

Abstract
Prion protein is known to have the ability to adopt a pathogenic conformation, which seems to be the basis for protein-only infectivity. The infectivity is based on self-replication of this pathogenic prion structure. One of possible mechanisms for such replication is the elongation of amyloid-like fibrils.
We measured elongation kinetics and thermodynamics of mouse prion amyloid-like fibrils at different guanidine hydrochloride (GuHCl) concentrations. Our data show that both increases in temperature and GuHCl concentration help unfold monomeric protein and thus accelerate elongation. Once the monomers are unfolded, further increases in temperature raise the rate of elongation, whereas the addition of GuHCl decreases it.
We demonstrated a possible way to determine different activation energies of amyloid-like fibril elongation by using folded and unfolded protein molecules. This approach separates thermodynamic data for fibril-assisted monomer unfolding and for refolding and formation of amyloid-like structure.
Introduction
Prion protein (PrP) plays a big role in a number of lethal neurological diseases, known as transmissible spongiform encephalopaties. These disorders are associated with aggregation of normal cellular prion protein (PrPC) into pathogenic beta-sheet-rich prion isoforms (PrPSc). Although the majority of suspected cases of human prion diseases are sporadic [1], prions are mostly known because of their infectivity. The infectious nature of prion diseases is based on the ability of PrPSc to self-replicate by converting PrPC into same pathogenic isoform. One of possible mechanisms of pathogenic prion structure replication is elongation of amyloid-like fibrils.
Since the discovery of prions, one of the main tasks in the field was to produce infectious PrP conformation in vitro, as this would finally prove the hypothesis of protein-only infection [2]. A number of attempts generated amyloid-like structures [3], [4]. Such prion protein fibrils share some properties of PrPSc (such as beta-sheet-rich secondary structure and ability to self-replicate by addition of native PrP) but have much shorter proteinase K (PK) resistant cores [5], and show slight infectivity only in mice which overexpress PrPC 16 fold [6]. Later studies showed that PK resistance can be extended by annealing at high temperature [7], or by using protein misfolding cyclic amplification (PMCA) [8]. When examined by hydrogen/deuterium (H/D) exchange, annealed fibrils showed only slight differences in deuterium incorporation, when compared to untreated amyloid-like fibrils [9]. Such fibrils induced disease in hamsters only after the second passage, thus they are not directly infective [10]. PMCA-generated recombinant PrP amyloid-like fibrils have an extended region, protected from H/D exchange [8], and can induce disease in hamsters, though not as effectively as PrPSc [11]. Studies of H/D exchange on brain-derived infective PrP show similar highly packed structure as in case of recombinant PrP amyloid-like fibrils, but in a much longer region (entire 90–230 region versus 160–220 region) [12]. It suggests the possibility that brain-derived PrPSc may be similar to amyloid-like fibrils, just with a much longer beta-sheet core region. Recently Ma and co-workers reported a protocol for de novo generation of infectious prions from recombinant PrP [13]. It has similar infectivity to brain-derived prions, but due to low efficiency of the protocol, the structure of these prions is still unknown.
The structure of recombinant PrP amyloid-like fibrils was extensively studied by a number of techniques all giving similar conclusions – parallel in-register beta-sheet in the C-terminal region (starting from residues 160–170, up to residues 220–225) and a disordered N-terminus [9], [14], [15]. The difference in structure between the native prion protein and the amyloid form requires a conversion of the native alpha-helices into beta-sheets. Usually, amyloid-like fibrils are formed in the presence of moderate denaturant concentration [3], [4], under conditions where the native protein is at least partially unfolded. However in vivo, the environment does not help in unfolding of native helices. We tried to test if prion protein amyloid-like fibrils self-replicate using stable native PrP, and how environmental factors (such as temperature and denaturant concentration) affect fibril elongation kinetics.
