

Abstract
Tumours of the spinal cord, although rare, are associated with high morbidity. Surgical resection remains the primary treatment for patients with this disease, and offers the best chance for cure. Such surgical procedures, however, carry substantial risks such as worsening of neurological deficit, paralysis and death. New therapeutic avenues for spinal cord tumours are needed, but genetic studies of the molecular mechanisms governing tumourigenesis in the spinal cord are limited by the scarcity of high-quality human tumour samples. Many spinal cord tumours have intracranial counterparts that have been extensively studied, but emerging data show that the tumours are genetically and biologically distinct. The differences between brain and spine tumours make extrapolation of data from one to the other difficult. In this Review, we describe the demographics, genetics and current treatment approaches for the most commonly encountered spinal cord tumours—namely, ependymomas, astrocytomas, haemangioblastomas and meningiomas. We highlight advances in understanding of the biological basis of these lesions, and explain how the latest progress in genetics and beyond are being translated to improve patient care.
Introduction
Spinal cord tumours can occur in the parenchyma of the cord (intramedullary lesions), in the thecal sac but external to the cord (extramedullary lesions), or outside of the thecal sac (extradural lesions). Symptoms related to tumour growth vary depending on tumour location, and include myelopathy, numbness, loss of pain and temperature sensation, and radiculopathy if the tumour encroaches on nerve roots as they exit the spinal canal. Surgical resection combined with radiotherapy is the treatment of choice for most patients with spinal cord tumours, as no significant improvement in survival has been observed with chemotherapy alone in small cohort studies.1–6 Given the limited efficacy of chemotherapy, rationally designed therapeutics for spinal cord tumours are urgently needed.
Intramedullary spinal cord tumours (IMSCTs) in adult patients account for only 5–10% of all spinal tumours, but are the most common spinal tumour in children.7 Approximately 850–1,700 cases of IMSCT are diagnosed annually in adults, with astrocytomas, ependymomas and haemangioblastomas comprising the majority of intra-medullary lesions.5 Ependymomas are the most common spinal lesions in adults, and occur in the cervical and thoracic cord or in the filum terminale.8–10 Astrocytomas in the spinal cord comprise about 40% of all IMSCTs, but only 3% of CNS astrocytomas.5,7 Astrocytomas and ependymomas most commonly affect patients with the neurocutaneous syndromes neurofibromatosis type 1 (NF1) and NF2, respectively.7,11–17
NF1 and NF2 have an autosomal dominant pattern of inheritance and no known risk factors. NF1 affects 1 in 3,000 people worldwide, whereas the prevalence of NF2 is approximately 1 in every 40,000–60,000 people.18,19 Patients with neurofibromatosis are at an increased risk of developing various lesions including IMSCTs, and mutations in the NF1 and NF2 genes have been isolated in sporadic IMSCTs.11,13,14,18,20,21 NF1 is a completely penetrant genetic disorder characterized by the presence of café-au-lait spots, axillary freckling, Lisch nodules on the iris, and nodular or plexiform neurofibromas that may lie beneath the skin or in deep tissue along peripheral nerves. NF2 is associated with bilateral vestibular schwannomas and the presence of a spinal cord lesion such as an ependymoma or meningioma.18
Haemangioblastomas are the third most common intramedullary lesion, and some of these tumours are associated with von Hippel–Lindau (VHL) disease—a disorder that leads to abnormal tumour growth in various regions of the body. 10–30% of patients with VHL disease present with haemangioblastoma in the spinal cord.5,22 Haemangioblastomas are most commonly treated with radiotherapy or surgery but, owing to their hypervascular nature, the efficacy of angiogenesis inhibitors in these lesions is currently under investigation.1,6
Intradural extramedullary spinal cord lesions include meningiomas, neurofibromas and schwannomas. Meningiomas are benign lesions that constitute 25% of all spinal cord tumours and occur at high frequency in patients with NF2.18,19 Schwannomas are nerve sheath tumours that occur sporadically and can also be associated with NF2. These lesions frequently occur at the dorsal root and can arise within the intradural space.18 Neurofibromas are benign tumours of the PNS that comprise multiple cell types and are the hallmark of NF1.19,23 Notably, plexiform neurofibromas can progress to become malignant peripheral nerve sheath tumours.
In this Review, we describe the genetics of spinal ependymomas, astrocytomas, haemangioblastomas, and meningiomas—the most common spinal cord tumours that are frequently encountered in clinical practice. Emerging targeted therapies for spinal cord tumours, which are in clinical trials for most of these pathologies, are also discussed. We highlight the lack of evidence for generalizability of intracranial tumour genetics to their spinal counterparts, particularly in the case of ependymoma. Research to improve our knowledge of the genetic differences between spinal and intracranial tumour subtypes will be critical in guiding novel diagnostic and therapeutic strategies for patients with spinal cord tumours.
Intramedullary tumours
Astrocytomas
Demographics
Astrocytomas are the most common IMSCT in children, and the second most common in adults.5,24,25 These lesions present as diffuse, heterogeneously enhancing masses in the parenchyma of the spinal cord (Figure 1). The WHO characterizes astrocytomas into four grades: pilocytic (grade I), diffuse or low grade (grade II), anaplastic (grade III) and glioblastoma (grade IV). Of note, pilocytic astrocytoma is clinically distinct from the grade II–III–IV spectrum of astrocytoma, and this type of astrocytoma predominates in children and adolescents.7 By contrast, astrocytomas in adults are more likely to be of the diffuse type.7
Figure 1.

Surgery and imaging in spinal cord astrocytoma. a–c | Intraoperative images showing the excision of a diffuse intramedullary astrocytoma in the spinal cord from a posterior approach. The spinal cord appears enlarged, and the surgical approach begins with a midline myelotomy to separate the dorsal columns (a). The tumour is then exposed and excised via careful dissection (b). Gliosis and hyperaemia can be seen in the resection cavity (c). d, e | Preoperative sagittal (d) and axial (e) T2-weighted MRI reveal hyperintensity and expansion of the spinal cord.
Glioblastoma multiforme (GBM) in the spinal column is aggressive, resistant to chemotherapeutic intervention, and has the worst prognosis of spinal cord astro-cytomas.26–29 Primary glioblastomas, or de novo GBMs, are the most common malignant tumour in the brain but are rare in the spinal cord. Advances in our understanding of brain GBM biology, however, have provided useful information for investigations into astrocytoma tumourigenesis and progression in the spinal cord.26,30
Genetics
Genomic studies dedicated exclusively to spinal astrocytoma are limited owing to the rarity of this lesion, the small size of the tumour (which limits availability of tissue for research purposes), and the risks of complications associated with surgical resection of infiltrative spinal cord tumours. Such complications include worsening of neurological deficit, paralysis, bowel and/ or bladder dysfunction, and death. Studies into the genetics of intracranial astrocytoma have paved the way to identification of candidate genes for spinal astrocytoma tumourigenesis (Table 1) and revealed the central role of BRAF and isocitrate dehydrogenase 1 (IDH1) and IDH2 genes.27,31–37
Table 1.
