMed] [Google Scholar]
- Hrycyna C. A. (2001) Molecular genetic analysis and biochemical characterization of mammalian P-glycoproteins involved in multidrug resistance. Semin. Cell Dev. Biol. 12, 247–256. [DOI] [PubMed] [Google Scholar]
- Kimura Y.; Morita S. Y.; Matsuo M.; Ueda K. (2007) Mechanism of multidrug recognition by MDR1/ABCB1. Cancer Sci. 98, 1303–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo T. W.; Clarke D. M. (2005) Recent progress in understanding the mechanism of P-glycoprotein-mediated drug efflux. J. Membr. Biol. 206, 173–185. [DOI] [PubMed] [Google Scholar]
- Sharom F. J. (2011) The P-glycoprotein multidrug transporter. Essays Biochem. 50, 161–178. [DOI] [PubMed] [Google Scholar]
- Eckford P. D.; Sharom F. J. (2009) ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 109, 2989–3011. [DOI] [PubMed] [Google Scholar]
- Gottesman M. M.; Fojo T.; Bates S. E. (2002) Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2, 48–58. [DOI] [PubMed] [Google Scholar]
- Loscher W.; Potschka H. (2005) Drug resistance in brain diseases and the role of drug efflux transporters. Nat. Rev. 6, 591–602. [DOI] [PubMed] [Google Scholar]
- Boulton D. W.; DeVane C. L.; Liston H. L.; Markowitz J. S. (2002) In vitro P-glycoprotein affinity for atypical and conventional antipsychotics. Life Sci. 71, 163–169. [DOI] [PubMed] [Google Scholar]
- Schmitt U.; Kirschbaum K. M.; Poller B.; Kusch-Poddar M.; Drewe J.; Hiemke C.; Gutmann H. (2012) In vitro P-glycoprotein efflux inhibition by atypical antipsychotics is in vivo nicely reflected by pharmacodynamic but less by pharmacokinetic changes. Pharmacol., Biochem. Behav. 102, 312–320. [DOI] [PubMed] [Google Scholar]
- Wang J. S.; Taylor R.; Ruan Y.; Donovan J. L.; Markowitz J. S.; Lindsay De Vane C. (2004) Olanzapine penetration into brain is greater in transgenic ABCB1a P-glycoprotein-deficient mice than FVB1 (wild-type) animals. Neuropsychopharmacology 29, 551–557. [DOI] [PubMed] [Google Scholar]
- Doran A.; Obach R. S.; Smith B. J.; Hosea N. A.; Becker S.; Callegari E.; Chen C.; Chen X.; Choo E.; Cianfrogna J.; Cox L. M.; Gibbs J. P.; Gibbs M. A.; Hatch H.; Hop C. E.; Kasman I. N.; Laperle J.; Liu J.; Liu X.; Logman M.; Maclin D.; Nedza F. M.; Nelson F.; Olson E.; Rahematpura S.; Raunig D.; Rogers S.; Schmidt K.; Spracklin D. K.; Szewc M.; Troutman M.; Tseng E.; Tu M.; Van Deusen J. W.; Venkatakrishnan K.; Walens G.; Wang E. Q.; Wong D.; Yasgar A. S.; Zhang C. (2005) The impact of P-glycoprotein on the disposition of drugs targeted for indications of the central nervous system: Evaluation using the MDR1a/1b knockout mouse model. Drug Metab. Dispos. 33, 165–174. [DOI] [PubMed] [Google Scholar]
- Kirschbaum K. M.; Uhr M.; Holthoewer D.; Namendorf C.; Pietrzik C.; Hiemke C.; Schmitt U. (2010) Pharmacokinetics of acute and sub-chronic aripiprazole in P-glycoprotein deficient mice. Neuropharmacology 59, 474–479. [DOI] [PubMed] [Google Scholar]
- Wang J. S.; Zhu H. J.; Donovan J. L.; Yuan H. J.; Markowitz J. S.; Geesey M. E.; Devane C. L. (2009) Aripiprazole brain concentration is altered in P-glycoprotein deficient mice. Schizophr. Res. 110, 90–94. [DOI] [PubMed] [Google Scholar]
