

Abstract
Centromeric Protein-F (Cenp-F) family members have been identified in organisms from yeast to human. Cenp-F proteins are a component of kinetochores during mitosis, bind to the Rb family of tumor suppressors, and have regulatory effects on the cell cycle and differentiation; however, their role in these processes has not been resolved. Here, we provide evidence that the role of murine Cenp-F (mCenp-F, also known as LEK1) remains largely conserved and that the domains within the C-terminus collectively function to regulate the G2/M cell cycle checkpoint. Overexpression of the C-terminal domain of mCenp-F decreases DNA synthesis. Analyses of deletion mutants of mCenp-F reveal that the complete C-terminal domain is required to delay cell cycle progression at G2/M. Signal transduction pathway profiling experiments indicate that the mCenp-F-mediated cell cycle delay does not involve transcriptional activity of key cell cycle regulators such as Rb, E2F, p53, or Myc. However, endogenous mCenp-F colocalizes with pRb and p107, which demonstrates in vivo protein-protein interaction during cell division. These observations suggest that the domains of the C-terminus of mCenp-F have a conserved function in control of mitotic progression through protein-protein interaction with pocket proteins, thus providing a direct connection between cell cycle regulation and mitotic progression.
Keywords: G2/M checkpoint, Cenp-F, LEK1, Pocket proteins
INTRODUCTION
Cenp-F was first discovered as a kinetochore-associated protein that was expressed in a cell cycle-dependent manner [1,2]. Mitosin, the product of the same gene as Cenp-F, was isolated from a screen for Rb-binding proteins [3,4]. Homologous proteins have been proposed and investigated in invertebrate and prokaryotic model organisms [5,6]. Cenp-F genes appear to be well conserved and are widely distributed among living organisms. We report here that vertebrate Cenp-F genes are orthologous members of a gene family that are found as single genes in the human, mouse, rat, and chicken genome, specifically human Cenp-F/Mitosin, murine Cenp-F (LEK1) and avian CMF-1 [1,3,7,8]. The Cenp-F family shares several well-conserved functional domains that are located in the C-terminal portion of the protein sequence. These domains include a bipartite nuclear localization signal, a pocket protein binding domain, and a farnesylation domain [3,9–11].
Genetic homology relates vertebrate Cenp-F genes; yet previous studies have described important differences in the temporal and spatial expression patterns of avian, rodent, and human Cenp-F proteins that distinguish each member of this family. The expression of avian and murine Cenp-F is regulated during development, where they are highly expressed early in development and down-regulated during differentiation [7,12]. Human Cenp-F (hCenp-F) has only been studied in cell lines and neoplastic tissues, and its temporal expression during development has not been characterized. Moreover, avian and murine Cenp-F proteins are expressed throughout the cell cycle, whereas hCenp-F expression is detected only during specific stages of the cell cycle. Human Cenp-F is not found in cells during interphase, but is apparent in the nucleus as cells progress into the S phase of the cell cycle [2]. hCenp-F has been used as a marker of the kinetochore and the G2 phase of the cell cycle in several studies [2,13–15]. Furthermore, hCenp-F has been shown to localize to distinct centromeric loci within the nucleus[3]. In contrast, there are conflicting reports on the subcellular localization of murine Cenp-F. Centromeric localization of mCenp-F was not observed in the C2C12 cell line[7]. However, a recent report provided evidence that murine Cenp-F localized to the kinetochore [16]. In this report, we sought to resolve these differences by examining the subcellular localization of endogenous mCenp-F during mitosis.
Vertebrate Cenp-F proteins have the capacity to bind to the retinoblastoma family of pocket proteins. Cenp-F/pocket protein binding has been described in human, murine, and avian model systems [10,17,18], which demonstrates the conservation of this Cenp-F function. Numerous studies have shown that members of the retinoblastoma family of transcriptional suppressors are critical in the regulation of cell cycle events [19]. This family of proteins, which includes Rb, p107, and p130, has been implicated in the regulation of the G1/S checkpoint in the cell cycle by suppressing transcription of cell cycle-regulatory genes through phosphorylation by cyclin/CDK complexes as well as interactions with various transcription factors [20,21]. Numerous proteins have the capacity to bind to and interact with pocket proteins; however, the significance of their association and the underlying function is not yet clarified for many of these proteins [22]. The interaction of pocket proteins and E2F transcription factors is well documented and demonstrates a clear pathway of cell cycle-dependent transcriptional regulation [19,21]. Although the pocket protein-binding domain of Cenp-F proteins bears some homology to similar domains of E2F transcription factors, Ashe et al demonstrated that mCenp-F could bind an Rb fusion protein that is not capable of binding E2F [23]; thus mCenp-F and E2F have distinct interactions with Rb. However, the significance of the association of pocket proteins and Cenp-F is not well defined. In order to determine the functional significance of mCenp-F/pocket protein interactions, we examined the possibility of altered cell cycle-dependent transcriptional activity.
Kinetochores are complex structures composed of many different proteins that assemble in a highly ordered manner at the centromere of chromosomes. They actively participate in chromosome segregation during mitosis and can delay onset of anaphase until all chromosomes are attached to microtubules [24]. Accurate transmission of chromosomes during cell division is dependent upon proper kinetochore assembly and function. Thus, kinetochores are a critical component of the G2/M checkpoint. Previous studies have shown that hCenp-F is recruited to the kinetochore in a cell cycle dependent manner and that this process is dependent on a particular domain within the C-terminus of the protein [3]. hCenp-F becomes associated with the outer kinetochore plate during the early stages of mitosis and persists throughout mitosis [1,3]. Overexpression of the C-terminal domain of hCenp-F leads to an accumulation of cells at the G2/M boundary [3,11]. However, the functional role of Cenp-F proteins at the kinetochore is unknown. Here, we examine the C-terminal domains of mCenp-F to determine if functional similarities exist between human and murine Cenp-F in the regulation of the G2/M checkpoint.
