

Abstract
Parkinson's disease is a neurological disorder which afflicts an increasing number of individuals. If the wider complex of extrapyramidal symptoms referred to as “age-related parkinsonism” is included, the incidence is near 50% of the population above 80 years of age. This review summarizes recent studies from our laboratories as well as other research groups in the quest to explore the multi-faceted etiology of age-related neurodegeneration, in general, and degeneration of the substantia nigra dopaminergic neurons, in particular. Our work during recent years has focused on assessment of potential interactive effects of a reduction in glial cell line-derived neurotrophic factor (GDNF) and the aging process (intrinsic factors) and early neurotoxin exposure (an extrinsic factor) on dopamine (DA) systems and the behaviors they mediate. The guiding hypothesis directing the research to be described was that a combination of the two factors would exacerbate the decline in the DA transmitter system function that occurs during aging. The results obtained were consistent with the well-established aging-related decline in function and structure of neurons utilizing DA as a transmitter and motor function, and extended knowledge by establishing that the genetic reduction of Gdnf exacerbated these aging related changes. Thus, Gdnf reduction appears to increase the vulnerability of the DA neurons to the many different challenges associated with the aging process. Assessment of methamphetamine effects on young Gdnf+/− mice indicated that reduced GDNF availability increased the vulnerability of DA systems to this well-established neurotoxin. The work discussed in this review is consistent with earlier work demonstrating the importance of GDNF for maintenance of DA neurons and also provides a novel model for progressive DA degeneration and motor dysfunction.
Keywords: Monoamines, Dopamine, Aging, Substantia Nigra, Striatum, Parkinsonism, Motor function, Drug abuse and aging, Neurodegeneration
1. Introduction
Idiopathic Parkinson' disease (PD) is the second most common movement disorder. It is characterized by a massive and progressive loss of dopamine (DA) neurons in the substantia nigra (SN) pars compacta with accompanying, severe motor symptoms including tremor, bradykinesia, rigidity and postural instability. Parkinsonian syndrome (PS) also includes extrapyramidal symptoms associated with DA cell loss. The incidence of this wider group of neurological conditions is even greater than PD, with more than 50% of individuals over the age of 80 affected (see Bennett et al., 1996). In this review, we will refer to PD and parkinsonism as parkinsonian syndrome (PS). Pathological studies of PS indicate the presence of a cascade of events including increased levels of iron and monoamine oxidase (MAO)-B activity, activation of microglial cells, enhanced cytokine response, oxidative stress, glutamatergic excitotoxicity, nitric oxide synthesis, cellular inclusion bodies containing alpha-synuclein, reduced expression of trophic factors, reduced oxidative stress scavengers such as glutathione, and altered calcium homeostasis (Mandel et al., 2003). The synuclein family was recently spotlighted when alpha-synuclein was demonstrated to be linked to parkinsonism, both neuropathologically and gentically (Surguchov, 2008; see Table 1). Recent studies by Braak and colleagues (Braak and Del Tredici, 2008) indicate that SN DA neurons are not the first to succumb; in fact, the disesase appears to affect central, peripheral, and enteric nervous systems, including brainstem motor neurons, sympathetic ganglia, olfactory systems, the pontine locus coeruleus noradrenergic neurons (LC NE), and then, finally, the SN DA neurons (Braak et al., 2007;2008; Braak and Del Tredici, 2008). This progression is paralled by by increasing symptamotology from tremor through akensia, cognitive dysfunction, etc. Thus, PS appears to progressively include an ever-increasing number of brain regions as well as the peripheral nervous system and spinal cord.
Table 1. Genes involved with Parkinsonian Syndrome.
| PARK loci | Gene | Gene Locus | Inheritance Pattern |
|---|---|---|---|
| PARK1/PARK4 | SNCA (alpha-synuclein) | 4q21 | Autosomal dominant |
| PARK8 | LRRK2 (Leucine-rich repeat kinase 2) | 12q12 | Autosomal dominant |
| PARK2 | Parkin | 6q25–q27 | Autosomal recessive |
| PARK6 | PINK1 (PTEN-induced putative kinase 1) | 1p35–p36 | Autosomal recessive |
| PARK7 | DJ-1 | 1p36 | Autosomal recessive |
| PARK9 | ATP13A2 | 1p36 | Autosomal recessive |
The etiology of PS remains unclear; however, both human and animal research suggests that a genetic vulnerability seems likely for both idiopathic PD and age-related parkinsonism. Studies of PD patients in the 1990s identified several genes that play a role in the disease, at least in some cases (see Table 1). These include PARK1/PARK4 (alpha-synuclein), PARK8 (leucine-rich repeat kinase 2 (LRRK2)), PARK2 (Parkin), PARK6 (PTEN-induced kinase 1 (PINK1)), PARK7 (DJ-1), and PARK9 (ATP13A2), many of which code for proteins implicated in mitochondrial function (see Terzioglu and Galter, 2008). Point mutations, duplications and triplications in the alpha-synuclein gene are associated with a rare familial dominant form of PS. Mutations in alpha-synuclein and LRRK2 genes are commonly associated with dominant PS, while mutations in Parkin, DJ-1, PINK1, and ATP13A2 genes are associated with early onset autosomal recessive PS (see Lesage and Brice, 2009). In contrast, other studies strongly indicate that extrinsic factors such as endo- or exotoxins, contribute to the age of onset, as well as the progression of the disease (see e.g. Gaenslen et al., 2008). In fact, studies on both rodents and non-human primates indicate that exposure to environmental toxins produces progressive damage well before the onset of clinical symptoms of PS.
With the aging baby boomer generation, the number of aging-related neurological disorders will increase exponentially over the next few decades. Because reduced agility and slower gait are associated with the normal aging process as well as PS, the differential diagnosis of movement disorders in the elderly is difficult (Baron, 2005). Although many potential causative factors for PS have been identified, they are overshadowed by aging itself, which appears to be the number one risk factor for parkinsonism and must be considered when designing appropriate animal models. In a normal human brain, the number of DA neurons declines by 3-6% every decade. The rate of decline is much greater in PD patients, suggesting that PD is not simply accelerated aging, as previously thought. Previous work indicates that the PD incidence rate in subjects over 85 years old was over 10 times greater than that of 56- to 65-year-old subjects, emphasizing that age represents the number one risk factor for this disease complex (Vanitallie, 2008). In addition, up to 50% of individuals aged 80 and older exhibit extrapyramidal symptoms consistent with age-related PS (Bennett et al., 1996). Despite the importance of age documented by these clinical studies, most animal models of PD utilize young animals exposed to a neurotoxin such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or 6-hydroxydopamine (6-OHDA; Terzioglu and Galter, 2008) and not enough emphasis is placed on aging itself as a factor.
Despite reported success with neurotrophic or neuroprotective therapeutic drugs or cell replacement therapies in animal models of PS, none have lead to a clinically effective treatment avenue which cures all aspects of the disease complex (see e.g. Toulouse and Sullivan, 2008). A possible explanation for the lack of developing effective treatments for PS from animal models is that the models have not factored in the aging process, which may prevent recovery after a toxic event. It is suggested that animal models which consider aging as well as the multifaceted genetic and environmental components represented in the disease process may be more effective in the development of potential therapeutic agents. Current animal models can be assigned to three different categories: 1) neurotoxins using acute or chronic administration models; 2) transgenic mouse models mimicking PD linkage studies or deletion of genes necessary for the development and survival of dopamine neurons; and 3) tissue-specific knockouts targeting DA neurons (Terzioglu and Galter, 2008). Although these current models are valuable and have told us much about the disease process, few studies have included the combination of genetic and environmental factors. In the experiments reviewed here, we have applied the general multifactorial, or “dual-hit”, hypothesis of neurodegeneration to a specific controlled internal genetic factor, Gdnf gene reduction, combined with a controlled external factor, drug exposure (methamphetamine (METH) or MPTP). Thus, the experiments form our laboratories reviewed below provide evidence that a genetic vulnerability combined with an environmental toxin renders DA neurons especially sensitive to age-related events such as oxidative stress or neuroinflammatory cascades. More spefically, we present evidence for a dual-hit model, combining a growth factor knockout model (Gdnf+/− or Gfrα1+/− mice) with a known neurotoxin (METH or MPTP, respectively) causing additive effects on the aging-related changes in DA systems and associated behavioral changes.
2. Development and Aging of SN DA neurons
2.1. Development of the DA transmitter system
Visualization of the central dopaminergic cell groups was first achieved by Annica Dahlström and Kjell Fuxe and was published in their seminal paper in the 1960s (Dahsltröm and Fuxe, 1964). Before this mapping study utilizing the then novel Falck-Hillarp Fluorescence method (Falck et al., 1959), visualization of DAergic cell bodies and nerve fibers was not possible, albeit DA and its metabolites had been detected in brain tissue and CSF using quantitative biochemical methods (Carlsson et al., 1957). Dahlström and Fuxe found three groups of DA nerve cell bodies in the midbrain, as well as some smaller cell groups in the midline of the hypothalamus. They utilized the nomenclature A8-10 for the midbrain nuclei, and A11-15 for the hypothalamic nuclei and a schematic presentation of these nuclei is shown in Figure 1A. Ascending as well as descending fiber bundles were detected, and it was established that the DA nuclei innervated both cortical and subcortical structures with a dense plexus of neurites (Dahlstrom and Fuxe, 1964). Figure 1B provides a visualization of DA neurites and cell bodies in the rodent brain and indicates that the DAergic neurites contain numerous collateral branches and varicosities along their course. Alterations in the DA transmitter systems extend from early in utero development throughout the life span, and challenges to these systems, for example by drugs, toxins, ablation, inflammation, etc, produce long-lasting or permanent changes (see e.g. Monahan et al., 2008). Early development of A8-A10 nuclei includes cell fate determination, differentiation and migration, and later on events such as neurite growth, guidance and synapse formation determine the branching and postsynaptic definition of the final terminal tree of this widespread transmitter system in the brain (Smits et al., 2006). As discussed in another review in this volume by Fuxe et al., DA can act via classical regulated synaptic release or volume transmission (see Fuxe et al current volume). Recent studies on rodents indicate that DA neurons undergo two significant postnatal waves of apoptosis which allow the fine-tuned connectivity in cortical and subcortical areas, which contribute to their significant roles in memory processing, reward, motor function, and motor coordination processes (Burke, 2004; Van den Heuvel et al., 2008). DA systems develop rapidly during the last week of gestation in the mouse, being detected by embryonic day (E) 13, with the entire nigrostriatal tract visualized by E16, and adult florescence pattern noted by E18 (see e.g. Burke, 2004; Van den Heuvel et al., 2008). Development of DA systems in mice continues postnatally with increasing functional capacity into adolescence, and then declines and remains relatively stable through early adulthood (Burke, 2004).
Figure 1.
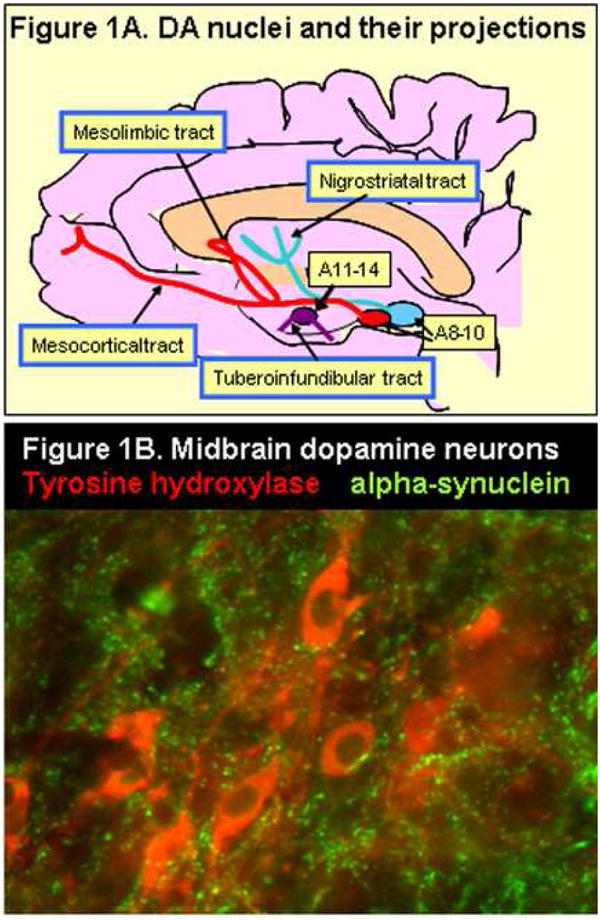
A = Schematic representation of the DA nuclei in midbrain (A8-10) and in the hypothalamus (A11-15). Note that there are both ascending and descending pathways. B = Fluorescence immunohistochemistry visualizing a small group of DA neurons in the pars compacta of the SN. The section was incubated with both tyrosine hydroxylase (TH; red stain) and alpha-synuclein antibodies (red) to demonstrate markers for both neurites and cell bodies in this region. Note the excessive branching and varicose appearance of the neurites.