Genetic mutations associated with astrocytoma
| Locus | Gene | Tumour location | Tumour grade or type | Study details and/or findings |
|---|---|---|---|---|
| 7q34 | BRAF | Spinal cord and brain | Pilocytic astrocytomas | Eight paediatric spine pilocytic astrocytomas39 |
| 9q21 | CDKN2A | Spinal cord and brain | Pilocytic astrocytomas | 147 pilocytic astrocytomas, including nine spinal cases34 |
| 17q25 | H3F3A | Spinal cord and brain | GBM | H3.3 gene identi ed as epigenetic marker of midline or spinal GBM27,28,46,47 |
| 2q33.3 | IDH1 | Brain > spinal cord | Intracranial astrocytoma and glioblastoma | Immunohistochemical evidence of tumour in spine; well-established as cranial tumour27,31,46,47 |
| 17q11.2 | NF1 | Brain > spinal cord | Pilocytic astrocytomas | 37 pilocytic astrocytomas, including one spinal case13 |
| 17p13.1 | TP53 | Brain | Diffuse astrocytoma | Mutation evident in 60% of patients with astrocytomas30,41,113 |
| Xq21.1 | ATRX | Brain | All astrocytoma | Associated with IDH1 in study of 363 brain tumours48 |
| 10q23.3 | PTEN | Unspecified | Pilocytic astrocytomas | 147 pilocytic astrocytomas, including nine spinal cases34,35 |
Abbreviations: ATRX, α-thalassaemia/mental retardation syndrome X-linked; CDKN2A, cyclin-dependent kinase inhibitor 2A; GBM, glioblastoma multiforme; H3F3A, histone 3 variant H3.3; IDH1, isocitrate dehydrogenase 1; NF1, neurofibromatosis type 1; PTEN, phosphatase and tensin homologue; TP53, tumour suppressor protein 53.
BRAF is a proto-oncogene that regulates cell growth and has been implicated in various tumour types. Fusion variants of the serine–threonine protein kinase BRAF have been reported in gliomas of the CNS. In the spinal cord, BRAF–KIAA1549 fusion genes are common in pilocytic astrocytomas33,37,38 (reported in up to 75% of these tumours, although this value was based only on small studies39). In addition, the canonical Val600Glu BRAF mutation has been found in a variety of low-grade astrocytomas including pleomorphic xanthoastrocytoma.32,35,38
Approximately 68% of intracranial astrocytomas and 12% of glioblastomas demonstrate mutations in IDH1 and/or IDH2.26,40 Mutations in the IDH gene promote tumourigenesis via disruption of α-ketoglutarate production and subsequent abnormal production of 2-hydroxyglutarate31—a metabolite that competitively inhibits multiple histone demethylases—resulting in abnormal DNA methylation. This sequence of events is a clear example of how one genetic alteration can have widespread epigenetic consequences. The rate of IDH mutations in spinal astrocytoma are not fully understood, but their frequency in spinal cord lesions is likely to be low given that most spinal astrocytomas occur in children, a population in whom IDH mutations are infrequent.37
Beyond these common mutations in astrocytoma, studies of spinal cord tumours have identified additional genes of interest. An analysis of nine cases of pilocytic astrocytomas revealed deletion of the tumour-suppressor gene cyclin-dependent kinase inhibitor 2A (CDKN2A; also known as p16) as the most common mutation.34 This study also found loss of heterozygosity (LOH) at 9p21 (which encompasses CDKN2A), or at 10q23 (which encompasses the phosphatase and tensin homologue gene), in 31.6% and 50.0% of pilocytic astrocytomas, respectively.34
Pilocytic and high-grade intramedullary astrocytomas have also been reported in association with familial NF1.41–43 Patients with NF1 have a mutation in the neurofibromin gene (17q11.2) that causes abnormal replication of nonmyelinating Schwann cells in the PNS. LOH of the NF1 gene was observed in 92% of pilocytic astrocytomas in patients with familial NF1 compared with only 4% of those in patients without NF1.13 Of note, however, only one case of definite spinal pilocytic astrocytoma was included in this cohort.
Data on intramedullary glioblastomas are scarce. Segmental loss of chromosome 8 has been described in one isolated case of paediatric spinal glioblastoma,44 whereas a histopathological and immunohistochemical study of six spinal GBMs revealed that five were immunoreactive for tumour suppressor protein 53 (TP53).45 Up to 60% of all diffuse astrocytomas have mutation of the TP53 gene, but further studies are needed to determine whether this mutation is as common in spinal astrocytoma.40
Two landmark papers, published in 2012, reported high frequency of alterations in the gene encoding the replication-independent histone 3 variant H3.3 (H3F3A) in paediatric glioblastoma, which implicated abnormal DNA methylation in the generation of intracranial and spinal astrocytoma.28,46 H3.3 is evolutionarily conserved throughout eukaryotes; however, two recurrent mutations (Lys27Met and Gly34Arg) in H3F3A were identified in 20–30% of nonbrainstem glioblastomas and nearly 80% of brainstem glioblastomas.28,46,47 The H3.3 amino acid residue Lys27 is also known to be abnormally methylated in the setting of mutant IDH1, further highlighting the importance of the H3.3 variant and abnormal DNA methlyation in CNS tumourigenesis.27,31
Interestingly, mutations in the ATRX (α-thalassaemia/mental retardation syndrome X-linked) gene were linked to IDH1 mutations in diffuse nonpilocytic astro-cytomas and secondary GBM in a cohort of 363 patients with brain tumours.48,49 The ATRX gene regulates incorporation of H3.3 into telomeres and, therefore, has a critical role in chromatin remodelling and genomic stability.48 Further research revealed that the H3.3 Lys27 mutation is the defining mutation of an epigenetic and biological subgroup of GBM that is characterized by proneural methylation, decreased expression of the gene encoding forkhead box protein G1 (a transcription factor involved in neuron growth and survival), younger patient age, and midline or spinal location.27 The exact frequency of H3F3A mutations in spinal cord GBMs remains to be elucidated, but emerging data suggest that intramedullary GBMs also have recurrent mutations in H3F3A.27
Treatment
Treatment of spinal astrocytoma in a symptomatic patient generally involves surgery (biopsy or resection) with or without radiation. In a review of surgical and adjuvant therapies for intramedullary lesions, Harrop and colleagues found that only low-quality evidence, in the form of case series, exists to guide clinical decision-making in patients with IMSCTs.50 Surgical resection is beneficial in patients with pilocytic astrocytoma, especially when surgery is performed prior to onset of severe neurological decline (Figure 1).50 High-grade lesions (WHO grade III and IV) are associated with a worse prognosis and have poorly demarcated boundaries for surgical resection. For these tumours, no significant survival benefit of aggressive surgical debulking has been reported, with 6-month mortality reported to be as high as 70%.5,51 Even with low-grade astrocytoma (WHO grade II), gross total resection is achieved in as few as 12% of tumours.52
Postoperative radiotherapy for low grade intramedullary astrocytoma is associated with 5-year survival rates of 60–90%.50 Adjuvant spinal radiotherapy has been advocated for patients who undergo biopsy only, for those with WHO grade II, III and IV lesions, and for those with advanced, progressive disease.5,50 In one series of 22 adult patients with recurrent spinal cord astrocytoma, treatment with temozolomide (TMZ) was associated with an overall survival rate of 23 months.2 Radiographical and clinical response rates of 37% have been reported with TMZ treatment in high-grade spinal cord astrocytoma.53 Chemotherapy in pilocytic astrocytomas of childhood has demonstrated variable efficacy.5
For GBMs, vascular endothelial growth factor (VEGF) inhibition with bevacizumab is the only targeted therapy that is currently approved by the FDA, and attempts to inhibit the epidermal growth factor receptor pathway have yet to translate to improved outcomes.54,55 Clinical trials of multikinase inhibitors, including cediranib, sunitinib and pazopanib, have uncovered drug-associated toxicities, with minimal or no improvement in patient survival.56–59 Drugs that target heat-shock proteins and proteasome inhibitors are also under investigation in recurrent GBM.54
Ependymomas
Demographics
Ependymomas are the most common IMSCT in adults.7,60 These lesions are thought to arise from ependymal cells of the central canal, but emerging evidence suggests that these tumours have similar histopathology to radial glial stem cells that have undergone malignant transformation.61–64 In children, 90% of ependymomas occur intracranially, but when they arise in the spinal cord these tumours are often of the myxopapillary variant, originating from the filum terminale or conus medullaris.7,8,10,65 In adults, 60% of ependymomas occur at either end of the spine—in the cervical spine, or in filum terminale as the myxopapillay variant.66
Ependymomas present as a centrally located mass in the spinal cord, grow slowly and have clearly demarcated borders (Figure 2). The WHO classifies ependymomas as subependymoma or myxopapillary (grade I), ependymoma (grade II) or anaplastic (grade III). Dissemination to the cerebrospinal fluid is considerably more likely with myxopapillary ependymomas than with other forms of ependymomas, and can be evident even at presentation.67 Subependymomas are slow growing and less common in the spinal cord than are other ependymomas (Figure 2).