METHODS
DNA sequence analysis, alignments, and phylogenetics
Independent genomes were queried with coding sequences for human Cenp-F/Mitosin, murine and rat Cenp-F (Lek1), chicken CMF1, and C. elegans HCP1 and 2 using the BLAST function of the NCBI suite of genomic biology tools (http://www.ncbi.nlm.nih.gov/Genomes/index.html). Results of these searches lead to pre-assembled gene maps that include intron/exon boundaries and predicted protein products for the Cenp-F gene family. Alignments of the predicted proteins were carried out using the AlignX program software of the Vector NTI 7.0 suite of analysis programs (Oxford). Cenp-F proteins were phylogenetically arranged by the Neighbor Joining method to build an unrooted Cenp-F gene tree, where the distance of the branches represents phylogenetic distance.
Generation of mCenp-F DNA constructs
Primers were designed to amplify specific sequences of mCenp-F using standard RT-PCR methods to create cDNA clones. The sequences of the primers used are as follows: For the N-terminal mCenp-F expression construct (5′mCenp-F/eGFP, amino acids 1–465) the 5′ primer was TGAGGGGGATCCTAAAAGGGAAAATCAAAGGTTGATGGAGATA, and the 3′ primer was TTTCCTGAAGATCTCGTAACATGG. The C-terminal constructs were generated with the same 5′ primer AGTCGAGTGTCAGAATTAGATG. The 3′ primer for the full length C-terminal expression construct was CTTCACTGGACCAGGCAGTTTGA. The 3′ primer for the NLS deletion was TGGCCAGATGCTGACTTCATCTCTGA. The 3′ primer for the pocket protein-binding domain deletion construct was CAGGGCGAGTGCTCTTACTGTCCTGA. The 3′ primer for the farnesylation domain deletion was CGTTGTTCGTGGGATCAGGCCTGA. The 3′ primer for the deleted pocket protein-binding domain with an added farnesylation domain was CTGGACCCGGCAAGGGCGAGTGCTCTTTGA. The amplified DNA was inserted, in-frame into expression vectors [pEGFP-C1, pEGFP-C2 (Invitrogen), or pcDNA3.1/NT-GFP-TOPO (Clontech)] such that N-terminal green fluorescent protein (GFP)/mCenp-F fusion proteins would be produced. Each construct was sequenced to ensure mCenp-F insert was in the proper reading frame and coding sequence was maintained.
Cell culture
NIH 3T3 and Cos-7 cell lines were obtained from ATCC. The NIH 3T3 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Sigma) supplemented with 10% neonatal bovine serum and 100ug/ml penicillin/streptomycin. Cos-7 cells were maintained in DMEM medium supplemented with 10% fetal bovine serum (Gibco-BRL) and 100ug/ml penicillin/streptomycin.
Transfections
NIH 3T3 or Cos-7 cells were seeded at a density of 7.5 × 105 or 4.0 × 105 respectively, per 1-well chamber slide (Nunc) 24 hours prior to transfection. The transfection solution was prepared using 4ug DNA and 10ul Lipofectamine 2000 (Invitrogen) according to manufacturer’s recommendations. 500ul Opti-MEM (Gibco) medium was used to dilute DNA and Lipofectamine 2000 reagent. Cells were washed briefly with Opti-MEM, and the transfection solution was placed on the cells for a period of 6 hours; then growth media was added back to the cultures. The transfected cells were analyzed 24 and 48 hours post transfection. Non-transfected cells or cells transfected with empty expression vector served as controls. Total cell lysates of transfected cell cultures were analyzed by Western blot using a GFP antibody (Clontech) to confirm proper fusion protein expression.
Western blot
Protein samples were separated by SDS-PAGE and transferred to immunoblot PVDF membrane (BioRad). Membranes were blocked with 5% non-fat milk, 2% BSA in PBS-T [0.1% Tween-20, PBS] for at least 2 hours. After blocking, membranes were incubated with primary antibody (1:1000 dilution) in 2%BSA in PBS-T overnight at 4°C on a rocker, then washed three times with PBS-T, and followed with an alkaline phosphatase conjugated secondary antibody (1:500 dilution) and subsequent washes with PBS-T. Membranes were developed using NBT/BCIP plus suppressor (Pierce) containing 1mM Levamisole.
Flow cytometry
Cells were transfected as described above. At 24 or 48 hours post-transfection, cells were trypsinized off culture dishes and transferred to microcentrifuge tubes. Media with serum was added to stop the trypsin reaction, and then cells were centrifuged at 200Xg for 8 minutes. The supernatant was removed and the cell pellet was resuspended in 50μl PBS. 450μl of ice-cold ethanol fixative [70% EtOH/PBS] was then pipetted into the cell solution and mixed well. The tubes were then placed on ice for at least 2 hours and stored at 4 °C. To run samples, cells were centrifuged at 200×g for 5 minutes to remove ethanol and then rinsed once with PBS and centrifuged again. The pellet was then resuspended with Propidium Iodide (PI) solution [0.1% Triton-X/PBS, 0.2 mg/ml Rnase-A (Sigma), 0.02 mg/ml PI (Molecular Probes)] and incubated for 30 minutes at room temperature. For each sample, 10,000 cells were acquired using a Beckman Coulter Epics XL-MCL flow cytometer and were analyzed using Expo32 ADC software. Results are the culmination of three independent experiments.
Luciferase assays
Firefly Luciferase expression vectors (pRb-TA-Luc, Pe2F-TA-Luc, pp53-TA-Luc, pMyc-TA-Luc, and Pta-Luc) were obtained from the Mercury cell cycle profiling system kit (Clontech). A Renilla luciferase vector (Prl-TK) was used as an internal control to measure transfection efficiency and normalize firefly luciferase expression levels (Promega). Each firefly luciferase vector (0.3ug) was combined with Prl-TK (0.3ug) and each mCenp-F/GFP (0.8ug) construct and transfected into NIH 3T3 cells in 24 well plates as described above. Expression levels of firefly luciferase and renilla luciferase were measured using a dual-luciferase reporter assay kit (DLR 1000, Promega) following manufacturer’s protocol. Assays were performed sequentially using one reaction tube per transfected cell lysate. Relative luciferase units were measured on a Luminometer and recorded for firefly and then renilla luciferase. Non-transfected control cell lysates were measured to normalize for background luminescence. Transfections were done in triplicate for each mCenp-F construct for each experiment and the results are from three independent experiments.
