

Summary
A major advance in adoptive T-cell therapy (ACT) is the ability to efficiently endow patient’s T cells with reactivity for tumor antigens through the stable or regulated introduction of genes that encode high affinity tumor-targeting T-cell receptors (TCRs) or synthetic chimeric antigen receptors (CARs). Case reports and small series of patients treated with TCR- or CAR-modified T cells have shown durable responses in a subset of patients, particularly with B-cell malignancies treated with T cells modified to express a CAR that targets the CD19 molecule (1-8). However, many patients do not respond to therapy and serious on and off-target toxicities have been observed with TCR- and CAR-modified T cells (9-13). Thus, challenges remain to make ACT with gene-modified T cells a reproducibly effective and safe therapy and to expand the breadth of patients that can be treated to include those with common epithelial malignancies. This review discusses research topics in our laboratories that focus on the design and implementation of ACT with CAR-modified T cells. These include cell intrinsic properties of distinct T-cell subsets that may facilitate preparing therapeutic T-cell products of defined composition for reproducible efficacy and safety, the design of tumor targeting receptors that optimize signaling of T-cell effector functions and facilitate tracking of migration of CAR-modified T cells in vivo, and novel CAR designs that have alternative ligand binding domains or confer regulated function and/or survival of transduced T cells.
Keywords: gene therapy, immunotherapies, T cells, cancer, T-cell receptors, chimeric antigen receptors
Introduction
A major advance in adoptive T-cell therapy (ACT) is the ability to efficiently endow patient’s T cells with reactivity for tumor antigens through the stable or regulated introduction of genes that encode high affinity tumor-targeting T-cell receptors (TCRs) or synthetic chimeric antigen receptors (CARs). Case reports and small series of patients treated with TCR- or CAR-modified T cells have shown durable responses in a subset of patients, particularly with B-cell malignancies treated with T cells modified to express a CAR that targets the CD19 molecule (1-8). However, many patients do not respond to therapy, and serious on and off-target toxicities have been observed with TCR and CAR-modified T cells (9-13). Thus, challenges remain to make ACT with gene-modified T cells a reproducibly effective and safe therapy, and to expand the breadth of patients that can be treated to include those with common epithelial malignancies. This review discusses research topics in our laboratories that focus on the design and implementation of ACT with CAR-modified T cells. These include cell intrinsic properties of distinct T-cell subsets that may facilitate preparing therapeutic T-cell products of defined composition for reproducible efficacy and safety, the design of tumor targeting receptors that optimize signaling of T-cell effector functions and facilitate tracking of migration of CAR-modified T cells in vivo, and novel CAR designs that have alternative ligand binding domains or confer regulated function and/or survival of transduced T cells.
Composition of T-cell products for ACT
The majority of clinical trials of ACT have been performed with T cells that are not gene modified, rather tumor-reactive or virus-specific T cells are derived from the blood or tumor infiltrates of cancer patients or from allogeneic donors in the setting of hematopoietic stem cell transplantation (14-19). ACT with unmodified autologous or allogeneic T cells has had a major impact in melanoma and in viral infections after allogeneic stem cell transplant. While presenting its own unique challenges, studies of ACT with unmodified T cells have been instructive for elucidating principles for effectively transferring T-cell immunity and providing insights that are relevant to ACT with genetically modified T cells.
Transfer of unmodified T cells
The adoptive transfer of tumor or virus-specific T cells that have been isolated ex vivo, expanded in culture for various periods of time, and administered to patients has demonstrated important therapeutic effects in a subset of patients with advanced solid tumors and recurrent viral infections (20-22). Unfortunately, ACT with unmodified T cells is not uniformly effective in eliminating tumors, and the mechanisms responsible for successful tumor eradication, or resistance to therapy when tumors only temporarily regress or do not respond to ACT, have remained elusive. The major challenge in the initial studies was simply isolating tumor-reactive T cells from the blood or infiltrates of most patients and then expanding these T cells to the large numbers (1010 – 1011) that were perceived to be required for therapeutic efficacy (23, 24). Tumor-specific cytolytic activity and cytokine production were the major measurements of potency of the ACT products used in these trials, and as a result there was marked heterogeneity in the source, phenotype, clonality, specificity, frequency, and affinity of T cells that were administered to patients, compounded by the variation in the methods and duration of T-cell culture.
The first studies of ACT in patients with melanoma administered tumor-reactive TILs or T-cell clones specific for melanocyte differentiation antigens without prior conditioning of the patient. Despite the infusion of 109 - 1011 T cells and the administration of interleukin-2 (IL-2) after T-cell transfer to promote their survival in vivo, the persistence of transferred T cells was dismally short in these trials; antitumor effects, if discernable, were often transient (25-27). The use of lymphodepleting chemotherapy or chemoradiotherapy prior to ACT to increase the levels of the homeostatic gamma chain cytokines IL-7 and IL-15 and to deplete regulatory cell subsets proved to be an important advance, and resulted both in improved T-cell persistence and complete tumor regression in a significant proportion of treated patients, particularly those that received ACT with TILs (28, 29). Of interest, the T cells that proliferated after the infusion of polyclonal TIL products into lymphodepleted patients were often highly oligoclonal (28, 30), which suggested that cell-intrinsic properties of only a subset of tumor-reactive T cells might underlie their ability to persist and proliferate in vivo. These observations focused further attention on characteristics of T cells that might be predictive of persistence and in vivo expansion after adoptive transfer, and several parameters of the transferred TIL including telomere length and expression of costimulatory molecules were shown to correlate with detection of transferred T cells for prolonged periods after ACT, and with superior antitumor responses (31, 32).
T-cell differentiation and lineage relationship
T cells consist of phenotypically and functionally distinct naïve and memory T-cell subsets that vary both in their longevity and frequency in the peripheral blood in normal individuals and patients. Naive T cells are antigen inexperienced and characterized by the expression of CD45RA, CD62L, and CD28 and CD27 costimulatory molecules, whereas the memory T-cell subset expresses CD45RO and contains CD62L+ central (Tcm) and CD62L- effector memory (Tem) subsets (33). CD8+ memory T-cell subsets can be further subdivided into those that express high levels of CD161, the majority of which express a restricted Vα TCR (Vα7.2) and recognize bacterial ligands presented by the MR1 class I molecule (34-38), and a CD45RA+CD62L+CD95+CD122+ subset that has a phenotype intermediate between that of Tn and Tcm and has been proposed as a memory stem cell (Tscm) (39). Each of these T-cell subsets express different transcription factors and gene expression profiles, and their role in host immunity and potential for use in ACT continue to be the subject of intense research.
Mouse models of viral infection have been instructive in defining the lineage relationships of individual CD8+ T-cell subsets, providing insights into the basis for longevity of T-cell memory, and elucidating features of T cells that are important to consider for ACT. Fate mapping of the differentiation of individual naive T cells in response to antigen supports a model in which naive T cells differentiate in a linear fashion to slowly proliferating long-lived Tcm and to rapidly expanding but shorter-lived Tem and Teff cells (40, 41) (Fig. 1). In a primary immune response, individual naive T cells were shown to contribute differently to the formation of the individual memory subsets and the degree of expansion in the primary response did not predict expansion potential in a secondary challenge (40, 41). Thus, large Tem subsets that were formed after a primary response typically failed to dominate the response to secondary challenge. This disparate capacity of different T-cell subsets to proliferate and survive is likely to influence their behavior when used in ACT, and has implications for the types of T cells to select for genetic modification prior to cell transfer.
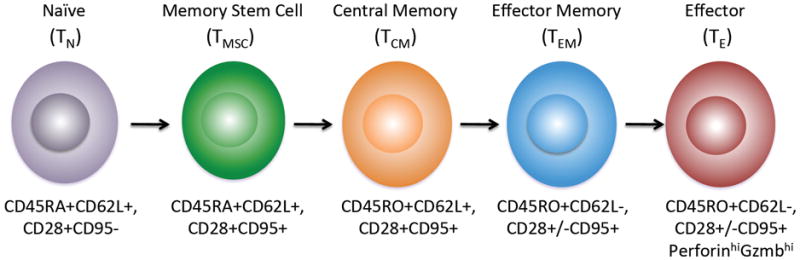
Fig. 1. Linear differentiation of T-cell subsets.
The phenotype of naive, memory, and effector subsets is shown and the linear pathway of differentiation from a naive T cell is based on recent data from fate mapping studies in murine models (40,41).
The frequency distribution of individual T-cell subsets in the blood, lymph node, and tissues is determined in large part by the expression of homing receptors that direct the migration of T cells (34, 42). Because CD8+ Tscm and Tcm express CD62L and CCR7, that directs these cells to lymph nodes, the frequency of each of these subsets in the blood is low in normal individuals compared with CD62L- Tem. In cancer patients, cytotoxic chemotherapy can reduce total lymphocyte numbers for very prolonged periods and further skew the distribution of CD4+ and CD8+ T cells and the proportions of naive and memory subsets (43, 44). Thus, if T cells that are present in the peripheral blood are simply genetically modified with tumor targeting CARs or TCRs without prior selection of subsets, there is little control over the phenotype of the cell product that is prepared, and consequently the migration, survival, and function of these cells after transfer could be quite different. Predictably, in CAR T cell trials in which the T-cell products were derived from peripheral blood, major differences in CD4 and CD8 content and in the proportion of T cells that express costimulatory molecules and lymph node homing receptors were observed (6).
Genetic modification of selected T-cell subsets for ACT
The availability of genetic approaches for rapidly deriving therapeutic T cells for cancer therapy provides the opportunity to utilize T-cell products in which the composition and function are defined to a much greater extent than currently. Moreover, in addition to introducing the tumor targeting receptor, the T cells can be genetically marked with cell surface receptors to facilitate systematic study of their persistence, proliferation, migration, and function in vivo after ACT.
We speculated that the differentiation state of T cells from which therapeutic effector cells were derived might confer heritable properties that determine their ability to persist long term after ACT, and establish durable memory populations in lymph nodes and bone marrow. To examine the behavior of Teff cells derived from different subsets, we developed a non-human primate model using M. nemestrina in which the culture methods for T-cell activation, expansion and gene transfer are similar to those employed in human ACT (45). In the initial experiments, we sort-purified CD8+ T cells from the Tcm and Tem subsets, derived Teff cell clones specific for cytomegalovirus (CMV) from each of these subsets by limiting dilution, gene marked the T-cell clones with a B-cell lineage surface marker to enable tracking in vivo, and then sequentially adoptively transferred these T cells back to the animal, without administering lymphodepleting chemotherapy prior to ACT or cytokines post ACT. We observed that antigen-specific CD8+ Teff clones derived from the Tem subset survived in the blood for less than 7 days after adoptive transfer and were not detected in lymph nodes, bone marrow, or tissue sites. The Teff clones derived from the Tcm subset had equivalent cytolytic activity and granzyme B and perforin expression as Teff derived from Tem cells, and as a consequence of in vitro activation, had downregulated expression of CD62L and CD127. However, the Tcm-derived Teff cells persisted in high frequency in the blood after adoptive transfer, migrated to lymph nodes and bone marrow, and reconstituted both Tcm and Tem phenotypes (46) (Fig. 2). Animals that received gene marked Tcm-derived Teff and were followed for > 4 years have persistence of large numbers of gene marked cells in the memory pool, demonstrating that the progeny of a single Tcm cells can provide long-lasting immunity in a primate.
