

Abstract
Microbes in soils and other environments produce extracellular enzymes to depolymerize and hydrolyze organic macromolecules so that they can be assimilated for energy and nutrients. Measuring soil microbial enzyme activity is crucial in understanding soil ecosystem functional dynamics. The general concept of the fluorescence enzyme assay is that synthetic C-, N-, or P-rich substrates bound with a fluorescent dye are added to soil samples. When intact, the labeled substrates do not fluoresce. Enzyme activity is measured as the increase in fluorescence as the fluorescent dyes are cleaved from their substrates, which allows them to fluoresce. Enzyme measurements can be expressed in units of molarity or activity. To perform this assay, soil slurries are prepared by combining soil with a pH buffer. The pH buffer (typically a 50 mM sodium acetate or 50 mM Tris buffer), is chosen for the buffer's particular acid dissociation constant (pKa) to best match the soil sample pH. The soil slurries are inoculated with a nonlimiting amount of fluorescently labeled (i.e. C-, N-, or P-rich) substrate. Using soil slurries in the assay serves to minimize limitations on enzyme and substrate diffusion. Therefore, this assay controls for differences in substrate limitation, diffusion rates, and soil pH conditions; thus detecting potential enzyme activity rates as a function of the difference in enzyme concentrations (per sample).
Fluorescence enzyme assays are typically more sensitive than spectrophotometric (i.e. colorimetric) assays, but can suffer from interference caused by impurities and the instability of many fluorescent compounds when exposed to light; so caution is required when handling fluorescent substrates. Likewise, this method only assesses potential enzyme activities under laboratory conditions when substrates are not limiting. Caution should be used when interpreting the data representing cross-site comparisons with differing temperatures or soil types, as in situ soil type and temperature can influence enzyme kinetics.
Keywords: Environmental Sciences, Issue 81, Ecological and Environmental Phenomena, Environment, Biochemistry, Environmental Microbiology, Soil Microbiology, Ecology, Eukaryota, Archaea, Bacteria, Soil extracellular enzyme activities (EEAs), fluorometric enzyme assays, substrate degradation, 4-methylumbelliferone (MUB), 7-amino-4-methylcoumarin (MUC), enzyme temperature kinetics, soil
Introduction
Extracellular enzymes (EEs) produced by soil bacteria, fungi, and archaea are involved in innumerable biogeochemical processes, and are central to the processing, stabilization, and destabilization of soil organic matter and nutrient cycling in terrestrial ecosystems 1. By producing EEs, soil microbes decompose and transform polymeric organic matter into smaller soluble molecules, thereby liberating previously bound micro- and macronutrients, which allows plants and microbes to assimilate available nutrients from the soil. EEs have been studied for decades, primarily by measuring their activities in laboratory assays2-4, since it is very difficult to directly detect and quantify enzymes.
Extracellular enzyme activity (EEA) is most strongly controlled by the concentration of enzymes and corresponding substrates. The abundance of different C-, N-, and P-degrading enzymes in soils is controlled by numerous factors including microbial biomass, community composition, substrate availability, microclimate, and stoichiometric demands5,6. However, in situ EEAs within the soil environment are also affected by temperature7,8 , the binding of enzymes to soil clays and humic properties2, and diffusion constraints 9, which ultimately regulate the active enzyme pool, in terms of size, substrate availability, and turnover rates10-12. Therefore, acknowledging in situ soil conditions is critical when using laboratory enzyme assays to interpret soil microbial function across different environmental sites.
Many different classes of EEA can be quantified in laboratory assays using a variety of synthetic substrates (please refer to "List of Reagents Table" for more detail). Some protocols utilize substrates in assays that are coupled to a colorimetric reaction that can be detected with a spectrophotometer, while others, including the protocol we describe here, utilize substrates that are bound to a fluorescent moiety. Fluorescence EE assays are typically more sensitive (by an order of magnitude) than colorimetric assays (which use a chromogenic moiety linked with a synthetic substrate)12-14. Sensitivity in EEA detection includes two aspects: one is related to the quantity of the compound of interest detected and the other related to the lowest detectable potential enzyme activity. Methods for colorimetric P-nitrophenol (pNP)-based assays can be found in past works15,16. In brief, soil (typically sieved to <2 mm and air-dried) is incubated at optimal or field-relevant temperature and pH. The rate at which the reaction product released is determined colorimetrically with a spectrophotometer14. The higher sensitivity of the fluorescence enzyme assays is due in part to the more sensitive detection of the fluorogenic moiety separation associated with substrate degradation rather than recording absorbance after the separation of a specific chromogenic moiety at a specific wavelength. The two most commonly used synthetic fluorescent indicators are 4-methylumbelliferone (MUB)17 and 7-amino-4-methylcoumarin (MUC)18,19. MUC-linked substrates are commonly associated with N-rich synthetic substrates such as proteins and/or amino acids. Fluorescent techniques were first developed for aquatic samples20,21, and their application to soils requires controls for signal quenching and interference22,23. Assays can either be conducted using traditional "bench top" chemistry with large volumes, or can be employed in microplate based protocols, with increased throughput but possibly higher measurement error. While there are several widely cited protocols for fluorescent detection of EEAs in soils24, many labs employ subtle variations on these protocols, often inadvertently or due to differences in laboratory equipment or reagents. Seemingly small differences in the details of protocols can strongly affect measured EEAs25,26 and the lack of standardized enzymes makes it challenging to calibrate assays between different laboratories. Thus, there is an important need for the dissemination of detailed protocols to encourage the standardization of EEA assays.
In our protocol, soil samples are prepared by combining soil samples with a pH buffer and homogenizing with a blender. The slurries are then inoculated with a nonlimiting amount of fluorescently labeled C-, N-, or P-rich substrate, chosen depending on the specific research question of interest. Using soil slurries in the enzyme assays serves as a control to minimize substrate diffusion limitations. The fluorescent moieties are quenched till they are cleaved from their respective substrates, and thus enzyme activity can be detected as the fluorescent dye is released from the substrate by an enzyme-catalyzed reaction. The increasing fluorescence intensity through time reflects the rate of the enzyme-catalyzed reaction.
The general concept of the fluorescence enzyme assay is that synthetic substrates bound with a fluorogenic moiety (fluorescent dye), are added to soil samples 27. During enzyme-catalyzed substrate degradation, the bond breaks between the fluorescent dye and the substrate. The fluorescent dye liberated from the substrate is consequently used as an indirect assessment of enzyme activity, and can be quantified using a microplate reader to detect the fluorescence intensity of the dye. In brief, fluorescence quantification is accomplished as the liberated dye emits light of one wavelength after absorbing light of a different wavelength. The fluorescence intensity is recorded by a plate reader capable of both excitation and detection. Enzyme activity can be subsequently quantified based on the known fluorescent-dye concentrations of the substrate (i.e. known quantities of synthetic substrate added to soil samples) along with referencing a standard dilution curve of fluorescence intensities for the specific fluorogenic moiety of the substrate used in the assay (i.e. 4-methylumbelliferone (MUB) or 7-amino-4-methylcoumarin (MUC)). (Please refer to the protocol section for specific details on enzyme activity quantification).
Laboratory soil enzyme assays are useful for assessing microbial community function, but there are several technical limitations that users should recognize10. Fluorescence assays can suffer from interference caused by impurities and/or instability of many fluorescent compounds when exposed to light, so caution is required when handling fluorescent substrates25. Soil particulates and/or organic material in the soil slurries can also interfere with fluorescence intensities, known as the quenching effect 26. Furthermore, laboratory enzyme assays only assesses potential EEAs under laboratory conditions. In vitro assays measure EEAs under conditions where substrate diffusion and abundance is nonlimiting. Therefore, the data provided by these assays may not be a good proxy for EEAs under in situ soil conditions10. Overall, enzyme activity is very useful for relative comparisons in which soil types are similar. However, when using this method to compare activities among soils that differ in physical or chemical properties, caution should be used. This is due to the fact that differences in soil type and temperature can drastically alter the state of in situ enzyme kinetics. Another limitation is that relatively few substrates are commercially available (compared to the natural environment). Furthermore, the synthetic substrates used for enzyme assays are relatively simple (easily soluble) may not accurately represent the soil substrates present or available in situ. Another factor to consider is that using soil slurries will incorporate the activity of some stabilized enzymes (i.e. immobilized by organic matter or clays) that may not be active under in situ conditions 2. Laboratory enzyme assays also do not provide information regarding the persistence of enzymes in the soil (enzyme turnover rates) or information regarding the specific microbial species that are producing soil enzymes.