Proliferation assay
5-bromo-2′-deoxyuridine (BrdU, Roche) was used as a marker of DNA synthesis. BrdU labeling medium (Growth media + 1unit/ml BrdU) was added to transfected cell cultures 24 hours post transfection. The cells were incubated in labeling medium for three hours, then washed twice with PBS, and prepared for immunostaining following manufacturer’s protocol. Cells were imaged and nuclei were counted per field for BrdU-positive (BrdU+) and GFP-positive cells (GFP+). A minimum of 150 transfected GFP+ cells were counted for each data point. The labeling index was determined by the number of BrdU+, GFP+ cells divided by the total number of GFP+ cells. Statistical significance was determined by a one-way analysis of variance (N=10) and multiple pairwise comparisons by using the Student-Newman-Keuls method between the three sample groups.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde in phosphate buffered saline (PBS), permeabilized with 0.25% Triton-X in PBS, and blocked with 2% Bovine Serum Albumin (BSA) in PBS prior to staining. All immunofluorescence experiments were carried out in conjunction with parallel samples using peptide competitors as controls for primary antibody specificity and controls lacking primary antibody incubation for secondary antibody specificity. Rb, p107, and p130 primary antibodies were obtained from Santa Cruz. Secondary antibodies conjugated to Alexa Fluor 488 or 568 were obtained from Molecular probes. The laboratory of Dr. David Bader (Vanderbilt University, Nashville, TN) generously provided the antibody used for endogenous mCenp-F (LEK1). This antibody was generated from a peptide sequence localized to the C-terminus of the protein (Ashe et al, 2003; Goodwin et al, 1999). Antibodies were diluted 1:1000 for primary and 1:500 for secondary antibodies in 1%BSA/PBS and incubated on cells for at least 1 hour at room temperature or overnight at 4°C in a humidity chamber. Between incubations, cells were washed 5 times with PBS. To-Pro III or Hoechst dye was used to counter stain nuclei. DAPCO was used as a mounting media for coverslips. Confocal images were collected using either a Bio-Rad MRC 1024 (Lasersharp software) or a Zeiss LSM 510 microscope.
RESULTS
Specific domains of Cenp-F proteins are highly conserved across diverse species
BLAST searches, using species-specific Cenp-F sequences as queries, were carried out on the human, mouse, rat, avian, worm, and yeast genomes using the BLASTN program with default settings (available via the NCBI website). A single Cenp-F gene was found in each of the human, mouse, rat, avian, and yeast genomes. Two Cenp-F-like genes were found in the C. elegans genome (HCP1, HCP2). Cenp-F related proteins were aligned to identify conserved protein domains and regions of homology. The N-terminus of Cenp-F proteins has a predicted series of four coiled coils that share homology to myosin-tail structures. Avian (1625aa), worm (1475aa) and yeast (406aa) Cenp-F proteins are considerably smaller than the human and rodent proteins (2997aa) and homology between them is limited to the termini of these proteins. The C-terminal domains of vertebrate Cenp-F proteins are predicted to be globular and contain functional domains including a nuclear localization signal (NLS), a pocket protein binding domain (PPBD), and a farnesylation domain (FD). The predicted structure and domains of vertebrate Cenp-F proteins is diagrammed in Figure 1A. The highest level of homology (>80%) is found within the C-terminus of vertebrate Cenp-F proteins (Fig. 1B). To determine the evolutionary relationship of the proposed Cenp-F protein family, a phylogenetic tree was constructed based on the alignment of Cenp-F-like proteins (Fig. 1C). Cenp-F related proteins are most homologous to the C-terminal domains of vertebrate Cenp-F. Avian CMF1 (1525–1625) is 72% similar to mouse Cenp-F (2139–2912). Regions of worm HCP1 (687–819; 1215–1475) are 45% and 42% similar, respectively. Multiple regions of yeast Okp-1 (74–116; 231–285; 283–306; 345–388) are 46%, 55%, 52%, and 60% similar, respectively. Therefore, further studies were focused on the most conserved regions of Cenp-F-like proteins and conducted using murine Cenp-F (mCenp-F).
Figure 1. Functional domains of Cenp-F-related proteins.

A: Schematic of the conserved protein motifs present in Cenp-F proteins. The majority of the N-terminal Cenp-F protein consists of a myosin-like coiled coil domain. The C-terminal region contains several functional domains conserved among Cenp-F family members including a bipartite nuclear localization signal (NLS), a pocket protein binding domain (PPBD), and a farnesylation domain (F). B: Diagram of the intron/exon boundries and regions of homology between human and rodent Cenp-F. The N terminus contains an alternatively spliced exon. Both the N and C terminal domains constitute the highly homologous regions; however, the human gene contains regions within the C-terminus that are deleted in rodent Cenp-F genes. C: Phylogenetic tree of known Cenp-F protein sequences constructed using the Neighbor Joining method. D: Diagram demonstrating similar sequences within C-terminal domains shared among Cenp-F family members. Avian CMF1 (1525–1625) is 72% similar to mouse Cenp-F (2139–2912). Regions of worm HCP1 (687–819; 1215–1475) are 45% and 42% similar, respectively. Multiple regions of yeast Okp-1 (74–116; 231–285; 283–306; 345–388) are 46%, 55%, 52%, and 60% similar, respectively.
mCenp-F/eGFP fusion proteins mimic localization of endogenous mCenp-F
Expression constructs were engineered to express the conserved domains of the N-terminal (5′C/eG) and C-terminal domains (3′C/eG) of mCenp-F fused to EGFP tag (Fig. 2A). The 5′C/eG and 3′C/eG constructs were constructed based on the most highly conserved regions of vertebrate Cenp-F family members and represent amino acids 1–465 and 2139–2912 of mCenp-F protein sequence respectively. To confirm the expression of these constructs, COS-7 cells were transiently transfected and total protein lysates were collected 24 hours post transfection. Western blot analysis using an anti-GFP antibody detected protein products of the predicted molecular weight for the 5′C/eG and 3′C/eG fusion proteins, as well as the C1 GFP vector-only construct (Fig. 2B).
Figure 2. Expression of the C-terminus of mCenp-F alters the rate of DNA synthesis.