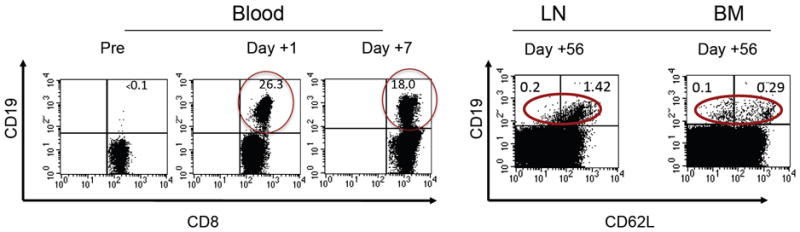
Fig. 2. High-level persistence and migration of adoptively transferred T cells derived from a single central memory T (Tcm) cell.
CD8+ Tcm cells were sort-purified based on expression of CD62L and CD9,5 and single T cells specific for cytomegalovirus were derived and expanded in limiting dilution cultures and retrovirally marked with a truncated CD19 molecule to facilitate detection in vivo. The expanded, gene marked T cells were adoptively transferred to the animal without preceding lymphodepletion or the administration of cytokines post infusion. The frequency of gene marked (CD19+) CD8+ T cells in the CD8+ T cell subset in blood, lymph nodes (LN), and bone marrow (BM) is shown.
Recent studies in the non-human primate model have examined the ability of Tn-derived Teff cells to persist after ACT and have shown that T-cell products derived from Tn cells also have the capacity to persist at high levels in non-lymphodepleted animals. However, it has not been possible to evaluate the ability of these cells to respond to antigen challenge. A potential advantage of using Tn is the ability to influence the differentiation of these cells during in vitro culture by modifying signaling pathways with small molecules or cytokines such as IL-7, IL-15, or IL-21 (47, 48).
Deriving CAR-modified T cells for ACT from selected T-cell subsets
Further support for the differential capacity of T cells from different CD8+ T-cell subsets that are genetically modified with tumor targeting receptors to function in ACT comes both from studies with murine T cells and with human T cells in immunodeficient Nod/Scid/γc-/- (NSG) mice (39, 49, 50). However, the compelling data in these preclinical models has not been implemented in clinical studies, and rigorous analysis of the importance of deriving T cells for ACT from defined subsets is only now beginning. This in part reflects the absence of robust, clinical grade methods for selecting defined T-cell subsets from peripheral blood or leukapheresis products obtained from patients. Cell selection can be particularly challenging for isolating rare subsets such as CD8+ Tcm that may often comprise <2% of PBMCs, and CD8+ Tscm that are of far lower frequency and require a constellation of markers for selection. Our laboratories have reported the development of clinical grade immunomagnetic selection methods for isolating CD8+ Tcm that rely on the use of multiple antibodies to remove CD4+, CD14+, and CD45RA+ cells from PBMC preparations and then positively selecting the CD62L+ fraction to enrich CD45RO+CD62L+CD8+ Tcm cells (51, 52). The selected Tcm cells can be readily modified with tumor-specific CARs and expanded for use in ACT. While effective, the purity and yield with this approach could be improved. A novel technology that is more facile for segregating cells based on a combination of markers and provides high purity and yield is the use of low affinity antibody derived Fab-fragments fused to Strep-tag to enable stable binding to a target cell surface molecule when the Fabs are multimerized with Streptactin-labeled beads, and rapid reversal of binding by dissociating the multimer complexes into monomers by the addition of D-biotin (53). This technology enables serial positive enrichment of T cells based on multiple markers, is amenable to selecting virtually any desired subset of T cells that can be distinguished by a combination of markers, and has been demonstrated to provide highly pure populations of human CD8+ Tcm cells for genetic modification (53).
We have examined the functional characteristics of human CD8+ and CD4+ T cells derived from Tn, Tcm, and Tem subsets and modified with CARs specific for several tumor associated molecules including CD19, CD20, ROR1, L1CAM, αVβ6 integrin, and IL-13Rα2. We have demonstrated that individual T-cell subsets alone and in combination have distinct antitumor potency in NSG mice engrafted with human tumor xenografts. Based on these studies, we initiated the first clinical trial using a CD19 CAR in patients with advanced B-cell malignancies in which the composition of the CAR-modified T-cell product that is infused into each patient consists of a defined proportion of CD4+ and CD8+ T cells derived from selected subsets. It is anticipated that this approach will permit more precise determination of the potential relationship between T-cell dose, toxicity, and antitumor efficacy, and serve as a foundation for comparative studies of therapeutic regimens that use T-cell products derived from other subsets and combinations.
Structural issues in CAR design using scFvs
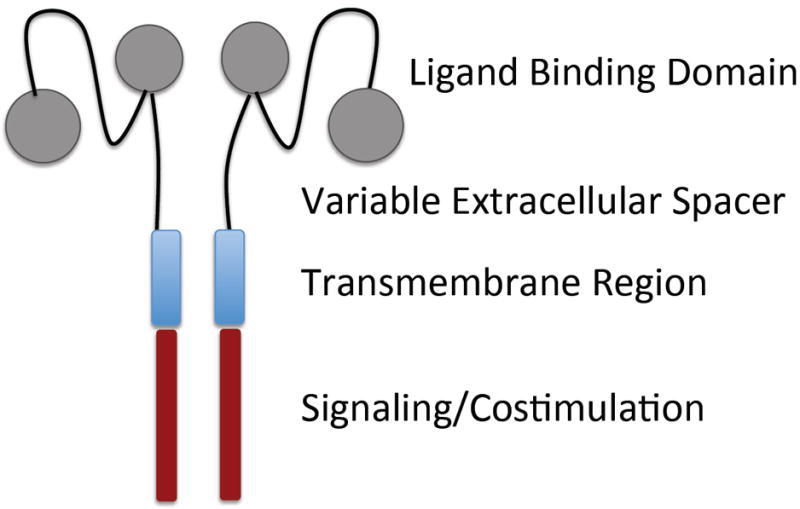
The general concept underlying the design of synthetic chimeric antigen receptors is to incorporate an extracellular polypeptide domain that selectively binds a molecule on the target cell and is linked through a transmembrane domain to signaling molecules that trigger T-cell effector functions (Fig. 3). CARs have a major advantage over conventional TCRs in that a single receptor construct can be used to treat all patients with a malignancy that expresses the target molecule, and tumor variants that have downregulated or mutated components of the antigen processing machinery for presenting tumor antigens on HLA molecules to escape endogenous immunity will remain susceptible. Within this general framework, the design of CARs for individual target molecules has largely been empiric, and various extracellular components have been used to link the ligand-binding domain to the transmembrane domain, and distinct constellations of intracellular signaling domains have been incorporated to activate effector functions in the T cell. For example, CARs designed to target CD19 on B-cell malignancies that are being used in the clinic have employed two different scFvs and been designed with different extracellular, transmembrane regions, and intracellular signaling domains. Although all have shown evidence of antitumor efficacy in ACT, it is premature to conclude that all will be equivalently effective in providing durable responses (3, 4, 6).
Fig. 3. Elements of synthetic chimeric antigen receptors.
Schematic of ligand binding, non-signaling, and signaling elements of a CAR that can be altered to optimize tumor cell recognition and signaling of T-cell function.
It is uncertain whether the design of CARs for other tumor-associated molecules that are not as abundantly expressed as CD19 on target cells will be as amenable to empiric design, or might benefit from structural modeling of T-cell/tumor cell interactions to promote effective T-cell signaling and tumor cell death. We have studied the design of CARs specific for the orphan tyrosine kinase receptor ROR1 that was initially identified as a signature gene in chronic lymphocytic leukemia (54, 55) and has since been implicated in the malignant phenotype of a variety of epithelial tumors, including non small cell lung cancer and triple negative breast cancer (56-59). The design issues encountered with developing ROR1 CARs illustrate some of the structural considerations for developing CARs. This review discusses research topics in our laboratories that focus on the design and implementation of ACT with CAR-modified T cells. These include cell intrinsic properties of distinct T-cell subsets that may facilitate preparing therapeutic T-cell products of defined composition for reproducible efficacy and safety, the design of tumor targeting receptors that optimize signaling of T-cell effector functions and facilitate tracking of migration of CAR-modified T cells in vivo, and novel CAR designs that have alternative ligand binding domains or confer regulated function and/or survival of transduced T cells.
Ligand binding
Because of the high specificity of monoclonal antibodies, the ligand binding domain is most often a single chain variable fragment (scFv) constructed from the variable heavy (VH) and variable light (VL) sequences of a monoclonal antibody (mAb) specific for a tumor cell surface molecule, which is then fused to a transmembrane domain, one or more intracellular costimulatory signaling modules, and CD3ζ (60, 61). The tumor-binding component of the CAR need not be an scFv, however, and other protein binding ligands that might impart greater tumor selectivity have been fused to T-cell signaling molecules and are discussed in greater detail later in this review. The prevalence of mAbs for tumor associated molecules has facilitated the design of a large number of CARs that redirect T-cell specificity to tumor cells in vitro, including CARs specific for CD19, CD20, ROR1, Her2, CAIX, folate receptor α, Lewis Y, and FAP (62-70). A concern with many of the candidate targets expressed on epithelial cancers or their stroma is the potential for CAR-modified T cells to cause toxicity to normal tissues that also express these molecules. This risk has unfortunately been born out in patients for Her2 and CAIX, and in animal models for FAP (10, 71, 72).
The biophysical properties of ligand binding with CAR T cells differ from that of a TCR in many respects, and it is remarkable that the majority of CARs that have been constructed appear to function effectively in vitro and in vivo. The number of target molecules on tumor cells that can engage a CAR is substantially greater than the number of MHC/peptide complexes. For example it is estimated that B-cell tumors express >104 CD19 molecules and 103-104 ROR1 molecules, which is far in excess of the estimated number of MHC molecules that would present a single tumor associated peptide (73-75). To compensate for low MHC/peptide density and establish synapse formation between the T cell and target cell, TCR signaling involves serial triggering and accumulation of signaling intermediates (76, 77), and it is unlikely that if serial triggering of CARs occurs, it would do so with similar kinetics as that of the TCR. Studies in which TCR affinity has been increased by introducing mutations in TCR sequences have revealed there is a threshold of receptor affinity beyond which antigen engagement results in activation induced T cell death and loss of therapeutic activity (78). The scFv component of a CAR typically has a much higher binding affinity for antigen than that of a TCR for its MHC-peptide ligand. Thus, there is the potential for sustained signaling after engagement of target cells by CAR-modified T cells with uncertain consequences for effector function, cell survival, and therapeutic efficacy.