2.2 Age-related effects on DA neurons in animal models
After the stable period throughout early adulthood, the DA system begins to decline during the aging process as evidenced by neurochemical, morphological, and accompanying behavioral changes. Several animal models substantiate the relationship of motor function and DA transmitter system deterioration with aging (see eg. Hebert and Gerhardt, 1998; Cass et al., 2002; Yurek and Fletcher-Turner, 2000). For example, aging-related reductions in DA uptake are reported for Rhesus monkeys (Dejesus et al., 2001), for DA release in F344 rats (Hebert and Gerhardt, 1997), and for DA receptor binding in mice (Ingram 2000). There are also reports of aging-related exacerbated response to DA-specific neurotoxins such as 6-OHDA (Cass et al., 2002) and MPTP (Mandavilli et al., 2000) in mice. Although the mechanisms for these aging-related increases in nigral neuron vulnerability are not established, reductions in support systems such as growth factors have been postulated to increase the susceptibility of SN DA neurons to external stressors and toxins (see eg. Yurek and Fletcher-Turner, 2000; 2001). Recent studies in non-human primates also suggest that the microenvironment in midbrain regions containing DA cell bodies may be especially susceptible to aging-related increased inflammation in the brain following neurotoxin administration (Kanaan et al., 2008). This observation again suggests that attention should be given to the aging process and its special challenges in designing animal models for the motor dysfunction and DA cell loss associated with PS. The midbrain SN region is especially rich in microglia (Zecca et al., 2008) and there is substantial evidence that neurotoxins and endotoxins activate these cells to release damaging cytokines (Sawada et al., 2008). The possibility of aging-related reduction in support systems is an important consideration for interpreting animal studies evaluating neuroprotective agents. For example, an agent may have bonafide protective effects against a neurotoxin when administered to young animals but be ineffective in aged animals because of an aging related decline in the support systems through which the agent acts. It is therefore important to take into account aging as the primary risk factor for PS, when designing studies of DA neurons and motor function in regards to neuroprotective or damaging agents.
2.3 Aging processes affecting motor function and DA neurons in humans
In humans, the incidence of extrapyramidal symptoms related to PS via multifactorial processes increases significantly with age, involving up to 50% of the population over 85 years of age (Bennett et al, 1996; Kluger et al., 1997), as discussed in the Introduction to this chapter. The DA nigrostriatal system, although not the only mediator of motor function, is clearly involved in aging-related decline in motor function (see eg. Volkow et al., 1998; Naoi and Maryama, 1999; Palmer and DeKosky, 1993; see also Ingram, 2000; Baron, 2005, Vanitalle et al., 2008). In humans, the number of midbrain dopaminergic neurons declines by 3-6.0% per decade after 50 years of age (Fearnley and Lees, 1991; Gibb and Lees, 1991). In addition, aging studies of the human brain also demonstrate that intensely stained DAT-ir SNpc neurons decline by 11.2% per decade (Ma et al., 1999). These data are in conjunction with previous studies demonstrating a dramatic reduction of DAT mRNA within SNpc neurons during the 5th decade of life (Bannon et al., 2005). Furthermore, imaging studies of the striatum in humans indicate substantial age-related declines in DA D2 receptors (Volkow et al., 1996), DA D1 receptors (Suhara et al., 1991), and DAT protein (Volkow et al., 1996; see also Kaasinen and Rinne 2002). Postmortem studies have shown that by the time PS is identified symptomatically, as much as 70-75% of the dopamine-containing neurons have been lost in PD patients, suggesting that the disease is not simply accelerated aging, but producing a much more severe and aggressive course of decline (Mandel et al., 2003). Thus, both normal aging and PS lead to a progressive loss of SN DA neurons, albeit at a different final degree and slope. Figure 4 summarizes our proposed model that the aging-related decline in DA system function is multifactorial including both genetic and external factors. The figure is general but reflects our focus on Gdnf reduction as the genetic factor. Since GDNF is required for DA development, and is considered a classical target-derived neurotrophic factor (Tomac et al., 1995), the possibility of a genetic factor which impacts only maintenance of the adult transmitter system is excluded from the model. As indicated by Figure 4, several scenarios for our mouse model are possible, including a parallel slope leading to earlier motor dysfunction (which would indicate additive effects of the genetic and environmental factors), or a steeper decline, which would suggest synergistic effects of our genetic plus external factors. Because both PS and PD are thought to be multifactorial, optimal animal models need to be developed to assess the interplay between genetic mutations and mitochondrial damage, oxidative stress, neuroinflammation, and growth factor loss, which have all been implicated in the disease (see Granholm et al., 2008 for review). Further, most neurotoxic models for PS have not included aging as a factor, even though this is the primary risk factor for the disease complex in humans (see e.g. Bennett et al., 1996; Eggers, 2009). The present review presents evidence for such a model, which takes into consideration both external (environmental) and internal (genetic) factors, as well as the aging process, which lead to progressive motor dysfunction and DA cell loss in a mouse model.
Figure 4.
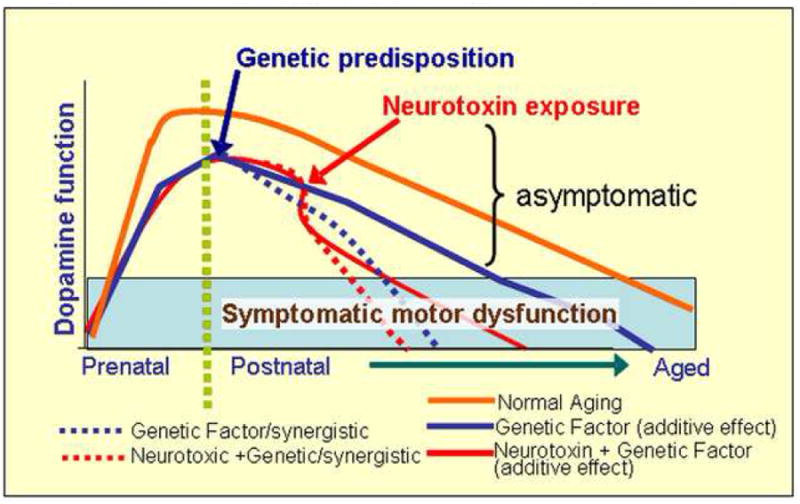
Schematic illustration of the dual-hit hypothesis combining a genetic with an environmental factor. While DA neuron function deteriorates slowly during the normal aging process, finally rendering symptoms of motor dysfunction with old age (Symptomatic motor dysfunction bar), this process can be exacerbated by both Gdnf partial deletion (genetic predisposition) and the postnatal METH administration (external neurotoxin), resulting in an additive or synergistic effect, as indicated by parallel versus steeper slopes. A steeper slope would indicate synergistic effects of the factors, while a parallel slope would indicate an additive effect, leading to the same rate of progression but an earlier entrance into the symptomatic phase. Both of these possible scenarios lead to earlier motor dysfunction, as evidenced by our findings described in the current review.
3. Neurotrophic factors and dopamine neurons
3.1 Neurturin, BDNF, and FGF
Many research groups have searched for the perfect neuroprotective factor for DA neurons since the discovery of the first neurotrophic factor named nerve growth factor (NGF) in the early 70s and 80s (Levi-Montalcini, 2003). Recent work indicates that overall trophic activity is greater in the striatum of middle-aged and aged non-human primates than in their younger counterparts, and that a neurotoxic lesion elevated trophic activity in the young but not in aged subjects (Collier et al., 2005). These studies suggest that the aged brain is unable to activate trophic factors as a defense mechanism, suggesting that exogenous treatment with trophic factors in lesion models or with normal aging may be a viable treatment strategy. Further, several investigators have reported decreased levels of neurotrophins, specifically brain-derived neurotrophic factor (BDNF) and glial cell line- derived neurotrophic factor (GDNF, discussed below) in the nigrostriatal DA region and cerebrospinal fluid of PD patients (Nagatsu et al., 2000).
Some growth factors are important for maintenance of function and for neurotransmission of DA neurons in adult or aged life, while others are specifically involved in differentiation during early development. Wnt5, sonic hedgehog (SHH) and fibroblast growth factor (FGF) have mostly been associated with differentiation of stem cells into DAergic cells as well as migration into appropriate brain regions during development (Sanchez-Pernaute et al., 2008; Papanikolaou et al., 2008), while other factors such as BDNF and GDNF are directly involved in DA neurotransmission and maintenance in the adult animal (Fumagalli et al., 2006). FGF appears unique in that this ubiquitous growth factor also plays a role in early development as well as in neuronal health in aging and PS. Recent work from Murase and collaborators (Murase and McKay, 2006) suggested that FGF signaling elevates DA levels in calbindin-negative SN neurons, which are those at most risk in PD patients (Murase and McKay, 2006). Neurturin, a member of the GDNF family of neurotrophic factors (see Saarma, 2000), is an interesting protein, which is currently in clinical trials for PD patients, through delivery via viral vectors (Herzog et al., 2008; Isacson and Kordower, 2008) and holds promise for future treatment of this disease as well. Although initial studies do not indicate that neurturin may be as successful in the clinic as originally hoped. with the current delivery method, it is likely that development of other delivery systems will yield results depending on the robust effects observed in rodent and primate models (see Herzog et al., 2008). Exogenous administration of BDNF into the SN or striatum of animal models of PD provides significant neuroprotection for DA neurons, both in non-human primates and in rodents (Tsukahara et al., 1995). In our research group, we recently examined long-term consequences of a partial genetic deletion of BDNF (see Boger et al 2009, in revision), and did not find any observable genotype effects of this growth factor deletion upon DA neuron morphology, even though some behavioral components for motor function were altered with aging in both BDNF heterozygous mice and age-matched WT mice (Boger et al., 2009). Thus, our continued studies on gene deletions remained focused on alterations in the GDNF system as detailed below. The data described above support findings that there are several important growth factors partaking in DA neuron induction, maintenance, and protection against injury. However, our previous and continuing work suggest that GDNF is one of the key players in DAergic function over the life span, at least in rodents.
3.2 Glial cell line-derived neurotrophic factor (GDNF)
GDNF is a member of the transforming growth factor-β superfamily of neurotrophic factors (Krieglstein et al., 1995; Saarma, 2000); is required for the development, survival, and maintenance of DA neurons (Burke, 2006); and is required for the survival of kidneys, spinal cord motor neurons, sensory neurons, and cranial nerve motor neurons (Pichel et al., 1996; Granholm et al., 1997; Bates, 2000; Saarma, 2000; Malcangio, 2003; Sah et al., 2005). In vitro studies have shown that neurturin and persephin (PSPN), which are two family members of the GDNF family, but not GDNF, were capable of DA neuron induction in vitro in the presence of TGF-beta (Roussa et al., 2008). The authors found that GDNF was capable of rescuing the TGF-beta neutralization-dependent loss of tyrosine hydroxylase (TH)-pos. neurons, suggesting a role for GDNF in survival but not induction of the DA cell type. In the adult brain, high levels of Gdnf mRNA are likely restricted to ventral and dorsal striatum, thalamus, olfactory bulbs, and cingulate cortex (Trupp et al., 1997; Ron et al., 2005). Early research established that GDNF is required for high-affinity DA uptake, expression of the DA synthetic enzyme, tyrosine hydroxylase (TH), and neurite outgrowth of cultured midbrain DAergic neurons (Lin et al., 1993). Importantly for studies in our laboratory, GDNF is lower in the SN of aged rodents (Yurek and Fletcher-Turner, 2000) and in PD patients (Jenner and Olanow, 1998). In addition, intra-cerebral GDNF infusion promotes recovery of abnormal DA system morphology and related behavioral deficits in rodent and nonhuman primate models of PD (Bowenkamp et al., 1995; Gash et al., 1996; Lindner et al., 1995; Mandel et al., 1997; Cass et al., 1999; Granholm et al., 2000). GDNF's involvement in motor dysfunction and DA neuron degeneration has been established to the extent that it has recently been used in a clinical trial as a potential therapeutic agent for PD patients (Gill et al., 2003; Slevin et al., 2007). These clinical trials utilizing administration of GDNF demonstrated a significant increase in DA uptake in the putamen and patients demonstrated an improvement on the Unified Parkinson's Disease Rating Scale (UPDRS), of 42, or 38%, of the off- and on-medication states, respectively (Gill et al., 2003; Slevin et al., 2007). In addition, infusion of GDNF resulted in an increase in local (at site of infusion) TH-ir nerve fibers, as well as possible sprouting of nigral nerve fibers (Love et al., 2005). However, due to differences in probe placement, probe size, GDNF doses administered, as well as side effects related to the high levels of GDNF in non-related brain regions, GDNF clinical trials have been halted, even though the clinical data suggest a definite benefit from this neurotrophin in patients (Gill et al., 2003; Slevin et al., 2007). Data from animal studies, as well as the sparse clinical data available, still indicate that GDNF has significant beneficial effects on the SN DA system, especially during aging or neurotoxic exposure. These studies warrant the further assessment of GDNF as a possible therapeutic for the treatment of PD.