- El Ela A. A.; Hartter S.; Schmitt U.; Hiemke C.; Spahn-Langguth H.; Langguth P. (2004) Identification of P-glycoprotein substrates and inhibitors among psychoactive compounds--implications for pharmacokinetics of selected substrates. J. Pharm. Pharmacol. 56, 967–975. [DOI] [PubMed] [Google Scholar]
- Schmitt U.; Abou El-Ela A.; Guo L. J.; Glavinas H.; Krajcsi P.; Baron J. M.; Tillmann C.; Hiemke C.; Langguth P.; Hartter S. (2006) Cyclosporine A (CsA) affects the pharmacodynamics and pharmacokinetics of the atypical antipsychotic amisulpride probably via inhibition of P-glycoprotein (P-gp). J. Neural Transm. 113, 787–801. [DOI] [PubMed] [Google Scholar]
- Pacchioni A. M.; Gabriele A.; Donovan J. L.; Lindsay Devane C.; See R. E. (2009) P-glycoprotein inhibition potentiates the behavioural and neurochemical actions of risperidone in rats. Int. J. Neuropsychopharmacol. 1–11. [DOI] [PubMed] [Google Scholar]
- Dey S.; Ramachandra M.; Pastan I.; Gottesman M. M.; Ambudkar S. V. (1997) Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 94, 10594–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C.; Berridge G.; Higgins C. F.; Mistry P.; Charlton P.; Callaghan R. (2000) Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharmacol. 58, 624–632. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B.; Ling V. (1997) Positively cooperative sites for drug transport by P-glycoprotein with distinct drug specificities. Eur. J. Biochem. 250, 130–137. [DOI] [PubMed] [Google Scholar]
- Lugo M. R.; Sharom F. J. (2005) Interaction of LDS-751 and rhodamine 123 with P-glycoprotein: Evidence for simultaneous binding of both drugs. Biochemistry 44, 14020–14029. [DOI] [PubMed] [Google Scholar]
- Qu Q.; Sharom F. J. (2002) Proximity of bound Hoechst 33342 to the ATPase catalytic sites places the drug binding site of P-glycoprotein within the cytoplasmic membrane leaflet. Biochemistry 41, 4744–4752. [DOI] [PubMed] [Google Scholar]
- Shapiro A. B.; Fox K.; Lam P.; Ling V. (1999) Stimulation of P-glycoprotein-mediated drug transport by prazosin and progesterone. Evidence for a third drug-binding site. Eur. J. Biochem. 259, 841–850. [DOI] [PubMed] [Google Scholar]
- Aller S. G.; Yu J.; Ward A.; Weng Y.; Chittaboina S.; Zhuo R.; Harrell P. M.; Trinh Y. T.; Zhang Q.; Urbatsch I. L.; Chang G. (2009) Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 323, 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M. S.; Oldham M. L.; Zhang Q.; Chen J. (2012) Crystal structure of the multidrug transporter P-glycoprotein from Caenorhabditis elegans. Nature 490, 566–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen P. C.; Verhalen B.; Wilkens S.; McHaourab H.; Tajkhorshid E. (2013) On the origin of large flexibility of P-glycoprotein in the inward-facing state. J. Biol. Chem. 288, 19211–19220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K. F.; Zhao Y.; Burkett B. A.; Wong I. L.; Chow L. M.; Chan T. H. (2006) Flavonoid dimers as bivalent modulators for p-glycoprotein-based multidrug resistance: Synthetic apigenin homodimers linked with defined-length poly(ethylene glycol) spacers increase drug retention and enhance chemosensitivity in resistant cancer cells. J. Med. Chem. 49, 6742–6759. [DOI] [PubMed] [Google Scholar]