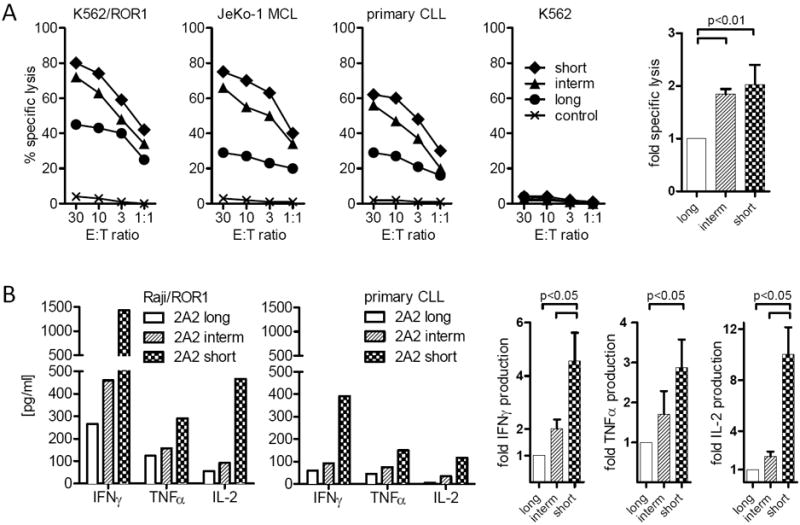
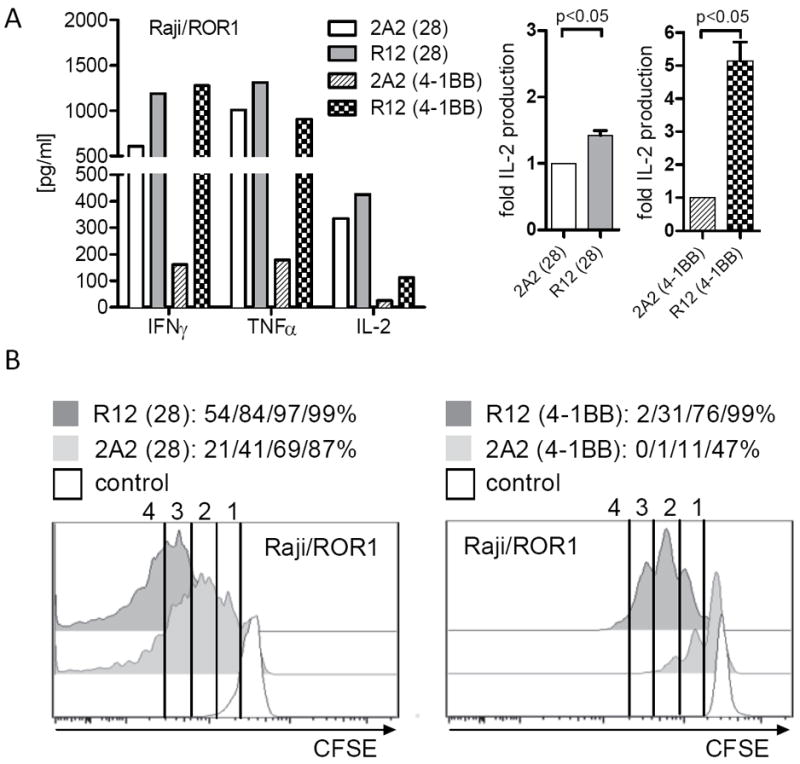
Using ROR1 as a model antigen, we examined the potential for the affinity of the mAb from which the scFv is derived to affect T-cell signaling and antitumor activity of a CAR. ROR1 is a 120-kDa cell surface glycoprotein containing extracellular immunoglobulin (Ig)–like, Frizzled, and Kringle domains and is expressed on CLL, B-ALL, and a variety of epithelial cancers, and implicated in tumor cell survival, proliferation, and metastasis (56-59). ROR1 is expressed during embryogenesis but absent from normal adult tissues, apart from a subset of immature B-cell precursors present in the bone marrow and low-level expression on adipocytes (79, 80). In collaboration with Christoph Rader, we derived ROR1-specific CARs from two mAbs (R12 and 2A2) that both target an epitope in the Ig-like/Frizzled region of ROR1 but have different affinities such that R12 is approximately 50 fold higher affinity than 2A2 (81). The R12 and 2A2 CARs were designed in identical formats that contained the same extracellular spacer, transmembrane and intracellular signaling domains, and the lentiviral vector used to express the CARs in T cells encoded a truncated EGFR marker to enable purification of transduced T cells (50, 82). Primary CD8+ T cells were transduced with lentiviral vectors encoding each of these CARs, selected for transgene expression and analyzed for their ability to recognize ROR1+ tumor cells. T cells modified with each of the 2A2 and R12 ROR1-CARs specifically lysed K562/ROR1 and Raji/ROR1 tumor cells with approximately equivalent efficiency. However, analysis of cytokine production showed that T cells expressing the high affinity R12 CAR that contained either a CD28 or 4-1BB costimulatory domain produced greater amounts of IFN-γ, TNF-α, and IL-2 production compared to T cells expressing the corresponding lower affinity 2A2 constructs. R12 ROR1-CAR T cells also underwent more cell divisions after co-culture with ROR1+ tumor cells compared to T cells expressing the respective 2A2 CARs (82) (Fig. 4).
Fig. 4. Effect of CAR ScFv affinity on T-cell effector function and proliferation.
(A) Multiplex cytokine analysis after a 24-hour stimulation of 5×104 CAR-modified T cells expressing a ROR-1 specific CAR constructed with the 2A2 scFv or with the higher affinity R12 scFv with primary Raji lymphoma cells that express ROR1. Data are shown for CAR designs that include either the CD28 or 4-1BB costimulatory domains. The right panels show the fold increase in IL-2 production. (B) Proliferation of carboxyfluorescein-labeled CD8+ T cells modified with the 2A2 ROR1 and high affinity R12 ROR1 CARs 72 h after stimulation with primary CLL cells. Numbers above each histogram indicate the number of cell divisions, and the fraction of T-cells in each gate that underwent ≥3/2/1 cell divisions is provided above each plot.
We were concerned that the slower dissociation of R12 from ROR1 could prolong T-cell activation and confer an increased susceptibility to activation induced cell death (AICD). However, after co-culture with ROR1+ tumor cells in vitro, we observed a lower rate of AICD in T cells modified with the R12 ROR1-CAR compared to 2A2. The superior signaling of effector functions and cell proliferation by the high affinity R12 ROR1 CAR translated into more effective elimination of human tumor xenografts in NSG mice (82). T cells transduced to express the R12 ROR1-specific CAR were more effective than T cells transduced to express the 2A2 ROR1-specific CAR in eliminating the ROR1+CD19+ mantle cell lymphoma cell line (JeKo-1), and this reflected more rapid proliferation and accumulation of higher numbers of R12 CAR T cells in vivo (82). Indeed in the JeKo-1 model, the R12 CAR was comparable to a CD19 CAR despite the lower number of ROR1 molecules on the tumor cells. The superiority of the high affinity R12 CAR has also been observed in a transplantable ROR1+ breast cancer model (D. Sommermeyer, S. Riddell, unpublished data). These studies identify scFv affinity as a key parameter that can be manipulated to improve CAR function, which may be especially relevant when targeting molecules that are expressed at lower density on tumor cells. It is unknown whether CARs with higher affinity than R12 would further improve therapeutic efficacy; it seems likely that like TCRs, there will be an affinity threshold beyond which T cells would be prone to AICD.
Extracellular non-signaling domains
The spatial interaction between T cells and target cells in HLA-restricted T-cell recognition is the product of natural evolution and is determined by the dimensions of the TCR and HLA molecules. By contrast, the interactions between CAR-modified T cells and target cells are governed by the structure of the respective target molecule on the tumor cell, the location of the epitope that is recognized by the scFv, and the design of the extracellular domain of the CAR. It had previously been appreciated that the optimal length of the CAR extracellular domain may vary for in vitro recognition of tumor cells, depending on the target molecule (83). However, the specific requirements in the design of the non-antigen binding components of the CAR extracellular domain to mediate potent tumor recognition and T-cell function in vitro and in vivo are understudied compared to the ongoing efforts in the field to evaluate different intracellular CAR signaling domains.
We have studied the role of extracellular domain spacer length on the recognition of ROR1+ tumors by T cells transduced with the 2A2 and R12 ROR1-specific CARs that target a region of ROR1 located at the membrane distal Ig-like/Frizzled domain interface. We constructed a library of CARs that linked the scFv via long (229 amino acid) IgG4 hinge-CH2-CH3, intermediate (119 amino acid) IgG4 hinge-CH3, and short (12 amino acid) IgG4 hinge only spacer sequences to CD28/CD3ζ or 4-1BB/CD3ζ signaling modules. T cells were transduced with each of the ROR1 CARs and selected for equivalent levels of CAR expression. ROR1 CAR T cells expressing long, intermediate, and short extracellular spacers sequences all lysed ROR1+ tumor cells, demonstrating that for ROR1, there is substantial leniency in CAR design to achieve measurable tumor cell lysis. However, there was a clear hierarchy in cytolytic function with the short spacer CAR conferring dramatically superior lysis of tumor cells (Fig. 5). Moreover, there were marked differences in cytokine production and T-cell proliferation with the short spacer configuration clearly superior (82). We then designed a similar panel of ROR1-specific CARs using an scFv that targets an epitope in the membrane proximal Kringle domain and observed that for this epitope only the long spacer format confers tumor recognition and signaling of T-cell cytokine production and proliferation (Sommermeyer D, Riddell SR, unpublished data). These data suggest that for many target molecules, spatial constraints are likely to govern the efficiency of T cell/tumor cell recognition and that the non-signaling extracellular spacer can be a critical determinant to optimize in CAR design.
Fig. 5. Effect of non-signaling extracellular spacer domain length on recognition of ROR1+ tumor cells with a ROR1 CAR specific for a membrane distal target epitope.
(A) Cytolytic activity of T cells expressing the 2A2 ROR1-CAR with a long, intermediate, or short extracellular spacer domain against ROR1+ tumor cells and control K562 cells. Control T cells are modified with the EGFRt marker gene only. Cytotoxicity data from 4 independent experiments (E:T = 30:1) were normalized (cytolytic activity by ROR1-CAR 2A2 = 1) and analyzed by Student’s t-test (bar diagram). (C) Multiplex cytokine analysis after a 24-h stimulation of 5×104 CAR T-cells with Raji/ROR1 cells and primary CLL cells.
Signaling and costimulation
A substantial amount of research in CAR design has focused on examining different intracellular signaling modules to activate T cell effector functions. Initial CAR designs incorporated either the CD3ζ chain or the signaling component of Fc receptor-γ or epsilon to activate T-cell cytolytic function (60). The first clinical translation of CAR-modified T cells as ACT for malignancy employed such ‘first generation’ CARs that targeted L1CAM, CD20, and CAIX, and significant antitumor activity was not observed (84-86). In these studies, CAR T cells typically did not proliferate in vivo and persistence was either transient or the T cells were present at a very low frequency. It is important to note that in these studies the CAR gene was introduced into polyclonal unselected T cells either by retroviral gene transfer or electroporation, which was inefficient and required long-term culture to generate T-cell products (87). In some studies the CAR cassette also contained an immunogenic selectable marker gene, and the patients did not typically receive lymphodepleting chemotherapy prior to T-cell infusions. Thus, characteristics of the T cells administered in ACT, inefficient methods of gene transfer and prolonged culture, and lack of patient conditioning likely also contributed to poor efficacy in these first attempts at clinical translation of ACT with CAR-modified T cells.
Effective T-cell signaling requires costimulation and it was logical to construct ‘second generation’ and ‘third generation’ CARs that include incorporate CD28, 4-1BB, or other costimulatory domains alone and in tandem with CD3ζ (63, 88-90). Alternative strategies for providing costimulation and supporting T-cell survival by co-expressing costimulatory receptors, their ligands, and cytokines have also been devised (91-93). Several groups have demonstrated that CARs that provide costimulatory signaling result in enhanced cytokine production, T-cell proliferation and survival, and enable sequential rounds of T-cell proliferation after antigen engagement in vitro and in vivo compared with CARs that contain CD3ζ alone (63, 88-90). Thus far, only one clinical trial has directly compared in the same patient T cells transduced with a CD19-specific CAR containing CD28/CD3ζ with those transduced with the same CAR containing only CD3ζ (94). The investigators observed superior initial expansion and persistence of T cells transduced with the CD28/CD3ζ construct, however antitumor activity was not as dramatic in this study as reported for other ACT trials with CD19-modified T cells, potentially reflecting other variations in CAR design and/or T-cell product composition. However, this study provides an important precedent for how future trials might be performed to directly compare the behavior of T cells transduced with distinct CAR designs.