Results and Discussion
Amyloid-like fibril formation includes several events – primary nucleation (de novo formation of amyloid-like particles), elongation (growth of fibrils via addition of new protein molecules) and secondary nucleation (increase of active fibril ends by breaking or branching of fibrils). Elongation is the main driving force for self-replication of amyloid-like fibrils. It can be initiated by adding preformed fibrils to a protein solution. Recombinant PrP can aggregate into fibrils under different conditions [3], [4], [14], [16], [17], but fibrils prepared by agitation at 37°C in presence of 2 M guanidine hydrochloride (GuHCl) [9], [14], [15] have been best characterized. Such fibrils were used as seeding material in our study. Ultrasonic treatment of fibrils is widely used in studies of amyloids and prions [18]–[22], and is known to break fibrils into shorter pieces [21], [22]. It helps both to homogenize fibril suspension, and to increase the rate of the process. Figure 1 shows a comparison of truncated mouse prion protein fibril elongation kinetics using seeds prepared using different sonication times. It is clear that sonication leads to several fold faster elongation rates, but every additional pulse is less efficient. Prolonged sonication may lead to minimum-sized amyloid-like fibrils [21], [23], and such fibrils seem to be the most efficient seeding material. Using sonicated fibrils as seeds under quiescent conditions gives the possibility of observing elongation without any visible nucleation events (Figure 1).
Figure 1. Different numbers of 30 second pulses were applied to fibrils before using them as seeds.
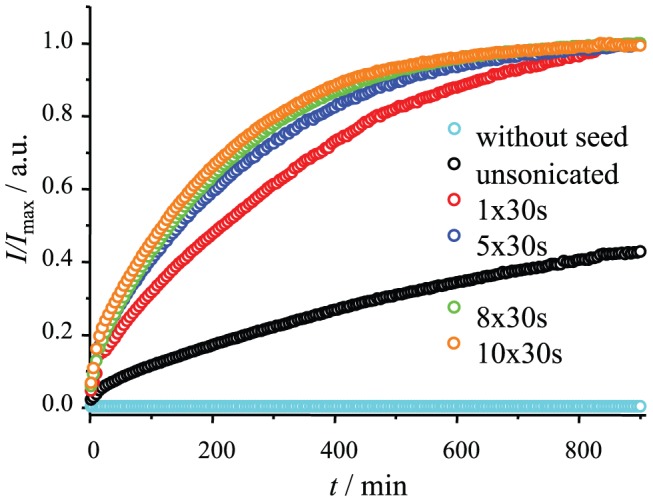
Kinetics of elongation was followed by Thioflavin T assay at 37°C in the presence of 2 M guanidine hydrochloride.
We followed elongation kinetics at six different temperatures in the presence of five different GuHCl concentrations (Figure 2). In all cases higher temperature leads to increased rates of elongation, however, the rate dependence on GuHCl concentration is more complicated. We summarized all measured elongation rates in Figure 3. At lower temperatures (40°C), the highest elongation rates are at moderate denaturant concentration (similar to previously reported moderate denaturant concentrations leading to the shortest lag times in spontaneous PrP fibrillation [24]), however, raising the temperature results in a decrease in optimal GuHCl concentrations.
Figure 2. Fibril elongation at different temperatures in the presence of 0.5 M (A), 1 M (B), 1.5 M (C), 2 M (D) and 2.5 M (E) GuHCl.
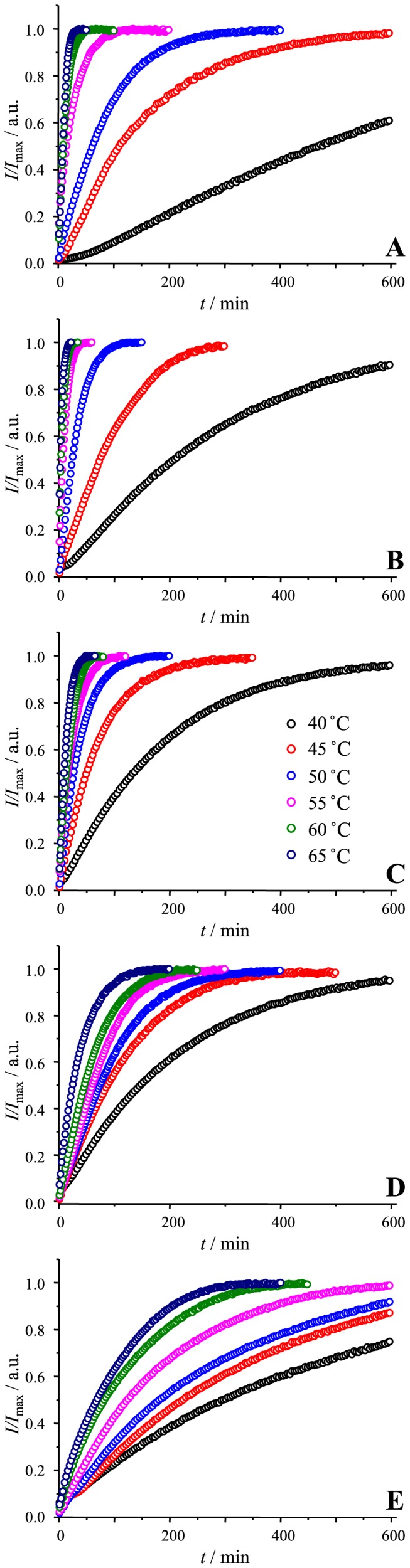
Figure 3. Three-dimensional landscape of fibril elongation rates at different temperatures and denaturant concentrations.