- Chan K. F.; Zhao Y.; Chow T. W.; Yan C. S.; Ma D. L.; Burkett B. A.; Wong I. L.; Chow L. M.; Chan T. H. (2009) Flavonoid dimers as bivalent modulators for P-glycoprotein-based multidrug resistance: Structure-activity relationships. ChemMedChem 4, 594–614. [DOI] [PubMed] [Google Scholar]
- Namanja H. A.; Emmert D.; Davis D. A.; Campos C.; Miller D. S.; Hrycyna C. A.; Chmielewski J. (2011) Toward eradicating hiv reservoirs in the brain: Inhibiting P-glycoprotein at the blood-brain barrier with prodrug abacavir dimers. J. Am. Chem. Soc. 134, 2976–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namanja H. A.; Emmert D.; Pires M. M.; Hrycyna C. A.; Chmielewski J. (2009) Inhibition of human P-glycoprotein transport and substrate binding using a galantamine dimer. Biochem. Biophys. Res. Commun. 388, 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires M. M.; Emmert D.; Hrycyna C. A.; Chmielewski J. (2009) Inhibition of P-glycoprotein-mediated paclitaxel resistance by reversibly linked quinine homodimers. Mol. Pharmacol. 75, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires M. M.; Hrycyna C. A.; Chmielewski J. (2006) Bivalent probes of the human multidrug transporter P-glycoprotein. Biochemistry 45, 11695–11702. [DOI] [PubMed] [Google Scholar]
- Sauna Z. E.; Andrus M. B.; Turner T. M.; Ambudkar S. V. (2004) Biochemical basis of polyvalency as a strategy for enhancing the efficacy of P-glycoprotein (ABCB1) modulators: Stipiamide homodimers separated with defined-length spacers reverse drug efflux with greater efficacy. Biochemistry 43, 2262–2271. [DOI] [PubMed] [Google Scholar]
- Stinchcomb A. L.; Paliwal A.; Dua R.; Imoto H.; Woodard R. W.; Flynn G. L. (1996) Permeation of buprenorphine and its 3-alkyl-ester prodrugs through human skin. Pharm. Res. 13, 1519–1523. [DOI] [PubMed] [Google Scholar]
- Wagner J.; Grill H.; Henschler D. (1980) Prodrugs of etilefrine: Synthesis and evaluation of 3′-(o-acyl) derivatives. J. Pharm. Sci. 69, 1423–1427. [DOI] [PubMed] [Google Scholar]
- Sharom F. J.; Liu R.; Romsicki Y.; Lu P. (1999) Insights into the structure and substrate interactions of the P-glycoprotein multidrug transporter from spectroscopic studies. Biochim. Biophys. Acta 1461, 327–345. [DOI] [PubMed] [Google Scholar]
- Dey S.; Ramachandra M.; Pastan I.; Gottesman M. M.; Ambudkar S. V. (1998) Photoaffinity labeling of human P-glycoprotein: Effect of modulator interaction and ATP hydrolysis on substrate binding. Methods Enzymol. 292, 318–328. [DOI] [PubMed] [Google Scholar]
- Safa A. R.; Roberts S.; Agresti M.; Fine R. L. (1994) Tamoxifen aziridine, a novel affinity probe for P-glycoprotein in multidrug resistant cells. Biochem. Biophys. Res. Commun. 202, 606–612. [DOI] [PubMed] [Google Scholar]
- Liu R.; Siemiarczuk A.; Sharom F. J. (2000) Intrinsic fluorescence of the P-glycoprotein multidrug transporter: Sensitivity of tryptophan residues to binding of drugs and nucleotides. Biochemistry 39, 14927–14938. [DOI] [PubMed] [Google Scholar]
- Liu R.; Sharom F. J. (1996) Site-directed fluorescence labeling of P-glycoprotein on cysteine residues in the nucleotide binding domains. Biochemistry 35, 11865–11873. [DOI] [PubMed] [Google Scholar]
- Sharom F. J.; Liu R.; Qu Q.; Romsicki Y. (2001) Exploring the structure and function of the P-glycoprotein multidrug transporter using fluorescence spectroscopic tools. Semin. Cell Dev. Biol. 12, 257–265. [DOI] [PubMed] [Google Scholar]