Novel CAR ligand-binding domain structures
Subsequent to the original description of a ‘T-body’ CAR design by Eshhar in 1993, the synthetic antigen receptor field has been dominated by scFv-based extracellular antigen binding domains (60, 66). Consequently, the evolution of CARs has relied heavily on constructing scFvs from murine monoclonal antibody VH and VL sequences cloned from a relatively small repertoire of already available hybridomas that were known to produce mAbs specific for tumor associated antigens. Many of the available targets on solid tumors are also expressed on some normal cells that are not as dispensable as B cells targeted with CD19-specific CARs. Moreover, because the scFv constructs are derived from murine rather than human sequences, they have a potentially greater risk of immunogenicity due to the recognition of murine components of the CAR. The advent of combinatorial scFv libraries, including human scFv phage display and ribosome displayed scFv libraries designed with parallel deep sequencing monitoring, should greatly expand the range of epitopes on the surface of tumor cells for which a scFv can be identified, and the utilization of human scFvs in CARs may reduce their potential immunogenicity (95, 96). A novel approach for identifying scFvs for tumor specific CARs is the direct screening and selection of ‘CAR bodies’ using scFv libraries formatted as CARs and enriched through lentiviral expression in T cells that are then expanded by stimulation with tumor cells of interest (97). This method directly selects for scFvs that bind tumor-associated molecules and function in a CAR format based on the ability to support T-cell activation and proliferation, and could potentially identify novel cell surface targets that might be derived from splice variants or alterations in glycosylation.
While scFv-targeting domains are likely to be a mainstay of CAR design, they are not the sole ligand binding polypeptides that can be adapted to CARs. Our group and others have investigated alternative ligand binding domains to design CARs. These include human proteins that serve as soluble ligands for tumor cell surface receptors. The targeting ligand can either be a soluble polypeptide such as a cytokine or growth factor that is tethered to the CAR extracellular spacer region, or an extracellular domain (ECD) of a membrane anchored ligand or receptor for which the binding pair counterpart is expressed on the tumor cell. The first design, termed a ‘zetakine’ CAR, uses a soluble cytokine as the ligand binding domain and is exemplified by a construct wherein the human IL-13 cytokine is linked to extracellular spacer, transmembrane and signaling domains, and the cell surface IL-13 receptor complex is the targeted ligand for the CAR (98). By taking advantage of IL-13 variants (muteins) that have modulated receptor selectivity and affinity, we demonstrated it was feasible to selectively target the IL13Rα2 receptor complex expressed on human gliomas with IL-13 zetakine CAR-modified T cells (99-102). A similar approach was used in the design of vascular endothelial growth factor (VEGF) zetakine CAR that targets the VEGF receptor (VEGFR) expressed on tumor vasculature, thus demonstrating the versatility of this strategy (103). The use of the ECD of a membrane anchored ligand or receptor for which the binding pair counterpart is expressed on the tumor cell has also been used successfully in CAR design. An example of a fully human ECD tumor cell recognition domain was described by the Gottschalk et al. who designed a CAR composed of the CD27 ECD for targeted recognition of CD70 expressing tumors (104). Sentman et al. (105, 106) have demonstrated that this design format is bioactive even if the targeting domain ECD being incorporated into the CAR is from a type II transmembrane protein. Their CAR ECD composed of human NKG2D places the cytoplasmic signaling domains of CD28 and zeta in an inverted configuration relative to their natural orientation to the T-cell plasma membrane. Remarkably, this type II transmembrane CAR signals in a nominal manner and the NKG2D CAR redirected T-cell specificity to tumors expressing a variety of cell surface NKG2D ligands, such as MICA and MICB. This laboratory has extended the repertoire of CARs based on conversion of NK activating receptors by creating a functionally active chNKp30 CAR for redirected targeting of B7-H6 expressing target cells (107).
In addition to targeting ligands using antibody and natural ligand-receptor pairs, affinity peptides culled from combinatorial libraries and de novo designed affinity proteins can serve as the targeting domains for CARs. Our group tested the hypothesis that a small 12-mer unconstrained linear peptide specific for αVβ6 integrin, a wound and carcinoma associated integrin, could serve as a CAR tumor recognition domain (108). Human T cells transduced to express such a peptide redirected CAR specifically lysed αVβ6+ human ovarian cancer cells (109). This ‘minimal’ targeting domain design may be amenable to the creation of strings of peptides arrayed in series to provide multivalent ligand binding and/or multiplexed to simultaneously engage multiple target epitopes on the tumor cell. In contrast to peptides derived from unconstrained combinatorial libraries, platforms that utilize diverse structured synthetic repertoires of affinity proteins in the format of combinatorial libraries have been described. One such platform uses designed anykrin repeat proteins (DARPins) that are amenable to extensive tuning of affinity, on and off rates, and multiplexed specificities (110). Prototype DARPins have been designed to target Her2 and EGFR, and could conceivably be designed to bind other tumor associated molecules. CARs have yet not been constructed using a DARPin as the ligand-binding domain, but it seems likely they would signal similar to CARs derived using an scFv, zetakine, or peptide as the ligand binding domain. Lastly, synthetic affinity proteins designed de novo through computational methodologies have been described and represents an opportunity to take a directed approach to create CARs specific for predefined tumor cell surface target structures with highly defined properties (111, 112). A potential caveat to the clinical use of these highly orthogonal binding proteins as CARs expressed in T cells is the potential for enhanced immunogenicity. Nevertheless, these examples illustrate the inherent flexibility in the design of synthetic CARs that can function in T cells and provide optimism that synthetic biology can overcome the problem of identifying targets on solid tumors. Novel approaches for measuring biophysical parameters of synthetic receptors, interrogating the quality of T-cell signaling through these receptors, and screening for on or off-target toxicities to normal cells are clearly necessary to facilitate clinical translation of ACT using this next generation of CAR designs.
Multiplexing CAR antigen recognition
Tumors are genetically unstable and by the time they become clinically evident, they are usually comprised of a very large number of clonally diverse populations of cells. Thus, therapies targeting a single pathway or molecule often fail to completely eradicate all tumor cells due to the selection and outgrowth of resistant variants. ACT with CAR T cells is currently being pursued in the clinic as a monospecific targeted therapeutic. This approach is essential in the first clinical trials to verify the safety of administering genetically modified T cells and of targeting the selected antigen/epitope. However, it must be anticipated that tumor cell variants that lack the target ligand or have altered expression of other molecules required for effective T-cell recognition and induction of tumor cell death are likely to emerge as a cause of treatment failure in some patients.
The risk of that a subset of tumor cells will escape ACT with CAR T cells is expected to diminish in a factorial manner if two or more antigens expressed on individual tumor cells are targeted (113). Moreover, providing multispecificity of therapeutic CAR T cells could allow for the simultaneous targeting of tumor stroma and/or angiogenic vasculature in a coordinated manner with the destruction of tumor cells, potentially removing environmental factors that are essential for tumor cell survival (114, 115). The application of ACT with CAR T cells that provide more than one target specificity can be approached by at least three distinct strategies: (i) preparing ACT products from T-cell lines that are separately transduced to express different CARs that recognize more than one molecule (‘mixing’); (ii) designing a single vector that expresses two or more CAR constructs in a tandem format using ribosomal skip elements (‘combining’); and (iii) construction and expression of a single CAR construct that houses more than one antigen binding domain (‘multiplexing’).
A multiplexed CAR design has potential practical advantages over mixing and combining approaches. Multiplexing would avoid the costs required to produce multiple GMP grade vectors and prepare multiple independently transduced T-cell lines necessary for the mixing approach, and could overcome limitations in vector capacity and propensity for vector recombination because of redundant sequences if retrovirus or lentiviruses were used in the combining approach. Recently, Ahmed et al. (116) described the design of a dual specific CAR designated a ‘TanCAR’ by arraying two scFvs in tandem separated by a flexible linker between each of the scFvs. The construct was a proof of concept dual specific CAR that consisted of a HER2-specific scFv and a CD19-specific scFv that has no practical direct clinical application; however, this work clearly demonstrated that dual targeting can be accomplished in a single CAR design that is permissive for the steric conformations required for binding to either of the scFv target antigens. Our group has constructed a dual specific CD19/CD20 TanCAR wherein the inter scFv linker was designed following the principles used for the composition of bi-specific antibody reagents to minimize cross pairing of the VL and VH sequences of the two scFvs (117). We achieved bi-specific targeting of CD19 and CD20 with our TanCAR. However, we had previously observed that targeting CD19 and CD20 was optimal with different extracellular spacer domain lengths, and we were not successful in providing optimal signaling outputs using the FMC63 anti-CD19 and the Leu16 anti-CD20 scFvs in a single construct. This obstacle is predicted by the studies demonstrating that the extracellular spacer domain length will differ depending on the target epitope and needs to be considered in the engineering and design of TanCAR constructs going forward.
Alternative approaches for achieving multiple target specificities have been proposed and prototype systems devised wherein the targeting moiety is a molecularly independent soluble molecule tagged with an epitope that is recognized by a ‘universal’ CAR. This strategy therefore allows for multiplexed targeting based on the complexity of a mixture of soluble tagged binding partners. This approach has been described by Tamada et al (118), who developed a CAR housing a fluorescein isothiocyanate (FITC) specific scFv which conferred recognition of tumor cells with bound FITC conjugated monoclonal antibodies. Urbanska et al (119) devised an alternative strategy whereby an avidin N-terminal domain of the universal CAR allows for binding to soluble biotinylated targeting molecules such as biotinylated antibodies. The in vivo utility of these systems await validation in relevant animal models, and their clinical utility would be limited both by the necessity to co-deliver both T cells and antibody reagents, and potentially by immunogenicity. Thus, this approach may share both the limitations of antibody/small molecule based therapy and ACT, since the T cell must co-localize both temporally and physically with both the tumor cell as well as the tumor bound targeting intermediate.
Conditional CAR function based on split receptor systems
Engineering a conditional functional output in response to two simultaneous input parameters is termed an ‘AND’ logic gate. Creating AND logic gates for conditional CAR T-cell antitumor activity would be a significant asset, particularly when a truly tumor specific target epitope is not available (Fig. 6A). The AND logic gated CAR can therefore create tumor specificity when the co expression of two (or more) antigens is unique to tumor cells. The challenge of creating CARs that operate as AND logic gates is to tune each component such that one target antigen is not sufficient to elicit a full activation output. Conceptually, this might be achieved by co-expressing two CARs with attenuated signaling outputs, either based on detuning of the scFv affinity, by employing a CAR extracellular spacer that is sized for suboptimal synapse formation, or by crippling signaling through CD3ζ by mutating one or more of the immune receptor tyrosine activation motifs (ITAMs), such that only when both of the CARs are engaged by ligand is the signaling threshold necessary for T-cell activation achieved (120). Kloss et al (121) achieved a conditional AND gated CAR system using a detuned CD3ζ based on screening scFvs, which when incorporated in a first generation CD3ζ only construct gave signaling outputs that were below the activation threshold for triggering T cell effector functions. By then co-expressing this detuned CAR with a chimeric costimulatory receptor (CCR) housing both CD28 and 4-1BB endodomains and activated via an scFv to a second tumor targeting epitope, a signaling threshold was achieved that resulted in T-cell activation and anti-tumor efficacy in a murine model system (121). Our group has repurposed PD-1, the receptor for PD-L1 that is frequently expressed on tumor cells and a mediator of T-cell exhaustion/anergy, to derive a chimeric costimulatory receptor that consists of the PD-1 extracellular domain linked to the cytoplasmic domain of CD28. Co-expressing such a PD-1 chimeric costimulatory receptor that can engage PD-L1 on tumor cells or tumor infiltrating myeloid cells with a detuned first generation scFv CAR for a tumor associated antigen can provide a generic strategy for designing AND gate split receptor CARs.