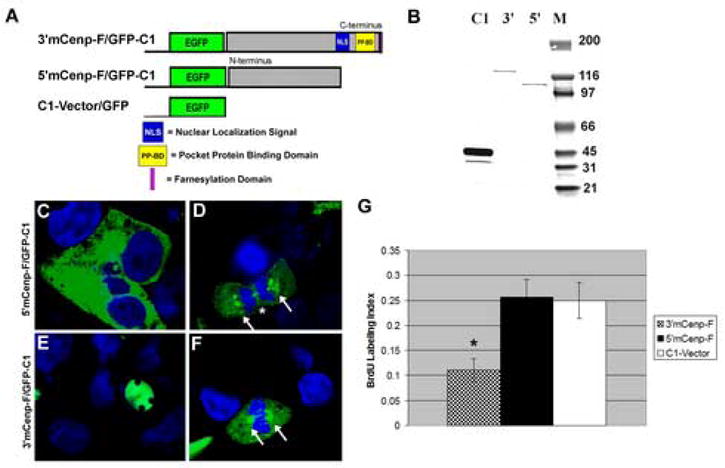
A. Cenp-F/eGFP fusion proteins were engineered to express N and C terminal regions of mCenp-F as depicted. The C1-Vector served as a control for transfection studies. B: Western blot analysis using an antibody against GFP shows expression of fusion proteins that migrate at the predicted molecular weight. C–F: Expression of constructs in transfected cells. C. Green fluorescence (eGFP) indication fusion protein expression was detected in the cytoplasm of cells transfected with the 5′C/eG construct. Blue staining regions are nuclei (To-Pro III). Whereas, the 3′C/eG fusion protein expression construct localized to the nucleus (E), which mimics the expression of endogenous mCenp-F protein. During mitosis, both 5′C/eG and 3′C/eG constructs localize to regions that include the kinetochore (D, F). G: Cells were transfected with 5′C/eG, 3′C/eG, and the C1-control vector and the rate of DNA synthesis was measured via BrdU assay. Cells transfected with the 5′C/eG and C1-vector constructs, had a labeling index of approximately 25%. Cells that expressed the 3′C/eG construct had a labeling index of 10%. Thus, cells expressing the construct containing the 3′ domains of mCenp-F had a significantly lower DNA synthesis (P<0.005, n=3).
Confocal analysis was carried out to determine the subcellular localization of 5′C/eG and 3′C/eG constructs. During interphase, the 5′C/eG construct was found to localize to the cytoplasm (Fig. 2C), while 3′C/eG construct localized to the nucleus, but is excluded from the nucleoli (Fig. 2E). The control GFP protein was found in both the nuclear and cytoplasmic compartments. To ensure that the GFP moiety was not altering the distribution of mCenp-F, cells stained with an antibody against the endogenous C-terminal mCenp-F were compared to cells transfected with 3′C/eG construct. The endogenous mCenp-F antibody staining was consistent with the distribution of 3′C/eG. During mitotic phases of the cell cycle, the distribution of 5′C/eG and 3′C/eG was similar in that both constructs were found to localize to areas that include the kinetochore and the intracellular bridge (Fig. 2D and F), which is also consistent with endogenous localization of human Cenp-F proteins during mitosis.
Overexpression of C-terminal mCenp-F decreases DNA synthesis
COS-7 cells were transiently transfected with 5′C/eG, 3′C/eG, or C1-vector/eG and analyzed for BrdU incorporation 24 hours post transfection. Fluorescence confocal microscopy was used to quantify the BrdU labeling index, which was calculated from the number of BrdU-positive, GFP-positive nuclei per total GFP-positive nuclei for each of the constructs. The results of these studies are summarized in figure 2G. Expression of the 3′C/eG construct significantly reduced the BrdU labeling index by approximately 50% as compared to the 5′C-eG construct and C1 control (11% versus 26% and 25%, p < 0.001). In contrast, the labeling index of the 5′C/eG expression construct was not significantly affected as compared to the C1 control (26% and 25%). These studies indicate that overexpression of C-terminal domains of mCenp-F significantly decreases DNA synthesis and thus negatively regulates cell cycle progression.
C-terminal domains of mCenp-F regulate subcellular and subnuclear localization
Deletion constructs were produced in which the nuclear localization signal (NLS), the pocket protein binding domain (PPBD), and the farnesylation domain (F) were serially removed from the parent 3′mCenp-F/eG construct (2139–2912aa). The resulting constructs are referred to as 3′dNLS/eG, 3′dPPBD/eG, and 3′dF/eG and consist of amino acids 2139–2723, 2139–2765, and 2139–2906 of mCenp-F protein sequence respectively (Figure 3A). Western blot analysis of total protein lysates confirmed fusion protein expression in transfected cells and each deletion mutant migrated to its predicted molecular weight (Figure 3B). Each deletion construct was transfected into Cos7 cells to examine the expression and subcellular localization of these constructs. The 3′C/eG full-length construct localized to the nucleus and mimicked endogenous expression of mCenp-F (Figure 3C). The deletion construct lacking the NLS (3′dNLS/eG) was largely localized to the cytoplasm, though some low level of green fluorescence was detected in the nucleus of some cells (Figure 3D). Bright, punctate spots of GFP were observed within the nucleus of the construct in which the pocket protein-binding domain was deleted (3′dPPBD/eG, Figure 3E). No significant difference was observed between cells transfected with the construct lacking the farnesylation domain (3′dF/eG, Figure 3F) and the full-length 3′C/eG construct (Figure 3C). To determine if the farnesylation domain would change the subnuclear localization of the PPBD mutant, an additional construct was generated in which the farnesylation domain was added to the C-terminus of the 3′dPPBD/eG construct (3′dPPBD+F/eG). Confocal analysis of the 3′dPPBD+F/G construct demonstrated nuclear punctate spots similar to those observed in the 3′dPPBD/eG deletion construct (Compare Figure 3E with 3G). These results indicate that the addition of the farnesylation domain did not change the subnuclear localization of the deleted PPBD. The C1-vector/eG construct was distributed throughout the cells with significant green fluorescence found in both the nuclear and cytoplasmic compartments of transfected cells (Figure 3H). Thus, the C-terminal domains of mCenp-F are critical for proper subcellular localization. In addition, the pocket protein binding domain of mCenp-F directs specific localization within the nucleus.