3.3 Effects of complete or partial Gdnf deletion on DA neurons
A Gdnf gene knockout murine model developed to explore the role of GDNF in fetal and postnatal development (see Pichel et al., 1996; Sanchez et al., 1996; Moore et al., 1996) was used in our studies to determine the role of the neurotrophic factor in aging- related changes in DA systems. Earlier studies indicated that in utero development of mesencephalic DA neurons was unaffected in homozygous Gdnf−/− mice (Pichel et al., 1996; Granholm et al., 1997). Although in vivo experiments on Gdnf−/− mice are not possible since they die postnatal day 1 due to renal agenesis, fetal grafts from Gdnf−/− mice indicate that postnatal survival of mesencephalic DA neurons is GDNF dependent (Granholm et al., 2000), suggesting that GDNF becomes important as the DA neuron establishes innervation and synaptic connections in striatal and cortical target regions. In fact, previous studies have established GDNF as a target-derived growth factor for the nigrostriatal system, with classical retrograde transport from the striatum back to SN DA cell bodies (Tomac et al., 1995). To examine the role of Gdnf reduction on DA systems throughout the lifespan, we utilized heterozygous mice (Gdnf+/−) which exhibit approximately 65% of GDNF levels seen in age-matched wildtype mice (Boger, et al., 2006; Griffin, et al., 2006).
In addition to its involvement in maintenance of neural function, GDNF is required for ureter bud formation and branching during metanephros development of the kidney, and is essential for proper innervation of the gastrointestinal tract (Pichel et al, 1996). Thus, mice lacking both copies of Gdnf lack kidneys and die at birth (Pichel et al., 1996). In contrast, Gdnf+/− mice with one Gdnf allele, although having 30% fewer glomeruli than WT controls, the glomeruli are of normal size (Cullen-McEwen et al., 2001) at 30 days of age, and are larger than those of age matched WT mice by 14 months of age. Important for studies utilizing Gdnf+/− mice, the compensatory increase in size of the glomeruli allowed normal kidney filtration rate (Cullen-McEwen et al., 2003; see also Boger et al., 2006).
To help establish whether or not general health problems contributed to the behavioral and DA system abnormalities observed in our studies on Gdnf+/− mice, body weights and kidney size, morphology, and function were evaluated at the ages assessed during the life span. Assessment of kidney size and BUN/creatinine levels in serum from mice of all ages examined in our experiments and discussed below confirmed these findings, and indicated normal kidney function up to at least 22 months of age. In addition, feeding behavior of Gdnf+/− mice was not impaired and they maintained body weights similar to WT mice raised on the same diet. Further, mortality rates for Gdnf+/− mice did not differ from WT littermates at least up to 24 months of age. Thus, in spite of the abnormal kidney function and enamel matrix, the peripheral dysfunctions appeared to be unrelated to the altered DA morphology and behavioral abnormalities noted in the studies discussed below (Boger et al., 2006).
The initial two studies to be discussed examined the interactive effects of genetic Gdnf reduction and the aging process (Zaman, et al., 2003; Boger, et al. 2006) and evaluated behavior (open field motor activity and rotorod performance), neurochemistry (monoamine neurotransmitter & metabolite levels, monoamine uptake), and morphology (TH immunohistochemistry with stereological cell count and sprouting density). The third study (Boger et al. 2007) discussed below introduced a methamphetamine (METH) challenge to examine the possibility of a METH × Gdnf Reduction × Aging interactive effect on the DA system, neuroinflammation and associated behaviors. The fourth study (Boger, et al., 2009) built upon the third study by further examining the role of inflammation as a possible mediator of the observed effects of a partial Gdnf gene deletion combined with a psychostimulant on DA systems and behavior, using minocyline treatment to reduce inflammation induced by METH administration and/or a partial Gdnf gene deletion (Boger et al., 2007). Two final studies (Zaman, et al., 2008; Boger, et al., 2008) to be discussed were to establish the role of the primary GDNF receptor GFRα1 in the abnormalities noted for the Gdnf+/− mice by including similar studies on Gfrα1+/− mice.
Results of the Boger, et al. (2006) study confirmed previous reports (e.g., Dean et al., 1981) that spontaneous motor activity, motor coordination, and SN TH-ir neurons declined with aging, from 4 – 20 months in our study. In addition, the study established that the aging-related decline occurred earlier for Gdnf+/− than for age-matched wildtype (WT) mice, suggesting that genetic reduction of GDNF enhances these aging-related changes. This result complements an earlier experiment (Zaman, et al., 2003) indicating that both horizontal and vertical movements declined more rapidly for Gdnf+/−compared to WT mice when tested over a one hour period at 18 months of age. Genotype effects on motor function and DA cell counts were first observed at 12 months of age in the Boger (2006) study. At later time periods, these values for WT mice declined to levels comparable to Gdnf+/− mice, suggesting that Gdnf reduction might accelerate the alterations observed with these parameters during the normal aging process in mice, or, alternatively, that early developmental problems in the Gdnf+/− mice related to the lower GDNF levels, gave rise to accelerated aging of these systems. Gdnf+/− mice, however, continued to have inferior performance on the motorically more complex rotorod task in comparison to age-matched WT controls noted below, again suggesting an accelerated aging process for motor systems. Importantly, the enhanced abnormalities for the Gdnf+/− mice were not associated with notable reductions in bodyweight, kidney function, or survival rate over the evaluation period negating a general health problem as causative for the observed effects. The decline in motor activity at the later age for WT mice suggested the possibility that striatal GDNF might become reduced in the normal aging process, thus accounting for the reduced motor function. Although we have not observed a significant aging-related reduction of GDNF in WT mice in our experiments (Griffin, et al., 2006), a trend towards lower GDNF levels was observed, and this interpretation is consistent with reported lower GDNF with age in the rat (Yurek et al., 2001) and with the rescue effect of administered GDNF on the health of DA neurons and aging-related motor deficits for both rats (Hebert and Gerhardt, 1997) and non-human primates (Gash et al., 1996). In addition, the absence of overall reduced GDNF levels with aging does not rule out the possibility of changes in its distribution, or receptor function with the aging process.
Performance on an accelerated rotorod by Gdnf+/− mice was inferior for aged (20 month) subjects, but not for young adult (8 month) mice compared with age-matched WT mice (Boger et al., 2006). The poorer performance by the aging Gdnf+/− mice was due to the more rapid fall time as the rotation speed increased from 8 to 40 revolutions per minute. It is important that performance of both genotypes improved across a three-day testing period, suggesting that the coordination deficit noted for the aged Gdnf+/− mice was not reflective of reduced motor learning ability. Interestingly, although the mean fall time for 20-month-old WT mice was less than that of their 8-month-old counterparts, the difference was not supported statistically under the conditions of the experiment. The absence of a deficit for the aging WT mice is somewhat surprising since motor coordination generally deteriorates relatively early in rodents (Bickford et al., 1992). Clearly, however, the aging Gdnf+/− mice exhibited a motor coordination deficit. The neuronal substrate for this deficit is unclear, but could be related to either noradrenergic or dopaminergic neurotransmission deficits, since both of these systems have been implicated in this particular behavioral task (Bickford, 1995; McEntee et al., 1987; Mason and Iversen, 1977), and both systems are abnormal in Gdnf+/− mice as noted below.
Consistent with the many studies indicating that DAergic pathways are susceptible to the aging process (Carlsson 1987; Seeman et al., 1987; Kish et al., 1992), the number of SNpc TH immunoreactive neurons declined with age (20%, 12 vs 20 month) as previously reported for C57BL/6 mice (McNeill and Koek, 1990; Tatton et al., 1991). In comparison, the aging related reduction in SNpc TH immunoreactive neurons occurred earlier and to a greater extent for Gdnf+/− mice (37.5%, 8 vs 12 month). Further morphological inspection revealed a far greater loss of fibers and terminals in this region for Gdnf+/− than WT mice suggesting that GDNF may be involved in the maintenance of the SN DAergic neuronal population, especially during aging. The loss of TH immunoreactive neurons and neurites in the SN region of 12 month old Gdnf+/− mice is illustrated in Figure 2. The reduction in DA availability in aging Gdnf+/− mice was consistent with a report (Airavaara et al., 2004) of increased striatal DAergic postsynaptic activity in Gdnf+/− mice. Even though Gdnf+/− mice demonstrated a greater reduction of SNpc TH immunoreactive neurons with age compared to WT mice, they did not exhibit degeneration of TH immunoreactive neurons in the VTA region of the midbrain. However, Zaman, et al (2003) reported a significant degeneration of NE neurons in the locus coeruleus (LC) and its projection areas in 18 month old Gdnf+/−mice compared to age-matched WT controls, suggesting that the LC-NE neurons are dependent upon GDNF for their maintenance as well. Supportive of LC NE neuron loss, NE levels were reduced in the locus coeruleus of 18-month-old Gdnf+/− mice compared to age-matched WT mice, as was NE uptake in tissue from the cerebellum or brain stem of the Gdnf+/− mice (Zaman et al., 2003). Since LC TH immunoreactive neurons (NE) and SN TH immunoreactive neurons (DA), are both involved with mediating motor activity, the impact of Gdnf reduction on either or both of the two neural systems could account for the motor deficits noted in the older Gdnf+/− mice as well as the greater cognitive deficits reported for aging mice of this genotype (Gerlai et al., 2003). Targeted delivery of GDNF to either LC or SN neurons in Gdnf+/− mice would reveal if one or both of these systems are involved in the motor deficit of aged Gdnf+/− mice, and should be addressed in future studies. Interestingly, LC NE neurons are known to deteriorate early on in patients with PS (see e.g. Mori et al., 2006; Braak and Del Tredici, 2008) and it has been suggested that these neurons may play an integral role in the subsequent loss of SN DA neurons. There is a clear connection between LC NE degeneration and alpha-synuclein-positive neuronal inclusion bodies in PD patients and a suggested connection between these two transmitters in the degenerative process seems likely (Mori et al., 2006). LC NE neurons are sensitive to neurotoxins in animal models just like the SN DA neurons, and are known to deteriorate along with DA neurons in e.g. rotenone models of PS (Lin et al., 2008). Our studies of the Gdnf+/− mice therefore suggest that this model may be useful also for studies of the connection between NE and DA transmitter systems during normal aging and degenerative disease in a systematic fashion.
Figure 2.
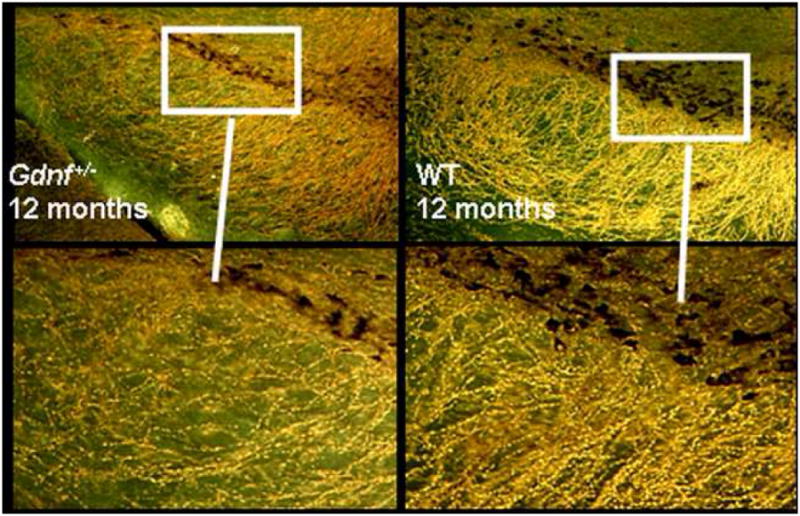
Darkfield illustration of the loss of TH-positive cell bodies and fibers in 12-month old Gdnf+/− mice (left side of panel) compared to age-matched WT controls (right side panel). Cell bodies in the Gdnf+/− mice are atrophied and a marked loss of TH-positive neurites in the pars reticulata can be seen.
Collectively, these studies indicate that Gdnf+/− mice have deficits in basal motor function and DA morphology common to the normal aging process, but occurring at an earlier time than noted for WT controls. We propose that this genetic reduction in Gdnf predisposes the DA system, and perhaps other monoamine systems, to an increased vulnerability to intrinsic and extrinsic derived insults which can occur during youth and can enhance the aging-related decline of midbrain dopamine neuronal function. Based on this information, a “dual-hit” hypothesis of neurodegeneration was proposed in which a combination of factors during early pre- or postnatal development can interact with Gdnf gene deletion and aging to enhance the vulnerability of DA neurons leading to compromised neuronal functioning during aging (see eg. Sarabi et al., 2003). The animal model used in these studies provides an assessment of whether genetic predisposition coupled with prenatal or postnatal neurotoxin exposure can enhance aging-related neurodegeneration resulting in motor disorders.