To understand the impact of denaturant, we plotted our kinetic data in an Arrhenius plot and compared them with MoPrP89-230 thermal denaturation curves, obtained from circular dichroism spectra (Figure 4). In the case of 0.5 M GuHCl (Figure 4A), the Arrhenius plot is linear over the range of 40–60°C (with calculated activation energy Ea = 165.8±10.0 kJ/mol); at this temperature range more than half of the protein is still folded. At higher temperature, most of the protein is unfolded and the rate of elongation does not follow the same line in the Arrhenius plot. Similar data can be observed for 1 M GuHCl (Figure 4B), but under such conditions, MoPrP89-230 unfolds at a lower temperature, thus, a valid linear fit for the Arrhenius plot is available only for the temperature range 40–55°C (Ea = 177.5±3.3 kJ/mol). At higher GuHCl concentrations most of the data were obtained at temperatures leading to a major fraction of unfolded protein, thus, a linear fit to the Arrhenius plot of fibril elongation at 1.5 M GuHCl (Figure 4C) is good for the temperature range 50–65°C (Ea = 58.3±6.3 kJ/mol), at 2 M GuHCl (Figure 4D) for 45–65°C (Ea = 44.4±4.9 kJ/mol), and at 2.5 M GuHCl (Figure 4E) for 40–65°C (Ea = 51.4±2.6 kJ/mol). Based on the data, two processes with different activation energies can be separated. Fibril elongation which assimilates folded PrP has greater energetic barrier (∼170 kJ/mol) than elongation using unfolded PrP (∼50 kJ/mol). Knowing that fibril formation requires complete rearrangement of native PrP secondary structure [9], [14], [15], it is easy to imagine that loss of secondary structure may significantly lower energetic barriers to the reaction.
Figure 4. Comparison of Arrhenius plots with protein thermal unfolding curves in the presence of 0.5 M (A), 1 M (B), 1.5 M (C), 2 M (D) and 2.5 M (E) GuHCl.
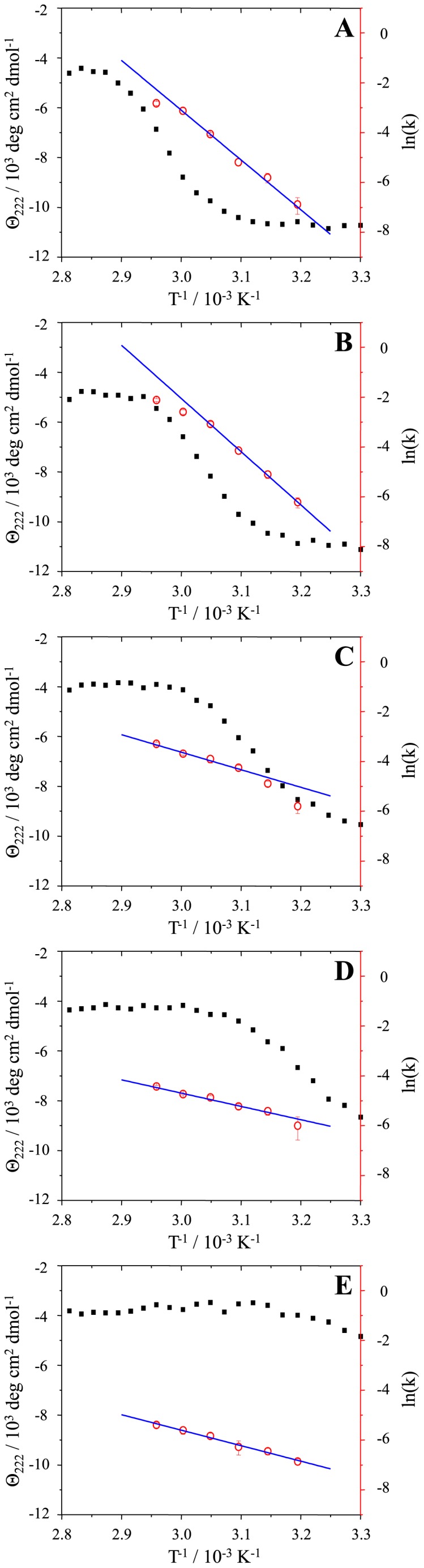
Black squares represent mean residue molar ellipticities, measured by circular dichroism, red circles represent the natural logarithm of elongation rates, and blue lines are the fits used to extract activation energies.