Figure 2.

Surgery and imaging in spinal cord ependymoma. a, b | Intraoperative pictures show the tan-coloured tissue that is often seen with ependymomas (arrow; a), and a resection cavity after tumour removal (b). c, d | Preoperative sagittal (c) and axial (d) T2-weighted MRI of thoracic ependymoma demonstrating a space-occupying lesion that is hyperintense to the cord in the thoracic spine. The lesion causes marked cord displacement on axial view; the cord can be seen as a hypointense sliver of tissue (arrow; d).
Genetics
Childhood and adult ependymomas are distinct clinical entities, and emerging evidence suggests that they arise from biologically distinct stem cell precursors.64 In a cross-species comparison of mouse and human ependymoma transcriptomes, human supratentorial ependymomas bore transcriptomic similarities to embryonic cerebral neural stem cells (NSCs) from mice that were deficient in CDKN2A, which is conserved between humans and mice and, as mentioned above, is a tumour-suppressor gene. Human spinal cord ependymoma cells, however, resembled adult mouse NSCs.64 This genetic signature reflects the clinical phenotype, with spinal ependymomas being more common in adults, and childhood ependymoma arising primarily in the intracranial compartment. Notably, however, this study has major limitations as the researchers drew conclusions on the basis of findings from a genetically modified mouse model and the investigation involved comparison of tissues from different species.
Studies of ependymoma genetics often include both spinal and intracranial cohorts (Table 2), but substantial evidence suggests that spinal ependymomas are genetically distinct from their cerebral counterparts.65,68,69 Application of consensus hierarchical clustering and non-negative matrix factorization to two nonoverlapping databases of grossly histologically similar ependymoma tissues enabled identification of three distinct groups of tumours on the basis of their localization—namely, supratentorial, posterior fossa and posterior fossa with spine.70 The spinal ependymomas clustered with a distinct subgroup of posterior fossa ependymomas, and demonstrated whole-chromosome anomalies, as well as mutations affecting the ciliogenesis, microtubule assembly, and mitochondrial and oxidative metabolism pathways.70 These robust results were replicated on multiple subgroup analyses.
Table 2.
Genetic mutations associated with ependymoma
| Locus | Gene | Tumour location | Tumour grade or type | Study details |
|---|---|---|---|---|
| 17q21.3 | HOXB5 | Spinal cord | Grade II–III | 39 ependymomas, including 10 spinal samples69 |
| 1p35 | PLA2G5 | Spinal cord | Grade II | 39 ependymomas, including 10 spinal samples69 |
| 10p15 | ITIH2 | Spinal cord | Grade II | 39 ependymomas, including 10 spinal samples69 |
| 22q12 | NF2 | Spinal cord | Sporadic and NF2-associated Grade II–III | 27ependymomas, including 15 spinal samples68 |
| 9q21 | CDKN2A | Brain | Grade II and III | 122 intracranial ependymomas71 |
| 1q | Multiple | Brain | Grade II and III | 122 intracranial ependymomas71 |
Abbreviations: CDKN2A, cyclin-dependent kinase inhibitor 2A; HOXB5, homeobox 5; ITIH2; inter-α-trypsin inhibitor heavy chain 2; NF2, neurofibromatosis type 2; PLA2G5; phospholipase A2 group 5.
Analysis of RNA expression in newly diagnosed lesions found overexpression of genes encoding homeobox B5 (HOXB5), phospholipase A2 group 5 (PLA2G5) and inter-α-trypsin inhibitor heavy chain 2 (ITIH2) in four of four grade I spinal ependymomas and three of six grade II spinal ependymomas.69 In an RNA microarray-based correspondence analysis, the HOX family of genes was further implicated in extracranial ependymoma development.65 This study included 14 spinal ependymomas but, notably, the researchers did not specify whether tumours in the extracranial cluster involved brainstem as well as spinal cord tumours.65 HOXB5 is a transcription factor implicated in lung and gut development, but in one study this gene was found to have a role in migration of neural crest cells and was associated with acute myeloid leukaemia and oesophageal neoplasms.69 In a larger study that used comparative genomic hybridization in 122 patients with intracranial ependymoma, poor prognosis was associated with amplification of chromosome 1q and homozygous deletion of CDKN2A.71
Further highlighting the heterogeneity of spinal and intracranial ependymomas, mutations in the NF2 gene are found almost exclusively within spinal cord ependymomas.21 Homozygous deletion, LOH and mutations in the NF2 gene have been found in both sporadic and NF2-associated spinal ependymomas, and monosomy or alteration of chromosome 22q is observed in 30% and 40% of spinal ependymomas, respectively.65,68,72 The NF2 gene is located on chromosome 22q12 and encodes the scaffolding protein merlin, also known as schwannomin. Merlin is a moesin–ezrin–radixin-like protein and is part of the 4.1 family of transmembrane-to-actin cytoskeleton linker proteins.73 In familial NF2 and various human cancers, abnormalities in the merlin protein prevents cells from responding to contact inhibition, leading to dysregulation of cell growth and proliferation.73,74
Treatment
Spinal cord ependymomas display variable growth characteristics, and treatment is dictated by the degree of cord compression and symptoms.20 Surgery is appropriate for symptomatic patients, and treatment with adjuvant radiation led to 10-year survival of 50–100% in reported cases.50 The effects of adjuvant chemotherapy, specifically etoposide and carboplatin, have been described in a few case reports and case cohorts in which the choice of agent used to treat the spinal cord lesion was determined by the treatment used for intracranial ependymoma.4 In a study of 10 patients with recurrent intramedullary ependymoma who were treated with etoposide, two experienced a partial response and five patients showed stabilization of disease following one cycle of drug administration.4 Another trial involving 25 patients with recurrent spinal ependymoma found no beneficial response to chemotherapy in patients who had failed a platinum-based regimen. The use of targeted therapeutics in spinal cord ependymomas has not been well-described; however, in one case in which the tumour expressed platelet-derived growth factor (PDGF) receptor, tumour regression was reported following administration of imatinib—a drug that targets the PDGF-β receptor.75
Haemangioblastoma
Demographics
Spinal cord haemangioblastomas are rare, benign, vascular lesions that can occur sporadically or as a manifestation of VHL disease (Figure 3). When these tumours arise in the spinal cord, bleeding can cause neurological impairment owing to the formation of space-occupying haematomas. Approximately 20–30% of haemangioblastomas occur in association with VHL disease, and they are also part of the clinical presentation of retinal angioma, renal cysts, pheochromocytoma, endolymphatic tumours and epididymal cystadenoma.22,76 VHL disease has an autosomal dominant inheritance pattern with 90% penetrance, and is caused by loss of the tumour suppressor gene located at 3p25–26. When the VHL gene is mutated or absent, cells constitutively express hypoxia inducible factors (HIFs), which stimulates the growth of new blood vessels.22,76,77
Figure 3.

Surgical and preoperative imaging of haemangioblastoma in the cervical spine. a | Sagittal postcontrast T1-weighted MRI shows a well-defined lesion (arrow) in the spinal cord at C2–C3. b | 3D CT angiogram reconstruction demonstrating a large vascular lesion (arrow). c | As haemangioblastomas are richly vascular, direct arterial injection can facilitate preoperative imaging, surgical planning, and preoperative embolization. Image depicts filling of ipsilateral vertebral artery and subsequent tumour filling. d | Intraoperative image of haemangioblastoma. Permission obtained from American Association of Neurological Surgeons © Sciubba, D. M. et al. J. Neurosurg. Spine 5, 96–100 (2006).