4. Genetic alterations combined with environmental toxins
4.1 The dual-hit hypothesis
Both genetic inheritance and sharing of a common environment in the same family can explain the increased risk of PD in relatives of PD patients compared with relatives of controls in familial aggregation studies (Logroscino, 2005). Therefore, the idea that PD and other neurodegenerative disorders are caused by a complex interaction between genetic predisposition and endotoxins or neurotoxins is not novel (Hawkes et al., 2008), and widely accepted in the field, as noted by a recent consensus statement by a group of scientists working in the fore-front of this field (see Bronstein et al., 2008). The dual-hit hypothesis states that multiple factors, which can be both external and internal, combined are responsible for the most common multiple system neurodegenerative disorders, in particular PS. Recently there has been great interest in developing animal models to test this dual-hit hypothesis, such as administering MPTP to alpha-synuclein, Parkin, and DJ-1 transgenic mice (Manning-Boğ and Langston, 2007). Langston and collaborators have minted the expression “fusion models” which recapitulate several pathological processes in the same animal model by integrating e.g. gene deletion and environmental factors to study gene-environment interactions (Manning-Bog and Langston, 2007). While most animal models for PS are associated with an acute loss of DA neurons following a massive dose of neurotoxins, Meredith and collaborators (Meredith et al., 2008) recently developed a chronic MPTP administration regimen leading to cellular inclusions, progressive DA cell loss, and other hallmarks for PS including oxidative stress, mitochondrial damage, and inflammatory processes. Carvey and his associates have developed a dual-hit rat model combining prenatal administration of an endotoxin, lipopolysaccharide (LPS) combined with postnatal neurotoxin administration, and discovered that the two challenges produced an additive effect on the SN DA system, resulting in almost all of the pathological hallmarks of PD (Zhu et al., 2007). The authors suggested that this increased vulnerability to the secondary neurotoxin exposure later in life could be the result of a life-long alteration in glutathione metabolism, an enzymatic pathway dysfunction implicated in PS. Another recent “dual-hit” model for PS (Richardson,et al., 2006) indicated that exposing pregnant and lactating mice to the pesticide dieldrin altered DA neurochemistry and exacerbated MPTP toxicity in the offspring. These are interesting models that can provide valuable information regarding additive effects of neurotoxic exposures, as well as identify biological mechanisms of the disease. However, to our knowledge, a dual-hit model had not been developed to investigate progressive PS in which a genetic alteration is combined with an environmental toxin.
4.2 Gdnf+/− combined with methamphetamine exposure
Because of its well-established toxic effects on DA systems, METH was used to evaluate the interactive effects of Gdnf reduction, psychostimulant exposure, and aging on DA system structure and function (Boger, et al., 2007). METH is a powerfully addictive psychostimulant that causes major damage to monoamine-containing axon terminals in the striatum (Ricaurte et al., 1982; Sonsalla et al. 1992). High METH doses adversely affect monoaminergic systems of rodents and primates reducing brain levels of DA and serotonin (5-HT), as well as their synthesis controlling enzymes, tyrosine and tryptophan hydroxylase, respectively (Hotchkiss and Gibb, 1980; Wagner et al., 1980; Bakhit et al., 1981; Davidson et al., 2001). METH is a substrate for monoamine transporters and is transported into DAergic terminals, initially increasing cytosolic DA release and ultimately depleting DA (Kita et al., 2003). Possible mediators contributing to METH toxicity on DA terminals include: 1) hyperthermia (Bowyer et al. 1992; O'Callaghan and Miller, 1994), 2) glutamate excitoxicity (Sonsalla et al. 1986, 1992; Mark et al., 2004), 3) increased production of reactive oxygen species (O'Dell et al., 1991; Yamamoto and Zhu, 1998; LaVoie and Hastings, 1999), and 4) microglia activation (LaVoie et al., 2004). The extensive METH abuse by young people has provided evidence that METH effects on DA and 5-HT are prolonged. For example, examination of the DA transporter protein (DAT) indicated only a 20% recovery in former METH abusers, who also demonstrated slower motor speed and deficits in verbal learning (Volkow et al., 2001a; 2001b). The Boger et al., (2007) study examined the interactive effects of METH exposure, a partial Gdnf gene deletion, and aging on DA system function using the procedures described above for the Genotype × AGE interaction. The general hypothesis guiding the study was that METH would exacerbate the more rapid decline of DA systems noted above for Gdnf+/− mice during the aging process.
The METH binge procedure used in this study was identical to that reported by Sonsalla, et al., (1992) and involved 4 IP injections of the drug (10 mg/kg) at two-hour intervals. The procedure elevated body temperature progressively across the four injections to the same extent for Gdnf+/− and WT mice when injected at 2.5 months of age. METH elevated motor activity after the first injection, but reduced activity after the fourth injection, to the same extent for both genotypes. Consistent with the similar hyperthermic and motor responses to the METH binge for the two genotypes, METH concentrations in blood and striatal tissue assessed at 30 min intervals for up to three hours after the final METH injection were similar for Gdnf+/− and WT mice. In spite of these genotypic similarities, the long-term toxic effects of METH in the Boger et al., (2007) study reflected in the behavioral, neurochemical, and morphological abnormalities noted below were substantially greater for Gdnf+/− mice.
Long-term behavioral effects of the METH binge were measured beginning two weeks after the binge. At this time, motor activity was unaffected by the METH binge for mice of either genotype. A pharmacological challenge with the selective DAT inhibitor, nomifensine, established that its stimulatory effect was attenuated for METH binge-treated mice, revealing a DA system dysfunction at two weeks after the METH binge, although not apparent in the baseline spontaneous locomotion parameter. At 9 months of age, total motor activity was significantly less in mice that had been treated with METH in early adulthood than in saline control mice, regardless of genotype. In contrast, at 12 months of age (Mid-Life), saline-treated Gdnf+/− mice displayed less motor activity than WT mice. Furthermore, METH-treated Gdnf+/− mice were less active than either Gdnf+/− mice treated with saline or WT mice treated with METH. Thus, there was an apparent interaction of partial Gdnf deletion, METH exposure, and age on motor activity. The interactive effects of the three factors on behavior were supplemented with a number of morphological and neurochemical effects noted below (Boger et al., 2007).
The METH binge procedure that reduced motor activity also reduced striatal tissue levels of serotonin (21%), DA (76%) and its metabolite, DOPAC (60%) when assessed in WT mice two weeks following the binge (Boger et al., 2007). Interestingly, at this younger age (3 months), DA and DOPAC levels in the striatum of mice not exposed to the METH binge were approximately 1.7 times greater for Gdnf+/− than WT mice. Two-weeks following METH administration, GDNF+/− mice demonstrated a proportionally greater reduction in striatal DA (88%) and DOPAC (75%) tissue content compared to METH-treated WT mice (DA: 76%, DOPAC: 60%). The result suggests that lower GDNF levels resulted in a greater METH-induced reduction of DA and its degradation metabolite. In contrast to the elevated striatal levels of DA and DOPAC for younger Gdnf+/− mice compared to the WT controls, when assessed at 12 months, both compounds, as well as serotonin, were lower in the striatum of Gdnf+/− mice. In addition, the METH-induced reductions of 5-HT, DA, and DOPAC noted for the three-month-old mice two weeks following the METH binge remained for mice assessed at 12 months of age, 6 months after the binge, and, importantly, the METH-induced reduction was greater in the Gdnf+/− mice, suggesting additive effects of the genetic and environmental factors (Boger et al., 2007).
As a follow-up to the monoamine changes associated with the METH binge and because METH is taken up by DA neurons via the dopamine transporter (DAT), DAT protein and its activity were examined in striatal tissue from the two genotypes at both 3 and 12 months of age. Although DAT protein levels (DAT-ir) did not differ according to either genotype or age, DAT uptake in synaptosomal preparations was greater in tissue from Gdnf+/− than WT mice at either age. Since METH gains entry to DA neurons via DAT, the enhanced DAT activity of Gdnf+/− mice provides a possible mechanism for the greater toxic effects of METH in this genotype and also supports the possibility that elevating GDNF might be protective against METH toxicity.
METH-induced morphological abnormalities were also greater in Gdnf+/−compared to WT mice, For example, tyrosine hydroxylase immunoreactivity (TH-ir) density in the medial striatum was lower in Gdnf+/− than WT mice at 3 months of age, and this genotypic difference was magnified two weeks after the METH binge (see Figure 3). In the lateral striatum, TH-ir was reduced to the same extent in both genotypes following METH treatment. At 12 months of age, striatal TH-ir density was lower in Gdnf+/− than WT mice regardless of the particular striatal area examined, and the METH-induced reduction in TH-ir density remained for the lateral, but not the medial striatum. Thus, METH toxicity was exacerbated in Gdnf+/− vs. WT mice. It is of interest to note, that no significant differences existed between the four treatment groups at either age in terms of TH-ir in the nucleus accumbens shell or core.
Figure 3.
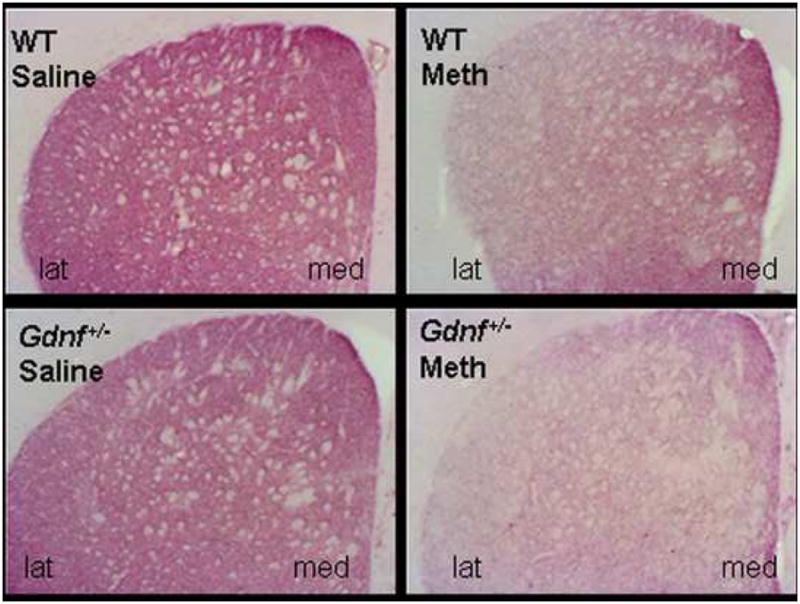
A METH binge at 2.5 months of age gave rise to a marked reduction in striatal TH immunostaining, as illustrated here two weeks following the toxin exposure. This effect was significantly increased in the Gdnf+/− mice compared to WT mice, especially in lateral striatum (lat) compared to medial striatum (med).
To test a possible mechanism for the reductions in TH-ir, striatal and SN were assessed for neuroinflammation using CD45 immunostaining two weeks following a METH binge to determine the extent of microglia activation (Boger et al., 2007). Microglial activation was greater in SN, but not striatum, of Gdnf+/− than WT mice regardless of whether they had been exposed to METH. The fact that METH resulted in the greatest elevation of CD45-ir in the SN of Gdnf+/− mice, suggests the possibility that lower GDNF levels might contribute to a greater inflammatory response to METH. The absence of METH-induced microglial activation in the striatum in this study differ from previous reports demonstrating that microglia were activated in the striatum, but not SN, within 1-6 days of a METH binge (LaVoie et al., 2004). The different outcomes for the two studies suggest that the microglial activation in the striatum had resolved 2 weeks following METH exposure whereas the SN microgliosis took longer than a week to develop.
4.3 Activated inflammatory pathways in Gdnf+/− mice and methamphetamine exposed mice are attenuated by minocycline
As mentioned above (section 4.2), mice with a partial genetic deletion of Gdnf displayed increased neuroinflammation in the SN as early as 3 months of age (Boger et al., 2007). Therefore, a third study (Boger, et al., 2009) was directed toward further assessment of biological mechanisms involved in the Gdnf+/− and METH enhanced reduction in TH-ir, specifically neuroinflammation. In this study, 2.5 month old Gdnf+/− and WT mice were exposed to the METH binge described above. Starting 24 hrs after the METH binge, half of the mice in each group were injected daily for two weeks with minocycline (45mg/kg, IP) providing a 2 (Genotype) × 2 (METH Exposure) × 2 (Minocycline Treatment) experimental design. Minocycline has established anti- inflammatory (Tikka & Koistinaho, 2000; Tikka, et al. 2001) and anti-apoptotic (Zhu, et al., 2002; Wang, et al., 2004) properties and was administered in a regimen demonstrated to be neuroprotective in the 6-OHDA lesion rodent model of PD (Quintero, et al., 2006).