- Sharom F. J.; DiDiodato G.; Yu X.; Ashbourne K. J. (1995) Interaction of the P-glycoprotein multidrug transporter with peptides and ionophores. J. Biol. Chem. 270, 10334–10341. [DOI] [PubMed] [Google Scholar]
- Sharom F. J.; Yu X.; DiDiodato G.; Chu J. W. (1996) Synthetic hydrophobic peptides are substrates for P-glycoprotein and stimulate drug transport. Biochem. J. 320, 421–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharom F. J.; Liu R.; Vinepal B. (2010) Fluorescence studies of drug binding and translocation by membrane transporters. Methods Mol. Biol. 637, 133–148. [DOI] [PubMed] [Google Scholar]
- Globisch C.; Pajeva I. K.; Wiese M. (2008) Identification of putative binding sites of P-glycoprotein based on its homology model. ChemMedChem 3, 280–295. [DOI] [PubMed] [Google Scholar]
- Klepsch F.; Chiba P.; Ecker G. F. (2011) Exhaustive sampling of docking poses reveals binding hypotheses for propafenone type inhibitors of P-glycoprotein. PLoS Comput. Biol. 7, e1002036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clay A. T.; Sharom F. J. (2013) Lipid bilayer properties control membrane partitioning, binding, and transport of P-glycoprotein substrates. Biochemistry 52, 343–354. [DOI] [PubMed] [Google Scholar]
- Poller B.; Gutmann H.; Krahenbuhl S.; Weksler B.; Romero I.; Couraud P. O.; Tuffin G.; Drewe J.; Huwyler J. (2008) The human brain endothelial cell line hCMEC/D3 as a human blood-brain barrier model for drug transport studies. J. Neurochem. 107, 1358–1368. [DOI] [PubMed] [Google Scholar]
- Weksler B. B.; Subileau E. A.; Perriere N.; Charneau P.; Holloway K.; Leveque M.; Tricoire-Leignel H.; Nicotra A.; Bourdoulous S.; Turowski P.; Male D. K.; Roux F.; Greenwood J.; Romero I. A.; Couraud P. O. (2005) Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 19, 1872–1874. [DOI] [PubMed] [Google Scholar]
- Fellner S.; Bauer B.; Miller D. S.; Schaffrik M.; Fankhanel M.; Spruss T.; Bernhardt G.; Graeff C.; Farber L.; Gschaidmeier H.; Buschauer A.; Fricker G. (2002) Transport of paclitaxel (taxol) across the blood-brain barrier in vitro and in vivo. J. Clin. Invest. 110, 1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D. S.; Nobmann S. N.; Gutmann H.; Toeroek M.; Drewe J.; Fricker G. (2000) Xenobiotic transport across isolated brain microvessels studied by confocal microscopy. Mol. Pharmacol. 58, 1357–1367. [DOI] [PubMed] [Google Scholar]
- Hawkins B. T.; Sykes D. B.; Miller D. S. (2010) Rapid, reversible modulation of blood-brain barrier P-glycoprotein transport activity by vascular endothelial growth factor. J. Neurosci. 30, 1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Hawkins B. T.; Miller D. S. (2011) Aryl hydrocarbon receptor-mediated up-regulation of ATP-driven xenobiotic efflux transporters at the blood-brain barrier. FASEB J. 25, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagami T.; Yasui-Furukori N.; Saito M.; Tateishi T.; Kaneo S. (2005) Effect of verapamil on pharmacokinetics and pharmacodynamics of risperidone: In vivo evidence of involvement of P-glycoprotein in risperidone disposition. Clin. Pharmacol. Ther. 78, 43–51. [DOI] [PubMed] [Google Scholar]