Genetics
The VHL gene encodes the substrate-binding subunit of the E3 ubiquitin ligase that degrades HIF-α—a transcription factor that regulates glucose transport, glycolysis, erythropoeisis and angiogenesis. When the VHL gene is mutated or absent, cells cannot target HIF-α for poly-ubiquitination and degradation.22,76,77 Constitutively active HIF-α causes proliferation of blood vessels and growth of benign vascular lesions.78 In a review of VHL gene functions, Bader and colleagues reported that VHL-mutant cells secrete high levels of growth factors including VEGF, transforming growth factor-β, PDGF-β and tumour necrosis factor-α.78 These findings highlight the role of VHL as a multifaceted tumour-suppressor gene.
In VHL-associated tumours, 94% express germline mutations in the VHL gene, and 62% exhibit LOH at the VHL locus. By contrast, only about 20% of sporadic lesions have mutations in VHL and 50% have LOH of the VHL locus.22,76,79 As with most other hereditary cancer syndromes, haemangioblastoma formation in patients with VHL disease probably occurs via the ‘two-hit’ mechanism, whereby patients harbour the germline mutation and also undergo a second somatic mutation, which results in biallelic inactivation.79 Diverse genetic aberrations have been uncovered in sporadic haemangioblastomas, with LOH at 22q13 and 3p21-23 found in subsets of individuals with haemangioblastomas without VHL disease.80 In a study of the cytogenetic profiles of haemangioblastomas, loss of chromosome 19, 6 or 22q was observed in 35%, 30% and 15% of samples, respectively.81
Treatment
Spinal cord haemangioblastomas are common in patients with VHL disease, and early MRI screening is recommended from age 10 in individuals with this disorder.77 Surgery has shown benefit both in patients with VHL-associated haemangioblastoma and in those with sporadic haemangioblastoma,82 and is recommended when the tumour becomes symptomatic and the patient experiences weakness, pain or difficulty walking. For individuals with asymptomatic lesions, serial MRI and clinical monitoring are recommended. Preoperative angiography can assist the surgeon in planning and diagnosis. Embolization is rarely performed, however, owing to the risk of ischaemic injury in the adjacent spinal cord parenchyma.
Anatomically, haemangioblastomas of the spinal cord arise from the pial layer and are considered juxta-medullary, but can exhibit an encapsulated, intra-medullary component (Figure 3).83 Surgical resection of spinal haemangioblastoma is complicated by the risk of bleeding and local ischaemia during surgery. Complete resection is possible with the use of microsurgical techniques,83 and radiosurgery is a less invasive option for local tumour control.84 In small case series, antiangiogenic therapy with SU5416 (an inhibitor of VEGF receptors) and thalidomide led to stabilization of disease.85,86 Bevacizumab, a pan-VEGF inhibitor, has been used to successfully treat retinal haemangioblastomas in patients with VHL.87
Extramedullary intradural tumours
Meningiomas
Demographics
Spinal meningiomas are extramedullary intradural tumours arising from the meningothelial cells that reside within the leptomeninges of the spinal cord. These lesions account for 25% of all spinal tumours and comprise a diverse array of histologically distinct subtypes, including angiomatous, fibrous, meningothelial, metaplastic, psammomatous, transitional, atypical and clear-cell variants.30 Psammomatous, meningothelial and transitional meningioma are most commonly reported in the spine,88,89 and although these tumours are usually benign, they can cause substantial spinal cord compression (Figure 4). Surgical resection with adjuvant radiation is often the first line of treatment and is recommended in cases in which disease is persistent or recurrent.90 Meningiomas often manifest in the thoracic spine and are most common in female patients and in the fourth and fifth decade of life.90,91 Unlike their intracranial counterparts, spinal meningiomas are unlikely to recur after surgical resection, with recurrence rates of 0–13% reported.24,88,92–94
Figure 4.
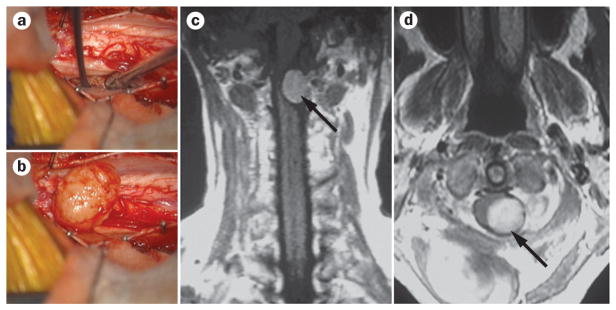
Surgery and imaging of spinal cord meningioma. a, b | Intraoperative images showing pre-exposure (a) and postexposure (b) of a spinal meningioma. c, d | Coronal (c) and axial (d) postgadolinium MRI demonstrating a cervical extramedullary meningioma (arrows) with avid enhancement.
Genetics
Chromosome 22q deletion and consequent loss of the gene is the most consistently described genetic abnormality associated with spinal meningiomas (Table 3).90,95–99 In a microarray-based study of spinal and intracranial meningiomas, Sayagues and colleagues reported that homozygous chromosome 22 deletions were more likely to be associated with spinal than with intracranial meningiomas, and lesions isolated from the spine were likely to originate from a single-cell clone.100
Table 3.
Genetic mutations associated with meningioma and haemangioblastoma
| Locus | Gene | Tumour location | Tumour grade or type | Study details |
|---|---|---|---|---|
| 22q12 | NF2 | Spinal cord | All meningiomas | 16 spinal meningiomas101 |
| 20q11.2 | MMP9 | Spinal cord | All meningiomas | 58 spinal meningiomas91 |
| 9q21 | CDKN2A | Brain > spinal cord | Grade III | 36 paired samples (18 patients)106 |
| Multiple | Hedgehog pathway* | Brain | All meningiomas | 36 intracranial meningiomas108 |
| 10p11.32 | DAL-1 | Unspecified | Malignant meningiomas | 63 sporadic meningiomas, locations unspecified96 |
| 22q13.1 | TIMP | Unspecified | All meningiomas | 30 meningiomas91 |
| 3p25-26 | VHL | Spinal cord | Sporadic and VHL-associated haemangioblastoma | 45 spinal haemangioblastomas76 |
Multiple members of the hedgehog signalling pathway are involved.
Abbreviations: CDKN2A, cyclin-dependent kinase inhibitor 2A; DAL-1, differentially expressed in adenocarcinoma of the lung; MMP9, matrix metalloproteinase 9; NF2, neurofibromatosis type 2; TIMP, tissue inhibitor of metalloproteinase; VHL, von Hippel–Lindau.
Homozygous deletion of the NF2 gene is evident in up to 80% of nonfamilial meningiomas and in 100% patients with NF2 and spinal meningioma.99 In a study of 16 cases of spinal meningioma, half of all patients had complete or partial deletion of chromosome 22q, and this mutation was associated with a more-aggressive anaplastic or atypical phenotype.101 LOH of DAL-1 (differentially expressed in adenocarcinoma of the lung, also known as EPB41L3) which, like merlin, is a member of the protein 4.1 family, has been found in up to 60% of sporadic meningiomas.95,96,102 In these studies, however, whether the mutation was found specifically in spinal meningiomas was not discussed.
The balance between overexpression and inhibition of matrix metalloproteinases (MMPs) has also been implicated in regulation of spinal meningioma growth and invasion.103–105 In a study of 58 spinal meningiomas, MMP-9 expression was identified in 73% of cases, and high levels of expression were observed in 46%.91 Immunoblot analysis has revealed low levels of tissue inhibitors of metalloproteinases 1 and 2 in multiple meningioma phenotypes.96,102,104 Of note, these studies did not specify whether spinal meningiomas were included in the analysis and, therefore, the role of tissue inhibitors of metalloproteinases in spinal tumourigenesis remains to be determined.