Confirming the earlier report noted above (Boger et al., 2007), the 3-month-old Gdnf+/− mice had greater microglial activation in SN and less TH-ir in medial striatum in comparison to age matched WT controls, and the METH binge produced similar SN microglia activation and striatal TH-ir reductions in both Gdnf+/− and WT mice. The earlier study was extended by examining the p38 MAPK pathway in the SN as a possible mediator of the impact of a partial Gdnf gene deletion and/or METH binge on the DA system. Results of the study indicated an increased phosphorylation of p38 MAPK in the SN of young adult Gdnf+/− compared to WT mice, an effect enhanced by the METH binge. The enhanced p38 MAPK phosphorylation in combination with the SN microglial activation (CD45 elevation) suggests that the p38 MAPK pathway may be activated by cell stressors such as the neuroinflammation induced by genetic Gdnf reduction and the neurotoxin, METH, in the study (Boger et al., 2009). These data, coupled with the previous studies, demonstrate that SN neuroinflammation precedes DA cell loss in Gdnf+/− mice, thus providing a possible mechanism for neurodegeneration in these mice.
Of particular note in this study was that the minocycline treatment paradigm, with demonstrated efficacy in animal models of neurotoxicity, attenuated the SN microglial activation and the phospho-p38 MAPK elevation associated with both the genetic Gdnf reduction in the Gdnf+/− mice and with the METH binge (Boger et al., 2009). Furthermore, this anti-inflammatory and anti- apoptotic antibiotic counteracted the lower TH-ir levels in the medial striatum noted in Gdnf+/ vs. WT mice; however, minocycline did not attenuate the striatal TH-ir loss induced by METH despite the reduced microglial activation. The finding that minocycline attenuated the TH-ir reduction associated with genetic Gdnf reduction, but not with METH treatment, suggests the likelihood of different mechanisms of TH-ir regulation and depletion. The neurotoxic action of METH on DA systems in spite of the minocycline-induced decrease in microglial activation is consistent with a report showing that METH- or MPTP-induced DA neurotoxicity was present in the absence of other inflammatory markers including interleukin-6, F4/80, & interleukin-1α (Sriram, et al., 2006). The latter study indicated a role for TNFα in the protective effects of minocycline against MPTP toxicity, suggesting that this or other pro-inflammatory cytokines might contribute to the METH-induced striatal damage. Thus, the Boger et al., (2009) study established that minocycline reduced microglial activation and phospho-p38 MAPK in the SN associated with either Gdnf reduction or the METH binge. However, minocycline attenuated the reduction in TH-ir immunoreactivity in only the Gdnf+/− mice, having no effect on the methamphetamine-induced TH-ir reduction. These data indicate that inflammation plays a crucial role in the loss of DA phenotype in Gdnf+/− mice, but does not mediate or exacerbate METH neurotoxicity. Thus, two different degenerative processes interact in this model, and future studies will seek to discover the multifactorial biological mechanisms underlying the interaction. .
An additional interesting observation regarding longterm effects of METH exposure recently reported by Melega et al (2007) indicated that even though TH immunoreadtivity recovered to base-line levels after exposing young non-human primates to METH, iron levels remained elevated to levels seen in aged monkeys. This result suggests the possibility of a continuing oxidative stress process which might prove important when degenerative processes continue with age. This possiblilty is being investigated in Gdnf+/− mice treated with METH. Recently, a large multi-center phase II study supported by the National Institute of Neurological Disorders and Stroke (NINDS) regarding minocycline treatment for patients with PD by the NET-PD Futility study group (Racettet et al., 2008) reported that minocycline was not futile in slowing down the progression of disability in PD subjects after 12-18 months of treatment. The dose of minocycline used for this multi-center study was 200 mg/d, and data from a small, 18-month follow-up study showed that minocycline does not demonstrate safety concerns that would preclude a large, phase III efficacy trial, even though some concerns regarding decreased tolerability of minocycline was noted. A possible reason for the lack of efficacy of minocycline in the trial goes back to the multi-faceted etiology of the disease. As in the case of Gdnf gene deletion combined with METH treatment, the disease is likely to present several, parallel degenerative processes in different individuals, which need to be addressed using a “cocktail” of different neuroprotective agents.
4.4 Effects of a partial GDNF receptor deletion
Given the established importance of GDNF in maintaining dopaminergic neurotransmitter systems noted above, two additional studies (Zaman, et al., 2008; Boger, et al., 2008) examined the nigrostriatal system and associated behaviors of mice with a genetic reduction of the GDNF high affinity receptor, Gfrα-1 (Gfrα-1+/−) to determine if they displayed abnormalities similar to those noted for the Gdnf+/− mice. In the initial experiment (Zaman, et al., 2008), motor activity and the stimulatory effects of a dopamine (DA) D1 receptor agonist (SKF 82958) were assessed in Gfrα-1+/− and WT mice at 8 and 18 months-of-age. In additional mice, striatal monoamine concentrations and dopaminergic nerve terminals and the number of DA neurons in substantia nigra (SN) were assessed at the same ages. Results supported the importance of the GFRα-1 receptor in maintaining normal function of the nigrostriatal DAergic system, with deficits observed for Gfrα-1+/− mice at both ages. Motor activity was lower for Gfrα-1+/− compared to their WT controls as was found for the Gdnf+/− mice in the experiment discussed above, and the stimulatory effects of the DA agonist were enhanced for the older Gfrα-1+/− mice. The greater motor activity for the aged Gfrα-1+/− mice injected with SKF 82958 rules out abnormal motor function as accountable for the reduced motor activity. Dopamine levels were lower in the striatum of the Gfrα-1+/− mice than WT controls at both ages, and the number of TH immunoreactive neurons in the SN was reduced most substantially in the older Gfrα-1+/− mice. The combined behavioral, pharmacological probe, neurochemical, and morphological measures provide evidence indicative of a more rapid aging-related decline in DAergic system function for Gfrα-1+/− mice as was noted for Gdnf+/− mice. The similar deficiencies for GFRα-1+/− and Gdnf+/− mice suggest that the GFRα-1 receptor is necessary for GDNF to maintain normal function of the nigrostriatal DAergic system. Although the study did not define the precise mechanism(s) for the aging-related changes in the DAergic system, it did establish that genetic reductions in the Gfrα-1 receptor can contribute to degenerative changes observed in this system during the aging process. The neural cell adhesion molecule (NCAM) has been identified as an alternative signaling receptor for GDNF (Cao et al., 2008) and was found to be involved in the promotive effect of GDNF on neurite outgrowth in DA neuron cultures. However, our findings presented here unequivocally establishe the role for the GFRα-1 receptor in maintenance of DA neuron health with aging.
To follow-up the GFRα1 experiment above, Boger, et al. (2008) examined the effect of a DA neurotoxic agent, MPTP, on DA system function of aged Gfrα-1+/− and WT mice similar to the METH effects on Gdnf+/− mice discussed above. In this study, an established neurotoxic regimen (4 daily injections) of MPTP was administered to 26-month-old male Gfrα1+/− and WT mice and its effects on locomotion, the number of TH-positive neurons in SN and striatum, and the expression of inflammation via CD45 and phospho-p38 MAPK immunostaining were determined. Control mice of each genotype received saline to form a 2 (Genotype) × 2 (MPTP) experimental design. As noted above, Gfrα1+/− mice were much less active than their WT littermates. When tested two days after the final injection, MPTP-treated mice had increased locomotion regardless of genotype. Gfrα1+/− mice had fewer TH immunoreactive neurons in the SN and reduced TH-ir in the striatum. Providing a possible mediation of the DA abnormalities, Gfrα1+/− mice exhibited a significantly increased expression of CD45 and phospho-p38 MAPK inflammatory markers in the SN. As noted above for METH effects on Gdnf+/−mice, MPTP exacerbated the DA and behavioral abnormalities of Gfrα1+/− mice with DA abnormalities and inflammatory response being greatest in MPTP treated Gfrα1+/− mice. The results of the two GFRα1 reduction experiments compliment the Gdnf gene deletion studies in establishing the importance of the GDNF system on maintenance of the DAergic system during the aging process, and further supported our overall dual-hit hypothesis as presented with two different neurotoxins (METH and MPTP) herein.
5. Conclusions and future directions
The above studies from our laboratories were consistent with the previously established aging-related decline in function and structure of SN DA neurons as well as aging-related reductions in motor function, and further established that a genetic reduction in GDNF or its receptor exacerbated these aging-related changes. Although not the focus of the studies, the reduction in GDNF was also accompanied by lower levels of additional monoamine neurotransmitters, norepinephrine and serotonin; however the reduction of GDNF had no impact on the VTA-nucleus accumbens DA pathway. These results indicate that a genetic reduction of Gdnf increases the vulnerability of SN DA neurons to many different challenges associated with the aging process, including oxidative stress, general growth factor loss, reduced neuroplasticity, and increased inflammatory markers. A challenge with the psychostimulant, METH, established that the GDNF reduction also increased the vulnerability of the nigrostriatal DA system to a well-established neurotoxin, again indicating the GDNF requirement for responding to neurotoxic events. The addition of the METH study also established the interactive effects of intrinsic (GDNF reduction) and extrinsic (METH exposure) challenges on an aging neurotransmitter system. A graphic summarization of this work is outlined in Fig 4 indicating that intrinsic challenges such as GDNF reduction and extrinsic challenges, such as early METH exposure, can interact to exacerbate the aging related declining function of the nigrostriatal DA system resulting in abnormal motor behavior, and also illustrates the fact that these multiple damages can have either additive or synergistic effects, although our studies suggest a “super-additive” effect of the genetic and environmental factors. The studies on mice with a deficiency of GDNF receptor, GFRα1, established similar abnormalities for the GDNF receptor-deprived animals, thus confirming the impact of reductions in the neurotrophic factor on maintaining the nigrostriatal DA system function during challenges. Finally, the studies suggest potential factors that might mediate the abnormalities associated with GDNF reduction, including enhanced inflammation and dopamine transporter activity. Studies are currently conducted in our laboratories to determine a possible role of oxidative stress and altered neuronal signaling that could result in accelerated age-related DAergic damage as observed in Gdnf+/− mice. These continued studies would aid in a better understanding of nigrostriatal dopamine system degeneration as well as help in the discovery of therapeutic strategies to aid in the treatment of motor disorders, including Parkinson's disease. We are hopeful that this dual-hit model can be utilized for high- and low-throughput drug discovery testing to explore drugs which may increase levels of endogenous GDNF in the brain, since the model represents a progressive genetic/environmental model for age-related PS. Further, our model exhibits LC NE degeneration as well, and can be utilized for studies of interaction between the NE and DA transmitter systems, especially during aging and degenerative stages.
Acknowledgments
The authors would like to thank Ms. Claudia Umphlet, Ms. Selena Sumner, and Mr. Alfred Moore for excellent technical assistance, and Dr. Baerbel Rohrer for directing the knockout animal core. The work was supported by a program project grant from the National Institutes on Aging (AG023630).