Figure 3. Subcellular localization of C-terminal domains of mCenp-F.
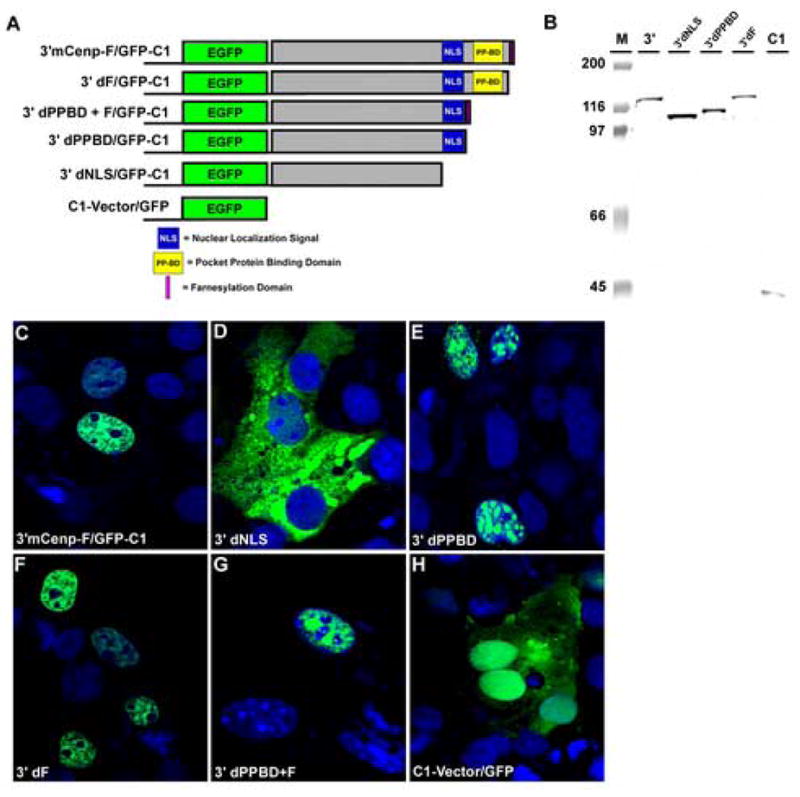
A. 3′C/G contained the full-length sequence for the C-terminus of mCenp-F (2139–2912aa) cloned into the C1 expression vector (Invitrogen). Deletion of the farnesylation domain created 3′dF/G (2139–2908aa). Deletion of the pocket protein-binding domain created 3′dPPBD/G (2139-–2765aa). Deletion of the bipartite nuclear localization signal created 3′dNLS/G (2139–2723aa). 3dPPBD+F/G consisted of the 3′dPPBD/G construct with the addition of the four amino acid farnesylation domain to the C-terminus of the construct. The C1 vector served as a control for transfection studies. B. Western blot analysis using a GFP antibody shows expression of deletion constructs migrating at their predicted molecular weights. M is the molecular weight markers. C-G: Confocal analysis of transfected cells was used determine the localization of the 3′mCenp-F deletion constructs. Green fluorescence indicative of GFP fusion protein expression was observed in the cytoplasm and occasionally in the nucleus of cells transfected with the 3′dNLS/G (D). The 3′C/G (C) and 3′dF/G (E) constructs mimic endogenous expression of mCenp-F. 3′dPPBD/G (F) and 3′dPPBD+F (G) displays punctuate subnuclear localization. In cells transfected with C1-vector, green fluorescence was observed throughout the entire cell (H).
C-terminal domains of mCenp-F regulate cell cycle progression
Flow cytometric analysis was carried out on the deletion constructs described above to determine the effect of mCenp-F C-terminal domains on cell cycle progression. NIH 3T3 cells were transfected with 3′C/eG, 3′dNLS/eG, 3′dPPBD/eG, 3′dF/eG, 3′dPPBD+F/eG constructs, or the control C1-vector/eG. Prior to flow cytometric analysis the transfection efficiency was determined for each culture via detection of GFP expression in living cells. At 24 and 48 hours post transfection, cultures that had a minimum of 70% GFP-positive cells were analyzed for DNA content via flow cytometry. In order to determine significance, at least 10,000 cells were analyzed per construct and three separate experiments were performed. At 24 and 48 hours, a consistent percentage of cells were observed to be in G1/G0 (2N) for all of the transfected constructs (data not shown). An increase in the percentage of cells in S phase was observed at the 48-hour time point (Fig 4A). This increase was slightly higher for 3′mCenp-F, but appeared relatively consistent for other constructs in comparison to the NT-vector and the mock-transfected controls. A significant increase (p<0.01) in the number of cells at G2/M was observed in cells transfected with the full-length C-terminal 3′mCenp-F construct at the 48 hour time point in comparison to controls (Fig. 4B). Detailed analysis of the various C-terminal deletions at the 48-hour time point indicated that overexpression of the 3′C/G construct consistently resulted in an accumulation of cells at the G2/M phase of the cell cycle that was greater than all other constructs tested. Deletion mutant constructs had less effect on G2/M accumulation at the 48 hour timepoint as the percentage of cells in G2/M decreased with each successive deletion (Fig. 4B).
Figure 4. Overexpression of C-terminus of mCenp-F results in G2/M delay.