In addition to the genes described above, many genomic alterations have been reported in intracranial meningiomas. Mutations in chromosome 9 at the CDKN2A–CDKN2B locus were identified as the most frequent mutation in meningiomas that progressed from grade II to grade III.105 The hedgehog signalling pathway has also been implicated in this transition: compared with high-grade meningiomas, low-grade meningiomas overexpressed several hedgehog pathway-related genes and had lower levels of transcripts of the gene that encodes protein patched homolog 1—a key ligand in the hedgehog pathway.95,106 Future research will be necessary to determine whether such intracranial mutations are also common to spinal meningioma.
Treatment
Whereas meningiomas are classically benign lesions with a good prognosis, patients with NF2 and a meningioma constitute a high-risk population who should be monitored closely for detection of distant and local recurrence of spinal tumours.107–109 The value of chemotherapy in meningioma is limited owing to the low mitotic activity of the tumour cells, and no effective therapy for meningioma following incomplete surgical resection is available. In vitro studies have demonstrated that the drug trabectedin exerts anti-meningioma activity via inhibition of angiogenesis.110 Trabectedin was tested in one patient with anaplastic intracranial meningioma and led to some radiographical response, but the study was terminated owing to drug toxicity in the fifth cycle of drug administration.110 AR-42, a histone deactylase inhibitor, has been tested in vitro and demonstrated marked antiapoptotic activity. A phase I trial of AR-42 is currently under way for patients with NF2 and meningioma.111 Everolimus, an mTOR inhibitor, is currently in phase II trials for patients with NF2 and meningioma,112 and two trials of PDGFR inhibitors, namely valatinib and sunitinib, are also ongoing.112
Conclusions and future directions
Recent advances have highlighted the unique genetic expression profiles of spinal haemangioblastoma and ependymoma, but much work remains to be done to understand differences between intracranial and spinal astrocytomas and meningiomas. By contrast, spinal astrocytomas are not substantially different from their supratentorial counterparts. Some evidence suggests that genetic alterations within spinal astrocytoma cohorts are more limited than in intracranial astrocytoma cohorts. One explanation for this phenomenon is that spinal astrocytomas become symptomatic at a smaller size than do intracranial lesions and, thus, become clinically apparent at an earlier stage. Consequently, spinal astrocytomas may not have time to accumulate as many genetic mutations as tumours that arise in noneloquent regions of CNS. The intracranial tumours would, therefore, present at a later stage of disease and would be much larger than spinal astrocytomas. This same logic may also apply to spinal and intracranial meningiomas.
For intracranial tumours, some targeted therapies have shown benefit, but such treatments may not be applicable to spinal tumour cohorts owing to the variable—or altogether different—genetic profile of spinal tumours compared with brain tumours. Clinical trials of targeted therapies in spinal tumour cohorts are, unfortunately, also limited owing to the rarity of spinal IMSCTs. Multi-institutional, multinational clinical trials are critically needed to enrol sufficient numbers of patients for comparisons between investigational drugs to reach statistical significance, and to enable research in spinal cord tumours to progress towards novel treatments.
Review criteria.
Articles indexed in PubMed, published after 1983, written in English, and concerning basic, translational or clinical studies of CNS tumours of the brain or spine were considered for inclusion. Search terms included “genetics AND spinal cord tumours”; “intramedullary spinal cord tumours”; “ependymoma genetics”; “astrocytoma AND spine”; “astrocytoma AND genetics”; and “haemangioblastoma AND genetics”. Articles were included only if full abstracts were available. Full-text papers were reviewed when available. The reference lists of identified papers were then searched for additional primary articles.
Key points.
Familial neurofibromatosis type 1 is associated with spinal astrocytoma and neurofibromas, whereas familial neurofibromatosis type 2 (NF2) is associated with spinal ependymoma and meningioma
von Hippel–Lindau (VHL) disease and mutations in the VHL gene are associated with spinal haemangioblastoma
Mutations in the gene encoding the histone 3 variant H3.3 (H3F3A) and in the fusion gene BRAF–KIAA1549 are evident in spinal astrocytomas
The genes associated with ependymoma (including mutations in NF2, HOXB5 and PLAG2) are heterogeneous, varying according to histological subtype, tumour location and patient age
Genes encoding protein 4.1 family members NF2 and DAL-1, members of the Hedgehog signalling pathway, matrix metalloproteinase-9, and tissue inhibitors of metalloproteinases, have been implicated in meningioma
Surgical resection and adjuvant radiotherapy remain the primary treatment modalities for spinal cord tumours, and targeted therapeutics are under investigation
Acknowledgments
This work was funded in part by the Burroughs Wellcome Career Award for Medical Scientists and the Johns Hopkins Clinician Scientist Award. Z. L. Gokaslan receives instructorship and research support from AO North America, AO Spine, American Association of Neurological Surgeons Neurosurgery Research and Education Foundation, and the Orthopaedic Research Foundation.
Footnotes
Competing interests
Z. L. Gokaslan declares associations with the following companies: US Spine, Spinal Kinetics. See the article online for full details of the relationships. The other authors declare no competing interests.
Author contributions
P. L. Zadnik and C. Bettegowda researched data for the article. All authors provided substantial contributions to discussions of the content, writing the article, and to reviewing and/or editing the manuscript before submission.
References
- 1.Benesch M, et al. Compassionate use of bevacizumab (Avastin) in children and young adults with refractory or recurrent solid tumors. Ann Oncol. 2008;19:807–813. doi: 10.1093/annonc/mdm510. [DOI] [PubMed] [Google Scholar]
- 2.Chamberlain MC. Temozolomide for recurrent low-grade spinal cord gliomas in adults. Cancer. 2008;113:1019–1024. doi: 10.1002/cncr.23677. [DOI] [PubMed] [Google Scholar]
- 3.Chamberlain MC. Salvage chemotherapy for recurrent spinal cord ependymona. Cancer. 2002;95:997–1002. doi: 10.1002/cncr.10826. [DOI] [PubMed] [Google Scholar]
- 4.Chamberlain MC. Etoposide for recurrent spinal cord ependymoma. Neurology. 2002;58:1310–1311. doi: 10.1212/wnl.58.8.1310. [DOI] [PubMed] [Google Scholar]
- 5.Chamberlain MC, Tredway TL. Adult primary intradural spinal cord tumors: a review. Curr Neurol Neurosci Rep. 2011;11:320–328. doi: 10.1007/s11910-011-0190-2. [DOI] [PubMed] [Google Scholar]
- 6.Schuch G, et al. Case 2. Hemangioblastomas: diagnosis of von Hippel–Lindau disease and antiangiogenic treatment with SU5416. J Clin Oncol. 2005;23:3624–3626. doi: 10.1200/JCO.2005.01.184. [DOI] [PubMed] [Google Scholar]
- 7.Parsa AT, Chi JH, Acosta FL, Jr, Ames CP, McCormick PC. Intramedullary spinal cord tumors: molecular insights and surgical innovation. Clin Neurosurg. 2005;52:76–84. [PubMed] [Google Scholar]