Protocol
1. Assay Set-up
Label 3 deep well plates: "Sample", "MUB standard", and "MUC standard".
Pour each standard (MUB of 0, 2.5, 5, 10, 25, 50, and 100 μM) and substrate (BG, CB, NAG, PHOS, XYL, AG, LAP) into separate clean, prelabeled reservoirs (oriented in rows).
- Pipette the appropriate MUB standard into corresponding wells of MUB standard plate (Table 1).
- Pipette 200 μl of 0 μM MUB into row A of MUB standard plate.
- Pipette 200 μl of 2.5 μM MUB into row B of MUB standard plate.
- Pipette 200 μl of 5 μM MUB into row C of MUB standard plate.
- Pipette 200 μl of 10 μM MUB into row D of MUB standard plate.
- Pipette 200 μl of 25 μM MUB into row E of MUB standard plate.
- Pipette 200 μl of 50 μM MUB into row F of MUB standard plate.
- Pipette 200 μl of 100 μM MUB into row G of MUB standard plate.
- Repeat steps 1.3.1 - 1.3.8 for MUC standards into corresponding wells of the MUC standard plate.
- Prep soil slurries for each soil sample.
- Weigh 2.75 g of field moist soil into blender.
- Add 91 ml of 50 mM buffer. Blend on high for 1 min.
- Pour blender contents into a clean glass bowl at least as wide as 8-channel pipette with a stir bar.
- Place on stir plate, mix soil slurry on low.
- Pipette 800 μl of soil slurry into first column of the Sample plate (Table 2).
- Repeat into 1st column of MUB standard and MUC standard plates (Table 1).
- Rinse blender, stir plate and stir bar with DI H2O or buffer between soil samples.
- Repeat steps 1.4.1 - 1.4.7)for each sample, moving to the next column with each subsequent soil sample (Tables 1 and 2).
- Pipette the appropriate substrate (in this example, 200 μM concentrations) into corresponding wells of the Sample plate (Table 2).
- Pipette 200 μl of BG substrate into row A of Sample plate.
- Pipette 200 μl of CB substrate into row B of Sample plate.
- Pipette 200 μl of NAG substrate into row C of Sample plate.
- Pipette 200 μl of PHOS substrate into row D of Sample plate.
- Pipette 200 μl of XYL substrate into row E of Sample plate.
- Pipette 200 μl of AG substrate into row F of Sample plate.
- Pipette 200 μl of LAP substrate into row G of Sample plate.
- Repeat steps 1.5.1 - 1.5.7 for each incubation temperature sample plate.
2. Incubation of Assay Plates
Seal the deep-well plates ("Sample", "MUB standard", and "MUC standard") with plate mats.
Invert each (sealed) plate by hand until the solution is thoroughly mixed (~10x).
Place plates in appropriate incubator for required incubation time period (Table 3).
Record initial time as Time 0. Also record the appropriate incubation times so you know when to remove the plate from the incubator (Table 3).
At the end of each incubation period, centrifuge the sealed plates for 3 min at ~2,900 x g.
After certification, transfer 250 μl from each well of the incubated deep well plates into corresponding wells in black flat-bottomed 96-well plates (one black plate will be used for each incubated deep well plate). It is important to transfer samples from the incubated deep well plates into the same wells (i.e. A1…A12, etc.) in the black flat-bottomed 96-well plates (Table 1, 2).
3. Fluorescent Measurements on a Microplate Fluorometer
Following manufacturer's instructions for the fluorometric plate reader used, set the excitation wavelength to 365 nm and emission wavelength to 450 nm (or use appropriate filters).
- Read the three plates on the fluorometer. Important: samples and standard plates (i.e. MUB or MUC) must be read using the same gain.
- Set the spectrophotometer to automatic gain and read the MUC and MUB Standard plates.
- Set gain to manual, and decrease the value from optimal to the next lowest number, rounded to the nearest five, for each Standard plate. Repeat 2x for each plate.
- Set the gain to automatic and run the Sample plate. Rerun the Sample plate manually to match the highest gain for each of the Standard plates (i.e. MUB and MUC).
4. Data Analysis
- Enter standard curve fluorescence data into spreadsheet for MUB (Table 4a) and MUC standard (Table 4b)
- Convert (μM) concentrations to (μmol) (Tables 4a and 4b).
For each sample's standard curve fluorescence data, calculate the slope, y-intercept, and R2 values for MUB and MUC (μmol) standard concentrations. Acceptable R2 values should exceed 0.98 for each sample (Tables 4a and 4b). It may be useful to visualize standard curves in a scatter plot; with fluorescence reads plotted as the dependent variable (y-axis) and standard concentration (μmol) plotted as the independent variable (x-axis) (Figure 1).
- You will use the MUB and MUC standard curve slope and y-intercept values to calculate the sample + substrate raw fluorescence data into potential EEAs. You must first enter the sample + substrate raw fluorescence data into a new spreadsheet (Table 5a). You will also need to enter the incubation time (hr) and soil dry weight for each sample (Table 5b). Note, the time-values entered in this example are 3 hr, because the samples were incubated at 25 °C (Table 3).
- Subtract the standard curve intercept values from the corresponding sample fluorescence values and then divide by the slope of the corresponding standard curve (Table 5c). To accomplish this, rearrange standard line equation to solve for sample enzyme concentration (x); x=(y-b)/m where: [y = sample fluorescence raw read; b = intercept from MUB or MUC Standard curve; m = slope from MUB or MUC Standard curve].
- Multiply sample μmol, from step 4.3.1 above, by 91 ml (buffer volume used in soil slurry) (Table 5d).
- Divide value obtained in step 4.3.2 by the sample-specific incubation time and dry soil mass (Table 5e): [enzyme amt. (μmol) x 91 ml x incubation (hr) x dry soil (g) x 0.8 ml x 1,000] Note: 0.8 ml is the volume of slurry used in each well, and 1,000 corrects for the actual soil amount in each well.
- Multiply the value obtained in step 4.3.3 by 1,000 to get the desired units (nmol activity/g dry soil/hr) (Table 5f).
Representative Results
Enzyme assays can be used to quantify potential EEAs and to compare activities among similar samples. Here, we present representative results from an experimental climate study comparing soils that experienced ambient climate conditions (ACN) to soils that were exposed to elevated CO2 and heating treatments (EHN) (Figures 2-6). Plant cover in all plots were characteristic of a northern mixed grass prairie dominated by the C4 grass Bouteloua gracilis (H.B.K) Lag. and two C3 grasses, Hesperostipa comata Trin and Rupr. and Pascopyrum smithii (Rydb.); about 20% of the vegetation is composed of sedges and forbs. More information regarding the site description and field experimental design can be found in references28-30. Soils were collected from two different depths (0-5 cm and 5-15 cm) within each of the treatment plots using a 1.5 cm diameter core to assess soil EEAs in response to altered climate conditions. In our examples (i.e. Figures 2-6), the sample size is (N = 3). Regardless of this minimal sample size, the variation is relatively small in most cases (as demonstrated by the error bars) and robustly reflects variability in potential EEAs among treatment plots. Analysis of Variance (ANOVA) and Tukey post hoc multiple comparisons were used to identify significant shifts in enzyme activity among treatment plots and soil depths.