Cells were transfected with the deletion constructs and analyzed for DNA content by flow cytometry to determine cell cycle phase distribution. A. An increase in the percentage of cells in S phase is observed for the 48-hour time point, but this occurs for all constructs including control. B. At the 48-hour time point, 3′mCenp-F causes a significant increase in the percentage of cells at G2/M in comparison to other constructs and controls (p<0.01, n=3). For each successive deletion of the C-terminal sequence of mCenp-F, the percentage of cells at G2/M is slightly lower.
mCenp-F does not activate cis-elements of cell cycle regulatory pathways
In order to investigate if the observed cell cycle regulatory effects caused by the 3′mCenp-F constructs were related to activation of known cell cycle regulation pathways, transcriptional activation assays were conducted on four distinct pathways: Rb, E2F, p53, and c-Myc. Expression vectors that contained a cis-acting enhancer element responsive to activation of each pathway upstream from a firefly luciferase reporter gene were used to assay the activity of each pathway. Therefore, expression of firefly luciferase in this system provides a quantitative assay for the induction or repression of each pathway as measured by standard luciferase-detection methods. A Renilla luciferase reporter vector was used as an internal control to monitor transfection efficiency. In addiction, the renilla vector contains an HSV-thymidine kinase promoter and provides neutral constitutive expression to which the experimental firefly luciferase levels were normalized. NIH 3T3 cells were co-transfected with a firefly luciferase reporter vector, a renilla reporter vector, and each of the mCenp-F deletion constructs used above. To ensure that the GFP vector containing the mCenp-F constructs was not having an effect, the GFP-only vector (NT-GFP) was transfected as a further control. Furthermore, a luciferase vector without an enhancer element (pTA-Luc) was used as a negative control. Comparison of luciferase expression levels from pTA-Luc transfected cultures to the expression levels in cells transfected with each vector containing an enhancer element indicated measurable activity for each of the regulatory pathways (Fig 5). Renilla luciferase activity demonstrated equivalent transfection efficiency among several experiments (n=3); however, induction of a particular pathway by the mCenp-F constructs was not observed since levels of activation were not significantly different for any of the constructs or enhancer elements tested (Fig 5). Since the cell cycle regulatory pathways are altered in immortalized cell lines, mCenp-F constructs were also transfected into primary mouse cell cultures. Data obtained from the primary cultures closely resembled data from the cell line (data not shown). These results indicate that the effects of mCenp-F on cell cycle regulation are not mediated by transcriptional activation of targets tested here via the major cell cycle regulatory pathways.
Figure 5. mCenp-F does not activate transcription in cell cycle regulatory pathways.
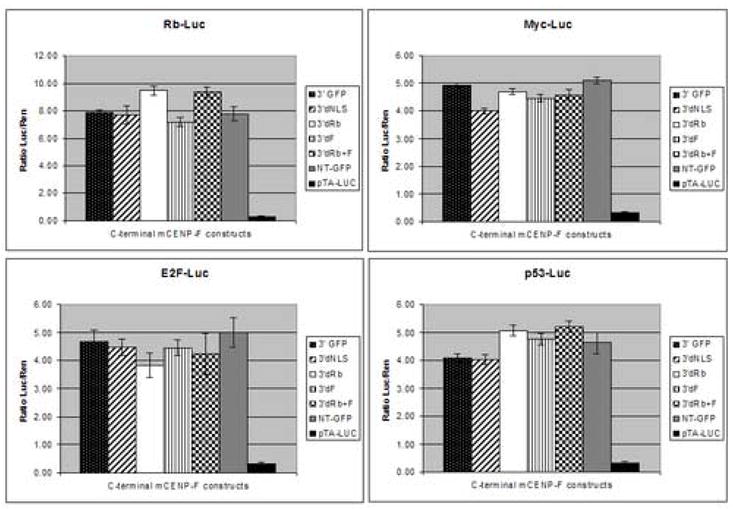
Luciferase assays were conducted on cells transfected with mCenp-F constructs. The activation of cell cycle regulatory pathways (Rb-Luc, E2F-Luc, p53-Luc, Myc-Luc) was examined by luciferase activity. A luciferase vector that did not contain a cis-acting element responsive to a particular pathway was used as a control (pTA-Luc). Increased levels of luciferase expression relative to control indicated measurable activity of each pathway; however, expression of mCenp-F constructs did not affect the activity level of the pathways measured. All results were normalized to control for transfection efficiency and background luciferase activity across experiments. No statistical differences were observed in NIH 3T3 cells or primary mouse cultures (n=3).
Co-localization of mCenp-F and pocket proteins
Biochemical studies have provided evidence that mCenp-F is capable of binding all three members of the Rb family of tumor suppressors, the pocket proteins pRb, p107, p130. However, spatial and temporal intracellular co-localization of mCenp-F and the pocket proteins has not been reported. Therefore, in vivo immunofluorescence studies were carried out using an antibody against endogenous mCenp-F and commercially available antibodies against the mouse pocket proteins pRb, p107, and p130. The distribution of pRb, p107, and p130 co-localization to mCenp-F were very similar, thus only the results for p107 are presented (Fig 6). Pocket protein and mCenp-F co-localization was observed in the nucleus during interphase. During metaphase, both pocket protein and mCenp-F are localized to areas that include the kinetochore and mitotic apparatus as well as evenly distributed throughout the cytoplasm. Both mCenp-F and pocket proteins become concentrated at the intracellular bridge during anaphase and at the midbody during cytokinesis. At the end of cell division, mCenp-F and pocket proteins become relocalized to the nucleus. These data provide evidence of colocalization of mCenp-F and pocket proteins during mitosis and suggest a role for the interaction of these proteins in the coordination of cell cycle regulation.
Figure 6. p107 co-localizes with mCenp-F during mitotic events.

During interphase (A), endogenous mCenp-F (green) and pocket proteins (p107, red) co-localize in nucleus and are excluded from nucleoli, DNA (To-ProIII, blue). At metaphase (B), mCenp-F and p107 are localized to areas that include the kinetochore (white arrow) at the outer edge of aligned chromosomes and appear to be associated with the mitotic spindle. During anaphase (C), the proteins become concentrated at the intracellular bridge and eventually become re-localized to daughter nuclei. Interestingly, mCenp-F and pocket proteins also co-localize at the midbody of dividing cells during cytokinesis (D, white arrowhead) where the remnants of polar microtubules and motor proteins are located.
DISCUSSION
Cenp-F proteins appear to occupy a unique position in cell physiology by physically interacting with proteins that assemble the mitotic apparatus and proteins that control cell cycle regulatory processes. Numerous kinetochore -associated proteins and cell cycle regulatory proteins, including the pocket protein family of transcriptional repressors, are conserved between diverse species [25]. Likewise, Cenp-F proteins are well conserved across numerous taxa (Fig. 1). Functional studies of prokaryotic and invertebrate C-terminal domains of Cenp-f related proteins in yeast (Okp1) and worms (Hcp1 and 2) support a conserved role for these proteins in the mitotic process ([6]; [5]. Our data expands knowledge of vertebrate Cenp-f family members by demonstrating that the C-terminal domains of murine Cenp-F have unique subcellular localization patterns and share a conserved function in control of mitotic progression through protein-protein interaction with pocket proteins.