- 8.McCormick PC, Torres R, Post KD, Stein BM. Intramedullary ependymoma of the spinal cord. J Neurosurg. 1990;72:523–532. doi: 10.3171/jns.1990.72.4.0523. [DOI] [PubMed] [Google Scholar]
- 9.Ruda R, Gilbert M, Soffietti R. Ependymomas of the adult: molecular biology and treatment. Curr Opin Neurol. 2008;21:754–761. doi: 10.1097/WCO.0b013e328317efe8. [DOI] [PubMed] [Google Scholar]
- 10.Sonneland PR, Scheithauer BW, Onofrio BM. Myxopapillary ependymoma. A clinicopathologic and immunocytochemical study of 77 cases. Cancer. 1985;56:883–893. doi: 10.1002/1097-0142(19850815)56:4<883::aid-cncr2820560431>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 11.Dow G, et al. Spinal tumors in neurofibromatosis type 2. Is emerging knowledge of genotype predictive of natural history? J Neurosurg Spine. 2005;2:574–579. doi: 10.3171/spi.2005.2.5.0574. [DOI] [PubMed] [Google Scholar]
- 12.Hagel C, et al. Clinical presentation, immunohistochemistry and electron microscopy indicate neurofibromatosis type 2-associated gliomas to be spinal ependymomas. Neuropathology. 2012;32:611–616. doi: 10.1111/j.1440-1789.2012.01306.x. [DOI] [PubMed] [Google Scholar]
- 13.Kluwe L, et al. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. J Neuropathol Exp Neurol. 2001;60:917–920. doi: 10.1093/jnen/60.9.917. [DOI] [PubMed] [Google Scholar]
- 14.Kluwe L, Tatagiba M, Funsterer C, Mautner VF. NF1 mutations and clinical spectrum in patients with spinal neurofibromas. J Med Genet. 2003;40:368–371. doi: 10.1136/jmg.40.5.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patronas NJ, et al. Intramedullary and spinal canal tumors in patients with neurofibromatosis 2: MR imaging findings and correlation with genotype. Radiology. 2001;218:434–442. doi: 10.1148/radiology.218.2.r01fe40434. [DOI] [PubMed] [Google Scholar]
- 16.Ueki K, Sasaki T, Ishida T, Kirino T. Spinal tanycytic ependymoma associated with neurofibromatosis type 2—case report. Neurol Med Chir (Tokyo) 2001;41:513–516. doi: 10.2176/nmc.41.513. [DOI] [PubMed] [Google Scholar]
- 17.Ebert C, et al. Molecular genetic analysis of ependymal tumors. NF2 mutations and chromosome 22q loss occur preferentially in intramedullary spinal ependymomas. Am J Pathol. 1999;155:627–632. doi: 10.1016/S0002-9440(10)65158-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoa M, Slattery WH., 3rd Neurofibromatosis 2. Otolaryngol Clin North Am. 2012;45:315–332. doi: 10.1016/j.otc.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Uhlmann EJ, Plotkin SR. Neurofibromatoses. Adv Exp Med Biol. 2012;724:266–277. doi: 10.1007/978-1-4614-0653-2_20. [DOI] [PubMed] [Google Scholar]
- 20.Plotkin SR, et al. Spinal ependymomas in neurofibromatosis type 2: a retrospective analysis of 55 patients. J Neurosurg Spine. 2011;14:543–547. doi: 10.3171/2010.11.SPINE10350. [DOI] [PubMed] [Google Scholar]
- 21.Lamszus K, et al. Molecular genetic alterations on chromosomes 11 and 22 in ependymomas. Int J Cancer. 2001;91:803–808. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1134>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 22.Glasker S. Central nervous system manifestations in VHL: genetics, pathology and clinical phenotypic features. Fam Cancer. 2005;4:37–42. doi: 10.1007/s10689-004-5347-6. [DOI] [PubMed] [Google Scholar]
- 23.Friedman JM. Epidemiology of neurofibromatosis type 1. Am J Med Genet. 1999;89:1–6. [PubMed] [Google Scholar]
- 24.Cohen-Gadol AA, Zikel OM, Koch CA, Scheithauer BW, Krauss WE. Spinal meningiomas in patients younger than 50 years of age: a 21-year experience. J Neurosurg. 2003;98:258–263. doi: 10.3171/spi.2003.98.3.0258. [DOI] [PubMed] [Google Scholar]
- 25.Epstein FJ, Farmer JP, Freed D. Adult intramedullary astrocytomas of the spinal cord. J Neurosurg. 1992;77:355–359. doi: 10.3171/jns.1992.77.3.0355. [DOI] [PubMed] [Google Scholar]
- 26.Parsons DW, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sturm D, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–437. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]
- 28.Wu G, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang R, et al. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:829–833. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- 30.Kleihues P, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002;61:215–225. doi: 10.1093/jnen/61.3.215. [DOI] [PubMed] [Google Scholar]
- 31.Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;18:5562–5571. doi: 10.1158/1078-0432.CCR-12-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim YH, et al. Frequent BRAF gain in low-grade diffuse gliomas with 1p/19q loss. Brain Pathol. 2012;22:834–840. doi: 10.1111/j.1750-3639.2012.00601.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin A, et al. BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549:BRAF fusion variants. J Neuropathol Exp Neurol. 2012;71:66–72. doi: 10.1097/NEN.0b013e31823f2cb0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horbinski C, Hamilton RL, Nikiforov Y, Pollack IF. Association of molecular alterations, including BRAF, with biology and outcome in pilocytic astrocytomas. Acta Neuropathol. 2010;119:641–649. doi: 10.1007/s00401-009-0634-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeuken JW, Wesseling P. MAPK pathway activation through BRAF gene fusion in pilocytic astrocytomas; a novel oncogenic fusion gene with diagnostic, prognostic, and therapeutic potential. J Pathol. 2010;222:324–328. doi: 10.1002/path.2780. [DOI] [PubMed] [Google Scholar]
- 36.Lv S, et al. Correlation between IDH1 gene mutation status and survival of patients treated for recurrent glioma. Anticancer Res. 2011;31:4457–4463. [PubMed] [Google Scholar]
- 37.Pollack IF, et al. IDH1 mutations are common in malignant gliomas arising in adolescents: a report from the Children’s Oncology Group. Childs Nerv Syst. 2011;27:87–94. doi: 10.1007/s00381-010-1264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ida CM, et al. BRAF alterations are frequent in cerebellar low-grade astrocytomas with diffuse growth pattern. J Neuropathol Exp Neurol. 2012;71:631–639. doi: 10.1097/NEN.0b013e31825c448a. [DOI] [PubMed] [Google Scholar]
- 39.Hawkins C, et al. BRAF-KIAA1549 fusion predicts better clinical outcome in pediatric low-grade astrocytoma. Clin Cancer Res. 2011;17:4790–4798. doi: 10.1158/1078-0432.CCR-11-0034. [DOI] [PubMed] [Google Scholar]
- 40.Walker C, Baborie A, Crooks D, Wilkins S, Jenkinson MD. Biology, genetics and imaging of glial cell tumours. Br J Radiol. 2011;84 (Spec 2):S90–S106. doi: 10.1259/bjr/23430927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chi JH, Cachola K, Parsa AT. Genetics and molecular biology of intramedullary spinal cord tumors. Neurosurg Clin N Am. 2006;17:1–5. doi: 10.1016/j.nec.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 42.Lee M, Rezai AR, Freed D, Epstein FJ. Intramedullary spinal cord tumors in neurofibromatosis. Neurosurgery. 1996;38:32–37. doi: 10.1097/00006123-199601000-00009. [DOI] [PubMed] [Google Scholar]
- 43.Yagi T, Ohata K, Haque M, Hakuba A. Intramedullary spinal cord tumour associated with neurofibromatosis type 1. Acta Neurochir (Wien) 1997;139:1055–1060. doi: 10.1007/BF01411560. [DOI] [PubMed] [Google Scholar]