The following representative results have been provided to demonstrate how this high-throughput, fluorometric assay can be used to test (1) overall EEA in soils, (2) how EEA stoichiometry can be indicative of ecosystem-level processes and (3) the relationship between incubation temperature and EEA. Soil EEA's are commonly studied to relate shifts in microbial function to soil nutrient cycling; useful indicators to assess microbial nutrient demands in response to climate change, plant community shifts, and more broadly ecosystem functioning31-33. EEA stoichiometry has been more recently adopted as an index to assess soil biochemical nutrient cycling by intersecting ecological stoichiometric theory and metabolic theory of ecology to assess potential microbial nutrient imbalances corresponding to environmental conditions5. Numerous studies have suggested that wide stoichiometric ratios are indicative of nutrient growth limitations34-36; and as soil nutrients become limited, microbes respond by allocating metabolic resources to produce specific enzymes to acquire deficient nutrients37. Ecoenzymatic C:N:P stoichiometry ratios are thus useful to identify relative shifts in potential microbial community nutrient demands in response to various environmental perturbations5. Lastly, temperature sensitivities of EEAs can be useful to assess how soil microbial community functional diversity is likely influenced by temperature shifts7,38. Enzyme temperature sensitivities can widely vary between soils for a single enzyme class, and microbial communities producing enzymes have demonstrated shifts in enzyme activity corresponding to shifts in climate from historic conditions7. Thus enzyme activity questions related to the thermal ecology of EEs can be a useful way to assess microbial functional dynamics and belowground ecosystem processes in response to climate changes39,40.
In this example, potential C-, N-, and P-enzyme acquisition activities assayed at 0-5 cm soil depths did not differ by experimental treatment (Figure 2a). However, at 5-15 cm soil depths, several potential EEAs did differ significantly (Figure 2b). For example, the C-degrading enzymes β-1,4-glucosidase and β-D-cellobiohydrolase were lower in the EHN plots (p ≤ 0.038; Figure 2b) compared to the ACN plots. The N and P mineralizing enzymes (β-1,4-N-acetylglucosaminidase and phosphatase, respectively) were also lower in the EHN plots (p ≤ 0.012; Figure 2b) compared to ACN at 5-15 cm soil depths.
Calculating and plotting the sum of all C-, N-, or P- cycling potential EEAs can be a useful approach to observe broader patterns regarding potential soil C-, N-, and/or P-cycles (Figure 3). In this example, the sum of β-1,4-glucosidase, β-D-cellobiohydrolase, β-Xylosidase, and α-1,4-glucosidase potential EEAs was calculated to represent potential C cycling activities. The sum of β-1,4-N-acetylglucosaminidase and L-leucine aminopeptidase was calculated to represent potential N cycling activities. Phosphatase was used to represent potential P cycling activities. In this example, potential EEAs for total C-, N- and P-cycling trended lower in the EHN plots compared to the ACN plots at the 5-15 cm soil depths (Figure 3). However, this trend was only significant for total N and P cycling activities (p ≤ 0.046; Figure 3). Soil EEAs did not significantly differ among the treatment plots at 0-5 cm soil depths (Figure 3). The findings for this example suggest contrasting trends in enzyme-activity functional group (i.e. C-, N-, or P-degrading enzymes) among the treatment plots (ACN vs. EHN) in response to soil depth. For example, C-, N- and P-degrading soil EEAs in the ambient plots (ACN) trended higher at lower depths compared to soils exposed to elevated CO2 and heating (EHN), which demonstrated an inverse pattern (Figure 3a-c).
Enzyme stoichiometry is another useful approach to assess potential enzyme activities in the environment (Figure 4). The microbial demand for nutrients is determined by the elemental stoichiometry of microbial biomass in relation to environmental nutrient availability32. Likewise, microbes produce specific enzymes (i.e. C-, N-, or P-degrading enzymes) to meet nutrient demands within their soil environments, also referred to as ecological stoichiometry41. The ratio of potential EEAs is one way to assess microbial nutrient demands. For example, a 1:1 ratio between two enzyme functional groups (for example in C:N nutrient acquisition) would suggest that the demand for N is high relative to the demand for C when considering microbial biomass C:N ratios at the community level is typically 8:142. In this example, soil enzyme stoichiometry C:N, C:P, or N:P activities significantly differ among the treatment plots at 0-5 cm soil depths (Figures 4a-c). However, potential enzyme C:P and N:P ratios were higher in the EHN plots compared to the ACN plots at the 5-15 cm soil depths (p = 0.05; Figures 4b and 4c). This observation suggests that there is relatively higher P mineralization EEAs compared to C and N EEA the ACN plots (compared to EHN) at the 5-15 cm soil depths. Enzyme C:N acquisition activity ratios demonstrated a similar trend to that demonstrated by total C EEAs with higher C:N due to higher potential C EEAs in the ACN plots at the lower depth compared to EHN (Figure 4a).
Temperature can strongly influence soil EEAs. Yet, in typical lab assays, soil enzymes are measured at a single temperature that may not correspond to in situ temperature conditions. Our fluorescence enzyme assay method allows us to consider in situ temperature effects by incorporating multiple laboratory incubation temperatures for comparison. Using laboratory incubations at multiple temperatures allows us to analyze temperature-dependent enzyme kinetics data using Arrhenius plot and Q10 calculations. Arrhenius plot are used to visualize activation energy; and are plotted using the logarithm of enzyme activity (y-axis dependent variable) as a function of the inverse temperature converted to degrees Kelvin (1/K) on the x-axis (i.e. independent variable; Figures 5a-c). Activation energy is commonly defined as the minimum energy required to catalyze a chemical reaction (i.e. degrade a given substrate into smaller products). For our purposes, activation energy serves as a proxy for the temperature sensitivity of enzyme catalyzed reactions. Higher activation energy indicates enzyme temperature sensitivity. Likewise, activation energies (i.e. Figures 5d-f) directly correspond to Q10 values (i.e. Figures 6a-c). A further explanation of formulas used to calculate activation energy and practical application can be found in many past works40,43-45. Arrhenius plots, activation energy, and Q10 plots provide redundant information and should not all be used in the same manuscript to present data for publication (Figures 5 and 6). Therefore, when using these techniques, it is necessary to choose the most appropriate plot type for your enzyme temperature - kinetics data10. All plot types (Arrhenius and Q10) were presented here for demonstration purposes; to provide visual examples of how to present enzyme results.
In our examples, we assessed potential enzyme kinetics for potential C-, N-, and P-EEAs among both treatment plots at the two soil depths (Figure 5; Figure 6). The finding demonstrated that the temperature sensitivity of EEAs was not significantly different among treatment plots at either soil depths for activation energy as demonstrated in the Arrhenius plots (Figures 5a-c) and corresponding activation energies (Figure d-f) and (not surprisingly) for Q10 plots (in this example between the 15 °C and 25 °C laboratory incubations; Figures 6a-c). This suggests that enzyme kinetics were not influenced by the field experimental treatments in this particular study.
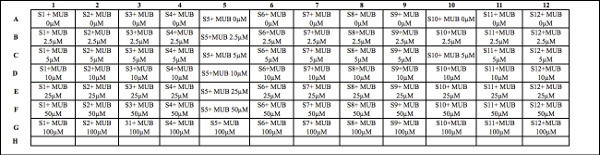 Table 1. Deep well plate design for fluorescence standard concentrations with soil samples. Template for organizing and loading fluorescence standards (MUB or MUC) with soil samples into the deep well plate prior to incubating. Note: The columns represent the same soil samples (800 μl of soil slurry additions). A gradient of fluorescence standard concentrations is loaded for each row (200 μl additions). Each cell represents fluorescence standard concentration plus soil slurry additions. For example, S1 - S12 represent soil samples (800 μl of soil slurry); MUB 0-100 μM = 4-Methylumbelliferone concentrations (200 μl additions). In this example, row H is open, and is consequently available to include a higher fluorescence standard concentration if needed. This decision to increase the standard curve concentrations may be necessary for higher potential enzyme activity (and/or the substrate concentrations used) for your specific soil samples. The potential enzyme activity for your samples should fall within the range of the standard curve values. Click here to view larger table.
Table 1. Deep well plate design for fluorescence standard concentrations with soil samples. Template for organizing and loading fluorescence standards (MUB or MUC) with soil samples into the deep well plate prior to incubating. Note: The columns represent the same soil samples (800 μl of soil slurry additions). A gradient of fluorescence standard concentrations is loaded for each row (200 μl additions). Each cell represents fluorescence standard concentration plus soil slurry additions. For example, S1 - S12 represent soil samples (800 μl of soil slurry); MUB 0-100 μM = 4-Methylumbelliferone concentrations (200 μl additions). In this example, row H is open, and is consequently available to include a higher fluorescence standard concentration if needed. This decision to increase the standard curve concentrations may be necessary for higher potential enzyme activity (and/or the substrate concentrations used) for your specific soil samples. The potential enzyme activity for your samples should fall within the range of the standard curve values. Click here to view larger table.
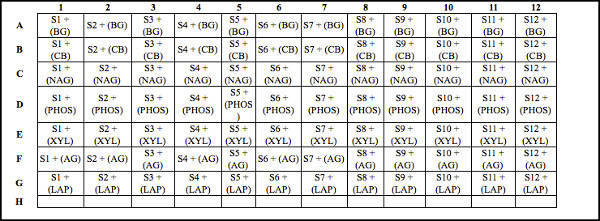 Table 2. Deep well plate design for soil samples with substrate. Template for organizing and loading soil samples and substrates into the deep well plate prior to incubating. Note: The columns represent the same soil samples (800 μl of soil slurry additions). Different substrates are loaded across each row (200 μl additions). Each cell represents soil samples plus substrate addition. For example, S1 - S12 represent unique soil samples (800 μl of soil slurry). Substrates (200 μl additions) include: BG = 4-Methylumbelliferyl β-D-glucopyranoside; CB = 4-Methylumbelliferyl β-D-cellobioside; NAG = 4-Methylumbelliferyl N-acetyl-β-D-glucosaminide; PHOS = 4-Methylumbelliferyl phosphate: XYL = 4-Methylumbelliferyl-β-D-xylopyranoside; AG = 4-Methylumbelliferyl α-D-glucopyranoside; and LAP = L-Leucine-7-amido-4-methylcoumarin hydrochloride. In this example, row H is open, and is consequently available to include another substrate to assess another potential enzyme activity if desired. Click here to view larger table.
Table 2. Deep well plate design for soil samples with substrate. Template for organizing and loading soil samples and substrates into the deep well plate prior to incubating. Note: The columns represent the same soil samples (800 μl of soil slurry additions). Different substrates are loaded across each row (200 μl additions). Each cell represents soil samples plus substrate addition. For example, S1 - S12 represent unique soil samples (800 μl of soil slurry). Substrates (200 μl additions) include: BG = 4-Methylumbelliferyl β-D-glucopyranoside; CB = 4-Methylumbelliferyl β-D-cellobioside; NAG = 4-Methylumbelliferyl N-acetyl-β-D-glucosaminide; PHOS = 4-Methylumbelliferyl phosphate: XYL = 4-Methylumbelliferyl-β-D-xylopyranoside; AG = 4-Methylumbelliferyl α-D-glucopyranoside; and LAP = L-Leucine-7-amido-4-methylcoumarin hydrochloride. In this example, row H is open, and is consequently available to include another substrate to assess another potential enzyme activity if desired. Click here to view larger table.
| Temperature | Time |
| 4 °C | ~23 hr |
| 15 °C | 6 hr |
| 25 °C | 3 hr |
| 35 °C | 1.5 hr |
Table 3. Incubator temperatures required for corresponding incubation time periods. Incubation temperatures and corresponding time periods for the enzyme assay procedure.
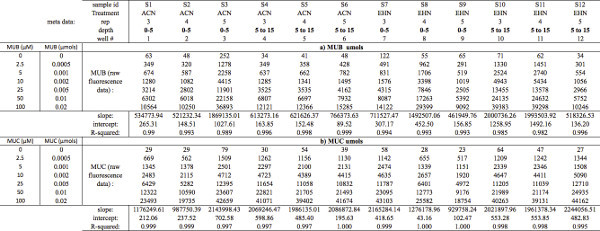 Table 4. MUB and MUC standard curve calculations. Demonstrates how to organize a) MUB and b) MUC raw fluorescence data (μmol) to subsequently calculate slope, y-intercept, and R-squared values. To calculate slope, y-intercept, and R-squared values from your standard concentration curves, select (or plot) the MUB and/or MUC raw fluorescence data as the dependent variable (y-axis) and standard concentration (μmol) as the independent variable (x-axis). Note: MUB = 4-Methylumbelliferone; MUC = 7-Amino-4-methylcoumarin. Click here to view larger table.
Table 4. MUB and MUC standard curve calculations. Demonstrates how to organize a) MUB and b) MUC raw fluorescence data (μmol) to subsequently calculate slope, y-intercept, and R-squared values. To calculate slope, y-intercept, and R-squared values from your standard concentration curves, select (or plot) the MUB and/or MUC raw fluorescence data as the dependent variable (y-axis) and standard concentration (μmol) as the independent variable (x-axis). Note: MUB = 4-Methylumbelliferone; MUC = 7-Amino-4-methylcoumarin. Click here to view larger table.
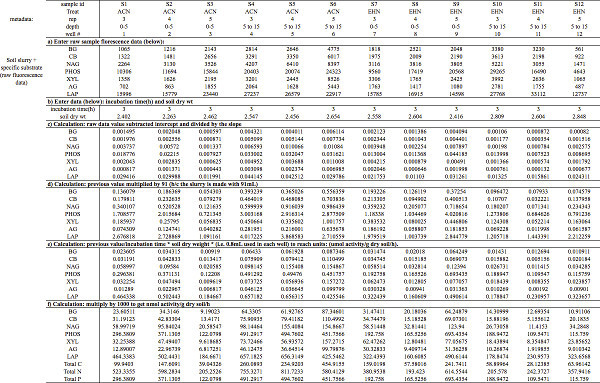 Table 5. Enzyme activity calculations. Demonstrates how to a) organize sample raw fluorescence data and subsequently perform the steps needed (b-f) to calculate EEAs into: μmol activity/g dry soil/hr. Note: S1 - S12 represent unique soil samples. Substrates include: BG = 4-Methylumbelliferyl β-D-glucopyranoside; CB = 4-Methylumbelliferyl β-D-cellobioside; NAG = 4-Methylumbelliferyl N-acetyl-β-D-glucosaminide; PHOS = 4-Methylumbelliferyl phosphate: XYL = 4-Methylumbelliferyl-β-D-xylopyranoside; AG = 4-Methylumbelliferyl α-D-glucopyranoside; and LAP = L-Leucine-7-amido-4-methylcoumarin hydrochloride. Click here to view larger table.
Table 5. Enzyme activity calculations. Demonstrates how to a) organize sample raw fluorescence data and subsequently perform the steps needed (b-f) to calculate EEAs into: μmol activity/g dry soil/hr. Note: S1 - S12 represent unique soil samples. Substrates include: BG = 4-Methylumbelliferyl β-D-glucopyranoside; CB = 4-Methylumbelliferyl β-D-cellobioside; NAG = 4-Methylumbelliferyl N-acetyl-β-D-glucosaminide; PHOS = 4-Methylumbelliferyl phosphate: XYL = 4-Methylumbelliferyl-β-D-xylopyranoside; AG = 4-Methylumbelliferyl α-D-glucopyranoside; and LAP = L-Leucine-7-amido-4-methylcoumarin hydrochloride. Click here to view larger table.
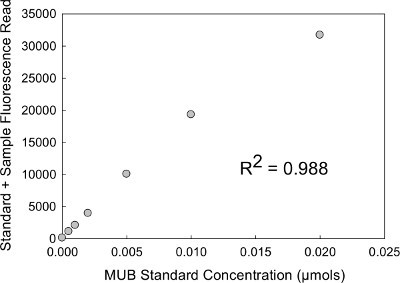 Figure 1. MUB standard curve plot. Scatterplot visualization of standard curves with raw fluorescence data as the dependent variable (y-axis) and standard concentration (μmol) as the independent variable (x-axis).
Figure 1. MUB standard curve plot. Scatterplot visualization of standard curves with raw fluorescence data as the dependent variable (y-axis) and standard concentration (μmol) as the independent variable (x-axis).
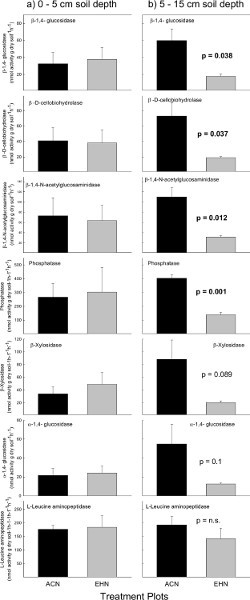 Figure 2. C, N and P cycling enzyme activities. Potential EEAs at the Prairie Heating and Elevated CO2 Enrichment (PHACE) site in control plots (ACN: exposed to ambient climate conditions) and treatment plots (EHN: exposed to increased temperature and elevated CO2). Soil enzymes were assayed at a) 0-5 cm soil depths and b) 5-15 cm soil depths. Vertical bars represent mean ± S.E. Note: ACN = ambient - climate plots; EHN = elevated CO2 and heating plots. Click here to view larger figure.
Figure 2. C, N and P cycling enzyme activities. Potential EEAs at the Prairie Heating and Elevated CO2 Enrichment (PHACE) site in control plots (ACN: exposed to ambient climate conditions) and treatment plots (EHN: exposed to increased temperature and elevated CO2). Soil enzymes were assayed at a) 0-5 cm soil depths and b) 5-15 cm soil depths. Vertical bars represent mean ± S.E. Note: ACN = ambient - climate plots; EHN = elevated CO2 and heating plots. Click here to view larger figure.
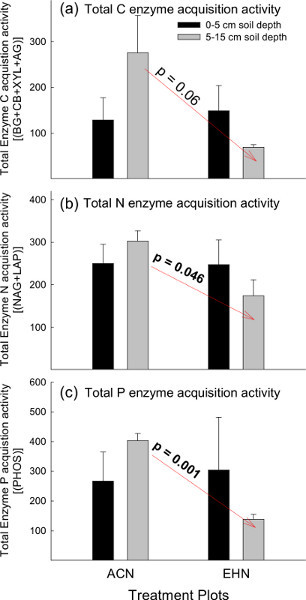 Figure 3. Total C, N and P cycling enzyme stoichiometric ratios. The sum of potential EEAs involved in the acquisition of a) C, b) N, and c) P- for both treatment groups in the PHACE experiment at 0-5 cm and 5-15 cm soil depths. Vertical bars represent mean ± S.E. Note: ACN = ambient - climate plots; EHN = elevated CO2 and heating plots. Click here to view larger figure.
Figure 3. Total C, N and P cycling enzyme stoichiometric ratios. The sum of potential EEAs involved in the acquisition of a) C, b) N, and c) P- for both treatment groups in the PHACE experiment at 0-5 cm and 5-15 cm soil depths. Vertical bars represent mean ± S.E. Note: ACN = ambient - climate plots; EHN = elevated CO2 and heating plots. Click here to view larger figure.
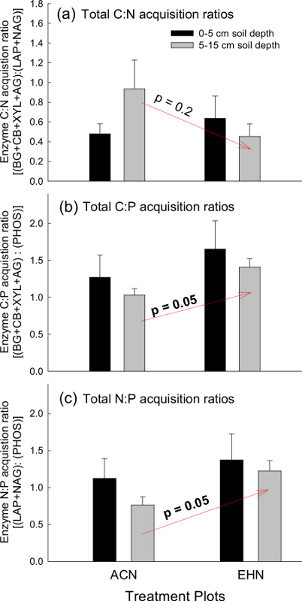 Figure 4. Enzyme stoichiometry for total C, N, and P cycling enzyme activities. Enzyme acquisition activity stoichiometric ratios at the Prairie Heating and Elevated CO2 Enrichment (PHACE) site in control plots (ACN: exposed to ambient climate conditions) and treatment plots (EHN: exposed to increased temperature and elevated CO2). Total soil a) C:N, b) C:P and c) N:P was plotted for both treatment groups at 0-5 cm and 5-15 cm soil depths. Vertical bars represent mean ± S.E. Note: ACN = ambient - climate plots; EHN = elevated CO2 and heating plots. Click here to view larger figure.
Figure 4. Enzyme stoichiometry for total C, N, and P cycling enzyme activities. Enzyme acquisition activity stoichiometric ratios at the Prairie Heating and Elevated CO2 Enrichment (PHACE) site in control plots (ACN: exposed to ambient climate conditions) and treatment plots (EHN: exposed to increased temperature and elevated CO2). Total soil a) C:N, b) C:P and c) N:P was plotted for both treatment groups at 0-5 cm and 5-15 cm soil depths. Vertical bars represent mean ± S.E. Note: ACN = ambient - climate plots; EHN = elevated CO2 and heating plots. Click here to view larger figure.
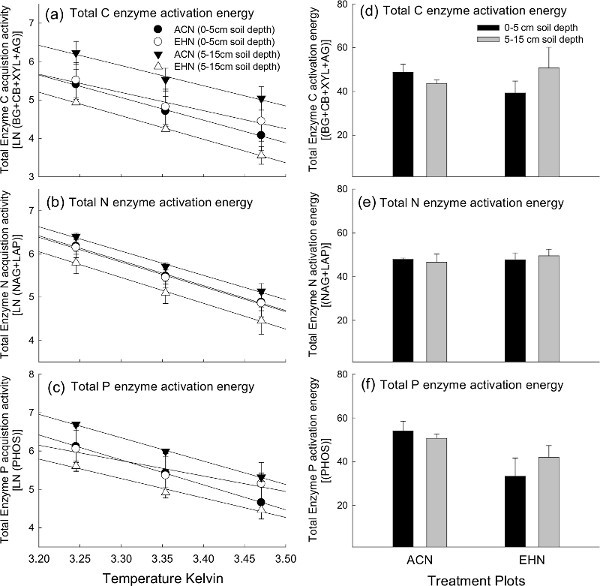 Figure 5. Total C, N and P cycling enzyme activation energies. Temperature sensitivity of potential EEAs displayed using Arrhenius plots for total a) C, b) N, and c) P degrading enzymes; as well as the corresponding activation energy values for total d) C, e) N, and f) P degrading enzymes among treatment plots and soil depth at the Prairie Heating and Elevated CO2 Enrichment (PHACE) site. Vertical bars for activation energy (i.e.
d-f) represent mean ± S.E. Note: ACN = ambient - climate plots; EHN = elevated CO2 and heating plots. For publication, it is important to note that the author would typically choose to present either Arrhenius plots (i.e.
a-c) or activation energy values (i.e.
d-f), but not both plot- types. Click here to view larger figure.
Figure 5. Total C, N and P cycling enzyme activation energies. Temperature sensitivity of potential EEAs displayed using Arrhenius plots for total a) C, b) N, and c) P degrading enzymes; as well as the corresponding activation energy values for total d) C, e) N, and f) P degrading enzymes among treatment plots and soil depth at the Prairie Heating and Elevated CO2 Enrichment (PHACE) site. Vertical bars for activation energy (i.e.
d-f) represent mean ± S.E. Note: ACN = ambient - climate plots; EHN = elevated CO2 and heating plots. For publication, it is important to note that the author would typically choose to present either Arrhenius plots (i.e.
a-c) or activation energy values (i.e.
d-f), but not both plot- types. Click here to view larger figure.
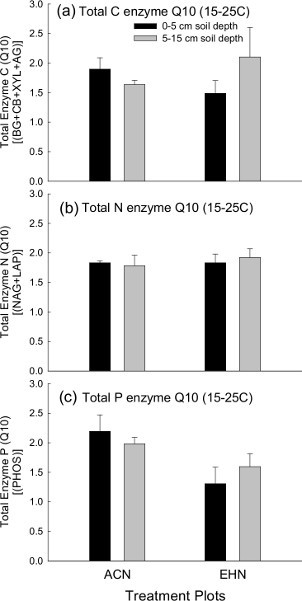 Figure 6. Total C, N and P cycling enzyme Q10 (15-25 °C). Temperature sensitivities of potential EEAs measured as Q10, based on laboratory incubations at 15 °C and 25 °C for total a) C, b) N, and c) P degrading enzymes among treatment plots and soil depth at the Prairie Heating and Elevated CO2 Enrichment (PHACE) site. Vertical bars for Q10 represent mean ± S.E. Note: ACN = ambient - climate plots; EHN = elevated CO2 and heating plots. Click here to view larger figure.
Figure 6. Total C, N and P cycling enzyme Q10 (15-25 °C). Temperature sensitivities of potential EEAs measured as Q10, based on laboratory incubations at 15 °C and 25 °C for total a) C, b) N, and c) P degrading enzymes among treatment plots and soil depth at the Prairie Heating and Elevated CO2 Enrichment (PHACE) site. Vertical bars for Q10 represent mean ± S.E. Note: ACN = ambient - climate plots; EHN = elevated CO2 and heating plots. Click here to view larger figure.
Discussion
The laboratory-based measurement of potential soil EEAs can provide important insights into microbial responses to their abiotic environment, and their consequences for ecosystem functioning. The results of this example data set suggest that minimal differences exist in soil enzyme activity or kinetics among climate treatment plots. However, inverse trends among plots encourage further investigation of covariates that may influence the production of microbial EEAs such as soil moisture, soil pH or plant growth. Overall, assessing EEAs in terms of (1) overall EEA in soils, (2) EEA stoichiometry (3) Arrhenius plots/activation energy, and (4) Q10 provides a wide spectrum of approaches that can be indicative of ecosystem-level processes from which to robustly characterize soil ecosystem functional dynamics.
High-throughput fluorescence-based EEA assays are a useful tool that is widely used to examine potential EEAs in soils and other environments. Importantly, potential activities reflect the enzyme pool size, but do not by themselves quantify enzyme production or turnover rates46. Although the technique is relatively straightforward, seemingly minor differences among lab protocols can hamper the comparability of results 13. Unfortunately, we do not currently have suitable standardized positive controls for EEAs. The use of stoichiometric ratios is one approach for overcoming these challenges. Otherwise, the advent of high-throughput techniques has advanced the study of enzymes in the environment4. Careful interpretation of the data generated by these assays may elucidate important trends in microbial activity.
The robustness of the protocol stems from the ability to select for conditions specific to individual samples, but this can also result in limitations. A series of modifications will be required to ensure samples are accurately measured for individual field sites:
Buffer
The buffer you select will depend on the pH of the soil. Buffering also stabilizes the fluorescence intensity of the fluorescent standards, which is highly pH dependent27,47. The pH buffer that we typically use to make the soil slurry is a 50 mM sodium acetate or Tris buffer which is chosen for the buffer's particular acid dissociation constant (pKa) to best match soil - sample pH levels. Sodium acetate has a pKa of 4.76, and Tris has a pKa of 8.06 so the amounts of these two buffers will vary in order to reach the desired pKa for the individual sample. Phosphate buffer (pKa = 7.2) has been suggested for neutral/slightly basic soils. However, we caution to test for analytical variability in preliminary studies before using this buffer, as high phosphate concentrations may interfere with enzyme activity.
Handling and storage of fluorescent substrates
Fluorescence assays can suffer from interference caused by impurities and/or instability of many fluorescent compounds when exposed to light, so caution is required when handling fluorescent substrates. We strongly recommend minimizing any light exposure to fluorescently labeled substrates and MUB and MUC standards. Using amber glass bottles or covering the glassware and containers used for making and storing fluorescent substrates and standards is highly recommended; aluminum foil to wrap glassware and containers works well. Likewise, efficiently inoculating plates and transferring to dark incubators is best practice. We recommend storing substrates and standards (-20 °C) for no longer than two months (while protecting them from light); and thawing substrates (5 °C) ~24-48 hr prior to beginning the enzyme assay(s).
Design and Replication
To best account for well to well sample variations, implementing negative assay controls and (if possible) assay replicates is recommended. Variation typically occurs due to differences in the amount of soil particles in each well and pipetting errors. Therefore, strong mixing and good pipetting technique will substantially minimize well to well variation. Furthermore, we strongly suggest implementing a negative assay control (buffer + substrate solution) to monitor substrate inconsistencies over time. This can be easily monitored when reading the enzyme plates by comparing negative control wells (we typically use the last column on the 96-well plates). Negative substrate controls are typically stable, therefore the signal increases well above detection limit, it is indicative of contamination or substrate instability, requiring replacement of the substrate and/or standard solutions.
To maximize throughput, our protocol includes several potential EEAs in a single deep well microplate, although other protocols perform one type of assay per plate (i.e. one substrate per plate) since different EEAs occur at different rates. Regardless, enzyme optimization must be performed on soils before performing this high throughout approach or the single plate approach. Multiple substrates can be used on a single plate if the reaction rates are relatively consistent for each enzyme within the time period of the incubation.
Our protocol recommends ~2.75 g soil to make the slurry solution, while ~1 gram is recommended in other protocols 24. We suggest that using more soil (if possible) is an effective approach to better capture within-sample soil variation in enzyme activity levels. In this protocol, we incubate 800 μl of soil slurry with 200 μl substrate, while others only incubate 200 μl of soil slurry with 50 μl of substrates. This is simply a function of scaling up that ultimately does not change the measured activity. There are also practical advantages for using larger volumes. One, it is easier to avoid soil particulates while pipetting into corresponding black flat-bottomed 96-well plates before recording intensities on the fluorometer. Second, the additional volume is useful in the case of accidental spills while transferring the black flat-bottomed 96-well plates to the fluorometer before recording enzyme-related fluorescence intensities. Even slight deviations in volume will significantly decrease fluorescence among wells. Lastly, due to the high throughput nature of this protocol, we commonly elect to rely on experimental replication to represent variation in enzyme activity levels rather than performing assay replicates. It is always best practice to include analytical replicates, but in practice, we feel our protocol provides a balanced tradeoff between analytical and experimental replicates given limited resources. Likewise, our approach uses relatively more well homogenized soil (2.75 g) per assay25, which inherently decreases within-soil variations. The decision to use analytical replicates should be carefully considered by testing for analytical error in preliminary studies48. However, we recommend that experimental designs with fewer than 4 treatment-group replicates should strongly consider using assay replicates.
Soil, Buffer Volumes, and Substrate concentration optimization
The soil buffer:substrate concentration ratio is an important variable which strongly influences the measured fluorescence when performing enzyme assays. The amount of soil in the slurry added to assay or the concentration of substrate may need to be adjusted depending on the activity of enzymes in the soil sample. The amounts selected for this example were based on previous testing of these soils to ensure that substrate availability was nonlimiting, and that we were measuring maximum potential rates under the assay conditions (Vmax) 44,45. However, for soil samples that have high concentrations of enzymes, the amount of soil or substrate concentration must be increased. We have found that increasing substrate concentrations (and adjusting the standard curve accordingly) can affect linearity of the curve. Therefore, we recommend reducing soil amounts and/or proportional adjustments to buffer volumes as necessary. Regardless, it is important to optimize substrate concentration for each soil type assayed because measured EEAs can differ by an order of magnitude greater between saturated and sub-saturated substrate concentrations 26. Therefore differences among experimental treatments (etc.) are susceptible to type II statistical error and less likely to be detected at sub-saturating conditions, and statistical error27,35. To optimize substrate concentrations, preliminary enzyme assays should performed on representative soil samples using a wide range of substrate concentrations. After recording the fluorescence, simply plot the data (similarly to the standard curve example; Figure 1) to identify the substrate concentration (x-axis) that corresponds to enzyme activity (y-axis) where the slope levels off (~0). Likewise, the substrate concentration corresponding to the point where the slope levels off (~0) is a good indication of the optimal substrate concentration for that particular soil.
This assay ultimately measures fluorescence over a given time produced by the fluorogenic moiety that is cleaved from the substrate as a result of enzyme-mediated substrate depolymerization. Therefore, step 5 (substrate addition) is critical and must be performed as efficiently as possible in order to minimize the time between when the substrate is added to the soils and when the assays are incubated. Likewise, once the substrate comes in contact with the sample, enzymatic reactions will start to occur. We recommend using a multi-channel pipette for this reason. It is strongly suggested to become efficient using the multi-channel pipette prior to the day you are performing your enzyme assays. To accomplish this, you can practice pipetting with water until you can easily transfer volumes into the 96-well plates with your pipettor.
Soil Quenching
Quenching refers to decreasing fluorescence intensity caused by particulates and/or organic material in the soil slurries-incubations26. Quenching can be influenced by adjusting the soil:buffer ratios25. Due to background fluorescence from individual samples, it is critical to run standards with samples to account for background (quenching) fluorescence. Although some protocols use a single concentration after testing that the signal is linear, we strongly recommend implementing a standard quench control for each sample to best control for the effects of quenching. Failure to do so will result in a standard curve not applicable to the sample, and an incorrect estimation of enzymatic activity. The addition of standards to soil slurries is not time-sensitive, as the standard addition does not affect the background fluorescence of the sample.
NaOH addition
The addition of NaOH is used in some protocols to optimize fluorometric enzyme activity measurements because the fluorescent dye released from the synthetic substrates exhibits peak fluorescence at pH>9.026,49. When considering this suggestion, the NaOH concentration necessary to the soil slurry pH (i.e. to pH>9) will vary depending on the particular soil and the pH buffer used26. However, others argue that NaOH may not be necessary because signal intensity is generally very high even at lower pH, and because it introduces an additional source of measurement error. For example, the effect of NaOH additions on slurry pH and thus MUB or MUC fluorescence changes over time25. MUB linked substrates have been shown to demonstrate consistent increased fluorescence for 20 min following NaOH additions before levels taper; while MUC has demonstrated steady decreased fluorescence for up to 60 min26. Therefore, it is important to standardize the time between NaOH addition and fluorescence measurement. Alternatively, if fluorescence levels are sufficiently detectable without addition, conducting the assays without adding NaOH has been suggested as an equally acceptable alternative26.
Temperature
Temperature sensitivity should be taken into account when deciding incubation temperature. If the primary interest is understanding enzyme kinetics, using three or more temperatures as illustrated using Arrhenius plots (in the results section) is a robust approach. If the sample site has characteristically low temperature such as permafrost soil, then the duration of incubation may need to be extended to allow for the enzymes to react in the colder incubation temperatures. While traditional enzyme kinetics suggests that an increase in temperature should result in increased enzyme activity, we have found that enzymes may be site specific in terms of temperature sensitivity50. Therefore to understand site-specific enzyme activity potential it is critical that incubation temperature and duration be adjusted to reflect field site values.
Conclusion
EEs are critical drivers of biogeochemical processes in soils, and thus we need to be able to measure their activities. There are many challenges to measuring EEAs in soils, including interference and inhibition. Despite these challenges, standardized protocols (as the one described here) can be universally applied to measure EEAs for a wide range of enzymes. While it is fairly easy to generate quality data following these protocols, the interpretation of this data in an ecological context requires careful consideration of what these assays are really measuring, and how EEAs under assay conditions may differ from those under in situ conditions.
Disclosures
No conflicts of interest declared.
Acknowledgments
This publication was funded by the Enzymes in the Environment Research Coordination Network Research supported by the US National Science Foundation (DEB# 1021559). This research was supported by the US National Science Foundation (DEB# 1021559), and the US Department of Energy's Office of Science (Biological and Environmental Research). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the US NSF.
References
- Burns RG, Dick RP. Enzymes in the environment: activity, ecology, and applications. Vol. 86. CRC; 2002. [Google Scholar]
- Burns RG. Enzyme activity in soil: Location and a possible role in microbial ecology. Soil Biology and Biochemistry. 1982;14(82):423–427. [Google Scholar]
- Bandick AK, Dick RP. Field management effects on soil enzyme activities. Soil Biology and Biochemistry. 1999;31:1471–1479. [Google Scholar]
- Burns RG, et al. Soil enzymes in a changing environment: Current knowledge and future directions. Soil Biology and Biochemistry. 2013;58:216–234. [Google Scholar]
- Sinsabaugh RL, Hill BH, Follstad Shah JJ. Ecoenzymatic stoichiometry of microbial organic nutrient acquisition in soil and sediment. Nature. 2009;462:795–798. doi: 10.1038/nature08632. [DOI] [PubMed] [Google Scholar]
- Sinsabaugh RL, et al. Stoichiometry of soil enzyme activity at global scale. Ecology Letters. 2008;11:1252–1264. doi: 10.1111/j.1461-0248.2008.01245.x. [DOI] [PubMed] [Google Scholar]
- Wallenstein M, Allison SD, Ernakovich J, Steinweg JM, Vol Sinsabaugh R. In: Soil Biology. Shukla G, Varma A, editors. Vol. 22. Berlin Heidelberg: Springer; 2011. pp. 245–258. [Google Scholar]
- Wallenstein MD, McMahon SK, Schimel JP. Seasonal variation in enzyme activities and temperature sensitivities in Arctic tundra soils. Global Change Biology. 2009;15:1631–1639. [Google Scholar]
- Steinweg JM, Dukes JS, Wallenstein MD. Modeling the effects of temperature and moisture on soil enzyme activity: Linking laboratory assays to continuous field data. Soil Biology and Biochemistry. 2012;55:85–92. [Google Scholar]
- Wallenstein MD, Weintraub MN. Emerging tools for measuring and modeling the in situ activity of soil extracellular enzymes. Soil Biology and Biochemistry. 2008;40:2098–2106. [Google Scholar]
- Sinsabaugh RL. Phenol oxidase, peroxidase and organic matter dynamics of soil. Soil Biology and Biochemistry. 2010;42:391–404. [Google Scholar]
- Allison S. Soil minerals and humic acids alter enzyme stability: implications for ecosystem processes. Biogeochemistry. 2006;81:361–373. [Google Scholar]
- Deng S, Popova IE, Dick L, Bench Dick R. scale and microplate format assay of soil enzyme activities using spectroscopic and fluorometric approaches. Applied Soil Ecology. 2013;64:84–90. [Google Scholar]
- Sinsabaugh R, Klug MJ, Collins HP, Yeager PE, Peterson SO. In: Standard Soil Methods For Long Term Ecological Research. Robertson GP, Coleman DC, Bledsoe CS, Sollins P, editors. Oxford University Press; 1999. [Google Scholar]
- Tabatabai MA. In: Methods of Soil Analysis. Page AL, Miller RH, Keeney DR, editors. Vol. 2. American Society of Agronomy, Inc; 1994. pp. 903–947. [Google Scholar]
- Parham JA, Detection Deng SP. quantification and characterization of B-glucosaminidase activity in soil. Soil Biology and Biochemistry. 2000;32:1183–1190. [Google Scholar]
- Jacks TJ, Kircher HW. Fluorometric assay for the hydrolytic activity of lipase using fatty acyl esters of 4-methylumbelliferone. Analytical Biochemistry. 1967;21(67):279–285. doi: 10.1016/0003-2697(67)90190-x. [DOI] [PubMed] [Google Scholar]
- Smith RE, Bissell ER, Mitchell AR, Pearson KW. Direct photometric or fluorometric assay of proteinases using substrates containing 7-amino-4-trifluoromethylcoumarin. Thrombosis Research. 1980;17(80):393–402. doi: 10.1016/0049-3848(80)90074-2. [DOI] [PubMed] [Google Scholar]
- Jasinski JP, Woudenberg RC. 7-Amino-4-methylcoumarin. Acta Crystallographica Section C. 1994;50:1954–1956. [Google Scholar]
- Hoppe HG. SIGNIFICANCE OF EXOENZYMATIC ACTIVITIES IN THE ECOLOGY OF BRACKISH WATER - MEASUREMENTS BY MEANS OF METHYLUMBELLIFERYL-SUBSTRATES. Mar. Ecol.-Prog. Ser. 1983;11:299–308. [Google Scholar]
- Somville M. Measurement and Study of Substrate Specificity of Exoglucosidase Activity in Eutrophic Water. Applied and Environmental Microbiology. 1984;48:1181–1185. doi: 10.1128/aem.48.6.1181-1185.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrah PR, Harris PJ. A fluorimetric method for measuring the activity of soil enzymes. Plant and Soil. 1986;92:81–88. [Google Scholar]
- Freeman C, Liska G, Ostle NJ, Jones SE, Lock MA. The use of fluorogenic substrates for measuring enzyme activity in peatlands. Plant and Soil. 1995;175:147–152. [Google Scholar]
- Saiya-Cork KR, Sinsabaugh RL, Zak DR. The effects of long term nitrogen deposition on extracellular enzyme activity in an Acer saccharum forest soil. Soil Biology and Biochemistry. 2002;34:1309–1315. [Google Scholar]
- DeForest JL. The influence of time, storage temperature, and substrate age on potential soil enzyme activity in acidic forest soils using MUB-linked substrates and l-DOPA. Soil Biology and Biochemistry. 2009;41:1180–1186. [Google Scholar]
- German DP, et al. Optimization of hydrolytic and oxidative enzyme methods for ecosystem studies. Soil Biology and Biochemistry. 2011;43:1387–1397. [Google Scholar]
- Marx MC, Wood M, Jarvis SC. A microplate fluorimetric assay for the study of enzyme diversity in soils. Soil Biology and Biochemistry. 2001;33:1633–1640. [Google Scholar]
- Parton WJ, Morgan JA, Wang G, Del Grosso S. Projected ecosystem impact of the Prairie Heating and CO2 Enrichment experiment. New Phytologist. 2007;174:823–834. doi: 10.1111/j.1469-8137.2007.02052.x. [DOI] [PubMed] [Google Scholar]
- Morgan J, et al. C4 grasses prosper as carbon dioxide eliminates desiccation in warmed semi-arid grassland. Nature. 2011;476 doi: 10.1038/nature10274. [DOI] [PubMed] [Google Scholar]
- Carrillo Y, Pendall E, Dijkstra FA, Morgan JA, Newcomb JM. Response of soil organic matter pools to elevated CO2 and warming in a semi-arid grassland. Plant and Soil. 2011.
- Allison SD, Vitousek PM. Responses of extracellular enzymes to simple and complex nutrient inputs. Soil Biology and Biochemistry. 2005;37:937–944. [Google Scholar]
- Moorhead DL, Sinsabaugh RL. A theoretical model of litter decay and microbial interaction. Ecological Monographs. 2006;76:151. [Google Scholar]
- Allison SD, Weintraub MN, Gartner TB, Waldrop MP. In: Soil Biology. Shukla G, Varma A, editors. Vol. 22. Berlin Heidelberg: Springer; 2011. pp. 229–243. [Google Scholar]
- Treseder KK, Vitousek PM. Effects of soil nutrient availability on investment in acquisition of N and P in Hawaiin rain forests. Ecology. 2001;82(2001):946–954. [Google Scholar]
- Fujita Y, de Ruiter P, Wassen M, Heil G. Time-dependent, species-specific effects of N:P stoichiometry on grassland plant growth. Plant and Soil. 2010;334:99–112. [Google Scholar]
- Elser JJ, Dobberfuhl DR, MacKay NA, Schampel JH. Organism Size, Life History, and N:P Stoichiometry. BioScience. 1996;46:674–684. [Google Scholar]
- Allison VJ, Condron LM, Peltzer DA, Richardson SJ, Turner BL. Changes in enzyme activities and soil microbial community composition along carbon and nutrient gradients at the Franz Josef chronosequence, New Zealand. Soil Biology and Biochemistry. 2007;39:1770–1781. [Google Scholar]
- German DP, Marcelo KRB, Stone MM, Allison SD. The Michaelis - Menten kinetics of soil extracellular enzymes in response to temperature: a cross-latitudinal study. Global Change Biology. 2012;18:1468–1479. [Google Scholar]
- German DP, Chacon SS, Allison SD. Substrate concentration and enzyme allocation can affect rates of microbial decomposition. Ecology. 2011;92:1471–1480. doi: 10.1890/10-2028.1. [DOI] [PubMed] [Google Scholar]
- Fierer N, Craine JM, McLauchlan K, Schimel JP. Liter quality and the temperature sensitivity of decomposition. Ecology. 2005;86:320–326. [Google Scholar]
- Sterner RW, Elser JJ. Ch. 5. In: Sterner RW, Elser JJ, editors. Ecological Stoichiometry: The Biology of Elements from Molecules to the Biosphere. Princeton, NJ: Princeton University Press; 2002. pp. 179–230. [Google Scholar]
- Cleveland C, Liptzin D. C:N:P stoichiometry in soil: is there a "Redfield ratio" for the microbial biomass. Biogeochemistry. 2007;85:235–252. [Google Scholar]
- Logan SR. The origin and status of the Arrhenius equation. Journal of Chemical Education. 1982;59:279. [Google Scholar]
- Trasar-Cepeda C, Gil-Sotres F, Leirós MC. Thermodynamic parameters of enzymes in grassland soils from Galicia, NW Spain. Soil Biology and Biochemistry. 2007;39:311–319. [Google Scholar]
- Menten L, Michaelis M. Die kinetik der invertinwirkung. Biochem Z. 1913;49:333–369. [Google Scholar]
- Wallenstein MD, Haddix ML, Lee DD, Conant RT, Paul EA. A litter-slurry technique elucidates the key role of enzyme production and microbial dynamics in temperature sensitivity of organic matter decomposition. Soil Biology and Biochemistry. 2012;47:18–26. [Google Scholar]
- Chrost J. Fluorescence correction for measurements of enzyme activity in natural waters using methylumbelliferyl-substrates. Archiv für Hydrobiologie. 1986;106:79. [Google Scholar]
- Reed GF, Lynn F, Meade BD. Use of Coefficient of Variation in Assessing Variability of Quantitative Assays. Clinical and Diagnostic Laboratory Immunology. 2002;9:1235–1239. doi: 10.1128/CDLI.9.6.1235-1239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead JAR, Smith JN, Williams RT. Studies in detoxification. 67. Biosynthesis of the glucuronides of umbelliferone and 4- Methylumbelliferone and their use in fluorimetric determination of b-glucuronidase. Biochemical Journal. 1955;61:569–574. doi: 10.1042/bj0610569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddix ML, et al. The Role of Soil Characteristics on Temperature Sensitivity of Soil Organic. Matter. Soil Sci. Soc. Am. J. 2011;75:56–68. [Google Scholar]