To check monomer-fibril equilibrium dependence on temperature and denaturant we performed a fibril denaturation assay [25]. At ambient temperature (Figure 5A) fibrils are a bit more stable compared to fibers at 60°C (Figure 5B). It is important to note that even at high temperature fibrils are relatively stable up to 2 M GuHCl concentration (>96% of ThT fluorescence is retained). Thus it may only slightly impact (within one standard deviation) the estimated rate of elongation and activation energies. At 2.5 M GuHCl equilibrium shifts towards depolymerization of fibrils (∼94% ThT fluorescence retained at ambient temperature and ∼88% at 60°C). As we normalized all curves to the same final level, it should lead to overestimation of elongation rate. However, at the same time some of fibrils could be completely depolymerized, leading to lower numbers of fibril ends and thus underestimation of the rate. We can't quantify effects of both possible events, thus we state that errors of our measurements at 2.5 M GuHCl may reach up to 12%. This error does not affect our findings.
Figure 5. Denaturation profiles of MoPrP amyloid-like fibrils at ambient temperature (A) and at 60°C (B).

These data were fitted to the two state depolymerization model.
Buell and coworkers [26] compared elongation enthalpies for a number of amyloidogenic proteins and peptides and found that the presence of tertiary structure generally increases the enthalpy of activation per residue [26]. One of the facts supporting this finding is the difference in fibril elongation activation enthalpies between native and reduced forms of human lysozyme [26]. Interestingly, MoPrP89-230 and human lysozyme have similar numbers of amino acids, similar amounts of alpha-helices (∼40%) and comparable amounts of beta-sheets (<10%). Further, activation enthalpy for native human lysozyme (167.7±14.7 kJ/mol) [26] is very similar to activation energy for folded MoPrP89-230. However, activation enthalpy for reduced human lysozyme (68±12 kJ/mol) [26] is greater than activation energy for unfolded MoPrP89-230. Reduced lysozyme still has some secondary structure, which may be the reason for this difference.
Recently the idea of the prion-like nature of amyloid fibrils is widely discussed and gets some support from experimental data [27]–[30]. At this point, understanding fibril elongation as one of the possible driving forces of infectivity is a very important goal. Surprisingly, thermodynamics of fibril elongation has not yet attracted much attention. Most of the available data on this topic is summarized in the work of Buell and co-workers [26]. We expanded current knowledge by describing elongation of mammalian prion fibrils under a variety of conditions, including both folded and unfolded protein states. In fact, we demonstrated a possible way to separate the energies of monomer unfolding and refolding into an amyloid structure. Our findings may help in better understanding the fibril growth process and relations between prions and amyloids.
Materials and Methods
Truncated mouse prion protein (MoPrP89-230) was expressed in E. coli and purified according to the previously described protocol [3]. After purification, the protein was stored frozen in 10 mM sodium acetate buffer, pH 4. In addition to the MoPrP sequence, the protein contains a 4-residue N-terminal extension (GSDP). Protein grade GuHCl (>99.7%) was obtained from Carl Roth. Thioflavin T (ThT) was obtained from Sigma. Sodium phosphates were obtained from Fisher Scientific UK.
To prepare fibrils, monomeric protein from a stock solution was diluted to a concentration of 0.5 mg/ml in 50 mM phosphate buffer (pH 6) containing 2 M GuHCl, and incubated for 3 days at 37°C with 220 rpm shaking (shaker incubator IKA KS 4000i). For seeding experiments fibrils were treated for 10 minutes using Bandelin Sonopuls 3100 ultrasonic homogenizer equipped with MS72 tip (using 20% power, cycles of 30 s/30 s sonication/rest, total energy applied to the sample per cycle ∼0.36 kJ). The sample was kept on ice during the sonication. Right after the treatment, 1 part fibrils was mixed with 19 parts 0.5 mg/ml of mouse prion solution containing 50 µM ThT and different concentrations of GuHCl in 50 mM phosphate buffer, pH 6. Elongation kinetics at different temperatures (40°C, 45°C, 50°C, 55°C, 60°C, 65°C) was monitored by ThT fluorescence assay (excitation at 470 nm, emission at 510 nm) using Qiagen Rotor-Gene Q real-time analyzer (see File S1 for broader description). ThT fluorescence curves were normalized by dividing each point by the maximum intensity of the curve. Rates of elongation were determined by linear fit of these curves in a range between 40–60% of the ordinate maxima (see File S1 for exponential fit comparison). Standard errors from 6 samples were calculated using Student's t-distribution at p = 0.05.