- 44.Sharma S, et al. Distinct molecular signatures in pediatric infratentorial glioblastomas defined by aCGH. Exp Mol Pathol. 2010;89:169–174. doi: 10.1016/j.yexmp.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 45.Govindan A, et al. Histopathologic and immunohistochemical profile of spinal glioblastoma: a study of six cases. Brain Tumor Pathol. 2011;28:297–303. doi: 10.1007/s10014-011-0041-5. [DOI] [PubMed] [Google Scholar]
- 46.Schwartzentruber J, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482:226–231. doi: 10.1038/nature10833. [DOI] [PubMed] [Google Scholar]
- 47.Khuong-Quang DA, et al. K27M mutation in histone H3.3 defines clinically and biologically distinct subgroups of pediatric diffuse intrinsic pontine gliomas. Acta Neuropathol. 2012;124:439–447. doi: 10.1007/s00401-012-0998-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiao Y, et al. Frequent ATRX, CIC, and FUBP1 mutations refine the classification of malignant gliomas. Oncotarget. 2012;3:709–722. doi: 10.18632/oncotarget.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kannan K, et al. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget. 2012;3:1194–1203. doi: 10.18632/oncotarget.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Harrop JS, Ganju A, Groff M, Bilsky M. Primary intramedullary tumors of the spinal cord. Spine (Phila Pa 1976) 2009;34:S69–S77. doi: 10.1097/BRS.0b013e3181b95c6f. [DOI] [PubMed] [Google Scholar]
- 51.Cohen AR, Wisoff JH, Allen JC, Epstein F. Malignant astrocytomas of the spinal cord. J Neurosurg. 1989;70:50–54. doi: 10.3171/jns.1989.70.1.0050. [DOI] [PubMed] [Google Scholar]
- 52.Raco A, et al. Long-term follow-up of intramedullary spinal cord tumors: a series of 202 cases. Neurosurgery. 2005;56:972–981. [PubMed] [Google Scholar]
- 53.Kaley TJ, Mondesire-Crump I, Gavrilovic IT. Temozolomide or bevacizumab for spinal cord high-grade gliomas. J Neurooncol. 2012;109:385–389. doi: 10.1007/s11060-012-0905-5. [DOI] [PubMed] [Google Scholar]
- 54.Patel M, Vogelbaum MA, Barnett GH, Jalali R, Ahluwalia MS. Molecular targeted therapy in recurrent glioblastoma: current challenges and future directions. Expert Opin Investig Drugs. 2012;21:1247–1266. doi: 10.1517/13543784.2012.703177. [DOI] [PubMed] [Google Scholar]
- 55.Olar A, Aldape KD. Biomarkers classification and therapeutic decision-making for malignant gliomas. Curr Treat Options Oncol. 2012;13:417–436. doi: 10.1007/s11864-012-0210-8. [DOI] [PubMed] [Google Scholar]
- 56.Neyns B, et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J Neurooncol. 2011;103:491–501. doi: 10.1007/s11060-010-0402-7. [DOI] [PubMed] [Google Scholar]
- 57.Batchelor TT, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. J Clin Oncol. 2010;28:2817–2823. doi: 10.1200/JCO.2009.26.3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Iwamoto FM, et al. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06–02) Neuro Oncol. 2010;12:855–861. doi: 10.1093/neuonc/noq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reardon DA, et al. Phase I study of sunitinib and irinotecan for patients with recurrent malignant glioma. J Neurooncol. 2011;105:621–627. doi: 10.1007/s11060-011-0631-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Milano MT, et al. Primary spinal cord glioma: a Surveillance, Epidemiology, and End Results database study. J Neurooncol. 2010;98:83–92. doi: 10.1007/s11060-009-0054-7. [DOI] [PubMed] [Google Scholar]
- 61.Poppleton H, Gilbertson RJ. Stem cells of ependymoma. Br J Cancer. 2007;96:6–10. doi: 10.1038/sj.bjc.6603519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taylor MD, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323–335. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 63.Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Ann Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson RA, et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature. 2010;466:632–636. doi: 10.1038/nature09173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Palm T, et al. Expression profiling of ependymomas unravels localization and tumor grade-specific tumorigenesis. Cancer. 2009;115:3955–3968. doi: 10.1002/cncr.24476. [DOI] [PubMed] [Google Scholar]
- 66.Hamilton RL, Pollack IF. The molecular biology of ependymomas. Brain Pathol. 1997;7:807–822. doi: 10.1111/j.1750-3639.1997.tb01066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakamura M, et al. Long-term surgical outcomes for myxopapillary ependymomas of the cauda equina. Spine (Phila Pa 1976) 2009;34:E756–E760. doi: 10.1097/BRS.0b013e3181b34d16. [DOI] [PubMed] [Google Scholar]
- 68.Singh PK, Gutmann DH, Fuller CE, Newsham IF, Perry A. Differential involvement of protein 4.1 family members DAL-1 and NF2 in intracranial and intraspinal ependymomas. Mod Pathol. 2002;15:526–531. doi: 10.1038/modpathol.3880558. [DOI] [PubMed] [Google Scholar]
- 69.Korshunov A, et al. Gene expression patterns in ependymomas correlate with tumor location, grade, and patient age. Am J Pathol. 2003;163:1721–1727. doi: 10.1016/S0002-9440(10)63530-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Witt H, et al. Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell. 2011;20:143–157. doi: 10.1016/j.ccr.2011.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Korshunov A, et al. Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol. 2010;28:3182–3190. doi: 10.1200/JCO.2009.27.3359. [DOI] [PubMed] [Google Scholar]
- 72.Scheil S, et al. Low frequency of chromosomal imbalances in anaplastic ependymomas as detected by comparative genomic hybridization. Brain Pathol. 2001;11:133–143. doi: 10.1111/j.1750-3639.2001.tb00386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Stamenkovic I, Yu Q. Merlin, a “magic” linker between extracellular cues and intracellular signaling pathways that regulate cell motility, proliferation, and survival. Curr Protein Pept Sci. 2010;11:471–484. doi: 10.2174/138920310791824011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li W, Cooper J, Karajannis MA, Giancotti FG. Merlin: a tumour suppressor with functions at the cell cortex and in the nucleus. EMBO Rep. 2012;13:204–215. doi: 10.1038/embor.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fakhrai N, et al. Recurrent spinal ependymoma showing partial remission under Imatimib. Acta Neurochir (Wien) 2004;146:1255–1258. doi: 10.1007/s00701-004-0374-5. [DOI] [PubMed] [Google Scholar]
- 76.Glasker S, et al. The impact of molecular genetic analysis of the VHL gene in patients with haemangioblastomas of the central nervous system. J Neurol Neurosurg Psychiatry. 1999;67:758–762. doi: 10.1136/jnnp.67.6.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shuin T, et al. Von Hippel–Lindau disease: molecular pathological basis, clinical criteria, genetic testing, clinical features of tumors and treatment. Jpn J Clin Oncol. 2006;36:337–343. doi: 10.1093/jjco/hyl052. [DOI] [PubMed] [Google Scholar]
- 78.Bader HL, Hsu T. Systemic VHL gene functions and the VHL disease. FEBS Lett. 2012;586:1562–1569. doi: 10.1016/j.febslet.2012.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Glasker S, et al. Reconsideration of biallelic inactivation of the VHL tumour suppressor gene in hemangioblastomas of the central nervous system. J Neurol Neurosurg Psychiatry. 2001;70:644–648. doi: 10.1136/jnnp.70.5.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Beckner ME, et al. Loss of heterozygosity reveals non-VHL allelic loss in hemangioblastomas at 22q13. Hum Pathol. 2004;35:1105–1111. doi: 10.1016/j.humpath.2004.05.014. [DOI] [PubMed] [Google Scholar]