Abbrevation List
- PD
Parkinson's disease
- PS
Parkinsonian syndrome
- DA
Dopamine
- NE
Norepinephrine
- SN
substantia nigra
- GDNF
Glial Cell line-derived neurotrophic factor
- NGF
Nerve Growth Factor
- METH
methamphetamine
- TH
tyrosine hydroxylase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Airavaara M, Planken A, Gäddnäs H, Piepponen TP, Saarma M, Ahtee L. Increased extracellular dopamine concentrations and FosB/DeltaFosB expression in striatal brain areas of heterozygous GDNF knockout mice. Eur J Neurosci. 2004;20:2336–2344. doi: 10.1111/j.1460-9568.2004.03700.x. [DOI] [PubMed] [Google Scholar]
- Bakhit C, Morgan ME, Peat MA, Gibb JW. Long-term effects of methamphetamine on the synthesis and metabolism of 5-hydroxytryptamine in various regions of the rat brain. Neuropharmacology. 1981;20:1135–1140. doi: 10.1016/0028-3908(81)90053-8. [DOI] [PubMed] [Google Scholar]
- Bannon MJ. The dopamine transporter: role in neurotoxicity and human disease. Toxicol Appl Pharmacol. 2005;204(3):355–60. doi: 10.1016/j.taap.2004.08.013. Review. [DOI] [PubMed] [Google Scholar]
- Baron MS. Movement disorders in the older patient: differential diagnosis and general management. Cleveland Clinic Journal of Medicine. 2005;72(3):S38–51. doi: 10.3949/ccjm.72.suppl_3.s38. [DOI] [PubMed] [Google Scholar]
- Bates CM. Kidney development: regulatory molecules crucial to both mice and men. Molecular Genetics & Metabolism. 2000;71:391–396. doi: 10.1006/mgme.2000.3072. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Beckett LA, Murray AM, Shannon KM, Goetz CG, Pilgrim DM, Evans DA. Prevalence of parkinsonian signs and associated mortality in a community population of older people. N Engl J Med. 1996;334:71–76. doi: 10.1056/NEJM199601113340202. [DOI] [PubMed] [Google Scholar]
- Bickford P, Heron C, Young DA, Gerhardt GA, De La Garza R. Impaired acquisition of novel locomotor tasks in aged and norepinephrine-depleted F344 rats. Neurobiol Aging. 1992;13:475–481. doi: 10.1016/0197-4580(92)90075-9. [DOI] [PubMed] [Google Scholar]
- Bickford P. Aging and motor learning: a possible role for norepinephrine in cerebellar plasticity. Rev Neurosci. 1995;6:35–46. doi: 10.1515/revneuro.1995.6.1.35. [DOI] [PubMed] [Google Scholar]
- Biskup S, Gerlach M, Kupsch A, Reichmann H, Riederer P, Vieregge P, Wullner U, Gasser T. Genes associated with Parkinson syndrome. J Neurol. 2008;255(5):8–17. doi: 10.1007/s00415-008-5005-2. [DOI] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Huang P, Zaman V, Smith AC, Hoffer BJ, Tomac AC, Granholm AC. A partial GDNF depletion leads to earlier age-related deterioration of motor function and tyrosine hydroxylase expression in the substantia nigra. Exp Neurol. 2006;202:336–347. doi: 10.1016/j.expneurol.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Patrick KS, Ramamoorthy S, Denehy ED, Zhu H, Pacchioni AM, Granholm AC, McGinty JF. Long-term consequences of methamphetamine exposure in young adults are exacerbated in glial cell line-derived neurotrophic factor heterozygous mice. J Neurosci. 2007;27:8816–8825. doi: 10.1523/JNEUROSCI.1067-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Zaman V, Hoffer B, Granholm AC. Differential effects of the dopamine neurotoxin MPTP in animals with a partial deletion of the GDNF receptor, GFR alpha1, gene. Brain Res. 2008;1241:18–28. doi: 10.1016/j.brainres.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boger HA, Middaugh LD, Granholm AC, McGinty JF. Minocycline restores striatal tyrosine hydroxylase in GDNF heterozygous mice but not in methamphetamine-treated mice. Neurobiol Dis. 2009;33:459–466. doi: 10.1016/j.nbd.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowenkamp KE, Hoffman AF, Gerhardt GA, Henry MA, Biddle PT, Hoffer BJ, Granholm AC. Glial cell line-derived neurotrophic factor supports survival of injured midbrain dopaminergic neurons. J Comp Neurol. 1995;355:479–489. doi: 10.1002/cne.903550402. [DOI] [PubMed] [Google Scholar]
- Bowyer JF, Tank AW, Newport GD, Slikker W, Jr, Ali SF, Holson RR. The influence of environmental temperature on the transient effects of methamphetamine on dopamine levels and dopamine release in rat striatum. J Pharmacol Exp Ther. 1992;260:817–824. [PubMed] [Google Scholar]
- Braak H, Rub U, Gai WP, Del Tredici K. Idiopathic Parkinson's disease: possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm. 2003;110:517–536. doi: 10.1007/s00702-002-0808-2. [DOI] [PubMed] [Google Scholar]
- Braak H, Sastre M, Bohl JR, de Vos RA, Del Tredici K. Parkinson's disease: lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathologica. 2007;113:421–429. doi: 10.1007/s00401-007-0193-x. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. Cortico-basal ganglia-cortical circuitry in Parkinson's disease reconsidered. Exp Neuro. 2008;212:226–229. doi: 10.1016/j.expneurol.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Bronstein J, Carvey P, Chen H, Cory-Slechta D, DiMonte D, Duda J, English P, Goldman S, Grate S, Hansen J, Hoppin J, Jewell S, Kamel F, Koroshetz W, Langston JW, Logroscino G, Nelson L, Ravina B, Rocca W, Ross GW, Schettler T, Schwarzschild M, Scott B, Seegal R, Singleton A, Steenland K, Tanner CM, Van Den Eeden S, Weisskopf M. 2008. Meeting report: consensus statement-Parkinson's disease and the environment: collaborative on health and the environment and Parkinson's Action Network (CHE PAN) conference 26-28 June 2007. Environ Health Perspect. 2009 Jan;117(1):117–21. doi: 10.1289/ehp.11702. Epub 2008 Aug 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke RE. Ontogenic cell death in the nigrostriatal system. Cell & Tissue Res. 2004;318:63–72. doi: 10.1007/s00441-004-0908-4. [DOI] [PubMed] [Google Scholar]
- Burke RE. GDNF as a candidate striatal target-derived neurotrophic factor for the development of substantia nigra dopamine neurons. J Neural Transm Suppl. 2006;(70):41–5. doi: 10.1007/978-3-211-45295-0_8. Review. [DOI] [PubMed] [Google Scholar]
- Cao JP, Wang HJ, Yu JK, Yang H, Xiao CH, Gao DS. Involvement of NCAM in the effects of GDNF on the neurite outgrowth in the dopamine neurons. Neuroscience Research. 2008;61(4):390–7. doi: 10.1016/j.neures.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Carlsson A, Lindquist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180:1200. doi: 10.1038/1801200a0. [DOI] [PubMed] [Google Scholar]
- Carlsson A. Development of new pharmacological approaches in Parkinson's disease. Adv Neurol. 1987;45:513–518. [PubMed] [Google Scholar]
- Cass WA, Harned ME, Bailey SL. Enhanced effects of 6-hydroxydopamine on evoked overflow of striatal dopamine in aged rats. Brain Res. 2002;938:29–37. doi: 10.1016/s0006-8993(02)02481-2. [DOI] [PubMed] [Google Scholar]
- Cass WA, Manning MW. GDNF protection against 6-OHDA-induced reductions in potassium-evoked overflow of striatal dopamine. J Neurosci. 1999;19:1416–1423. doi: 10.1523/JNEUROSCI.19-04-01416.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier TJ, Dung Ling Z, Carvey PM, Fletcher-Turner A, Yurek DM, Sladek JR, Kordower JH. Striatal trophic factor activity in aging monkeys with unilateral MPTP-induced parkinsonism. Exp Neurol. 2005;191(1):S60–S67. doi: 10.1016/j.expneurol.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Cullen-McEwen LA, Drago J, Bertram JF. Nephron endowment in glial cell line-derived neurotrophic factor (GDNF) heterozygous mice. Kidney Int. 2001;60:31–36. doi: 10.1046/j.1523-1755.2001.00767.x. [DOI] [PubMed] [Google Scholar]
- Cullen-McEwen LA, Kett MM, Dowling J, Anderson WP, Bertram JF. Nephron number, renal function, and arterial pressure in aged GDNF heterozygous mice. Hypertension. 2003;41:335–340. doi: 10.1161/01.hyp.0000050961.70182.56. [DOI] [PubMed] [Google Scholar]
- Dahlström A, Fuxe K. Evidence for the existence of monoamine-containing neurons in the central nervous system. I: Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol Scand. 1964;62(Suppl 232):1–55. [PubMed] [Google Scholar]
- Davidson C, Gow AJ, Lee TH, Ellinwood EH. Methamphetamine neurotoxicity: necrotic and apoptotic mechanisms and relevance to human abuse and treatment. Brain Res Brain Res Rev. 2001;36:1–22. doi: 10.1016/s0165-0173(01)00054-6. [DOI] [PubMed] [Google Scholar]
- Dejesus OT, Endres CJ, Shelton SE, Nickles RJ, Holden JE. Noninvasive assessment of aromatic L-amino acid decarboxylase activity in aging rhesus monkey brain in vivo. Synapse. 2001;39:58–63. doi: 10.1002/1098-2396(20010101)39:1<58::AID-SYN8>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- de Vicente JC, Cabo R, Ciriaco E, Laurà R, Naves FJ, Silos-Santiago I, Vega JA. Impaired dental cytodifferentiation in glial cell-line derived growth factor (GDNF) deficient mice. Ann Anat. 2002;184:85–92. doi: 10.1016/S0940-9602(02)80041-3. [DOI] [PubMed] [Google Scholar]
- Eggers AE. Why do Alzheimer's disease and Parkinson's disease target the same neurons? Med Hypotheses. 2009 Jun;72(6):698–700. doi: 10.1016/j.mehy.2008.12.047. Epub 2009 Feb 27. [DOI] [PubMed] [Google Scholar]
- Falck B, Hillarp NA, Torp A. Some observations on the histology and histochemistry of the chromaffin cells probably storing dopamine. J Histochem Cytochem. 1959;7:323–328. doi: 10.1177/7.5.323. [DOI] [PubMed] [Google Scholar]
- Fearnley JM, Lees AJ. Ageing and Parkinson's disease: substantia nigra regional selectivity. Brain. 1991;114:2283–2301. doi: 10.1093/brain/114.5.2283. [DOI] [PubMed] [Google Scholar]
- Fumagalli F, Racagni G, Riva MA. Shedding light into the role of BDNF in the pharmacotherapy of Parkinson's disease. Pharmacogenomics Journal. 2006;6:95–104. doi: 10.1038/sj.tpj.6500360. [DOI] [PubMed] [Google Scholar]
- Gaenslen A, Gasser T, Berg D. Nutrition and the risk for Parkinson's disease: review of the literature. J Neural Trans. 2008;115:703–13. doi: 10.1007/s00702-007-0005-4. [DOI] [PubMed] [Google Scholar]
- Gash DM, Zhang Z, Ovadia A, Cass WA, Yi A, Simmerman L, Russell D, Martin D, Lapchak PA, Collins F, Hoffer BJ, Gerhardt GA. Functional recovery in parkinsonian monkeys treated with GDNF. Nature. 1996;380:252–255. doi: 10.1038/380252a0. [DOI] [PubMed] [Google Scholar]
- Gerlai R, McNamara A, Choi-Lundberg DL, Armanini M, Ross J, Powell-Braxton L, Phillips HS. Impaired water maze learning performance without altered dopaminergic function in mice heterozygous for the GDNF mutation. Eur J Neurosci. 2003;14:1153–1163. doi: 10.1046/j.0953-816x.2001.01724.x. [DOI] [PubMed] [Google Scholar]
- Gibb WR, Lees AJ. Anatomy, pigmentation, ventral and dorsal subpopulations of the substantia nigra, and differential cell death in Parkinson's disease. J Neurol Neurosurg Psychiatry. 1991;54:388–396. doi: 10.1136/jnnp.54.5.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill SS, Patel NK, Hotton GR, O'Sullivan K, McCarter R, Bunnage M, Brooks DJ, Svendsen CN, Heywood P. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med. 2003;9:589–595. doi: 10.1038/nm850. [DOI] [PubMed] [Google Scholar]
- Granholm ACh, Srivastava N, Mott JL, Henry S, Henry M, Westphal H, Pichel JP, Shen L, Hoffer BJ. Morphological alterations in the peripheral and central nervous systems of mice lacking glial cell line-derived neurotrophic factor (GDNF): Immunohistochemical studies. J Neurosci. 1997;17(3):1168–1178. doi: 10.1523/JNEUROSCI.17-03-01168.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granholm AC, Reyland M, Albeck D, Sanders L, Gerhardt G, Hoernig G, Shen L, Westphal H, Hoffer B. Glial cell line-derived neurotrophic factor is essential for postnatal survival of midbrain dopamine neurons. J Neurosci. 2000;20:3182–3190. doi: 10.1523/JNEUROSCI.20-09-03182.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granholm ACh, Boger HA, Emborg ME. Mood, memory, and movement: An age-related neurodegenerative complex? Current Aging Science. 2008;1:133–139. doi: 10.2174/1874609810801020133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WC, 3rd, Boger HA, Granholm AC, Middaugh LD. Partial deletion of glial cell line-derived neurotrophic factor (GDNF) in mice: Effects on sucrose reward and striatal GDNF concentrations. Brain Res. 2006;1068:257–260. doi: 10.1016/j.brainres.2005.10.080. [DOI] [PubMed] [Google Scholar]
- Hawkes CH, Del Tredici K, Braak H. Parkinson's disease: a dual-hit hypothesis. Neuropathology & Applied Neurobiology. 2008;33:599–614. doi: 10.1111/j.1365-2990.2007.00874.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert MA, Gerhardt GA. Behaivoral and neurochemical effects of intranigral administration of glial cell line-derived neurotrophic factor on aged Fischer 344 rats. J Pharm Exp Ther. 1997;282:760–768. [PubMed] [Google Scholar]
- Hebert MA, Gerhardt GA. Normal and drug-induced locomotor behavior in aging: comparison to evoked DA release and tissue content in fischer 344 rats. Brain Res. 1998;797:42–54. doi: 10.1016/s0006-8993(98)00370-9. [DOI] [PubMed] [Google Scholar]
- Herzog CD, Dass B, Gasmi M, Bakay R, Stansell JE, Tuszynski M, Bankiewicz K, Chen EY, Chu Y, Bishop K, Kordower JH, Bartus RT. Transgene expression, bioactivity, and safety of CERE-120 (AAV2-neurturin) following delivery to the monkey striatum. Mol Ther. 2008 Oct;16(10):1737–44. doi: 10.1038/mt.2008.170. Epub 2008 Aug 26. [DOI] [PubMed] [Google Scholar]
- Hotchkiss A, Gibb JW. Blockade of methamphetamine-induced depression of tyrosine hydroxylase by GABA transaminase inhibitors. Eur J Pharmacol. 1980;66:201–205. doi: 10.1016/0014-2999(80)90143-0. [DOI] [PubMed] [Google Scholar]
- Ingram DK. Age-related decline in physical activity: generalization to nonhumans. Med Sci Sports Exerc. 2000;32:1623–1629. doi: 10.1097/00005768-200009000-00016. [DOI] [PubMed] [Google Scholar]
- Isacson O, Kordower JH. Future of cell and gene therapies for Parkinson's disease. Ann Neurol. 2008 Dec;64(2):S122–38. doi: 10.1002/ana.21473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenner P, Olanow CW. Understanding cell death in Parkinson's disease. Ann Neurol. 1998;44:S72–S84. doi: 10.1002/ana.410440712. [DOI] [PubMed] [Google Scholar]
- Kaasinen V, Rinne JO. Functional imaging studies of dopamine system and cognition in normal aging and Parkinson's disease. Neurosci Biobehav Rev. 2002;26:785–793. doi: 10.1016/s0149-7634(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Kordower JH, Collier TJ. Age and region-specific responses of microglia, but not astrocytes, suggest a role in selective vulnerability of dopamine neurons after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure in monkeys. GLIA. 2008;56:1199–1214. doi: 10.1002/glia.20690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, Rajput A, Deck JH, Hornykiewicz O. Aging produces a specific pattern of striatal dopamine loss: implications for the etiology of idiopathic Parkinson's disease. J Neurochem. 1992;58:642–648. doi: 10.1111/j.1471-4159.1992.tb09766.x. [DOI] [PubMed] [Google Scholar]
- Kita T, Wagner GC, Nakashima T. Current research on methamphetamine-induced neurotoxicity: animal models of monoamine disruption. J Pharmacol Sci. 2003;92:178–195. doi: 10.1254/jphs.92.178. Review. [DOI] [PubMed] [Google Scholar]
- Kluger A, Gianutsos JG, Golomb J, Ferris SH, Reisberg B. Motor/psychomotor dysfunction in normal aging, mild cognitive decline, and early Alzheimer's disease: diagnostic and differential diagnostic features. Int Psychogeriatr. 1997;9(1):307–316. doi: 10.1017/s1041610297005048. discussion 317-321. [DOI] [PubMed] [Google Scholar]
- Krieglstein K, Suter-Crazzolara C, Unsicker K. Development of mesencephalic dopaminergic neurons and the transforming growth factor-beta superfamily. J Neural Transm Suppl. 1995;46:209–216. [PubMed] [Google Scholar]
- LaVoie MJ, Card JP, Hastings TG. Microglial activation preceded dopamine terminal pathology in methamphetamine-induced neurotoxicity. Exp Neurol. 2004;187:47–57. doi: 10.1016/j.expneurol.2004.01.010. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Brice A. Parkinson's disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet. 2009;18(R1):R48–59. doi: 10.1093/hmg/ddp012. Review. [DOI] [PubMed] [Google Scholar]
- Levi-Montalcini R. The nerve growth factor and the neuroscience chess board. Archives Italiennes de Biologie. 2003;141:85–88. [PubMed] [Google Scholar]
- Lin LF, Doherty DH, Lile JD, Bektesh S, Collins F. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 1993;260:1130–1132. doi: 10.1126/science.8493557. [DOI] [PubMed] [Google Scholar]
- Lin CH, Huang JY, Ching CH, Chuang JI. Melatonin reduces the neuronal loss, downregulation of dopamine transporter, and upregulation of D2 receptor in rotenone- induced parkinsonian rats. Journal of Pineal Research. 2008;44(2):205–13. doi: 10.1111/j.1600-079X.2007.00510.x. [DOI] [PubMed] [Google Scholar]
- Lindner MD, Winn SR, Baetge EE, Hammang JP, Gentile FT, Doherty E, McDermott PE, Frydel B, Ullman MD, Schallert T, et al. Implantation of encapsulated catecholamine and GDNF-producing cells in rats with unilateral dopamine depletions and parkinsonian symptoms. Exp Neurol. 1995;132:62–76. doi: 10.1016/0014-4886(95)90059-4. [DOI] [PubMed] [Google Scholar]
- Logroscino G. The role of early life environmental risk factors in Parkinson disease: what is the evidence? Environmental Health Perspectives. 2005;113(9):1234–8. doi: 10.1289/ehp.7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahalik TJ, Hahn WE, Clayton GH, Owens GP. Programmed cell death in developing grafts of fetal substantia nigra. Exp Neurol. 1994;129:27–36. doi: 10.1006/exnr.1994.1144. [DOI] [PubMed] [Google Scholar]
- Love S, Plaha P, Patel NK, Hotton GR, Brooks DJ, Gill SS. 2005 Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat Med. 2005 Jul;11(7):703–4. doi: 10.1038/nm0705-703. [DOI] [PubMed] [Google Scholar]
- Ma SY, Ciliax BJ, Stebbins G, Jaffar S, Joyce JN, Cochran EJ, Kordower JH, Mash DC, Levey AI, Mufson EJ. Dopamine transporter-immunoreactive neurons decrease with age in the human substantia nigra. J Comp Neurol. 1999;409(1):25–37. doi: 10.1002/(sici)1096-9861(19990621)409:1<25::aid-cne3>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Malcangio M. GDNF and somatostatin in sensory neurones. Current Opinion in Pharmacology. 2003;3:41–45. doi: 10.1016/s1471-4892(02)00007-3. [DOI] [PubMed] [Google Scholar]
- Mandavilli BS, Ali SF, Van Houten B. DNA damage in brain mitochondria caused by aging and MPTP treatment. Brain Res. 2000;885:45–52. doi: 10.1016/s0006-8993(00)02926-7. [DOI] [PubMed] [Google Scholar]
- Mandel RJ, Spratt SK, Snyder RO, Leff SE. Midbrain injection of recombinant adeno-associated virus encoding rat glial cell line-derived neurotrophic factor protects nigral neurons in a progressive 6-hydroxydopamine-induced degeneration model of Parkinson's disease in rats. Proc Natl Acad Sci U S A. 1997;94:14083–14088. doi: 10.1073/pnas.94.25.14083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandel S, Grunblatt E, Riederer P, Gerlach M, Levites Y, Youdim MB. Neuroprotective strategies in Parkinson's disease: an update on progress. CNS Drugs. 2003;17:729–762. doi: 10.2165/00023210-200317100-00004. [DOI] [PubMed] [Google Scholar]
- Manning-Boğ AB, Langston JW. Model fusion, the next phase in developing animal models for Parkinson's disease. Neurotox Res. 2007;11(3-4):219–40. doi: 10.1007/BF03033569. Review. [DOI] [PubMed] [Google Scholar]
- Mark KA, Soghomonian JJ, Yamamoto BK. High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J Neurosci. 2004;24:11449–11456. doi: 10.1523/JNEUROSCI.3597-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason ST, Iversen SD. An investigation of the role of cortical and cerebellar noradrenaline in associative motor learning in the rat. Brain Res. 1977;134:513–527. doi: 10.1016/0006-8993(77)90826-5. [DOI] [PubMed] [Google Scholar]
- McEntee WJ, Mair RG, Langlais PJ. Neurochemical specificity of learning: dopamine and motor learning. Yale Journal of Biology & Medicine. 1987;60:187–193. [PMC free article] [PubMed] [Google Scholar]
- McNeill TH, Koek LL. Differential effects of advancing age on neurotransmitter cell loss in the substantia nigra and striatum of C57BL/6N mice. Brain Res. 1990;521:107–117. doi: 10.1016/0006-8993(90)91530-t. [DOI] [PubMed] [Google Scholar]
- Melega WP, Lacan G, Harvey DC, Way BM. Methamphetamine increases basal ganglia iron to levels observed in aging. Neuroreport. 2007;18(16):1741–5. doi: 10.1097/WNR.0b013e3282f0d4f4. [DOI] [PubMed] [Google Scholar]
- Meredith GE, Totterdell S, Potashkin JA, Surmeier DJ. Modeling PD pathogenesis in mice: advantages of a chronic MPTP protocol. Parkinsonism & Related Disorders. 2008;14(2):S112–5. doi: 10.1016/j.parkreldis.2008.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan AJ, Warren M, Carvey PM. Neuroinflammation and peripheral immune infiltration in Parkinson's disease: an autoimmune hypothesis. Cell Transplantation. 2008;17:363–372. [PubMed] [Google Scholar]
- Moore MW, Klein RD, Farinas I, Sauer H, Armanini M, Phillips H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- Mori F, Nishie M, Kakita A, Yoshimoto M, Takahashi H, Wakabayashi K. Relationship among alpha-synuclein accumulation, dopamine synthesis, and neurodegeneration in Parkinson disease substantia nigra. Journal of Neuropathology & Experimental Neurology. 2006;65(8):808–15. doi: 10.1097/01.jnen.0000230520.47768.1a. [DOI] [PubMed] [Google Scholar]
- Murase S, McKay RD. A specific survival response in dopamine neurons at most risk in Parkinson's disease. J Neurosci. 2006;26:975097–60. doi: 10.1523/JNEUROSCI.2745-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson's disease. J Neural Trans Supplementum. 2000;60:277–290. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
- Naoi M, Maruyama W. Cell death of dopamine neurons in aging and Parkinson's disease. Mech Ageing Dev. 1999;111:175–188. doi: 10.1016/s0047-6374(99)00064-0. [DOI] [PubMed] [Google Scholar]
- O'Callaghan JP, Miller DB. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:741–751. [PubMed] [Google Scholar]
- O'Dell SJ, Weihmuller FB, Marshall JF. Multiple methamphetamine injections induce marked increases in extracellular striatal dopamine which correlate with subsequent neurotoxicity. Brain Res. 1991;564:256–260. doi: 10.1016/0006-8993(91)91461-9. [DOI] [PubMed] [Google Scholar]
- Oo TF, Burke RE. The time course of developmental cell death in phenotypically defined dopaminergic neurons of the substantia nigra. Brain Res Dev Brain Res. 1997;20:19191–6. doi: 10.1016/s0165-3806(96)00173-3. [DOI] [PubMed] [Google Scholar]
- Palmer AM, DeKosky ST. Monoamine neurons in aging and Alzheimer's disease. J Neural Trans-Gen Sect. 1993;91:135–159. doi: 10.1007/BF01245229. [DOI] [PubMed] [Google Scholar]
- Papanikolaou T, Lennington JB, Betz A, Figueiredo C, Salamone JD, Conover JC. In vitro generation of dopaminergic neurons from adult subventricular zone neural progenitor cells. Stem Cells & Development. 2008;17:157–172. doi: 10.1089/scd.2007.0090. [DOI] [PubMed] [Google Scholar]
- Pichel JG, Shen L, Sheng HZ, Granholm AC, Drago J, Grinberg A, Lee EJ, Huang SP, Saarma M, Hoffer BJ, Sariola H, Westphal H. Defects in enteric innervation and kidney development in mice lacking GDNF. Nature. 1996;382:73–76. doi: 10.1038/382073a0. [DOI] [PubMed] [Google Scholar]
- Quintero EM, Willis L, Singleton R, Harris N, Huang P, Bhat N, Granholm AC. Behavioral and morphological effects of minocycline in the 6-hydroxydopamine rat model of Parkinson's disease. Brain Res. 2006;1093:198–207. doi: 10.1016/j.brainres.2006.03.104. [DOI] [PubMed] [Google Scholar]
- Racette B, Deppen P, Dewey RB, Scott B, Field J, Carter J, Brodsky M, Andrews P, Manyam B, Whetteckey J, Rao J, Cook M, Aminoff MJ, Christine C, Roth J, Nance M, Parashos S, Peterson S, Shannon K, Jaglin J, Singer C, Perez MA, Blenke A, Hauser R, McClain T, Dawson T, Dunlop B, Pahwa R, Lyons K, Parsons A, Leehey M, Bainbridge J, Shulman L, Weiner W, Pabst K, Elble R, Young C, Sethi K, Dill B, Martin W, Calabrese VP, Roberge P, Hamill R, Bodis-Wollner IG, Hayes E, Ross GW, Terashita S, Wooten GF, Trugman J, Keller MF, Sage JI, Caputo D, Fang J, Shearon D, Jennings D, Wojcieszek J, Belden J, Feigin A, Cox MM, Watts RL, Stover N, McMurray R, Albin R, Wernette K, Siderowf A, Reichwein S, Simon D, Tarsy D, Scollins L, Tintner R, Hunter C, LeWitt P, Beaumont W, Burns RS, Adler C, Lind M, Gorell J, Krstevska S, Flewellen M, Schneider J, Sendek S, Gollomp S, Vernon G, Coffey D, LeBlanc P, Lew MF, Wu A, Kawai C, Uitti R, Turk M, Bower J, Torgrimson S, Handler J, Sabbagh M, Obradov Z. A pilot clinical trial of creatine and minocycline in early Parkinson disease: 18-month results. Clin Neuropharmacol. 2008 May-Jun;31(3):141–50. doi: 10.1097/WNF.0b013e3181342f32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricaurte GA, Guillery RW, Seiden LS, Schuster CR, Moore RY. Dopamine nerve terminal degeneration produced by high doses of methylamphetamine in the rat brain. Brain Res. 1982;235:93–103. doi: 10.1016/0006-8993(82)90198-6. [DOI] [PubMed] [Google Scholar]
- Richardson JR, Caudle WM, Wang M, Dean ED, Pennell KD, Miller GW. Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of Parkinson's disease. FASEB J. 2006;20(10):1695–7. doi: 10.1096/fj.06-5864fje. [DOI] [PubMed] [Google Scholar]
- Ron D, Janak PH. GDNF and addiction. Rev Neurosci. 2005;16(4):277–85. doi: 10.1515/revneuro.2005.16.4.277. Review. [DOI] [PubMed] [Google Scholar]
- Roussa E, Oehlke O, Rahhal B, Heermann S, Heidrich S, Wiehle M, Krieglstein K. Transforming growth factor beta cooperates with persephin for dopaminergic phenotype induction. Stem Cells. 2008;26(7):1683–94. doi: 10.1634/stemcells.2007-0805. Journal Article. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- Saarma M. GDNF - a stranger in the TGF-beta superfamily? Eur J Biochem. 2000;267:6968–6971. doi: 10.1046/j.1432-1327.2000.01826.x. Review. [DOI] [PubMed] [Google Scholar]
- Sah DW, Ossipov MH, Rossomando A, Silvian L, Porreca F. New approaches for the treatment of pain: the GDNF family of neurotrophic growth factors. Current Topics in Medicinal Chemistry. 2005;5:577–583. doi: 10.2174/1568026054367593. [DOI] [PubMed] [Google Scholar]
- Sanchez MP, Silos-Santiago I, Frisen J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382:70–73. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- Sanchez-Pernaute R, Lee H, Patterson M, Reske-Nielsen C, Yoshizaki T, Sonntag KC, Studer L, Isacson O. Parthenogenetic dopamine neurons from primate embryonic stem cells restore function in experimental Parkinson's disease. Brain. 2008;131:2127–2139. doi: 10.1093/brain/awn144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarabi A, Chang CF, Wang Y, Tomac AC, Hoffer BJ, Morales M. Differential expression of the glial cell line-derived neurotrophic factor (GDNF) receptor GFRα-1 in heterozygous GFRα-1 null-mutant mice after stroke. Neurosci Lett. 2003;341:241–245. doi: 10.1016/s0304-3940(03)00195-2. [DOI] [PubMed] [Google Scholar]
- Sawada M, Sawada H, Nagatsu T. Effects of aging on neuroprotective and neurotoxic properties of microglia in neurodegenerative diseases. Neurodegenerative Diseases. 2008;5:254–256. doi: 10.1159/000113717. [DOI] [PubMed] [Google Scholar]
- Seeman P, Bzowej NH, Guan HC, Bergeron C, Reynolds GP, Bird ED, Riederer P, Jellinger K, Tourtellotte WW. Human brain D1 and D2 dopamine receptors in schizophrenia, Alzheimer's, Parkinson's, and Huntington's diseases. Neuropsychopharm. 1987;1:5–15. doi: 10.1016/0893-133x(87)90004-2. [DOI] [PubMed] [Google Scholar]
- Slevin JT, Gash DM, Smith CD, Gerhardt GA, Kryscio R, Chebrolu H, Walton A, Wagner R, Young AB. Unilateral intraputamenal glial cell line-derived neurotrophic factor in patients with Parkinson disease: response to 1 year of treatment and 1 year of withdrawal. J Neurosurg. 2007;106(4):614–20. doi: 10.3171/jns.2007.106.4.614. [DOI] [PubMed] [Google Scholar]
- Smits SM, Burbach JP, Smidt MP. Developmental origin and fate of mesodiencephalic dopamine neurons. Prog Neurobiol. 2006;78:1–16. doi: 10.1016/j.pneurobio.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Giovanni A, Sieber BA, Donne KD, Manzino L. Characteristics of dopaminergic neurotoxicity produced by MPTP and methamphetamine. Ann NY Acad Sci. 1992;648:229–238. doi: 10.1111/j.1749-6632.1992.tb24542.x. [DOI] [PubMed] [Google Scholar]
- Sonsalla PK, Nicklas WJ, Heikkila RE. Role for excitatory amino acids in methamphetamine-induced toxicity. Science. 1986;243:398–400. doi: 10.1126/science.2563176. [DOI] [PubMed] [Google Scholar]
- Sriram K, Miller DB, O'Callaghan JP. Minocycline attenuates microglial activation but fails to mitigate striatal dopaminergic neurotoxicity: role of tumor necrosis factor-alpha. J Neurochem. 2006;96:706–718. doi: 10.1111/j.1471-4159.2005.03566.x. [DOI] [PubMed] [Google Scholar]
- Suhara T, Fukuda H, Inoue O, Itoh T, Suzuki K, Yamasaki T, Tateno Y. Age-related changes in human D1 dopamine receptors measured by positron emission tomography. Psychopharmacology. 1991;103(1):41–45. doi: 10.1007/BF02244071. [DOI] [PubMed] [Google Scholar]
- Surguchov A. Molecular and cellular biology of synucleins. International Review of Cell & Molecular Biology. 2008;270:225–317. doi: 10.1016/S1937-6448(08)01406-8. [DOI] [PubMed] [Google Scholar]
- Tatton WG, Greenwood CE, Verrier MC, Holland DP, Kwan MM, Biddle FE. Different rates of age-related loss for four murine monoaminergic neuronal populations. Neurobiol Aging. 1991;12:543–556. doi: 10.1016/0197-4580(91)90086-y. [DOI] [PubMed] [Google Scholar]
- Terzioglu M, Galter D. Parkinson's disease: genetic versus toxin-induced rodent models. FEBS Journal. 2008;275:1384–1391. doi: 10.1111/j.1742-4658.2008.06302.x. [DOI] [PubMed] [Google Scholar]
- Tikka T, Fiebich B, Goldsteins G, Keinanen R, Koistinaho J. Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci. 2001a;21:2580–2588. doi: 10.1523/JNEUROSCI.21-08-02580.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikka T, Koistinaho J. Minocycline provides neuroprotection against NMDA neurotoxicity by inhibiting microglia. J Immunol. 2001b;166:7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- Tomac A, Lindqvist E, Lin LF, Ogren SO, Young D, Hoffer BJ, Olson L. Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature. 1995;373(6512):335–9. doi: 10.1038/373335a0. [DOI] [PubMed] [Google Scholar]
- Toulouse A, Sullivan AM. Progress in Parkinson's disease-where do we stand? Progress in Neurobiology. 2008;85:376–392. doi: 10.1016/j.pneurobio.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Trupp M, Belluardo N, Funakoshi H, Ibáñez CF. Complementary and overlapping expression of glial cell line-derived neurotrophic factor (GDNF), c-ret proto-oncogene, and GDNF receptor-alpha indicates multiple mechanisms of trophic actions in the adult rat CNS. J Neurosci. 1997;17:3554–3567. doi: 10.1523/JNEUROSCI.17-10-03554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukahara T, Takeda M, Shimohama S, Ohara O, Hashimoto N. Effects of brain-derived neurotrophic factor on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in monkeys. Neurosurgery. 1995;37:7337–41. doi: 10.1227/00006123-199510000-00018. [DOI] [PubMed] [Google Scholar]
- Van den Heuvel DM, Pasterkamp RJ. Getting connected in the dopamine system. Progress in Neurobiology. 2008;85:75–93. doi: 10.1016/j.pneurobio.2008.01.003. [DOI] [PubMed] [Google Scholar]
- Vanitallie TB. Parkinson disease: primacy of age as a risk factor for mitochondrial dysfunction. Metabolism: Clinical & Experimental. 2008;57(2):S50–S55. doi: 10.1016/j.metabol.2008.07.015. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Franceschi D, Sedler M, Gatley SJ, Miller E, Hitzemann R, Ding YS, Logan J. Loss of dopamine transporters in methamphetamine abusers recovers with protracted abstinence. J Neurosci. 2001a;21:9414–9418. doi: 10.1523/JNEUROSCI.21-23-09414.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Chang L, Wang GJ, Fowler JS, Leonido-Yee M, Franceschi D, Sedler MJ, Gatley SJ, Hitzemann R, Ding YS, Logan J, Wong C, Miller EN. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am J Psychiatry. 2001b;158:377–382. doi: 10.1176/appi.ajp.158.3.377. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Gur RC, Wang GJ, Fowler JS, Moberg PJ, Ding YS, Hitzemann R, Smith G, Logan J. Association between decline in brain dopamine activity with age and cognitive and motor impairment in healthy individuals. Am J Psychiatry. 1998;155:344–349. doi: 10.1176/ajp.155.3.344. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Fowler JS, Gatley SJ, Logan J, Wang GJ, Ding YS, Dewey S. PET evaluation of the dopamine system of the human brain. J Nucl Med. 1996;37:1242–1256. Review. [PubMed] [Google Scholar]
- Wagner GC, Ricuarte GA, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- Wang J, Wei Q, Wang CY, Hill WD, Hess DC, Dong Z. Minocycline up-regulates Bcl-2 and protects against cell death in mitochondria. J Biol Chem. 2004;279:19948–19954. doi: 10.1074/jbc.M313629200. [DOI] [PubMed] [Google Scholar]
- Wang M, Dean ED, Pennell KD, Miller GW. Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of Parkinson's disease. FASEB Journal. 2006;20(10):1695–7. doi: 10.1096/fj.06-5864fje. [DOI] [PubMed] [Google Scholar]
- Yamamoto BK, Zhu W. The effects of methamphetamine on the production of free radicals and oxidative stress. J Pharmacol Exp Ther. 1998;287:107–114. [PubMed] [Google Scholar]
- Yurek DM, Fletcher-Turner A. Lesion-induced increase of BDNF is greater in the striatum of young versus old rat brain. Exp Neurol. 2000;161:392–396. doi: 10.1006/exnr.1999.7274. [DOI] [PubMed] [Google Scholar]
- Yurek DM, Fletcher-Turner A. Differential expression of GDNF, BDNF, and NT-3 in the aging nigrostriatal system following a neurotoxic lesion. Brain Res. 2001;891:228–335. doi: 10.1016/s0006-8993(00)03217-0. [DOI] [PubMed] [Google Scholar]
- Zaman V, Li Z, Middaugh L, Ramamoorthy S, Rohrer B, Nelson ME, Tomac AC, Hoffer BJ, Gerhardt GA, Granholm ACh. The noradrenergic system of aged GDNF heterozygous mice. Cell Transplant. 2003;12:291–303. [PubMed] [Google Scholar]
- Zaman V, Boger HA, Granholm AC, Rohrer B, Moore A, Buhusi M, Gerhardt GA, Hoffer BJ, Middaugh LD. The nigrostriatal dopamine system of aging GFRalpha-1 heterozygous mice: neurochemistry, morphology and behavior. Euro J Neuro. 2008;28:1557–1568. doi: 10.1111/j.1460-9568.2008.06456.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zecca L, Casella L, Albertini A, Bellei C, Zucca FA, Engelen M, Zadlo A, Szewczyk G, Zareba M, Sarna T. Neuromelanin can protect against iron-mediated oxidative damage in system modeling iron overload of brain aging and Parkinson's disease. Journal of Neurochemistry. 2008;106:1866–1875. doi: 10.1111/j.1471-4159.2008.05541.x. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stravroskaya IG, Drozda M, Kim BYS, Ona V, Li M, et al. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Carvey PM, Ling Z. Altered glutathione homeostasis in animals prenatally exposed to lipopolysaccharide. Neurochemistry International. 2007;50(4):671–80. doi: 10.1016/j.neuint.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]