The GFP-fusion proteins generated in this study provide a useful study tool to investigate the dynamic subcellular distribution and function of the specific domains of mCenp-F. Overexpression of the C-terminal mCenp-F/GFP fusion protein significantly decreased DNA synthesis as compared to N-terminal mCenp-F/GFP or GFP vector-only constructs (Fig. 2). Furthermore, we found that overexpression of the C-terminal mCenp-F/GFP fusion protein also caused an increase in the number of cells at the G2/M phase of the cell cycle (Fig 4). These results are similar to those obtained when the homologous region of hCenp-F is overexpressed [11], demonstrating the ability to regulate cell cycle progression is conserved between rodent and human Cenp-F proteins. Moreover, we demonstrate that the complete C-terminal domain is required for the initiation of a cell cycle delay (Fig. 4). Sequence analysis of Cenp-F-related proteins suggests that the conservation of this function may exist in the cell cycle control mechanism of many more species.
In order to determine the functional significance of mCenp-F/pocket protein interactions, we examined the possibility of altered cell cycle transcription. Our study is the first to investigate and show that the effects on cell cycle regulation by mCenp-F are not due to activation of four main cell-cycle regulatory pathways (c-myc, Rb, p53, and E2F) at the level of transcriptional regulation. This result was surprising given the observation that C-terminal domains of mCenp-F bind to the long pocket of Rb-family members, which is responsible for binding E2F transcription factors. Our results indicate that the interaction of mCenp-F and pocket proteins does not result in transcriptional activation of an E2F-dependent promoter as would be predicted by the Cenp-F/long pocket interaction. These data indicate that Cenp-F proteins do not act as simple negative regulators of pocket protein-mediated events in cell cycle regulation. Nonetheless, mCenp-F/pocket protein colocolization studies demonstrate that protein-protein interaction between mCenp-F and pocket proteins is dynamic during mitosis (Fig. 6). These observations suggest a role for Cenp-F in intracellular localization of pocket proteins during cell division, thus providing a connection between cell cycle regulatory proteins and kinetochore assembly to regulate mitotic progression.
Kinetochore assembly and the subsequent regulation of mitosis by this process represent a poorly characterized but clearly important regulatory element of the cell cycle. Several proteins localize to the kinetochore throughout the cell cycle, whereas others are only present in mitotic kinetochores [24]. Cenp-F is one of the proteins that are recruited to the kinetochore in a cell cycle dependent manner [2,26], but its role within the complex is not yet known. As a centromeric protein, Cenp-F joins other evolutionarily conserved proteins such as Mad2, Skp1, and Bub1[27–29]. These proteins are also transiently associated with kinetochores and have been shown to be essential mitotic checkpoint components. Recently, Sgt1 was shown to be essential for kinetochore assembly and to affect the localization of Cenp-F to the kinetochore during mitosis [30]. Sgt1 was found to delay mitotic cells at G1, which suggests that the function of Sgt1 is upstream of Cenp-F activity at G2. However, like Sgt1, Bub1, and many other proteins associated with the kinetochore, the C-terminal domains of Cenp-F proteins are also highly conserved.
The present biochemical, cytological, and functional data suggest that Cenp-F connects cell cycle regulatory mechanisms through interactions with pocket proteins with mitotic checkpoints associated with kinetochore attachement and assembly of the mitotic apparatus. As the link between two critical and well-conserved processes, Cenp-F genes and their functional roles have been conserved across numerous taxa during evolution. A more complete understanding of Cenp-F function is necessary to fully determine the extent of Cenp-F involvement in cell cycle regulation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Rattner JB, Rao A, Fritzler MJ, Valencia DW, Yen TJ. CENP-F is a.ca 400 kDa kinetochore protein that exhibits a cell-cycle dependent localization. Cell Motil Cytoskeleton. 1993;26:214–226. doi: 10.1002/cm.970260305. [DOI] [PubMed] [Google Scholar]
- 2.Liao H, Winkfein RJ, Mack G, Rattner JB, Yen TJ. CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J Cell Biol. 1995;130:507–518. doi: 10.1083/jcb.130.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu X, Chang KH, He D, Mancini MA, Brinkley WR, Lee WH. The C terminus of mitosin is essential for its nuclear localization, centromere/kinetochore targeting, and dimerization. J Biol Chem. 1995;270:19545–19550. doi: 10.1074/jbc.270.33.19545. [DOI] [PubMed] [Google Scholar]
- 4.Shan B, Zhu X, Chen PL, Durfee T, Yang Y, Sharp D, Lee WH. Molecular cloning of cellular genes encoding retinoblastoma-associated proteins: Identification of a gene with properties of the transcription factor E2F. Mol Cell Biol. 1992;12:5620–5631. doi: 10.1128/mcb.12.12.5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore LL, Morrison M, Roth MB. HCP-1, a protein involved in chromosome segregation, is localized to the centromere of mitotic chromosomes in Caenorhabditis elegans. J Cell Biol. 1999;147:471–480. doi: 10.1083/jcb.147.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ortiz J, Stemmann O, Rank S, Lechner J. A putative protein complex consisting of Ctf19, Mcm21, and Okp1 represents a missing link in the budding yeast kinetochore. Genes Dev. 1999;13:1140–1155. doi: 10.1101/gad.13.9.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goodwin RL, Pabon-Pena LM, Foster GC, Bader D. The cloning and analysis of LEK1 identifies variations in the LEK/centromere protein F/mitosin gene family. J Biol Chem. 1999;274:18597–18604. doi: 10.1074/jbc.274.26.18597. [DOI] [PubMed] [Google Scholar]
- 8.Litvin J, Montgomery MO, Goldhamer DJ, Emerson CP, Jr, Bader DM. Identification of DNA-binding protein(s) in the developing heart. Dev Biol. 1993;156:409–417. doi: 10.1006/dbio.1993.1088. [DOI] [PubMed] [Google Scholar]
- 9.Ashe M, Pabon-Pena LM, Dees E, Price KL, Bader D. LEK1 is a potential inhibitor of pocket protein-mediated cellular processes. J Biol Chem. 2004;279:664–676. doi: 10.1074/jbc.M308810200. [DOI] [PubMed] [Google Scholar]
- 10.Redkar A, deRiel JK, Xu YS, Montgomery M, Patwardhan V, Litvin J. Characterization of cardiac muscle factor 1 sequence motifs: retinoblastoma protein binding and nuclear localization. Gene. 2002;282:53–64. doi: 10.1016/s0378-1119(01)00789-2. [DOI] [PubMed] [Google Scholar]
- 11.Hussein D, Taylor SS. Farnesylation of Cenp-F is required for G2/M progression and degradation after mitosis. J Cell Sci. 2002;115:3403–3414. doi: 10.1242/jcs.115.17.3403. [DOI] [PubMed] [Google Scholar]
- 12.Dees E, Pabon-Pena LM, Goodwin RL, Bader D. Characterization of CMF1 in avian skeletal muscle. Dev Dyn. 2000;219:169–181. doi: 10.1002/1097-0177(2000)9999:9999<::aid-dvdy1055>3.3.co;2-2. [DOI] [PubMed] [Google Scholar]
- 13.Blajeski AL, Kottke TJ, Kaufmann SH. A multistep model for paclitaxel-induced apoptosis in human breast cancer cell lines. Exp Cell Res. 2001;270:277–288. doi: 10.1006/excr.2001.5349. [DOI] [PubMed] [Google Scholar]
- 14.Kao GD, McKenna WG, Yen TJ. Detection of repair activity during the DNA damage-induced G2 delay in human cancer cells. Oncogene. 2001;20:3486–3496. doi: 10.1038/sj.onc.1204445. [DOI] [PubMed] [Google Scholar]
- 15.Sorensen CS, Lukas C, Kramer ER, Peters JM, Bartek J, Lukas J. Nonperiodic activity of the human anaphase-promoting complex-Cdh1 ubiquitin ligase results in continuous DNA synthesis uncoupled from mitosis. Mol Cell Biol. 2000;20:7613–7623. doi: 10.1128/mcb.20.20.7613-7623.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang ZY, Guo J, Li N, Qian M, Wang SN, Zhu XL. Mitosin/CENP-F is a conserved kinetochore protein subjected to cytoplasmic dynein-mediated poleward transport. Cell Research. 2003;13:275–284. doi: 10.1038/sj.cr.7290172. [DOI] [PubMed] [Google Scholar]
- 17.Zhu X, Mancini MA, Chang KH, Liu CY, Chen CF, Shan B, Jones D, Yang-Feng TL, Lee WH. Characterization of a novel 350-kilodalton nuclear phosphoprotein that is specifically involved in mitotic-phase progression. Mol Cell Biol. 1995;15:5017–5029. doi: 10.1128/mcb.15.9.5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashe M, Pabon-Pena LM, Dees E, Price KL, Bader D. LEK1 is a potential inhibitor of pocket protein-mediated cellular processes. J Biol Chem. 2004;279:664–676. doi: 10.1074/jbc.M308810200. [DOI] [PubMed] [Google Scholar]
- 19.Classon M, Salama S, Gorka C, Mulloy R, Braun P, Harlow E. Combinatorial roles for pRB, p107, and p130 in E2F-mediated cell cycle control. Proc Natl Acad Sci USA. 2000;97:10820–10825. doi: 10.1073/pnas.190343497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene. 1999;18:7873–7882. doi: 10.1038/sj.onc.1203244. [DOI] [PubMed] [Google Scholar]
- 21.Classon M, Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nature Reviews Cancer. 2002;2:910–917. doi: 10.1038/nrc950. [DOI] [PubMed] [Google Scholar]
- 22.Morris EJ, Dyson NJ. Retinoblastoma Protein Partners. Advances in Cancer Research. 2001;82:1–54. doi: 10.1016/s0065-230x(01)82001-7. [DOI] [PubMed] [Google Scholar]
- 23.Ashe M, Pabon-Pena LM, Dees E, Price KL, Bader D. LEK1 is a potential inhibitor of pocket protein-mediated cellular processes. J Biol Chem. 2004;279:664–676. doi: 10.1074/jbc.M308810200. [DOI] [PubMed] [Google Scholar]
- 24.Cleveland DW, Mao Y, Sullivan KF. Centromeres and Kinetochores: From Epigenetics to Mitotic Checkpoint Signaling. Cell. 2003;112:407–421. doi: 10.1016/s0092-8674(03)00115-6. [DOI] [PubMed] [Google Scholar]
- 25.Stark GR, Taylor WR. Analyzing the G2/M checkpoint. Methods Mol Biol. 2004;280:51–82. doi: 10.1385/1-59259-788-2:051. [DOI] [PubMed] [Google Scholar]
- 26.Johnson VL, Scott MIF, Holt SV, Hussein D, Taylor SS. Bub1 is required for kinetochore localization of BubR1, Cenp-E, Cenp-F and Mad2, and chrmosome congression. J Cell Sci. 2003;117:1577–1589. doi: 10.1242/jcs.01006. [DOI] [PubMed] [Google Scholar]
- 27.Taylor SS, McKeon F. Kinetochore localization of murine Bub1 is required for normal mitotic timing and checkpoint response to spindle damage. Cell. 1997;89:727–735. doi: 10.1016/s0092-8674(00)80255-x. [DOI] [PubMed] [Google Scholar]
- 28.Connelly C, Hieter P. Budding yeast SKP1 encodes an evolutionaryily conserved kinetochore protein required for cell cycle progression. Cell. 1996;86:275–285. doi: 10.1016/S0092-8674(00)80099-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y, Benezra R. Identification of a human mitotic checkpoint gene: hsMAD2. Science. 1996;274:246–248. doi: 10.1126/science.274.5285.246. [DOI] [PubMed] [Google Scholar]
- 30.Steensgaard P, Garre M, Muradore I, Transidico P, Nigg EA, Kitagawa K, Earnshaw W, Faretta M, Musacchio A. Sgt1 is required for human kinetochore assembly. EMBO reports. 2004;5:626–631. doi: 10.1038/sj.embor.7400154. [DOI] [PMC free article] [PubMed] [Google Scholar]