- 81.Rickert CH, Hasselblatt M, Jeibmann A, Paulus W. Cellular and reticular variants of hemangioblastoma differ in their cytogenetic profiles. Hum Pathol. 2006;37:1452–1457. doi: 10.1016/j.humpath.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 82.Na JH, et al. Spinal cord hemangioblastoma: diagnosis and clinical outcome after surgical treatment. J Korean Neurosurg Soc. 2007;42:436–440. doi: 10.3340/jkns.2007.42.6.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mandigo CE, Ogden AT, Angevine PD, McCormick PC. Operative management of spinal hemangioblastoma. Neurosurgery. 2009;65:1166–1177. doi: 10.1227/01.NEU.0000359306.74674.C4. [DOI] [PubMed] [Google Scholar]
- 84.Selch MT, et al. Image-guided linear accelerator-based spinal radiosurgery for hemangioblastoma. Surg Neurol Int. 2012;3:73. doi: 10.4103/2152-7806.98386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sardi I, et al. Monotherapy with thalidomide for treatment of spinal cord hemangioblastomas in a patient with von Hippel–Lindau disease. Pediatr Blood Cancer. 2009;53:464–467. doi: 10.1002/pbc.22065. [DOI] [PubMed] [Google Scholar]
- 86.Madhusudan S, et al. Antiangiogenic therapy for von Hippel–Lindau disease. JAMA. 2004;291:943–944. doi: 10.1001/jama.291.8.943. [DOI] [PubMed] [Google Scholar]
- 87.Hrisomalos FN, Maturi RK, Pata V. Long-term use of intravitreal bevacizumab (avastin) for the treatment of von hippel-lindau associated retinal hemangioblastomas. Open Ophthalmol J. 2010;4:66–69. doi: 10.2174/1874364101004010066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sandalcioglu IE, et al. Spinal meningiomas: critical review of 131 surgically treated patients. Eur Spine J. 2008;17:1035–1041. doi: 10.1007/s00586-008-0685-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Setzer M, Vatter H, Marquardt G, Seifert V, Vrionis FD. Management of spinal meningiomas: surgical results and a review of the literature. Neurosurg Focus. 2007;23:E14. doi: 10.3171/FOC-07/10/E14. [DOI] [PubMed] [Google Scholar]
- 90.Vranic A, Peyre M, Kalamarides M. New insights into meningioma: from genetics to trials. Curr Opin Oncol. 2012;24:660–665. doi: 10.1097/CCO.0b013e3283571a06. [DOI] [PubMed] [Google Scholar]
- 91.Barresi V, Alafaci C, Caffo M, Barresi G, Tuccari G. Clinicopathological characteristics, hormone receptor status and matrix metallo-proteinase-9 (MMP-9) immunohistochemical expression in spinal meningiomas. Pathol Res Pract. 2012;208:350–355. doi: 10.1016/j.prp.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 92.Maiuri F, De Caro ML, de Divitiis O, Vergara P, Mariniello G. Spinal meningiomas: age-related features. Clin Neurol Neurosurg. 2011;113:34–38. doi: 10.1016/j.clineuro.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 93.Mirimanoff RO, Dosoretz DE, Linggood RM, Ojemann RG, Martuza RL. Meningioma: analysis of recurrence and progression following neurosurgical resection. J Neurosurg. 1985;62:18–24. doi: 10.3171/jns.1985.62.1.0018. [DOI] [PubMed] [Google Scholar]
- 94.Solero CL, et al. Spinal meningiomas: review of 174 operated cases. Neurosurgery. 1989;25:153–160. [PubMed] [Google Scholar]
- 95.Pham MH, et al. Molecular genetics of meningiomas: a systematic review of the current literature and potential basis for future treatment paradigms. Neurosurg Focus. 2011;30:E7. doi: 10.3171/2011.2.FOCUS1117. [DOI] [PubMed] [Google Scholar]
- 96.Nunes F, et al. Inactivation patterns of NF2 and DAL-1/4.1B (EPB41L3) in sporadic meningioma. Cancer Genet Cytogenet. 2005;162:135–139. doi: 10.1016/j.cancergencyto.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 97.Weber RG, et al. Analysis of genomic alterations in benign, atypical, and anaplastic meningiomas: toward a genetic model of meningioma progression. Proc Natl Acad Sci USA. 1997;94:14719–14724. doi: 10.1073/pnas.94.26.14719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Carlson KM, Bruder C, Nordenskjold M, Dumanski JP. 1p and 3p deletions in meningiomas without detectable aberrations of chromosome 22 identified by comparative genomic hybridization. Genes Chromosomes Cancer. 1997;20:419–424. [PubMed] [Google Scholar]
- 99.Goutagny S, Kalamarides M. Meningiomas and neurofibromatosis. J Neurooncol. 2010;99:341–347. doi: 10.1007/s11060-010-0339-x. [DOI] [PubMed] [Google Scholar]
- 100.Sayagues JM, et al. Microarray-based analysis of spinal versus intracranial meningiomas: different clinical, biological, and genetic characteristics associated with distinct patterns of gene expression. J Neuropathol Exp Neurol. 2006;65:445–454. doi: 10.1097/01.jnen.0000229234.13372.d8. [DOI] [PubMed] [Google Scholar]
- 101.Arslantas A, et al. Detection of chromosomal imbalances in spinal meningiomas by comparative genomic hybridization. Neurol Med Chir (Tokyo) 2003;43:12–18. doi: 10.2176/nmc.43.12. [DOI] [PubMed] [Google Scholar]
- 102.Gutmann DH, et al. Loss of DAL-1, a protein 4.1-related tumor suppressor, is an important early event in the pathogenesis of meningiomas. Hum Mol Genet. 2000;9:1495–1500. doi: 10.1093/hmg/9.10.1495. [DOI] [PubMed] [Google Scholar]
- 103.Halaka AN, Bunning RA, Bird CC, Gibson M, Reynolds JJ. Production of collagenase and inhibitor (TIMP) by intracranial tumors and dura in vitro. J Neurosurg. 1983;59:461–466. doi: 10.3171/jns.1983.59.3.0461. [DOI] [PubMed] [Google Scholar]
- 104.Mizoue T, Kawamoto H, Arita K, Tominaga A, Kurisu K. Secretion of matrix metalloproteinase-9 and tissue inhibitor of metalloproteinase-1 by meningiomas detected by cell immunoblot analysis. Acta Neurochir (Wien) 1999;141:481–486. doi: 10.1007/s007010050328. [DOI] [PubMed] [Google Scholar]
- 105.Paek SH, et al. The role of matrix metalloproteinases and tissue inhibitors of matrix metalloproteinase in microcystic meningiomas. Oncol Rep. 2006;16:49–56. [PubMed] [Google Scholar]
- 106.Goutagny S, et al. Genomic profiling reveals alternative genetic pathways of meningioma malignant progression dependent on the underlying NF2 status. Clin Cancer Res. 2010;16:4155–4164. doi: 10.1158/1078-0432.CCR-10-0891. [DOI] [PubMed] [Google Scholar]
- 107.Horowitz P, et al. Novel oncogene discovery in meningiomas. Presented at the Congress of Neurological Surgeons, Annual Meeting; 2012. [Google Scholar]
- 108.Laurendeau I, et al. Gene expression profiling of the hedgehog signaling pathway in human meningiomas. Mol Med. 2010;16:262–270. doi: 10.2119/molmed.2010.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–2472. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 110.Kotecha RS, et al. Meningiomas in children and adolescents: a meta-analysis of individual patient data. Lancet Oncol. 2011;12:1229–1239. doi: 10.1016/S1470-2045(11)70275-3. [DOI] [PubMed] [Google Scholar]
- 111.Preusser M, et al. Trabectedin has promising antineoplastic activity in high-grade meningioma. Cancer. 2012;118:5038–5049. doi: 10.1002/cncr.27460. [DOI] [PubMed] [Google Scholar]
- 112.Blakeley J. Development of drug treatments for neurofibromatosis type 2-associated vestibular schwannoma. Curr Opin Otolaryngol Head Neck Surg. 2012;20:372–379. doi: 10.1097/MOO.0b013e328357d2ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Reifenberger G, Collins VP. Pathology and molecular genetics of astrocytic gliomas. J Mol Med (Berl) 2004;82:656–670. doi: 10.1007/s00109-004-0564-x. [DOI] [PubMed] [Google Scholar]