Thermal unfolding transition curves were measured on a Jasco J-815 circular dichroism spectropolarimeter For each experiment a sample of 0.05 mg/ml PrP was prepared in 50 mM phosphate buffer (pH 6) containing different amounts of GuHCl, and transferred to a 2 mm path length quartz cuvette. Ten CD spectra (in range 221–223 nm) were averaged for each temperature point. Temperature was raised by 2.5°C increments with an average rate of 1°C/min.
For chemical denaturation assays, amyloid fibrils were resuspended to a concentration of 25 µM in 50 mM phosphate buffer, pH 6, containing 0.5 M GuHCl and homogenized by sonication. These solutions were diluted 1∶4 in a buffer containing varying concentrations of GuHCl, and incubated for 60 min at 25°C or 60°C. Samples were then mixed 1∶20 with 50 µM ThT. Immediately after mixing, ThT fluorescence was measured at 480 nm using the excitation wavelength of 440 nm. The data was fit using a two state depolymerization model (each protein molecule would be either in fibrillar state and contribute to ThT fluorescence, or in monomer state). In this case the equilibrium constant is
| (1) |
where ff is fraction of protein molecules in fibrillar state. Assuming that the free energy of depolymerization (ΔGd) has a linear dependence on the concentration of the denaturant [31]
| (2) |
and using its relation to equilibrium constant
| (3) |
it is possible to get the dependence of the fraction of fibrils on the concentration of GuHCl
| (4) |
Supporting Information
(PDF)
Acknowledgments
The authors thank Professor Witold Surewicz for providing MoPrP89-230 plasmid, and Dr. Jonathan Cannon for improving the language of the manuscript.
Funding Statement
This research was funded by a grant (No. MIP-030/2012) from the Research Council of Lithuania. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Klug GMJa, Wand H, Simpson M, Boyd A, Law M, et al. (2013) Intensity of human prion disease surveillance predicts observed disease incidence. J Neurol, Neurosurg Psychiatry 84: 1372–7. [DOI] [PubMed] [Google Scholar]
- 2. Colby DW, Prusiner SB (2011) Prions. Cold Spring Harbor Perspect Biol 3: a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Swietnicki W, Morillas M, Chen SG, Gambetti P, Surewicz WK (2000) Aggregation and fibrillization of the recombinant human prion protein huPrP90-231. Biochemistry 39: 424–31. [DOI] [PubMed] [Google Scholar]
- 4. Baskakov IV, Legname G, Baldwin Ma, Prusiner SB, Cohen FE (2002) Pathway complexity of prion protein assembly into amyloid. J Biol Chem 277: 21140–8. [DOI] [PubMed] [Google Scholar]
- 5. Bocharova OV, Breydo L, Salnikov VV, Gill AC, Baskakov IV (2005) Synthetic prions generated in vitro are similar to a newly identified subpopulation of PrPSc from sporadic Creutzfeldt-Jakob Disease. Protein Sci 14: 1222–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Legname G, Baskakov IV, Nguyen HOB, Riesner D, Cohen FE, et al. (2004) Synthetic mammalian prions. Science 305: 673–6. [DOI] [PubMed] [Google Scholar]
- 7. Bocharova OV, Makarava N, Breydo L, Anderson M, Salnikov VV, et al. (2006) Annealing prion protein amyloid fibrils at high temperature results in extension of a proteinase K-resistant core. J Biol Chem 281: 2373–9. [DOI] [PubMed] [Google Scholar]
- 8. Smirnovas V, Kim JI, Lu X, Atarashi R, Caughey B, et al. (2009) Distinct structures of scrapie prion protein (PrPSc)-seeded versus spontaneous recombinant prion protein fibrils revealed by hydrogen/deuterium exchange. J Biol Chem 284: 24233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lu X, Wintrode PL, Surewicz WK (2007) Beta-sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc Natl Acad Sci U S A 104: 1510–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Makarava N, Kovacs GG, Bocharova OV, Savtchenko R, Alexeeva I, et al. (2010) Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol 119: 177–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim JI, Cali I, Surewicz K, Kong Q, Raymond GJ, et al. (2010) Mammalian prions generated from bacterially expressed prion protein in the absence of any mammalian cofactors. J Biol Chem 285: 