- Wang J. S.; Zhu H. J.; Markowitz J. S.; Donovan J. L.; DeVane C. L. (2006) Evaluation of antipsychotic drugs as inhibitors of multidrug resistance transporter P-glycoprotein. Psychopharmacology (Berlin, Ger.) 187, 415–423. [DOI] [PubMed] [Google Scholar]
- Kalvass J. C., Polli J. W., Bourdet D. L., Feng B., Huang S. M., Liu X., Smith Q. R., Zhang L. K., Zamek-Gliszczynski M. J., and the International Transporter Consortium. 2013Why clinical modulation of efflux transport at the human blood-brain barrier is unlikely: The ITC evidence-based position. Clin. Pharmacol. Ther. 94, 80–94. [DOI] [PubMed] [Google Scholar]
- Conley R. R.; Kelly D. L. (2007) Drug-drug interactions associated with second-generation antipsychotics: Considerations for clinicians and patients. Psychopharmacol. Bull. 40, 77–97. [PubMed] [Google Scholar]
- Cope M. B.; Nagy T. R.; Fernandez J. R.; Geary N.; Casey D. E.; Allison D. B. (2005) Antipsychotic drug-induced weight gain: Development of an animal model. Int. J. Obes. 29, 607–614. [DOI] [PubMed] [Google Scholar]
- Newman M. J.; Dixon R.; Toyonaga B. (2002) OC144–093, a novel P-glycoprotein inhibitor for the enhancement of anti-epileptic therapy. Novartis Found. Symp. 243, 213–226, discussion 226–230, 231–235. [PubMed] [Google Scholar]
- Germann U. A.; Willingham M. C.; Pastan I.; Gottesman M. M. (1990) Expression of the human multidrug transporter in insect cells by a recombinant baculovirus. Biochemistry 29, 2295–2303. [DOI] [PubMed] [Google Scholar]
- Hrycyna C. A.; Ramachandra M.; Pastan I.; Gottesman M. M. (1998) Functional expression of human P-glycoprotein from plasmids using vaccinia virus-bacteriophage T7 RNA polymerase system. Methods Enzymol. 292, 456–473. [DOI] [PubMed] [Google Scholar]
- Mayur Y. C.; Peters G. J.; Prasad V. V.; Lemo C.; Sathish N. K. (2009) Design of new drug molecules to be used in reversing multidrug resistance in cancer cells. Curr. Cancer Drug Targets 9, 298–306. [DOI] [PubMed] [Google Scholar]
- Ostergaard J.; Larsen C. (2007) Bioreversible derivatives of phenol. 1. The role of human serum albumin as related to the stability and binding properties of carbonate esters with fatty acid-like structures in aqueous solution and biological media. Molecules 12, 2380–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254. [DOI] [PubMed] [Google Scholar]
- Chifflet S.; Torriglia A.; Chiesa R.; Tolosa S. (1988) A method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: Application to lens ATPases. Anal. Biochem. 168, 1–4. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R. (2006) Principles of fluorescence spectroscopy, 3rd ed., Springer Science+Business Media, New York. [Google Scholar]
- Eckford P. D.; Sharom F. J. (2006) P-glycoprotein (ABCB1) interacts directly with lipid-based anti-cancer drugs and platelet-activating factors. Biochem. Cell Biol. 84, 1022–1033. [DOI] [PubMed] [Google Scholar]
- Eckford P. D.; Sharom F. J. (2008) Interaction of the P-glycoprotein multidrug efflux pump with cholesterol: Effects on ATPase activity, drug binding and transport. Biochemistry 47, 13686–13698. [DOI] [PubMed] [Google Scholar]
- Hartz A. M.; Bauer B.; Fricker G.; Miller D. S. (2004) Rapid regulation of P-glycoprotein at the blood-brain barrier by endothelin-1. Mol. Pharmacol. 66, 387–394. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.