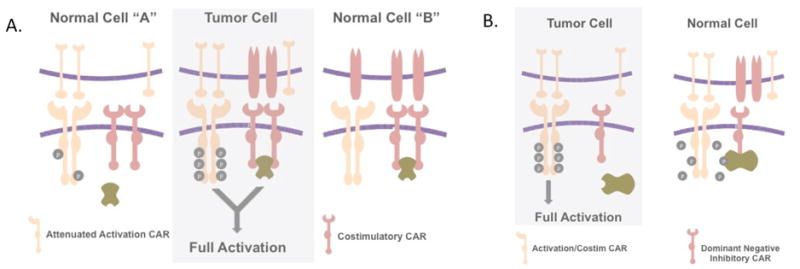
Fig. 6. Split receptor systems for conditional tumor recognition.
(A) Conceptual schematic of a split receptor that functions as an ‘AND’ logic gate. Tumor specificity is achieved when two cell surface antigens are uniquely co-expressed on tumor cells. The split receptor AND logic system targets the combination of the two antigens utilizing both an attenuated activation receptor CAR housing a CD3-ζ domain and a second receptor housing costimulatory intracellular signaling domains. Only the combination of the two receptors engaging antigen results in the activation of the T cell. (B) The concept of a system that is permissive of T-cell activation when antigen is present on tumor cells and a second antigen is absent, wherein the second antigen is present on normal cells. In this split receptor system the chimeric receptor for the inhibiting second receptor acts as in a dominant negative manner to prevent T-cell activation when both antigens are encountered on the same cell.
The principle of an AND gated CAR system has been established, and it is interesting to speculate whether logic gated split CAR systems can similarly be engineered to activate T cells based on the absence of a target antigen on tumor cells that is otherwise ubiquitously expressed on normal tissues. In such a gated system, activation through one CAR would be conditional on the absence of a dominant negative inactivation signal delivered by a second co-expressed CAR (Fig. 6B). Such a theoretical system could provide greater selectivity to tumor targeting if the activating CAR recognizes a ligand that is also expressed at some level on non-malignant cells. In this scenario, signaling after recognition of normal cells could be silenced by engagement of an inhibitory CAR by a second ligand that is expressed only on normal and not on tumor cells. This system could allow for effective targeting of a much more promiscuous set of targets for activating CARs. The challenge for such a dnCAR construct is to identify a cytoplasmic domain for the inhibitory CAR that is capable of suppressing the activating CAR’s endodomain upon co-localization. An appealing candidate for a NOT gated inhibitory CAR endodomain is that of the CD45 phosphatase (122).
CAR T-cell selection systems for clinical applications
The potency of ACT with CAR T cells is likely to be proportional to the frequency of transgene expressing cells in the infused product, their level of CAR protein expression, and the ability of the cells to proliferate and persist in vivo. Thus, the ability to enrich transduced populations of T cells to near homogeneity of CAR expressing T cells, and the calibration of CAR expression levels to ensure a sufficient threshold for and magnitude of signaling to engage effector functions, induce proliferation, and ensure T-cell viability are important variables that impact on the potency of CAR-modified T-cell products. Controlling the uniformity of transgene expression in products by in vitro selection can be approached by biochemical or physical means. Older technologies that imposed drug selection using xenogeneic enzymes such as neomycin, hygromycin. Or puromycin phosphotransferases and their corresponding mammalian antibiotic selection drugs for enriching transduced T cells have been largely abandoned because these methods required a relatively prolonged in vitro exposure time to select transgene positive cells from transgene negative cells and often resulted in poor outgrowth of T cells due to the low expression levels of the selectable marker. Moreover, these enzymes were not of human origin and clinical trials that have employed these systems to select for transgene expressing T cells that were used in ACT including early clinical trials of CAR-modified T cells, demonstrated the often rapid induction of immune responses to the epitopes derived from the selectable marker protein that resulted in rejection of the transferred T cells, even in immunologically compromised patient populations (123, 124).
Our group is evaluating alternate enzyme systems that confer resistance to drugs that are cytotoxic to activated proliferating human T cells but are less likely to be immunogenic. The desired attributes of these second generation cell selection systems include the use of minimally altered human enzyme proteins, selection reagents that are commercially available as pharmaceutical grade drugs for use in cGMP compliant platforms; rapid selection (<5 days), and the requirement for high level gene expression to achieve drug resistance, such that molecularly linked CAR constructs are also co-expressed at high levels. We have demonstrated that at least two enzyme-drug combinations fulfill these criteria. These include the dihydrofolate reductase (DHFR) that confers resistance to methotrexate (MTX) and inosine monophosphate dehydrogenase (IMPDH2) that confers resistance to mycophenolate mofetil (MMF). In vitro, MTX and mycophenolic acid (MPA), the active metabolite of MMF, are cytocidal to activated, proliferating human T cells. However, the expression of mutants of human DHFR and IMPDH2, each altered by two amino acid substitutions, can render transduced T cells resistant to these drugs (125, 126). Experiments wherein the expression of one of these selection enzymes is linked to CAR expression using a T2A ribosome skip element have confirmed that drug resistance enables selection of T cells that have enforced high-level CAR expression and mediate potent CAR redirected T-cell effector functions. Moreover, the concentrations of MTX and MPA required for selection of transduced T cells in vitro is within the range of serum peak and trough levels that are routinely tolerated in patients that receive these drugs for other indications. Thus, in vitro selected CAR-modified T cells that express DHFR or IMPDH2 transgenes may be capable of engrafting and functioning in patients receiving immunosuppressive regimens that include MTX and MMF, such as recipients of allogeneic hematopoietic stem cell transplant. Indeed, we have recently demonstrated that MTX and MMF resistance can be exploited to select for gene modified T cells in vivo rather than in vitro (126). This raises the possibility that in the future CAR-modified T cells could be rapidly derived ex vivo and then immediately reinfused followed by in vivo selection and expansion in the host by drug administration, rather than in a GMP facility. There is a growing list of alternative human muteins that confer T-cell resistance to the calcineurin inhibitors cyclosporine and FK-506, and the mTOR inhibitor rapamycin (127, 128), and our group has also rendered human T cell resistant to glucocorticoids through zinc finger nuclease genome editing of the glucocorticoid receptor. Each of these strategies acting alone or in combination can modulate the resistance of CAR-modified T cells and could find applications in selected clinical settings.
In vivo regulation of CAR T-cell function
Although strategies for regulating the survival of transferred T cells have advanced as discussed later in this review, their utility is largely confined to addressing potential late effects mediated by recognition by genetically modified T cells such as B-cell aplasia induced by ACT with CD19-modified CAR T cells. The activation of effective suicide gene ablation for mitigating early toxicities, such as the cytokine storm syndrome that is observed after CD19 CAR T-cell therapy, would be predicted to negate subsequent therapeutic benefit. Thus, it would be ideal to provide CAR-modified T cells with near real-time responsive regulatory elements that are controlled by clinician inputs, such as ‘ON’ or ‘OFF’ drug regulated transgene transcriptional systems, or context specific transcriptional regulation as exemplified by NFAT minimal promoter as a sensor of T-cell activation. This is an engineering frontier in synthetic biology with potentially significant benefits to ACT for cancer with genetically modified T cells that are redirected with a CAR or TCR. Unfortunately, as yet there is no ‘off-the shelf’ transgene expression regulation system that is compatible for use in humans. Drug-inducible transgene expression control platforms are used extensively in research laboratories but typically utilize xenogeneic chimeric transcriptional activators, require multiple vectors for implementation, and are responsive to a relatively limited set of drug inputs, each of which having features that hamper clinical use (129). Second generation transcriptional systems that utilize human component chimeric transcription factors, such as the mTOR-based chimeric transcription factor/promoter system that is activated by rapamycin and rapamycin derivatives with attenuated immunosuppressive activity may provide opportunities to regulate CAR expression in vivo, but have not yet been evaluated for this application (130).
Unlike transcriptional control systems that are cumbersome to incorporate into currently available vector systems, riboswitches are synthetic regulatory modular mRNA elements that can act in cis or trans, and affect either transgene or endogenous gene expression as ON or OFF switches (131-134). A prototype cis-acting ON riboswitch system for regulating transgene expression in primary T cells was described by Jensen and Smolke (134) and has potential applications for regulating CAR expression. An alternative approach in which protein stability is regulated has been proposed as a method to modulate transgene expression levels. This strategy is exemplified by the Shield-1 system in which engineered protein degradation domains are incorporated into transgene encoded proteins (135). The degradation domains are designed to be regulated by a small molecule input to achieve controlled levels of transgene-encoded proteins. The major challenges that remain before applying these novel technologies to regulating CAR expression will be to ensure sufficient stringency such that the basal level expression of the CAR is below that necessary to trigger T-cell activation in the presence of ligand, and sufficient ON state up regulation of CAR expression to allow T cells to be fully functional when ligand is engaged. Additionally, the drugs that are used to impose gene regulation with these systems will need to be sufficiently non-toxic over the periods of administration that are necessary to allow CAR-modified T cells to promote complete tumor eradication and exhibit pharmacodynamic and biodistribution properties that enable regulation of CAR expression in T cells that must migrate to diverse anatomic locations.
Context-specific regulation of transgene expression has begun to be advanced in the field of ACT. Activation dependent minimal promoter systems have been applied to regulate transgene expression in T cells. One example is the use of synthetic promoters that are linked to TCR and/or CAR activation status through NFAT minimal promoter (mp) constructs. This approach has been used to restrict transgenic IL-12 secretion by adoptively transferred melanoma TIL to the tumor microenvironment or associated tumor draining lymph nodes as a strategy to circumvent the toxicity of systemic IL-12 exposure (136). The NFATmp regulation of IL-12 synthesis was also described where the activation signal is generated by a third generation CAR specific for VEGFR (137). Adapting these systems to CAR-modified T cells for additional regulated output of engineered effector function, transgenes that modulate survival and proliferation, and/or modified responsiveness to the immunosuppressive microenvironment, represent a significant area of future engineering of CAR T cells.
Tracking and terminating engraftment of CAR T cells
An important feature to engineer into CAR-modified T cells for ACT is a mechanism that allows their ablation in vivo in response to a clinician prescribed drug. Several clinical circumstances can arise following the adoptive transfer of CAR-modified T cells and would warrant their elimination. These may include acute persistent toxicities due to cytokine release by the T cells, or harmful side effects from ‘off’ tumor CAR-mediated recognition of normal tissues. It is unclear how effective the administration of immunosuppressive drugs such as corticosteroids will be to completely ameliorate such toxicities particularly because CARs are often engineered to provide robust costimulation that promotes the expression of anti-apoptotic molecules. Even when CAR-modified T cells successfully eradicate the tumor, such as observed in some patients treated with CD19-specific CARs, later elimination of CAR T cells to allow reconstitution of a functional B cell repertoire from hematopoietic progenitor cells will be an important component to ensure the long term safety of this promising therapeutic modality.
Most viral vector and non-viral gene transfer vectors can accommodate both the coding sequence of a CAR and that of a suicide gene. Prodrug enzyme systems that trigger cell death, such as HSV-thymidine kinase (HSV-TK) that confers sensitivity of dividing T cells to ganciclovir. The use of HSV-TK as a suicide switch for adoptively transferred donor T cells is effective in attenuating GVHD in the setting of allogeneic hematopoietic stem cell transplant regimens that are highly immunosuppressive (138). However, the immunogenicity of HSV-TK is a liability in immunocompetent hosts. Recently, a dimerizable caspase suicide construct base on human proteins that confers sensitivity to a synthetic dimerizer drug (AP1903) has been introduced into donor T cells that were administered to recipients of haploidentical stem cell transplant. The administration of AP1903 was highly effective in rapidly and completely reversing clinical manifestations of GVHD that occurred after T-cell administration (139). Clearly, this suicide platform has general applicability for integration into CAR T-cell technologies, although including both CAR and iCASP9 constructs in a lentiviral or retroviral vector pushes the upper limits of payload capacity, especially if a cell tracking transgene is also included. Moreover, the widespread use of this highly active suicide system hinges on the availability of the dimerizer agent AP1903, which is not yet an FDA approved or commercially available drug. Thus, while the proof of concept function of the system is robust, applicability constraints on the basis of these collateral issues persist.
Our group has developed an alternate safety switch system based on the engineering of the human epidermal growth factor receptor (EGFR) to a small truncated derivative that is devoid of the N-terminal ligand binding/dimerization domains I and II, and the cytoplasmic signaling tail. This truncated EGFR, designated EGFRt, contains the transmembrane and extracellular domains III and IV and can be efficiently expressed on the cell surface. EGFRt is biologically inert when expressed by human T cells and can function as a cell marker for immunomagnetic purification of transduced T cells in vitro, a tracking tag for analysis of cell persistence and tissue localization by flow and immunohistochemistry, and a target for cell ablation by antibody dependent cell mediated cytotoxicity (ADCC) when bound by cetuximab (52, 140). The EGFRt transgene is expressed as a T2A linked polypeptide linked to our CD19 CAR in a third generation lentiviral vector that is currently in use in our adoptive therapy trials in Seattle in patients with CD19 expressing B cell malignancies. These trials either prescribe or allow cetuximab administration for either acute life threatening toxicities or for the ablation of CAR T cells in patients who have prolonged remissions, persisting CAR T cells, and B-cell aplasia with opportunistic infections.
Summary
The design and implementation of ACT with T cells modified with CARs represents an example of the translation of synthetic biology to the treatment of human malignancy. The future of this approach is extremely promising but achieving its potential is likely to require integration of several disciplines in biology and clinical medicine, including defining the optimal cell composition of therapeutic products, integrating structural biology and immune cell signaling, utilizing emerging approaches in regulating gene expression and cell behavior, and combining ACT with novel checkpoint inhibitors, cytokines, and tumor modulating agents. The merging of these disciplines will ensure that ACT enters the mainstream of cancer therapeutics.
Acknowledgments
The authors thank past and present members of our laboratories and the support of NIH grants CA136551; CA114536, AI053193, and P50CA132393.
Footnotes
The authors have no conflicts of interest to declare.
References
- 1.Morgan RA, Dudley ME, Wunderlich JR, Hughes MS. Cancer Regression in Patients After Transfer of Genetically Engineered Lymphocytes. Science. 2006;314(6):126–129. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robbins PF, Morgan RA, Feldman SA, Yang JC, Sherry RM, Dudley ME, Wunderlich JR, et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J Clin Oncol. 2011;29(7):917–924. doi: 10.1200/JCO.2010.32.2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A, June CH. T Cells with Chimeric Antigen Receptors Have Potent Antitumor Effects and Can Establish Memory in Patients with Advanced Leukemia. Science Translational Medicine. 2011;3(95):1–11. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kochenderfer JN, Dudley ME, Feldman SA, Wilson WH, Spaner DE, Maric I, Stetler-Stevenson M, et al. B-cell depletion and remissions of malignancy along with cytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor–transduced T cells. Blood. 2012;119(12):2709–2720. doi: 10.1182/blood-2011-10-384388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N Engl J Med. 2011;365(8):725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brentjens RJ, Riviere I, Park JH, Davila ML, Wang X, Stefanski J, Taylor C, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118(18):4817–4828. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brentjens RJR, Davila MLM, Riviere II, Park JJ, Wang XX, Cowell LGL, Bartido SS, et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Science Translational Medicine. 2013;5(177):1–9. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, Teachey DT, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–1518. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36(2):133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol Ther. 2010;18(4):843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan D-AN, Feldman SA, Davis JL, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011;19(3):620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brentjens R, Yeh R, Bernal Y, Riviere I, Sadelain M. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010;18(4):666–668. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, et al. Identification of a Titin-Derived HLA-A1-Presented Peptide as a Cross-Reactive Target for Engineered MAGE A3-Directed T Cells. Science Translational Medicine. 2013;5(197):1–11. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Riddell, Watanabe K, Goodrich J, Li C, Agha M, Greenberg P. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. 1992;257(5067):238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 15.Heslop HE, Ng CY, Li C, Smith CA, Loftin SK, Krance RA, Brenner MK, Rooney CM. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med. 1996;2(5):551–555. doi: 10.1038/nm0596-551. [DOI] [PubMed] [Google Scholar]
- 16.Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. Journal of Immunotherapy. 2003;26(4):332–342. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yee C, Thompson JA, Roche P, Byrd DR, Lee PP, Piepkorn M, Kenyon K, Davis MM, Riddell SR, Greenberg PD. Melanocyte Destruction after Antigen-Specific Immunotherapy of Melanoma: Direct Evidence of T Cell-Mediated Vitiligo. Journal of Experimental Medicine. 2000;192(11):1637–1644. doi: 10.1084/jem.192.11.1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warren EH, Fujii N, Akatsuka Y, Chaney CN, Mito JK, Loeb KR, Gooley TA, et al. Therapy of relapsed leukemia after allogeneic hematopoietic cell transplantation with T cells specific for minor histocompatibility antigens. Blood. 2010;115(19):3869–3878. doi: 10.1182/blood-2009-10-248997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, Duerkopp N, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Science Translational Medicine. 2013;5(174):1–12. doi: 10.1126/scitranslmed.3004916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Walter EA, Greenberg PD, Gilbert MJ, Finch RJ, Watanabe KS, Thomas ED, Riddell SR. Reconstitution of Cellular Immunity against Cytomegalovirus in Recipients of Allogeneic Bone Marrow by Transfer of T-Cell Clones from the Donor. N Engl J Med. 1995;333(16):1038–1044. doi: 10.1056/NEJM199510193331603. [DOI] [PubMed] [Google Scholar]
- 21.Melenhorst JJ, Leen AM, Bollard CM, Quigley MF, Price DA, Rooney CM, Brenner MK, Barrett AJ, Heslop HE. Allogeneic virus-specific T cells with HLA alloreactivity do not produce GVHD in human subjects. Blood. 2010;116(22):4700–4702. doi: 10.1182/blood-2010-06-289991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Restifo NP, Dudley ME, Rosenberg SA. Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol. 2012;12(4):269–281. doi: 10.1038/nri3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007;117(6):1466–1476. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Friedman KM, Devillier LE, Feldman SA, Rosenberg SA, Dudley ME. Augmented lymphocyte expansion from solid tumors with engineered cells for costimulatory enhancement. J Immunother. 2011;34(9):651–661. doi: 10.1097/CJI.0b013e31823284c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319(25):1676–1680. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 26.Rosenberg SA, Aebersold P, Cornetta K, Kasid A, Morgan RA, Moen R, Karson EM, Lotze MT, Yang JC, Topalian SL. Gene transfer into humans--immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990;323(9):570–578. doi: 10.1056/NEJM199008303230904. [DOI] [PubMed] [Google Scholar]
- 27.Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, Greenberg PD. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred T cells. Proceedings of the National Academy of Sciences. 2002;99(25):16168–16173. doi: 10.1073/pnas.242600099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, et al. Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science. 2002;298(5594):850–854. doi: 10.1126/science.1076514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17(13):4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou J, Dudley ME, Rosenberg SA, Robbins PF. Selective growth, in vitro and in vivo, of individual T cell clones from tumor-infiltrating lymphocytes obtained from patients with melanoma. J Immunol. 2004;173(12):7622–7629. doi: 10.4049/jimmunol.173.12.7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shen X, Zhou J, Hathcock KS, Robbins P, Powell DJ, Jr, Rosenberg SA, Hodes RJ. Persistence of Tumor Infiltrating Lymphocytes in Adoptive Immunotherapy Correlates With Telomere Length. Journal of Immunotherapy. 2007;30(1):123–129. doi: 10.1097/01.cji.0000211321.07654.b8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Powell DJ, Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105(1):241–250. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 34.Turtle CJ, Swanson HM, Fujii N, Estey EH, Riddell SR. A Distinct Subset of Self-Renewing Human Memory CD8+ T Cells Survives Cytotoxic Chemotherapy. Immunity. 2009;31(5):834–844. doi: 10.1016/j.immuni.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Le Bourhis L, Martin E, Péguillet I, Guihot A, Froux N, Coré M, Lévy E, et al. Antimicrobial activity of mucosal-associated invariant T cells. Nat Immunol. 2010;11(8):701–708. doi: 10.1038/ni.1890. [DOI] [PubMed] [Google Scholar]
- 36.Turtle CJ, Delrow J, Joslyn RC, Swanson HM, Basom R, Tabellini L, Delaney C, Heimfeld S, Hansen JA, Riddell SR. Innate signals overcome acquired TCR signaling pathway regulation and govern the fate of human CD161(hi) CD8α+ semi-invariant T cells. Blood. 2011;118(10):2752–2762. doi: 10.1182/blood-2011-02-334698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walker LJ, Kang Y-H, Smith MO, Tharmalingham H, Ramamurthy N, Fleming VM, Sahgal N, et al. Human MAIT and CD8αα cells develop from a pool of type-17 precommitted CD8+ T cells. Blood. 2012;119(2):422–433. doi: 10.1182/blood-2011-05-353789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, Bhati M, et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature. 2012;491(7426):717–723. doi: 10.1038/nature11605. [DOI] [PubMed] [Google Scholar]
- 39.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gerlach C, Rohr JC, Perié L, van Rooij N, van Heijst JWJ, Velds A, Urbanus J, et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science. 2013;340(6132):635–639. doi: 10.1126/science.1235487. [DOI] [PubMed] [Google Scholar]
- 41.Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Gräf P, Verschoor A, Schiemann M, Höfer T, Busch DH. Disparate individual fates compose robust CD8+ T cell immunity. Science. 2013;340(6132):630–635. doi: 10.1126/science.1235454. [DOI] [PubMed] [Google Scholar]
- 42.Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJC, Bickham KL, et al. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity. 2013;38(1):187–197. doi: 10.1016/j.immuni.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mackall CL, Fleisher TA, Brown MR, Andrich MP, Chen CC, Feuerstein IM, Magrath IT, Wexler LH, Dimitrov DS, Gress RE. Distinctions Between CD8+ and CD4+ T-Cell Regenerative Pathways Result in Prolonged T-Cell Subset Imbalance After Intensive Chemotherapy. Blood. 1997;89(10):3700–3707. [PubMed] [Google Scholar]
- 44.Hakim FT, Cepeda R, Kaimei S, Mackall CL, McAtee N, Zujewski J, Cowan K, Gress RE. Constraints on CD4 Recovery Postchemotherapy in Adults: Thymic Insufficiency and Apoptotic Decline of Expanded Peripheral CD4 Cells. Blood. 1997;90(9):3789–3798. [PubMed] [Google Scholar]
- 45.Berger C, Berger M, Anderson D, Riddell SR. A non-human primate model for analysis of safety, persistence, and function of adoptively transferred T cells. J Med Primatol. 2011;40(2):88–103. doi: 10.1111/j.1600-0684.2010.00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118(1):294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gattinoni L, Zhong X-S, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, et al. Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med. 2009;15(7):808–813. doi: 10.1038/nm.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klebanoff CA, Gattinoni L, Palmer DC, Muranski P, Ji Y, Hinrichs CS, Borman ZA, et al. Determinants of successful CD8+ T-cell adoptive immunotherapy for large established tumors in mice. Clin Cancer Res. 2011;17(16):5343–5352. doi: 10.1158/1078-0432.CCR-11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hinrichs CS, Borman ZA, Cassard L, Gattinoni L, Spolski R, Yu Z, Sanchez-Perez L, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proceedings of the National Academy of Sciences. 2009;106(41):17469–17474. doi: 10.1073/pnas.0907448106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Berger C, Wong CW, Forman SJ, Riddell SR, Jensen MC. Engraftment of human central memory-derived effector CD8+ T cells in immunodeficient mice. Blood. 2011;117(6):1888–1898. doi: 10.1182/blood-2010-10-310599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood. 2012;119(1):72–82. doi: 10.1182/blood-2011-07-366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Naranjo A, Brown CE, Bautista C, Wong CW, Chang W-C, Aguilar B, et al. Phenotypic and functional attributes of lentivirus-modified CD19-specific human CD8+ central memory T cells manufactured at clinical scale. J Immunother. 2012;35(9):689–701. doi: 10.1097/CJI.0b013e318270dec7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stemberger C, Dreher S, Tschulik C, Piossek C, Bet J, Yamamoto TN, Schiemann M, et al. Novel serial positive enrichment technology enables clinical multiparameter cell sorting. PLoS ONE. 2012;7(4):1–11. doi: 10.1371/journal.pone.0035798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klein U, Tu Y, Stolovitzky GA, Mattioli M, Cattoretti G, Husson H, Freedman A, et al. Gene expression profiling of B cell chronic lymphocytic leukemia reveals a homogeneous phenotype related to memory B cells. J Exp Med. 2001;194(11):1625–1638. doi: 10.1084/jem.194.11.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rosenwald A, Alizadeh AA, Widhopf G, Simon R, Davis RE, Yu X, Yang L, et al. Relation of gene expression phenotype to immunoglobulin mutation genotype in B cell chronic lymphocytic leukemia. J Exp Med. 2001;194(11):1639–1647. doi: 10.1084/jem.194.11.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamaguchi T, Yanagisawa K, Sugiyama R, Hosono Y, Shimada Y, Arima C, Kato S, et al. NKX2-1/TITF1/TTF-1-Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell. 2012;21(3):348–361. doi: 10.1016/j.ccr.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 57.Zhang S, Chen L, Cui B, Chuang H-Y, Yu J, Wang-Rodriguez J, Tang L, Chen G, Basak GW, Kipps TJ. ROR1 is expressed in human breast cancer and associated with enhanced tumor-cell growth. PLoS ONE. 2012;7(3):e31127. doi: 10.1371/journal.pone.0031127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bicocca VT, Chang BH, Masouleh BK, Muschen M, Loriaux MM, Druker BJ, Tyner JW. Crosstalk between ROR1 and the Pre-B cell receptor promotes survival of t(1;19) acute lymphoblastic leukemia. Cancer Cell. 2012;22(5):656–667. doi: 10.1016/j.ccr.2012.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cui B, Zhang S, Chen L, Yu J, Widhopf GF, Fecteau J-F, Rassenti LZ, Kipps TJ. Targeting ROR1 inhibits epithelial-mesenchymal transition and metastasis. Cancer Res. 2013;73(12):3649–3660. doi: 10.1158/0008-5472.CAN-12-3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eshhar Z. Specific Activation and Targeting of Cytotoxic Lymphocytes Through Chimeric Single Chains Consisting of Antibody-Binding Domains and the or Subunits of the Immunoglobulin and T-Cell Receptors. Proceedings of the National Academy of Sciences. 1993;90(2):720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sadelain M, Riviere I, Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat Rev Cancer. 2003;3(1):35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 62.Cooper LJN, Topp MS, Serrano LM, Gonzalez S, Chang W-C, Naranjo A, Wright C, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101(4):1637–1644. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 63.Imai C, Mihara K, Andreansky M, Nicholson IC, Pui C-H, Geiger TL, Campana D. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18(4):676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 64.Jensen M. CD20 is a molecular target for scFvFc:zeta receptor redirected T cells: Implications for cellular immunotherapy of CD20+ malignancy. Biology of Blood and Marrow Transplantation. 1998;4(2):75–83. doi: 10.1053/bbmt.1998.v4.pm9763110. [DOI] [PubMed] [Google Scholar]
- 65.Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN, Bleakley M, et al. The B-cell tumor–associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010;116(22):4532–4541. doi: 10.1182/blood-2010-05-283309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stancovski I, Schindler DG, Waks T, Yarden Y, Sela M, Eshhar Z. Targeting of T lymphocytes to Neu/HER2-expressing cells using chimeric single chain Fv receptors. J Immunol. 1993;151(11):6577–6582. [PubMed] [Google Scholar]
- 67.Lamers CHJ, Willemsen R, van Elzakker P, van Steenbergen-Langeveld S, Broertjes M, Oosterwijk-Wakka J, Oosterwijk E, Sleijfer S, Debets R, Gratama JW. Immune responses to transgene and retroviral vector in patients treated with ex vivo-engineered T cells. Blood. 2011;117(1):72–82. doi: 10.1182/blood-2010-07-294520. [DOI] [PubMed] [Google Scholar]
- 68.Parker LL, Do MT, Westwood JA. Expansion and characterization of T cells transduced with a chimeric receptor against ovarian cancer. Human Gene Ther. 2000;11:2377–2387. doi: 10.1089/104303400750038480. [DOI] [PubMed] [Google Scholar]
- 69.Westwood JA. Adoptive transfer of T cells modified with a humanized chimeric receptor gene inhibits growth of Lewis-Y-expressing tumors in mice. Proceedings of the National Academy of Sciences. 2005;102(52):19051–19056. doi: 10.1073/pnas.0504312102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kakarla S, Chow KK, Mata M, Shaffer DR, Song X-T, Wu M-F, Liu H, et al. Antitumor Effects of Chimeric Receptor Engineered Human T Cells Directed to Tumor Stroma. Mol Ther. 2013;21(8):1611–1620. doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lamers CH, Sleijfer S, van Steenbergen S, van Elzakker P, van Krimpen B, Groot C, Vulto A, et al. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol Ther. 2013;21(4):904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee C-CR, Restifo NP, Rosenberg SA. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. Journal of Experimental Medicine. 2013;210(6):1125–1135. doi: 10.1084/jem.20130110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Du X, Beers R, FitzGerald DJ, Pastan I. Differential cellular internalization of anti-CD19 and -CD22 immunotoxins results in different cytotoxic activity. Cancer Res. 2008;68(15):6300–6305. doi: 10.1158/0008-5472.CAN-08-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Baskar S, Kwong KY, Hofer T, Levy JM, Kennedy MG, Lee E, Staudt LM, Wilson WH, Wiestner A, Rader C. Unique Cell Surface Expression of Receptor Tyrosine Kinase ROR1 in Human B-Cell Chronic Lymphocytic Leukemia. Clinical Cancer Research. 2008;14(2):396–404. doi: 10.1158/1078-0432.CCR-07-1823. [DOI] [PubMed] [Google Scholar]
- 75.Hunt DF, Michel H, Dickinson TA, Shabanowitz J, Cox AL, Sakaguchi K, Appella E, Grey HM, Sette A. Peptides presented to the immune system by the murine class II major histocompatibility complex molecule I-Ad. Science. 1992;256(5065):1817–1820. doi: 10.1126/science.1319610. [DOI] [PubMed] [Google Scholar]
- 76.Borovsky Z, Mishan-Eisenberg G, Yaniv E, Rachmilewitz J. Serial triggering of T cell receptors results in incremental accumulation of signaling intermediates. J Biol Chem. 2002;277(24):21529–21536. doi: 10.1074/jbc.M201613200. [DOI] [PubMed] [Google Scholar]
- 77.Huppa JB, Axmann M, Mörtelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schütz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463(7283):963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Engels B, Chervin AS, Sant AJ, Kranz DM, Schreiber H. Long-term Persistence of CD4+ but Rapid Disappearance of CD8+ T Cells Expressing an MHC Class I-restricted TCR of Nanomolar Affinity. Mol Ther. 2012;20(3):652–660. doi: 10.1038/mt.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN, Bleakley M, et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010;116(22):4532–4541. doi: 10.1182/blood-2010-05-283309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dave H, Anver MR, Butcher DO, Brown P, Khan J, Wayne AS, Baskar S, Rader C. Restricted Cell Surface Expression of Receptor Tyrosine Kinase ROR1 in Pediatric B-Lineage Acute Lymphoblastic Leukemia Suggests Targetability with Therapeutic Monoclonal Antibodies. PLoS ONE. 2012;7(12):1–10. doi: 10.1371/journal.pone.0052655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Baskar S, Wiestner A, Wilson WH, Pastan I, Rader C. Targeting malignant B cells with an immunotoxin against ROR1. MAbs. 2012;4(3):349–361. doi: 10.4161/mabs.19870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hudecek M, Lupo-Stanghellini MT, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, Riddell SR. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res. 2013;19(12):3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Guest RD, Hawkins RE, Kirillova N, Cheadle EJ, Arnold J, O’Neill A, Irlam J, et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. Journal of Immunotherapy. 2005;28(3):203–211. doi: 10.1097/01.cji.0000161397.96582.59. [DOI] [PubMed] [Google Scholar]
- 84.Park JR, DiGiusto DL, Slovak M, Wright C, Naranjo A, Wagner J, Meechoovet HB, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15(4):825–833. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 85.Till BG, Jensen MC, Wang J, Chen EY, Wood BL, Greisman HA, Qian X, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112(6):2261–2271. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24(13):e20–2. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 87.Jensen MC, Clarke P, Tan G, Wright C, Chung-Chang W, Clark TN, Zhang F, et al. Human T lymphocyte genetic modification with naked DNA. Mol Ther. 2000;1(1):49–55. doi: 10.1006/mthe.1999.0012. [DOI] [PubMed] [Google Scholar]
- 88.Maher J, Brentjens RJ, Gunset G, Riviere I, Sadelain M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ /CD28 receptor. Nat Biotechnol. 2002;20(1):70–75. doi: 10.1038/nbt0102-70. [DOI] [PubMed] [Google Scholar]
- 89.Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N, Smith DD, Forman SJ, Jensen MC, Cooper LJN. CD28 Costimulation Provided through a CD19-Specific Chimeric Antigen Receptor Enhances In vivo Persistence and Antitumor Efficacy of Adoptively Transferred T Cells. Cancer Res. 2006;66(22):10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 90.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, Varela-Rohena A, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proceedings of the National Academy of Sciences. 2009;106(9):3360–3365. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stephan MT, Ponomarev V, Brentjens RJ, Chang AH, Dobrenkov KV, Heller G, Sadelain M. T cell–encoded CD80 and 4-1BBL induce auto- and transcostimulation, resulting in potent tumor rejection. Nat Med. 2007;13(12):1440–1449. doi: 10.1038/nm1676. [DOI] [PubMed] [Google Scholar]
- 92.Wilkie S, Schalkwyk MCI, Hobbs S, Davies DM, Stegen SJC, Pereira ACP, Burbridge SE, Box C, Eccles SA, Maher J. Dual Targeting of ErbB2 and MUC1 in Breast Cancer Using Chimeric Antigen Receptors Engineered to Provide Complementary Signaling. J Clin Immunol. 2012;32(5):1059–1070. doi: 10.1007/s10875-012-9689-9. [DOI] [PubMed] [Google Scholar]
- 93.Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M, Brentjens RJ. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119(18):4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G, Kamble RT, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121(5):1822–1826. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sotelo P, Collazo N, Zuñiga R, Gutiérrez-González M, Catalán D, Ribeiro CH, Aguillón JC, Molina MC. An efficient method for variable region assembly in the construction of scFv phage display libraries using independent strand amplification. MAbs. 2012;4(4):542–550. doi: 10.4161/mabs.20653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Larman HB, Xu GJ, Pavlova NN, Elledge SJ. Construction of a rationally designed antibody platform for sequencing-assisted selection. Proceedings of the National Academy of Sciences. 2012;109(45):18523–18528. doi: 10.1073/pnas.1215549109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Alonso-Camino V, Sánchez-Martín D, Compte M, Nuñez-Prado N, Diaz RM, Vile R, Alvarez-Vallina L. CARbodies: Human Antibodies Against Cell Surface Tumor Antigens Selected From Repertoires Displayed on T Cell Chimeric Antigen Receptors. Mol Ther Nucleic Acids. 2013;2:e93. doi: 10.1038/mtna.2013.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kahlon KS, Brown C, Cooper LJN, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160–9166. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- 99.Madhankumar AB, Mintz A, Debinski W. Interleukin 13 mutants of enhanced avidity toward the glioma-associated receptor, IL13Ralpha2. Neoplasia. 2004;6(1):15–22. doi: 10.1016/s1476-5586(04)80049-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Brown CE, Starr R, Martinez C, Aguilar B, D’Apuzzo M, Todorov I, Shih C-C, et al. Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells. Cancer Res. 2009;69(23):8886–8893. doi: 10.1158/0008-5472.CAN-09-2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D’Apuzzo M, Barish ME, Forman SJ, Jensen MC. Stem-like tumor-initiating cells isolated from IL13Rα2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin Cancer Res. 2012;18(8):2199–2209. doi: 10.1158/1078-0432.CCR-11-1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kong S, Sengupta S, Tyler B, Bais AJ, Ma Q, Doucette S, Zhou J, et al. Suppression of Human Glioma Xenografts with Second-Generation IL13R-Specific Chimeric Antigen Receptor-Modified T Cells. Clin Cancer Res. 2012;18(21):5949–5960. doi: 10.1158/1078-0432.CCR-12-0319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Niederman TMJ, Ghogawala Z, Carter BS, Tompkins HS, Russell MM, Mulligan RC. Antitumor activity of cytotoxic T lymphocytes engineered to target vascular endothelial growth factor receptors. Proc Natl Acad Sci U S A. 2002;99(10):7009–7014. doi: 10.1073/pnas.092562399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shaffer DR, Savoldo B, Yi Z, Chow KKH, Kakarla S, Spencer DM, Dotti G, et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood. 2011;117(16):4304–4314. doi: 10.1182/blood-2010-04-278218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. 2005;106(5):1544–1551. doi: 10.1182/blood-2004-11-4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang T, Barber A, Sentman CL. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res. 2006;66(11):5927–5933. doi: 10.1158/0008-5472.CAN-06-0130. [DOI] [PubMed] [Google Scholar]
- 107.Zhang T, Wu M-R, Sentman CL. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. The Journal of Immunology. 2012;189(5):2290–2299. doi: 10.4049/jimmunol.1103495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kraft S, Diefenbach B, Mehta R, Jonczyk A, Luckenbach GA, Goodman SL. Definition of an unexpected ligand recognition motif for alphav beta6 integrin. J Biol Chem. 1999;274(4):1979–1985. doi: 10.1074/jbc.274.4.1979. [DOI] [PubMed] [Google Scholar]
- 109.Pameijer CRJ, Navanjo A, Meechoovet B, Wagner JR, Aguilar B, Wright CL, Chang WC, Brown CE, Jensen MC. Conversion of a tumor-binding peptide identified by phage display to a functional chimeric T cell antigen receptor. Cancer Gene Ther. 2007;14(1):91–97. doi: 10.1038/sj.cgt.7700993. [DOI] [PubMed] [Google Scholar]
- 110.Tamaskovic R, Simon M, Stefan N, Schwill M, Plückthun A. Designed ankyrin repeat proteins (DARPins) from research to therapy. Meth Enzymol. 2012;503:101–134. doi: 10.1016/B978-0-12-396962-0.00005-7. [DOI] [PubMed] [Google Scholar]
- 111.Whitehead TA, Baker D, Fleishman SJ. Computational design of novel protein binders and experimental affinity maturation. Meth Enzymol. 2013;523:1–19. doi: 10.1016/B978-0-12-394292-0.00001-1. [DOI] [PubMed] [Google Scholar]
- 112.Koga N, Tatsumi-Koga R, Liu G, Xiao R, Acton TB, Montelione GT, Baker D. Principles for designing ideal protein structures. Nature. 2012;491(7423):222–227. doi: 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, Zhang J, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in Glioblastoma. Mol Ther. 2013 doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chinnasamy D, Tran E, Yu Z, Morgan RA, Restifo NP, Rosenberg SA. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 2013;73(11):3371–3380. doi: 10.1158/0008-5472.CAN-12-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Petrausch U, Schuberth PC, Hagedorn C, Soltermann A, Tomaszek S, Stahel R, Weder W, Renner C. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1) BMC Cancer. 2012;12(1):615. doi: 10.1186/1471-2407-12-615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Grada Z, Hegde M, Byrd T, Shaffer DR, Ghazi A, Brawley VS, Corder A, et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids. 2013;2:e105. doi: 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Baeuerle PA, Reinhardt C. Bispecific T-Cell Engaging Antibodies for Cancer Therapy. Cancer Res. 2009;69(12):4941–4944. doi: 10.1158/0008-5472.CAN-09-0547. [DOI] [PubMed] [Google Scholar]
- 118.Tamada K, Geng D, Sakoda Y, Bansal N, Srivastava R, Li Z, Davila E. Redirecting Gene-Modified T Cells toward Various Cancer Types Using Tagged Antibodies. Clin Cancer Res. 2012;18(23):6436–6445. doi: 10.1158/1078-0432.CCR-12-1449. [DOI] [PubMed] [Google Scholar]
- 119.Urbanska K, Lanitis E, Poussin M, Lynn RC, Gavin BP, Kelderman S, Yu J, Scholler N, Powell DJ. A Universal Strategy for Adoptive Immunotherapy of Cancer through Use of a Novel T-cell Antigen Receptor. Cancer Res. 2012;72(7):1844–1852. doi: 10.1158/0008-5472.CAN-11-3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Irving BA, Chan AC, Weiss A. Functional characterization of a signal transducing motif present in the T cell antigen receptor zeta chain. J Exp Med. 1993;177:1093–1103. doi: 10.1084/jem.177.4.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31(1):71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rhee I, Veillette A. Protein tyrosine phosphatases in lymphocyte activation and autoimmunity. Nat Immunol. 2012;13(5):439–447. doi: 10.1038/ni.2246. [DOI] [PubMed] [Google Scholar]
- 123.Riddell SR, Elliott M, Lewinsohn DA, Gilbert MJ, Wilson L, Manley SA, Lupton SD, et al. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat Med. 1996;2(2):216–223. doi: 10.1038/nm0296-216. [DOI] [PubMed] [Google Scholar]
- 124.Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, Forman SJ. Antitransgene Rejection Responses Contribute to Attenuated Persistence of Adoptively Transferred CD20/CD19-Specific Chimeric Antigen Receptor Redirected T Cells in Humans. Biology of Blood and Marrow Transplantation. 2010;16(9):1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Jonnalagadda M, Brown CE, Chang WC, Ostberg JR, Forman SJ, Jensen MC. Efficient selection of genetically modified human T cells using methotrexate-resistant human dihydrofolate reductase. Gene Ther. 2013;(2013):1–8. doi: 10.1038/gt.2012.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jonnalagadda M, Brown CE, Chang W-C, Ostberg JR, Forman SJ, Jensen MC. Engineering human T cells for resistance to methotrexate and mycophenolate mofetil as an in vivo cell selection strategy. PLoS ONE. 2013;8(6):e65519. doi: 10.1371/journal.pone.0065519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Brewin J, Mancao C, Straathof K, Karlsson H, Samarasinghe S, Amrolia PJ, Pule M. Generation of EBV-specific cytotoxic T cells that are resistant to calcineurin inhibitors for the treatment of posttransplantation lymphoproliferative disease. Blood. 2009;114(23):4792–4803. doi: 10.1182/blood-2009-07-228387. [DOI] [PubMed] [Google Scholar]
- 128.Huye LE, Nakazawa Y, Patel MP, Yvon E, Sun J, Savoldo B, Wilson MH, Dotti G, Rooney CM. Combining mTor Inhibitors With Rapamycin-resistant T Cells: A Two-pronged Approach to Tumor Elimination. Mol Ther. 2011;19(12):2239–2248. doi: 10.1038/mt.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Toniatti C, Bujard H, Cortese R, Al E. Gene therapy progress and prospects: transcription regulatory systems. Gene Ther. 2004;11:649–657. doi: 10.1038/sj.gt.3302251. [DOI] [PubMed] [Google Scholar]
- 130.Johnston J, Tazelaar J, Rivera VM, Clackson T, Gao G-P, Wilson JM. Regulated expression of erythropoietin from an AAV vector safely improves the anemia of beta-thalassemia in a mouse model. Mol Ther. 2003;7(4):493–497. doi: 10.1016/s1525-0016(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 131.Win MN, Smolke CD. A modular and extensible RNA-based gene-regulatory platform for engineering cellular function. Proceedings of the National Academy of Sciences. 2007;104(36):14283–14288. doi: 10.1073/pnas.0703961104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Beisel CL, Smolke CD. Design Principles for Riboswitch Function. PLoS Comput Biol. 2009;5(4):e1000363. doi: 10.1371/journal.pcbi.1000363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bayer TS, Smolke CD. Programmable ligand-controlled riboregulators of eukaryotic gene expression. Nat Biotechnol. 2005;23(3):337–343. doi: 10.1038/nbt1069. [DOI] [PubMed] [Google Scholar]
- 134.Chen YY, Jensen MC, Smolke CD. Genetic control of mammalian T-cell proliferation with synthetic RNA regulatory systems. Proc Natl Acad Sci U S A. 2010;107(19):8531–8536. doi: 10.1073/pnas.1001721107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bonger KM, Chen L-C, Liu CW, Wandless TJ. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nat Chem Biol. 2011;7(8):531–537. doi: 10.1038/nchembio.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP, Rosenberg SA, Morgan RA. Improving Adoptive T Cell Therapy by Targeting and Controlling IL-12 Expression to the Tumor Environment. Mol Ther. 2011;19(4):751–759. doi: 10.1038/mt.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, Rosenberg SA. Local Delivery of lnterleukin-12 Using T Cells Targeting VEGF Receptor-2 Eradicates Multiple Vascularized Tumors in Mice. Clinical Cancer Research. 2012;18(6):1672–1683. doi: 10.1158/1078-0432.CCR-11-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Traversari C, Marktel S, Magnani Z, Mangia P, Russo V, Ciceri F, Bonini C, Bordignon C. The potential immunogenicity of the TK suicide gene does not prevent full clinical benefit associated with the use of TK-transduced donor lymphocytes in HSCT for hematologic malignancies. Blood. 2007;109(11):4708–4715. doi: 10.1182/blood-2006-04-015230. [DOI] [PubMed] [Google Scholar]
- 139.Di Stasi A, Tey S-K, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, Straathof K, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365(18):1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wang X, Chang W-C, Wong CW, Colcher D, Sherman M, Ostberg JR, Forman SJ, Riddell SR, Jensen MC. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118(5):1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]