14083–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smirnovas V, Baron GS, Offerdahl DK, Raymond GJ, Caughey B, et al. (2011) Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat Struct Mol Biol 18: 504–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang F, Wang X, Yuan CG, Ma J (2010) Generating a prion with bacterially expressed recombinant prion protein. Science 327: 1132–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cobb NJ, Sönnichsen FD, McHaourab H, Surewicz WK (2007) Molecular architecture of human prion protein amyloid: a parallel, in-register beta-structure. Proc Natl Acad Sci U S A 104: 18946–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tycko R, Savtchenko R, Ostapchenko VG, Makarava N, Baskakov IV (2010) The α-helical C-terminal domain of full-length recombinant PrP converts to an in-register parallel β-sheet structure in PrP fibrils: Evidence from solid state NMR. Biochemistry 9488–9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cobb NJ, Apetri AC, Surewicz WK (2008) Prion protein amyloid formation under native-like conditions involves refolding of the C-terminal alpha-helical domain. J Biol Chem 283: 34704–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Colby DW, Giles K, Legname G, Wille H, Baskakov IV, et al. (2009) Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci U S A 106: 20417–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Serio TR, Cashikar AG, Kowal AS, Sawicki GJ, Moslehi JJ, et al. (2000) Nucleated conformational conversion and the replication of conformational information by a prion determinant. Science 289: 1317–21. [DOI] [PubMed] [Google Scholar]
- 19. Saborío GP, Permanne B, Spagnolo S (2001) Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411: 810–3. [DOI] [PubMed] [Google Scholar]
- 20. Dzwolak W, Smirnovas V, Jansen R, Winter R (2004) Insulin forms amyloid in a strain-dependent manner: an FT-IR spectroscopic study. Protein Sci 13: 1927–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chatani E, Lee YH, Yagi H, Yoshimura Y, Naiki H, et al. (2009) Ultrasonication-dependent production and breakdown lead to minimum-sized amyloid fibrils. Proc Natl Acad Sci U S A 106: 11119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Milto K, Botyriute A, Smirnovas V (2013) Amyloid-Like Fibril Elongation Follows Michaelis-Menten Kinetics. PLoS One 8: e68684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang YY, Knowles TPJ, Terentjev EM (2009) Strength of Nanotubes, Filaments, and Nanowires From Sonication-Induced Scission. Adv Mat 21: 3945–3948. [Google Scholar]
- 24. Baskakov IV, Legname G, Gryczynski Z, Prusiner SB (2004) The peculiar nature of unfolding of the human prion protein. Protein Sci 13: 586–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cobb NJ, Apostol MI, Chen S, Smirnovas V, Surewicz WK (2014) Conformational Stability of Mammalian Prion Protein Amyloid Fibrils Is Dictated by a Packing Polymorphism within the Core Region. J Biol Chem 289: 2643–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Buell AK, Dhulesia A, White Da, Knowles TPJ, Dobson CM, et al. (2012) Detailed analysis of the energy barriers for amyloid fibril growth. Angew Chem, Int Ed Engl 51: 5247–51. [DOI] [PubMed] [Google Scholar]
- 27. Eisele YS, Obermüller U, Heilbronner G, Baumann F, Kaeser Sa, et al. (2010) Peripherally Applied A{beta}-Containing Inoculates Induce Cerebral {beta}-Amyloidosis. Science 980: 10–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Westermark GT, Westermark P (2010) Prion-like aggregates: infectious agents in human disease. Trends Mol Med 16: 501–7. [DOI] [PubMed] [Google Scholar]
- 29. Brundin P, Melki R, Kopito R (2010) Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol 11: 301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frost B, Diamond MI (2010) Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci 11: 155–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Greene RF, Pace CN (1974) Urea and guanidine hydrochloride denaturation of ribonuclease, lysozyme, alpha-chymotrypsin, and beta-lactoglobulin. J Biol Chem 249: 5388–93. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF)