

Abstract
Identifying key mediators of cancer invasion and metastasis is crucial to the development of new and more effective therapies. We previously identified Filamin A interacting protein 1-like (FILIP1L) as an important inhibitor of cell migration and invasion. FILIP1L expression was inversely correlated with the invasive potential of ovarian tumors. In our present study, we established an orthotopic ovarian cancer model, wherein FILIP1L expression can be regulated in vivo. Using this model, we observed that expression of FILIP1L in ovarian cancer cells inhibited spontaneous lung metastasis. Experimental lung metastases (established via tail vein injection of cancer cells) as well as the extravasation step of metastasis were not inhibited by FILIP1L, suggesting that FILIP1L inhibits the earlier steps of metastasis such as invasion and intravasation. FILIP1L inhibited matrix metalloproteinase (MMP)-dependent invasion in vivo. MMP3, -7 and -9 were transcriptionally down-regulated, and MMP9 protein expression and activity were inhibited in FILIP1L-expressing tumors. Importantly, overexpression of MMP9 compensated for the anti-invasive activity of FILIP1L. Furthermore, our studies suggest that FILIP1L regulates invasion and metastasis by inhibiting components of the WNT signaling pathway. FILIP1L expression reduced the induction of WNT target genes such as MMP3, -7 and -9, and β-catenin-directed transcriptional activity, suggesting inhibition of the canonical WNT pathway. Nuclear β-catenin, an indicator of an active canonical WNT pathway, was reduced in FILIP1L-expressing tumors. Overall, these findings suggest that FILIP1L reduces β-catenin levels, which may lead to the transcriptional down-regulation of WNT target genes such as MMPs, resulting in inhibition of metastasis. Modulation of FILIP1L expression has the potential to be a target for cancer therapy.
Keywords: Metastasis, Invasion, FILIP1L, MMP, WNT
Introduction
Invasion is a critical first step in tumor metastasis, and invasive potential is correlated with poor outcomes in patients with a variety of cancers (1). Characterization of the cellular mechanisms involved in invasion will allow for the development of more effective cancer therapies. We recently identified Filamin A interacting protein 1-like (FILIP1L; previously known as down-regulated in ovarian cancer 1 [DOC1]) as an important inhibitor of cell migration and invasion. Increased expression of FILIP1L resulted in inhibition of migration in endothelial cells (2) and inhibition of invasion in ovarian cancer cells (3). FILIP1L expression was inversely correlated with the invasive potential of ovarian cancer cell lines and ovarian cancer specimens (3). In a recent study, we have shown that FILIP1L expression is inversely correlated with the invasive potential of various cancer cell lines such as breast, colon, lung and pancreatic cancer (4). In addition, FILIP1L expression was shown to be down-regulated in prostate cancers compared with normal tissues (5). Furthermore, others have shown that intraperitoneal delivery of the FILIP1L gene resulted in inhibition of metastatic ovarian cancer spread into the peritoneum and intra-abdominal organs (6). Overall, these findings suggest that FILIP1L may be an important inhibitor of cancer cell invasion and metastasis.
FILIP1L mRNA was originally characterized by its presence in human ovarian surface epithelial (HOSE) cells and its absence in ovarian carcinoma cells (7). FILIP1L down-regulation was confirmed by cDNA microarray analysis in ovarian carcinoma cells from patients with late-stage disease (8). Differential gene expression analysis revealed that the FILIP1L gene in ovarian cancer cells presents several tagging single nucleotide polymorphisms (9). FILIP1L was shown to be one of nine genes associated with functional suppression of tumorigenicity in ovarian cancer cell lines (10). Differential expression of FILIP1L was also observed in other types of cells, including prostate cancer and endothelial cells infected with herpes virus (11, 12). Recently, we and others have demonstrated that DNA methylation was the mechanism by which FILIP1L was down-regulated in ovarian and prostate cancer cells (3, 5).
Although these observations demonstrate that FILIP1L inhibits metastasis, it is not clear which step(s) of metastasis are inhibited by FILIP1L. To this end, we chose an orthotopic ovarian cancer mouse model in which cancer cells metastasize to distant organs such as lungs, where lung metastasis can occur through vessels, not by exfoliation and peritoneal spread. In addition, FILIP1L expression was controlled by a doxycycline (DOX)-inducible expression system which enabled us to determine the direct effect of FILIP1L expression in vivo. Our results demonstrate that expression of FILIP1L in ovarian cancer cells prevents spontaneous metastasis at the invasion and intravasation steps. We observed that expression of FILIP1L inhibited the expression and activity of matrix metalloproteinases (MMPs). FILIP1L expression reduced the induction of WNT target genes such as MMP3, -7 and -9, β-catenin-directed transcriptional activity and the amount of nuclear β-catenin, suggesting inhibition of the canonical WNT signaling pathway. Taken together, these data suggest that FILIP1L reduces β-catenin levels, leading to the transcriptional down-regulation of WNT target genes such as MMPs and resulting in inhibition of metastasis. A more thorough understanding of FILIP1L-mediated inhibition of tumor cell invasion may lead to novel methods of controlling this process.
Materials and Methods
Cell culture and development of inducible clones expressing FILIP1L
Human ovarian cancer cell line ES2 was purchased from American Type Culture Collection (Manassas). Inducible clones were generated using Tet-On 3G tetracycline-inducible expression system (Clontech), as recommended by the manufacturer. Briefly, ES2 cells were transfected with pCMV-Tet3G, a vector encoding Tet-On 3G transactivator. Tet-On 3G transactivator-expressing clones were then transfected with either pTRE3G-mCherry, a control vector encoding mCherry, or pTRE3G-mCherry-FILIP1L, a vector encoding both mCherry and FILIP1LΔC103 (amino acid 1-790) (2). Induction of protein expression in the presence of DOX was tested by either mCherry fluorescence or immunoblot using anti-mCherry or anti-FILIP1L antibodies as described in the following sections.
mCherry fluorescence
Cells were seeded in 96-well plates and treated with DOX for 48 h. mCherry fluorescence was measured using a Synergy Mx microplate reader (BioTek).
WST1 cell proliferation assay
Cells were seeded in 96-well plates, treated with DOX for 48 h and treated with WST1 (Roche) for 2 h. Cell proliferation by WST1 incorporation was measured using a Synergy Mx microplate reader.
Immunoblot
Whole cell lysates and tumor lysates were prepared from radioimmunoprecipitation assay (RIPA) buffer and T-PER (Fisher) buffer, respectively, separated on SDS-PAGE and transferred to nitrocellulose membrane. The membranes were blotted with antibodies against FILIP1L (2), mCherry (Clontech), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Chemicon), MMP9 (Abcam) and phospho-β-catenin (Ser33/37/Thr41) (Cell Signaling) followed by incubation with anti-mouse (for the first four) or anti-rabbit (for phospho-β-catenin) antibody conjugated to horseradish peroxidase. The signal was detected using chemilluminescence (Millipore). GAPDH blot was used as the loading control.
Quantitative real-time RT-PCR
Total RNA was prepared by RNeasy kit (Qiagen) and cDNA was prepared by SuperScript VILO cDNA Synthesis Kit (Invitrogen). qRT-PCR was performed using an ABI 7900HT SDS and ViiA7 real-time PCR instrument as recommended by the manufacturer (Applied Biosystems). The TaqMan primers used were described previously (3). For MMPs, WNT ligands and WNT receptors, SYBR Green reagent was used. The gene-specific primers used are described in Supplementary Table 1.
Ovarian orthotopic model
All use of vertebrate animals described in this study was conducted in accordance with NIH regulations and was approved by the Albert Einstein College of Medicine Animal Use Committee. Eight week-old female SCID mice (National Cancer Institute) were anesthetized with ketamine/xylazine/acepromazine (3:3:1) and ovaries were externalized from a small dorsal incision (1–2 cm), and 5×105 cells of control or FILIP1L clones were injected under the bursal membrane. The injection methods were adapted from http://www.jove.com/video/2125/in-vivo-imaging-therapeutic-treatments-an-orthotopic-mouse-model?id=2125. Twelve mice were used per clone. Following surgery, mice were treated with either regular water or water containing 0.1 mg/ml DOX (6 mice per group).
Spontaneous and experimental metastasis
For spontaneous metastasis assay, mice were euthanized at 2 weeks after orthotopic injection, and the lungs were removed and subjected to histologic examination. The excised primary tumors were weighed and snap frozen for histological, RNA and protein analysis. For experimental lung metastasis assay, 1×106 cells were injected into tail veins of 6-8 week-old female SCID mice. Four weeks after injection, mice were euthanized, and lungs were removed and subjected to histologic examination. Lungs were fixed in formalin and embedded in paraffin, and 5 μm sections were stained with H&E. For each lung sample, all micro-metastases were counted under a light microscope at 10× magnification by an observer blinded to the experimental conditions. Six sections, at 50 μm intervals (covering more than 80 % of the lung), were counted per mouse sample, so the represented data indicate the total number of micro-metastases for the entire lung.
ECIS transendothelial migration assay
In vitro extravasation was monitored by quantitative real-time transendothelial migration assay using ECIS (13) (Applied Biophysics). Briefly, human umbilical vein endothelial cells (HUVECs) (1×105) were plated in 8W10E plus electrode arrays precoated with 200 μg/mL gelatin and allowed to form complete confluence. The monolayers were then challenged with FILIP1L clones ±DOX (1×105). Impedance changes of the challenged HUVECs were monitored for the next 24 h to determine the effect of FILIP1L on transendothelial migration activity.
In vivo invasion
Ovarian orthotopic tumors were grown for 17-18 days after injection of either control or FILIP1L clone followed by ±DOX treatment. In vivo invasion assay with ovarian orthotopic tumors was performed with a modified method from the one previously described (14). Briefly, in vivo invasion assay uses microneedles filled with Matrigel and ±10% FBS to collect the invasive tumor cells from primary tumors. To test if MMP activity was involved in the in vivo invasion, either vehicle or the inhibitor GM6001 was also included in the microneedles. Ovarian tumors were externalized and microneedles were positioned in the primary tumor with a micromanipulator. Cells were collected for 4 h while animals were anesthetized with 2–5% isoflurane throughout. The number of tumor cells collected was counted on a widefield microscope (Olympus) after expelling them on a glass slide and incubating them for 10 minutes with DAPI.
Inverted invasion assay
Inverted invasion assays were performed as described previously (15). Collagen I (2 mg/ml) or matrigel (6 mg/ml) supplemented with fibronectin (50 μg/ml) was allowed to polymerize in transwell inserts (Corning) for 1 h at 37°C. Inserts were then inverted, and either control or FILIP1L clone ±DOX (1×105) were seeded directly onto the opposite side of the filter. Transwell inserts were placed in serum-free medium or medium supplemented with 10% FBS, and 50 ng/ml EGF was placed on top of the matrix. Forty-eight hours after incubation, invading cells moving toward the three-dimensional matrix were stained with Calcein-AM and visualized by spinning disc confocal microscopy (Zeiss). Images were analyzed by AxioVision LE software (Zeiss).
Transfection of Cells with MMP9 plasmids or FILIP1L siRNA
MMP9 cDNA was obtained from GeneCopoeia. FILIP1L clone was transfected with equimolar amounts of control empty plasmid or plasmid encoding MMP9 using X-fect solution following the manufacturer's protocols (Clontech). After a 24 h transfection, the cells were subjected to a cell invasion assay. ON-TARGETplus Non-Targeting siRNA Pool and SMARTpool of ON-TARGETplus FILIP1L siRNA was purchased from Thermo Scientific. HEYA8 ovarian cancer cells were transfected with equimolar amounts of either non-Targeting or FILIP1L siRNA using Dharmafect solution following the manufacturer's protocols (Thermo Scientific). After a 48 h transfection, the cells were subjected to a cell invasion assay.
Cell invasion assay
Cells transfected with MMP9 cDNA or FILIP1L siRNA were cultured at ∼80% confluence. Cells were starved in basal medium containing 0.2% bovine serum albumin for 16 h. Matrigel invasion was measured using the BD BioCoat Tumor Invasion System (BD Biosciences #354165) as recommended by the manufacturer. Seeding 4.5 × 104 of starved cells into the apical chambers was followed by adding a chemoattractant (10% FBS) to the basal chambers. After a 20 h incubation, quantification of cell invasion was achieved by post-cell invasion labeling with a fluorescent dye, calcein AM (BD Biosciences), and measuring the fluorescence of invading cells of the underside of the membrane at 494/517 nm (excitation/emission). Synergy Mx microplate reader (BioTek) was used to measure fluorescence, and Gen5 software (BioTek) was used to analyzed the data.
Gelatin zymography
Primary tumors from either control or FILIP1L clone were lysed in T-PER buffer. Tumor lysates were analyzed by SDS-PAGE on gels containing 0.1% gelatin. Novex 10% zymogram (Gelatin) gels, and running, sample, wash and developing buffers were purchased from Invitrogen. The gels were washed, incubated in developing buffer for 8 h, 15 h and 24 h at 37°C and stained with 0.2% Coomassie blue R250. Bands of lysis representing gelatinase activity were then visualized against a dark background. Developing for 15 h resulted in the optimum range of activity. To quantify MMP9 activity, pro-MMP9 activity represented by 92 kDa-band in zymogram gels was normalized by GAPDH bands on separate SDS-PAGE gels from the same tumor lysates. Quantification was performed using AlphaView SA software (ProteinSimple).
Immunofluorescence
Ovarian orthotopic tumors from FILIP1L clone-injected mice were removed at day 14 after injection, snap frozen, and cut into 5 μm tumor sections. These tumor sections were fixed with 4% paraformaldehyde for 10 min followed by permeabilization with 0.3% Triton X-100 for 15 min. The sections were blocked with 5% donkey serum in PBS, and treated with mouse anti-MMP9 antibody (abcam) or rabbit anti-β catenin antibody (Cell Signaling). The sections were then incubated with 2 μg/mL Alexa Fluor 488 anti-mouse IgG or anti-rabbit IgG (Invitrogen), respectively, and treated with 4,6-diamidino-2-phenylindole (DAPI) mounting media (Vector Laboratories). Images were acquired on an Axio Imager A2 microscope and analyzed by AxioVision LE software (Zeiss). To quantify MMP9- or β catenin-positive signals, Cell Profiler software (16) was used.
Cultured FILIP1L clone ±DOX were permeabilized with methanol, blocked and treated with mouse anti-FILIP1L antibody and rabbit anti-γ tubulin (Sigma Aldrich) or pericentrin antibody (Cell Signaling) followed by Alexa Fluor 568 anti-mouse IgG and Alexa Fluor 488 anti-rabbit IgG. Images were acquired on a Spinning disc confocal microscope and analyzed by AxioVision LE software (Zeiss).
Gelatin degradation
Analysis of gelatin degradation in cell culture was performed as described previously (17). Alexa 405 protein labeling kit (Invitrogen) was used for labeling gelatin. Either control or FILIP1L clone were cultured on Alexa Fluor 405–gelatin-coated MatTek dishes (MatTek Corporation) for 10 h and fixed in 4% paraformaldehyde for 15 minutes. Fluorescent images were taken by a widefield microscope. ImageJ software was used for image processing and quantification of invadopodial degradation areas. Images were processed by filtering, thresholding and contrast inversion.
TCF/LEF reporter assay
Dual-luciferase reporter assay was performed as recommended by the manufacturer (Cignal TCF/LEF Reporter kit; Qiagen CCS-018L). Briefly, FILIP1L clones were plated in 96-well plates and transfected with reporter constructs using attractene (Qiagen) as a transfection reagent. Twenty-four hours after transfection, cells were pretreated with either 10 μM pan-WNT agonist (Calbiochem) or 1 μM GSK-3β inhibitor BIO (Calbiochem) followed by ±DOX treatment. The luciferase reporter activity was measured by luminescence at 8 h or 16 h after DOX treatment following the manufacturer's instruction (Dual-Glo luciferase assay; Promega).
Statistical analysis
Data are presented as the mean±SEM for the indicated number of separate experiments. Statistical analyses were performed using a two-tailed Student's t test (GraphPad Prism 3.0), and differences were considered statistically significant at P < 0.05.
Results
Development of inducible stable clones and the ovarian orthotopic model
We have demonstrated that the amount of FILIP1L expression is least in ES2 ovarian carcinoma cells among the ovarian cell lines tested and that FILIP1L expression in transiently transfected ES2 cells results in inhibition of cell adhesion, migration and invasion (3). To study the phenotype of FILIP1L expression in vivo, we developed stable ES2 cell lines, which express FILIP1L and the fluorescent marker mCherry in a DOX-inducible manner (FILIP1L clone). We also developed a control clone, which expresses mCherry alone (control clone). mCherry fluorescence was detected in both control and FILIP1L clones in a DOX dose-dependent manner (Figure 1A). mCherry protein expression was also confirmed by immunoblot (Supplementary Figure S1A). FILIP1L clone, but not control clone, showed FILIP1L expression following DOX treatment (Supplementary Figure S1B). FILIP1L was expressed in a DOX dose- and time-dependent manner (Figure 1B). Additionally, FILIP1L expression at biologically relevant levels was confirmed by qRT-PCR data (Supplementary Figure S1C). To test if FILIP1L expression inhibits cell proliferation, we measured cell proliferation in both clones. As shown in Figure 1C, FILIP1L expression did not affect cell proliferation. Next, to test whether FILIP1L expression results in inhibition of cancer metastasis in vivo, we developed an ovarian orthotopic model in mice. When injected into the ovary of SCID mice, these clones developed very aggressive ovarian cancer, including significant ascites by day 17 and lethality in 90% of mice by day 19 (Figure 1D-E). Metastatic ovarian cancer spread into the peritoneum and intra-abdominal organs was prevalent by day 14. In addition, paraaortic and pelvic lymph nodes as well as liver demonstrated massive metastatic tumor growth by day 14 (data not shown). FILIP1L was expressed in the orthotopic tumors from FILIP1L clone-injected mice in a DOX dose-dependent manner (Figure 1F). We subsequently chose to use 0.1 mg/ml DOX to induce FILIP1L expression in mice. To identify spontaneous lung metastasis, we examined lungs from these tumor-bearing mice following DOX treatment. Lungs from DOX-treated, but not from untreated, mice showed mCherry fluorescence (Figure 1G), and H&E staining of lungs showed the presence of micro-metastases at 14 days after orthotopic injection (Figure 1H). These lungs rarely demonstrated macroscopic metastases. In this model, lung metastasis appears to occur mainly through vessels, not by pleural effusion as micro-metastases were evenly distributed throughout the lung sections (Figure 1H and data not shown). Thus, these data suggest that we successfully developed inducible stable clones and an ovarian orthotopic model that expresses FILIP1L and exhibits spontaneous lung metastasis that we could utilize for our subsequent studies to dissect the FILIP1L-inhibiting step(s) of metastasis.
Figure 1. Development of inducible stable clones and the ovarian orthotopic model.
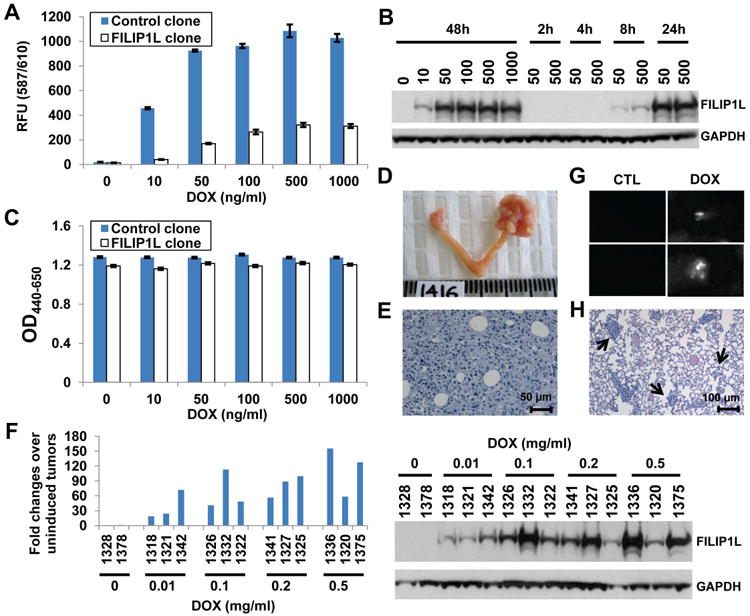
A, mCherry fluorescence measurement for control and FILIP1L clones treated with DOX for the indicated concentration. The y axis represents mCherry fluorescence measured at 587/610 nm (excitation/emission) (n=3). B, Immunoblot analysis for FILIP1L in FILIP1L clone treated with DOX for the indicated concentration (ng/ml) and time. C, WST1 cell proliferation assay for the same cells used in section A. The y axis represents cell proliferation measured with OD440-650nm (n=3). D, Ovarian orthotopic model. Mouse ovary from uninjected control (left) and FILIP1L clone-injected (right) 17 days after injection. E, H&E staining of ovarian orthotopic tumor shown in section D. F, qRT-PCR analysis for FILIP1L conducted on cDNA from FILIP1L clone-derived tumors (top). Drinking water containing the indicated concentration of DOX (mg/ml) was used to induce FILIP1L expression in mice. The numbers on the x axis indicate each mouse ID. The y axis represents fold change of each tumor over uninduced tumors (n=2), where each value was standardized with the housekeeping gene hRPL7. Immunoblot analysis for FILIP1L in the same tumor lysates used for qRT-PCR analysis (bottom). G, mCherry fluorescence of lungs from control clone-derived tumor-bearing mice 14 days after injection. Lungs were taken two days after giving mice drinking water containing 0.1 mg/ml DOX. White signals indicate lung metastases. H, H&E staining of the same lungs used in section G. Arrows indicate large (more than 50 cells)-, medium (6-50 cells)- and small (2-5 cells)-sized lung metastases, which were subjected to counting.
Inhibition of spontaneous lung metastasis by FILIP1L
First, we examined whether FILIP1L inhibits spontaneous lung metastasis in this orthotopic tumor model. Four groups were tested: either control or FILIP1L clone following ±DOX treatment. We analyzed both mCherry and FILIP1L expression in these tumors (Figure 2A-B). In accordance with in vitro cell proliferation data (Figure 1C), primary tumor growth was not inhibited by FILIP1L (Figure 2C and Supplementary Figure S2A-B). However, spontaneous lung metastases, detectable by day 14, were inhibited in the DOX-induced FILIP1L clone (Figure 2D). DOX itself had no effect, as a control clone did not show inhibition of lung metastases. These findings suggest that spontaneous lung metastasis, but not primary tumor growth, is inhibited by FILIP1L in this ovarian orthotopic tumor model.
Figure 2. Inhibition of spontaneous lung metastasis by FILIP1L.
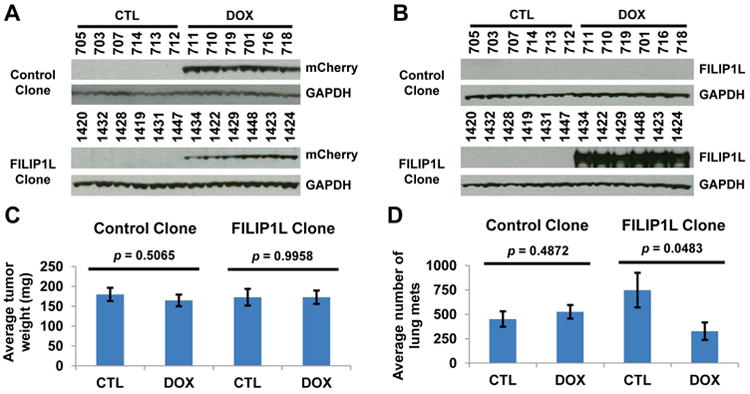
A-B, Immunoblot analysis for mCherry (A) and FILIP1L (B) in the tumor lysates from tumors used in spontaneous lung metastasis assay. The numbers indicate each mouse ID. C-D, Primary tumor growth (C) and spontaneous lung metastasis (D) were measured from the mice injected with either control or FILIP1L clone following ±DOX treatment (n=6). Images for ovarian orthotopic tumors are shown in Supplementary Figure S2A-D.
Extravasation is not inhibited by FILIP1L
Cancer metastasis consists of a series of steps including invasion, intravasation, extravasation and tumor growth at the metastatic site (1). Having observed that FILIP1L expression inhibits spontaneous lung metastasis, we sought to determine which step(s) of metastasis are inhibited by FILIP1L. To test whether extravasation and tumor growth at the metastatic site are inhibited by FILIP1L, we evaluated the effect of FILIP1L expression on the establishment of experimental lung metastases, those created by injecting cancer cells systemically via tail vein. This effectively bypasses the initial invasion and intravasation steps and evaluates extravasation and growth at a distant site. Experimental lung metastases measured by counting micro-metastases, detectable by day 28, were not inhibited by FILIP1L (Figure 3A). As with spontaneous lung metastasis, these lungs rarely demonstrated macroscopic metastases. To confirm that the extravasation step is not inhibited by FILIP1L, we performed an in vitro extravasation assay using Electric Cell-Substrate Impedance Sensing (ECIS) system. In this assay, when endothelial cell monolayers are challenged with metastatic cells, real-time observations by impedance are recorded that show binding of cells to endothelial cell layers, retraction of endothelial cell junctions and penetration of cells through endothelial monolayers. This type of assay has been used extensively to monitor transendothelial migration of tumor cells in vitro (13, 18, 19). The impedance rapidly dropped as FILIP1L clones interacted with HUVECs (Figure 3B). However, kinetics between untreated and DOX-treated cells were virtually the same, suggesting that the extravasation step is not inhibited by FILIP1L. Together, these data suggest that FILIP1L inhibits the earlier steps of metastasis such as invasion and intravasation, but not dissemination, extravasation and tumor growth at the metastatic site.
Figure 3. Extravasation is not inhibited by FILIP1L.
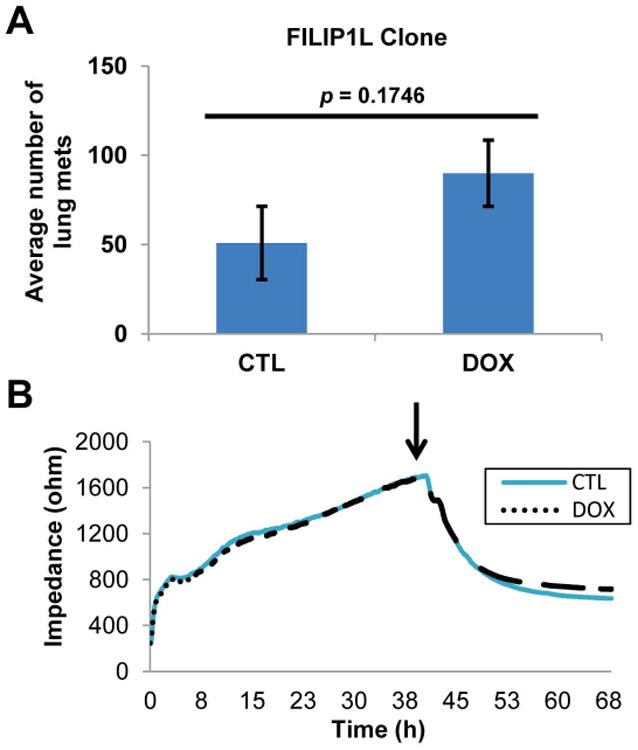
A, Experimental lung metastasis is not inhibited by FILIP1L. FILIP1L clones were injected via tail vein and lungs were procured and processed as in Figure 2D (n=10). B, Transendothelial migration is not inhibited by FILIP1L. Resistance changes in the impedance as confluent layers of HUVECs were challenged with FILIP1L clones ±DOX (indicated by arrow). Real-time impedance changes were recorded, wherein each curve represents the average of impedance change measured from four electrodes. 0.5 μg/ml DOX was used to induce FILIP1L expression for all in vitro experiments.
In vivo tumor cell invasion is inhibited by FILIP1L and requires MMPs
To confirm that an early invasion step of metastasis is inhibited by FILIP1L expression, we performed an in vivo invasion assay using ovarian orthotopic tumors. The number of invading cells in tumors from control clone with and without FBS was not different (Figure 4A). By contrast, FILIP1L expression in FILIP1L clones showed a significantly decreased invasive ability in vivo compared with controls (Figure 4B). Next, we were interested in whether the invasive ability, which was inhibited by FILIP1L, was also MMP dependent. If so, a similar decrease in invasive ability would be observed by inhibiting MMPs. To this end, we introduced a pan-MMP inhibitor GM6001. The addition of GM6001 resulted in a completely blocked invasive ability in vivo compared with controls (Figure 4C). In addition, the anti-invasive activity by GM6001 treatment in uninduced FILIP1L clone was not significantly different from that by FILIP1L in induced FILIP1L clone (Figure 4C). We previously showed that ES2 cells transiently transfected with FILIP1L invaded two-dimensional matrigel significantly less than those transfected with control (3). To definitively prove that FILIP1L inversely regulates the invasive properties of the ovarian cancer cells, we tested if knockdown of FILIP1L in FILIP1L-expressing ovarian cancer cell lines resulted in increased cell invasion. We transfected HEYA8 ovarian cancer cells with either non-targeting or FILIP1L siRNA and measured invasion. As shown in Supplementary Figure S4, HEYA8 cells transfected with FILIP1L siRNA demonstrated a reduction of FILIP1L compared with control and invaded Matrigel significantly more than those transfected with control siRNA. It has been shown that there are key differences between the characteristics of cells migrating on three-dimensional versus on two-dimensional matrices (20), and experimental systems measuring migration across plastic surfaces may not accurately model the type of motility that would be required by a tumor cell in order to move away from the primary tumor and form metastases at distant sites. To elucidate the potential contribution of FILIP1L in inhibiting the invasive phenotype of aggressive tumors, we performed an inverted invasion assay (15), in which cells must migrate upward through three-dimensional matrices. The median distance of invading cells in three-dimensional matrices from control clones ±DOX was not different, whereas invasion was significantly inhibited by FILIP1L expression in FILIP1L clone (Figure 4D and Supplementary Figure S3A-D). These results demonstrate that FILIP1L inhibits MMP-dependent in vivo invasion as well as invasion toward a three-dimensional matrix in our model.
Figure 4. In vivo tumor cell invasion is inhibited by FILIP1L and requires MMPs.
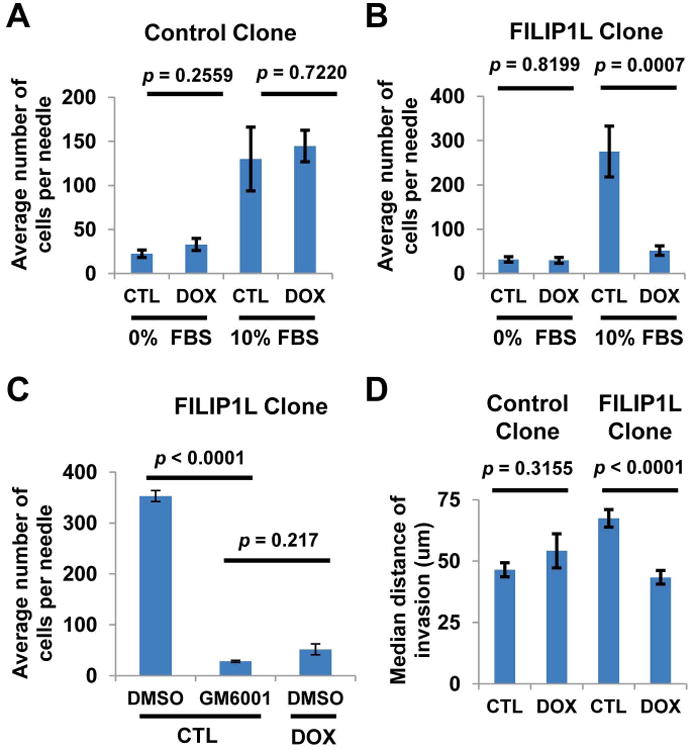
A-C, Ovarian orthotopic tumors were established as described in Figure 1 from control (A) or FILIP1L clone (B). The ability of tumor cells to invade from primary tumors into microneedles filled with Matrigel with or without 10% FBS was measured (n=10). Invasion was inhibited in the presence of 10 μM GM6001 (C). Error bars indicate SEM. D. Inverted invasion toward a 3D matrix consisting of Collagen I and fibronectin was measured (n=24). Representative image of serial optical sections captured at 15 μm intervals with confocal microscopy are shown in Supplementary Figure S3A-D. Similar data were obtained using a 3D matrix consisting of matrigel and fibronectin (data not shown).
FILIP1L down-regulates MMPs
Having observed that FILIP1L inhibits MMP-dependent in vivo invasion, we tested if FILIP1L expression is associated with down-regulation of MMPs. Using qRT-PCR analysis with species-specific primers which detect human but not mouse MMPs, we measured the expression of various MMPs in tumors from control and FILIP1L clones. Among the MMPs we tested, we observed that MMP3, -7 and -9 were transcriptionally down-regulated in FILIP1L-expressing tumors (Figure 5A). By contrast, MMP expression in tumors from control clones ±DOX treatment was not different (data not shown). We chose to further study MMP9 since, of the three, it has been more implicated in cancer cell invasion and metastasis, including in ovarian cancer. To test if protein expression of MMPs is also reduced in FILIP1L-expressing tumors, we measured MMP9 protein expression by immunofluoresence. MMP9 protein expression was significantly reduced in tumors from FILIP1L clones following DOX treatment (Figure 5B). Immunoblot against MMP9 also confirmed that MMP9 protein expression was reduced in FILIP1L-expressing tumors (Supplementary Figure S5). We then investigated if MMP activity itself is also reduced. To this end, we performed gelatin zymography using tumor lysates. MMP9 activity was not changed in tumors from control clones following DOX treatment, whereas it was significantly reduced in tumors from FILIP1L clones following DOX treatment (Figure 5C). Furthermore, we measured gelatin degradation in vitro. Gelatin degradation was not changed in control clones following DOX treatment, whereas it was significantly reduced in FILIP1L clones following DOX treatment (Figure 5D). We then tested if MMP9 overexpression could compensate for the effect of FILIP1L expression in FILIP1L clone. We transfected FILIP1L clone with a plasmid encoding control or MMP9 cDNA and measured invasion. As shown in Figure 5E, FILIP1L clone transfected with MMP9 cDNA demonstrated MMP9 overexpression. FILIP1L-induced cells transfected with MMP9 demonstrated significantly more invasion than those transfected with control. In contrast, FILIP1L-uninduced cells did not show differences in invasion with MMP9 overexpression. These data suggest that FILIP1L inhibits cell invasion through down-regulation of MMP expression.
Figure 5. FILIP1L down-regulates MMPs.

A, qRT-PCR analysis for MMPs conducted on cDNA from FILIP1L clone-derived tumors. The y axis represents fold change of DOX-induced tumors (n=3) over uninduced tumors (n=3), where each value was standardized with the housekeeping gene GAPDH. * indicates P<0.05. B, Immunofluorescent staining of MMP9 (green) in FILIP1L clone-derived tumors. Nuclei counterstained with DAPI (blue). A merged image is shown. Shown at right is the quantified data from four random fields per tumor (n=12). C, MMP9 activity of total tumor lysates from control and FILIP1L clones was measured using gelatin zymography. Each well represents a different tumor. The arrows indicate pro-MMP9 activity. The quantified data, where each pixel value of pro-MMP9 was standardized with that of GAPDH, is also shown. D, Gelatin matrix degradation in vitro from cultured control and FILIP1L clones was measured (n=50). E, Matrigel cell invasion assay 24 h after transfection of FILIP1L clones with control empty plasmid or plasmid encoding MMP9. Immunoblot analysis for MMP9 in the transfected cells is also shown. GAPDH blot is shown as the loading control.
FILIP1L inhibits the canonical WNT signaling pathway
MMPs are transcriptional targets of the canonical WNT pathway (21, 22). We showed that WNT-target MMPs such as MMP3, -7 and -9 were transcriptionally down-regulated in FILIP1L-expressing tumors (Figure 5A). We tested if WNT ligands are also transcriptionally down-regulated in FILIP1L-expressing tumors. We found that WNT ligands WNT2, -3A, -4, -5A, -7A and -11 were highly expressed in tumors compared with cultured cells (data not shown), and they were significantly down-regulated in FILIP1L-expressing tumors (Figure 6A). By contrast, WNT expression in tumors from a control clone ±DOX treatment was not different (data not shown). Interestingly, expression of WNT receptors, such as frizzled (FZD) 2, -3, -4 and -9 and low-density lipoprotein receptor-related protein (LRP) 5 and -6, was not different in tumors from FILIP1L clone ±DOX treatment (data not shown). We then tested if treatment of FILIP1L clone with canonical WNT resulted in induction of MMPs. WNT3A treatment resulted in induction of MMPs such as MMP3, -7 and -9 in uninduced cells (Vehicle-CTL compared with WNT3A-CTL), where this induction was significantly reduced in DOX-induced cells (WNT3A-CTL compared with WNT3A-DOX) (Figure 6B). WNT signaling comprises both canonical and non-canonical pathways (23). The canonical pathway is regulated by the stability of β-catenin where stabilized β-catenin can activate the canonical pathway through the transcription of WNT/β-catenin target genes (23). Thus, we next asked if FILIP1L inhibits β-catenin-directed transcriptional activity. The canonical WNT signaling pathway was activated by either pan-WNT agonist or GSK-3β inhibitor BIO. We observed that this activity was significantly reduced in FILIP1L-expressing cells compared with uninduced control (Figure 6C), suggesting that the canonical WNT pathway is inhibited by FILIP1L expression. Non-canonical WNT expression can be regulated by canonical WNT signals. For example, the activation of the WNT3A pathway increases the expression of non-canonical WNTs such as WNT5A and WNT11 (24, 25). As shown in Supplementary Figure S6, WNT3A treatment resulted in induction of WNT5A and WNT11 in uninduced cells (Vehicle-CTL compared with WNT3A-CTL), where this induction was significantly reduced in DOX-induced cells (WNT3A-CTL compared with WNT3A-DOX). However, WNT3A treatment did not result in induction of β-catenin in uninduced cells, nor was β-catenin expression inhibited by FILIP1L expression (Supplementary Figure S6). To elucidate how FILIP1L inhibits β-catenin-directed transcriptional activity, we analyzed the amount of N-terminally phosphorylated β-catenin, which is destined for proteasomal degradation, by immunoblot. The amount of N-terminally phosphorylated β-catenin was decreased following pan-WNT agonist treatment in uninduced cells. However, it remained unchanged in DOX-induced FILIP1L expressing cells (Figure 6D). Endostatin, a FILIP1L inducer (26), was previously shown to inhibit WNT signaling through proteasome-mediated β-catenin degradation (27). In addition, FILIP1L was shown to promote Heat shock factor 1 (HSF1) ubiquitination and degradation through the ubiquitin-proteasome system, leading to a reduction in HSF1-mediated transcription (28). Treatment with MG132, a proteasome inhibitor considerably increased phosphorylated β-catenin regardless of FILIP1L expression, suggesting that FILIP1L may regulate proteasome-mediated β-catenin degradation (Figure 6D). N-terminally phosphorylated β-catenin, which is destined to undergo proteasomal degradation, has been shown to associate with the centrosome (29-31). In addition, it has been shown that the centrosome also functions as the proteolytic center of the cell (32-35). Thus, we tested if FILIP1L localizes in the centrosome. As shown in Figure 6E, endogenous FILIP1L in induced FILIP1L clone colocalized with centrosomal marker proteins, such as γ-tubulin and pericentrin. Next, we measured the amount of β-catenin in the nucleus, which is an indicator of an active canonical WNT pathway, in the tumors resulting from FILIP1L clone by immunofluorescence. As shown in Figure 6E, the amount of β-catenin in the nucleus was significantly reduced in FILIP1L-expressing tumors. Thus, these data collectively suggest that FILIP1L operates downstream of, or parallel to, GSK-3β, possibly at the level of β-catenin, and that reduced β-catenin levels lead to the transcriptional down-regulation of WNT target genes such as MMPs, resulting in inhibition of metastasis.
Figure 6. FILIP1L inhibits the canonical WNT signaling pathway.

A, qRT-PCR analysis for WNT ligands conducted on cDNA from FILIP1L clone-derived tumors. The y axis represents fold change of DOX-induced tumors (n=3) over uninduced tumors (n=3), where each value was standardized with the housekeeping gene GAPDH. * indicates P<0.05. B, qRT-PCR analysis for MMPs conducted on cDNA from cultured FILIP1L clone, which was treated with either vehicle or WNT3A following ±DOX treatment. The y axis represents fold change over vehicle-treated, uninduced cells (Vehicle-CTL), where each value was standardized with the housekeeping gene GAPDH (n=3). C, β-catenin-regulated transcriptional activity is decreased by FILIP1L. A cultured FILIP1L clone was transfected with a TCF/LEF reporter construct, treated with either pan-WNT agonist or BIO overnight followed by ±DOX treatment. Reporter activity was measured 8 h after DOX treatment. The y axis represents a percentage of uninduced cells where firefly luciferase activity was standardized with Renilla luciferase activity (n=4). Measurement at 16 h after DOX treatment yielded similar results (data not shown). D, FILIP1L facilitates β-catenin inactivation following WNT activation. Cultured FILIP1L clone was treated with vehicle, pan-WNT agonist or 10 μM MG132 following ±DOX treatment. Cell lysates were immunoblotted for phospho-β-catenin (Ser33/37/Thr41). E, FILIP1L localizes at centrosomes. Cultured FILIP1L clone ±DOX treatment was immunofluorescently stained for FILIP1L (red), γ-tubulin or pericentrin (green) and nuclei counterstained with DAPI (blue). Merged images are also shown. F, Immunofluorescent staining of β-catenin (green) in FILIP1L clone-derived tumors. Nuclei counterstained with DAPI (blue). A merged image is shown. An inset indicates the magnified image for the selected region, showing DAPI and β-catenin staining separately. The quantified data from four random fields per tumor is also shown (n=12).
Discussion
Cancer metastasis is a multistep processes, involving migration and invasion through tumor stroma, intravasation, tumor cell dissemination, extravasation and cell growth at the metastatic sites (1). Our data demonstrate that FILIP1L inhibits spontaneous metastasis, but not experimental (tumor cells injected via tail vein) metastasis, suggesting that FILIP1L inhibits the earlier steps — migration, invasion and intravasation. In addition, FILIP1L inhibits MMP-dependent in vivo invasion, and its expression is associated with the down-regulation of MMPs. Expression of MMP3, -7 and -9 has been shown to be involved in promoting cancer metastasis (36-38). Although MMPs such as MMP9 have been shown to promote in vivo intravasation (39), it is unclear at this time whether MMPs are directly involved in the intravasation step in this tumor model. Further studies using in vivo intravasation assays and multiphoton intravital imaging will address this question.
Experimental lung metastasis would be expected to develop faster than spontaneous lung metastasis in most animal models. In our animal model, spontaneous lung metastases were routinely detected by 14 days from the orthotopically injected mice (5×105 cells per mouse were injected). However, experimental lung metastasis, which does not need initial invasion and intravasation, was detected by 28 days (1×106 cells per mouse were injected). A similar phenomenon was reported using prostate cancer cells, PCai1 (40). We speculate that the ovarian microenvironment may facilitate lung metastasis for cell lines such as ES2 by promoting tumor growth or secreting hormones.
In our gelatin zymography experiment, we mainly detected pro-MMP9 gelatinase activity, but not active MMP9 activity. This is consistent with studies which also showed only pro-MMP9 bands from ovarian tumor lysates (41, 42). In fact, pro-MMP9 gelatinolytic activity, but not active MMP-2 or MMP-9, has shown to serve as a useful statistically independent prognostic factor in ovarian cancer (42). Because FILIP1L expression reduces pro-MMP9 activity in gelatin zymography and gelatin degradation in vitro, this suggests that FILIP1L reduces MMP activity.
Our data demonstrate that WNT-target MMPs such as MMP3, -7 and -9 are down-regulated in FILIP1L-expressing tumors. In addition, β-catenin-mediated transcriptional activity is inhibited, and canonical WNT-induced transcriptional up-regulation of these MMPs is also inhibited in FILIP1L-expressing cells, indicating an effect of FILIP1L on the canonical WNT signaling pathway. The WNT pathway regulates many developmental processes, including cell-fate specification and cell polarity (43). Pathologically, WNT signaling has been implicated in cancer cell proliferation and metastasis (44). In the canonical WNT pathway, β-catenin is the key effector responsible for the pathway signal; β-catenin's translocation to the nucleus triggers transcription of WNT-specific genes that control cell fate. The final transcriptional output of β-catenin is regulated by cross-talk with other transcription factors and signaling pathways in a context-dependent, tissue-specific or temporally-restricted manner (23). TCF/LEF transcription factors serve as the main nuclear partners of β-catenin, guiding it to specific DNA loci. One example of a context-dependent WNT target is cyclin D1. Its transcript was found to be up-regulated upon WNT pathway activation in many cell types (45). However, other studies have failed to detect elevated cyclin D1 levels during WNT activation (46, 47). In our FILIP1L-expressing tumors, β-catenin transcriptional targets such as cyclin D1, cMYC and PCNA, which drive cell proliferation, did not show reduced expression (data not shown). This correlated with our data that the expression of FILIP1L in ovarian cancer inhibited spontaneous lung metastasis, whereas primary tumor growth was not inhibited. Thus, a subset of β-catenin targets promoting metastasis must be selectively inhibited in this model. Future experiments will employ ChIP-Seq and RNA-Seq analyses to identify occupied DNA binding sites for β-catenin as well as target genes that are regulated by β-catenin in tumors expressing low or high levels of FILIP1L.
It is not clear whether directing therapy against β-catenin or MMPs would result in the same effect as FILIP1L. As discussed, a subset of β-catenin targets promoting metastasis seems to be selectively inhibited by FILIP1L in our model. Thus, inhibiting β-catenin would result in inhibition of all the targets of β-catenin, which are different from those of FILIP1L inhibition. Ten of 24 murine MMPs, including MMP3, -7 and -9, have been shown to have antitumorigenic and anti-inflammatory roles, making them potential drug antitargets (48).
The non-canonical pathway is β-catenin-independent and is thought to regulate cytoskeletal rearrangements, thereby coordinating cell adhesion, migration and polarity (49). WNTs 5A and 11 are classified as typical non-canonical WNTs (49). Knockdown of WNT5A suppresses focal adhesion turnover and cell migration in various cells (50, 51). The expression of WNT5A was correlated with aggressiveness and poor prognosis of tumors including ovarian, breast, lung and gastric cancer as well as melanoma (50, 52-55), suggesting that WNT5A has oncogenic properties. In addition, knockdown of WNT5A was shown to suppress gastric cancer metastasis (56). WNT11 increases the migration of numerous types of cancer cells (57-59). As described earlier, non-canonical WNT expression can be regulated by canonical WNT signals. In this study, we observed that non-canonical WNT ligands WNT5A, -11 and -4 are considerably down-regulated in FILIP1L-expressing tumors. We also observed that WNT3A treatment resulted in induction of WNT5A and WNT11 where this induction was significantly reduced by FILIP1L expression. As these WNTs are transcriptional targets of β-catenin, a positive feedback loop may be active in this tumor model. Therefore, it will be of interest to confirm if the inhibition of the non-canonical WNT pathway through transcriptional down-regulation of non-canonical WNTs can also inhibit cancer cell invasion and metastasis.
We have previously shown that both mRNA and protein expression of FILIP1L were down-regulated in ovarian cancer cells compared to normal ovarian epithelial cells (3). In addition, FILIP1L expression was significantly lower in invasive serous carcinoma than in non-invasive serous borderline tumors (3). Activating mutations as well as missense mutations of β-catenin that render the mutant protein resistant to proteosomal degradation have been implicated in ovarian cancer, mainly in endometrioid subtype (60). Emerging data suggest that despite not having mutations, the Wnt/β-catenin pathway plays a role in carcinogenesis of all ovarian cancer subtypes (61-63).
In this study, we showed that in uninduced cells pan-WNT agonist treatment reduced N-terminally phosphorylated β-catenin, which is destined for proteasomal degradation. However, it remained unchanged in DOX-induced FILIP1L expressing cells. In addition, treatment of a proteasome inhibitor considerably increased phosphorylated β-catenin. Furthermore, we showed that FILIP1L localized in the centrosome in which N-terminally phosphorylated β-catenin has been shown to localize. Thus it is important for future studies to test whether centrosomal FILIP1L promotes β-catenin degradation and whether reduced β-catenin levels lead to down-regulation of WNT target genes, such as MMPs, and non-canonical WNTs, resulting in inhibition of metastasis.
In summary, we have demonstrated that expression of FILIP1L is associated with inhibition of spontaneous metastasis, which is mediated by down-regulation of MMPs through inhibition of the WNT signaling pathway. Further characterization of the mechanism of FILIP1L's inhibition of metastasis may improve understanding of the role played by FILIP1L in cancer metastasis and lead to the development of more effective anticancer agents.
Supplementary Material
What's new: Identifying key mediators of cancer metastasis is crucial to the development of more effective therapies. Filamin A interacting protein 1-like (FILIP1L) was recently identified as an important inhibitor of cancer metastasis. However, its mechanism of action is not known. Using our doxycycline-inducible ovarian orthotopic mouse model, we demonstrated that FILIP1L inhibits the earlier steps of metastasis such as invasion and intravasation. We also showed that FILIP1L inhibits WNT signaling, resulting in inhibition of metastasis.
Acknowledgments
This work was supported in part by a generous research gift from Linda and Earle Altman. YW and JSC were supported by National Institutes of Health (CA163131, CA1664468). BTB was supported by National Institutes of Health (CA150344).
Footnotes
Conflict of interest: Authors declare no conflict of interest.
References
- 1.Bravo-Cordero JJ, Hodgson L, Condeelis J. Directed cell invasion and migration during metastasis. Curr Opin Cell Biol. 2012;24:277–83. doi: 10.1016/j.ceb.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwon M, Hanna E, Lorang D, He M, Quick JS, Adem A, Stevenson C, Chung JY, Hewitt SM, Zudaire E, Esposito D, Cuttitta F, et al. Functional characterization of filamin a interacting protein 1-like, a novel candidate for antivascular cancer therapy. Cancer Res. 2008;68:7332–41. doi: 10.1158/0008-5472.CAN-08-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burton ER, Gaffar A, Lee SJ, Adeshuko F, Whitney KD, Chung JY, Hewitt SM, Huang GS, Goldberg GL, Libutti SK, Kwon M. Downregulation of Filamin A interacting protein 1-like is associated with promoter methylation and induces an invasive phenotype in ovarian cancer. Mol Cancer Res. 2011;9:1126–38. doi: 10.1158/1541-7786.MCR-11-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwon M, Lee SJ, Reddy S, Rybak Y, Adem A, Libutti SK. Down-regulation of filamin A interacting protein 1-like is associated with promoter methylation and an invasive phenotype in breast, colon, lung and pancreatic cancers. PLoS One. doi: 10.1371/journal.pone.0082620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Desotelle J, Truong M, Ewald J, Weeratunga P, Yang B, Huang W, Jarrard D. CpG Island Hypermethylation Frequently Silences FILIP1L Isoform 2 Expression in Prostate Cancer. J Urol. 2013;189:329–35. doi: 10.1016/j.juro.2012.08.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie C, Gou ML, Yi T, Deng H, Li ZY, Liu P, Qi XR, He X, Wei Y, Zhao X. Efficient inhibition of ovarian cancer by truncation mutant of FILIP1L gene delivered by novel biodegradable cationic heparin-polyethyleneimine nanogels. Hum Gene Ther. 2011;22:1413–22. doi: 10.1089/hum.2011.047. [DOI] [PubMed] [Google Scholar]
- 7.Mok SC, Wong KK, Chan RK, Lau CC, Tsao SW, Knapp RC, Berkowitz RS. Molecular cloning of differentially expressed genes in human epithelial ovarian cancer. Gynecologic oncology. 1994;52:247–52. doi: 10.1006/gyno.1994.1040. [DOI] [PubMed] [Google Scholar]
- 8.Matei D, Graeber TG, Baldwin RL, Karlan BY, Rao J, Chang DD. Gene expression in epithelial ovarian carcinoma. Oncogene. 2002;21:6289–98. doi: 10.1038/sj.onc.1205785. [DOI] [PubMed] [Google Scholar]
- 9.Quaye L, Dafou D, Ramus SJ, Song H, Gentry-Maharaj A, Notaridou M, Hogdall E, Kjaer SK, Christensen L, Hogdall C, Easton DF, Jacobs I, et al. Functional complementation studies identify candidate genes and common genetic variants associated with ovarian cancer survival. Hum Mol Genet. 2009;18:1869–78. doi: 10.1093/hmg/ddp107. [DOI] [PubMed] [Google Scholar]
- 10.Notaridou M, Quaye L, Dafou D, Jones C, Song H, Hogdall E, Kjaer SK, Christensen L, Hogdall C, Blaakaer J, McGuire V, Wu AH, et al. Common alleles in candidate susceptibility genes associated with risk and development of epithelial ovarian cancer. International journal of cancer Journal international du cancer. 2010 doi: 10.1002/ijc.25554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarze SR, DePrimo SE, Grabert LM, Fu VX, Brooks JD, Jarrard DF. Novel pathways associated with bypassing cellular senescence in human prostate epithelial cells. J Biol Chem. 2002;277:14877–83. doi: 10.1074/jbc.M200373200. [DOI] [PubMed] [Google Scholar]
- 12.Poole LJ, Yu Y, Kim PS, Zheng QZ, Pevsner J, Hayward GS. Altered patterns of cellular gene expression in dermal microvascular endothelial cells infected with Kaposi's sarcoma-associated herpesvirus. J Virol. 2002;76:3395–420. doi: 10.1128/JVI.76.7.3395-3420.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keese CR, Bhawe K, Wegener J, Giaever I. Real-time impedance assay to follow the invasive activities of metastatic cells in culture. BioTechniques. 2002;33:842–4. 6, 8–50. doi: 10.2144/02334rr01. [DOI] [PubMed] [Google Scholar]
- 14.Wyckoff JB, Segall JE, Condeelis JS. The collection of the motile population of cells from a living tumor. Cancer Res. 2000;60:5401–4. [PubMed] [Google Scholar]
- 15.Caswell PT, Spence HJ, Parsons M, White DP, Clark K, Cheng KW, Mills GB, Humphries MJ, Messent AJ, Anderson KI, McCaffrey MW, Ozanne BW, et al. Rab25 associates with alpha5beta1 integrin to promote invasive migration in 3D microenvironments. Dev Cell. 2007;13:496–510. doi: 10.1016/j.devcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 16.Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome biology. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yamaguchi H, Lorenz M, Kempiak S, Sarmiento C, Coniglio S, Symons M, Segall J, Eddy R, Miki H, Takenawa T, Condeelis J. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–52. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saxena NK, Sharma D, Ding X, Lin S, Marra F, Merlin D, Anania FA. Concomitant activation of the JAK/STAT, PI3K/AKT, and ERK signaling is involved in leptin-mediated promotion of invasion and migration of hepatocellular carcinoma cells. Cancer Res. 2007;67:2497–507. doi: 10.1158/0008-5472.CAN-06-3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang HS, Hung Y, Su CH, Peng ST, Guo YJ, Lai MC, Liu CY, Hsu JW. CD44 cross-linking induces integrin-mediated adhesion and transendothelial migration in breast cancer cell line by up-regulation of LFA-1 (alpha L beta2) and VLA-4 (alpha4beta1) Exp Cell Res. 2005;304:116–26. doi: 10.1016/j.yexcr.2004.10.015. [DOI] [PubMed] [Google Scholar]
- 20.Even-Ram S, Yamada KM. Cell migration in 3D matrix. Curr Opin Cell Biol. 2005;17:524–32. doi: 10.1016/j.ceb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 21.Song Y, Yang QX, Zhang F, Meng F, Li H, Dong Y, Han A. Suppression of nasopharyngeal carcinoma cell by targeting beta-catenin signaling pathway. Cancer epidemiology. 2012;36:e116–21. doi: 10.1016/j.canep.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 22.Denys H, De Wever O, Nusgens B, Kong Y, Sciot R, Le AT, Van Dam K, Jadidizadeh A, Tejpar S, Mareel M, Alman B, Cassiman JJ. Invasion and MMP expression profile in desmoid tumours. British journal of cancer. 2004;90:1443–9. doi: 10.1038/sj.bjc.6601661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valenta T, Hausmann G, Basler K. The many faces and functions of beta-catenin. EMBO J. 2012;31:2714–36. doi: 10.1038/emboj.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Railo A, Pajunen A, Itaranta P, Naillat F, Vuoristo J, Kilpelainen P, Vainio S. Genomic response to Wnt signalling is highly context-dependent--evidence from DNA microarray and chromatin immunoprecipitation screens of Wnt/TCF targets. Exp Cell Res. 2009;315:2690–704. doi: 10.1016/j.yexcr.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 25.Ueno S, Weidinger G, Osugi T, Kohn AD, Golob JL, Pabon L, Reinecke H, Moon RT, Murry CE. Biphasic role for Wnt/beta-catenin signaling in cardiac specification in zebrafish and embryonic stem cells. Proc Natl Acad Sci U S A. 2007;104:9685–90. doi: 10.1073/pnas.0702859104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mazzanti CM, Tandle A, Lorang D, Costouros N, Roberts D, Bevilacqua G, Libutti SK. Early genetic mechanisms underlying the inhibitory effects of endostatin and fumagillin on human endothelial cells. Genome Res. 2004;14:1585–93. doi: 10.1101/gr.2552804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanai J, Gloy J, Karumanchi SA, Kale S, Tang J, Hu G, Chan B, Ramchandran R, Jha V, Sukhatme VP, Sokol S. Endostatin is a potential inhibitor of Wnt signaling. J Cell Biol. 2002;158:529–39. doi: 10.1083/jcb.200203064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hu Y, Mivechi NF. Promotion of heat shock factor Hsf1 degradation via adaptor protein filamin A-interacting protein 1-like (FILIP-1L) J Biol Chem. 2011;286:31397–408. doi: 10.1074/jbc.M111.255851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang P, Senga T, Hamaguchi M. A novel role of phospho-beta-catenin in microtubule regrowth at centrosome. Oncogene. 2007;26:4357–71. doi: 10.1038/sj.onc.1210217. [DOI] [PubMed] [Google Scholar]
- 30.Hadjihannas MV, Bruckner M, Behrens J. Conductin/axin2 and Wnt signalling regulates centrosome cohesion. EMBO Rep. 2010;11:317–24. doi: 10.1038/embor.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fuentealba LC, Eivers E, Geissert D, Taelman V, De Robertis EM. Asymmetric mitosis: Unequal segregation of proteins destined for degradation. Proc Natl Acad Sci U S A. 2008;105:7732–7. doi: 10.1073/pnas.0803027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Badano JL, Teslovich TM, Katsanis N. The centrosome in human genetic disease. Nat Rev Genet. 2005;6:194–205. doi: 10.1038/nrg1557. [DOI] [PubMed] [Google Scholar]
- 33.Bahmanyar S, Kaplan DD, Deluca JG, Giddings TH, Jr, O'Toole ET, Winey M, Salmon ED, Casey PJ, Nelson WJ, Barth AI. beta-Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev. 2008;22:91–105. doi: 10.1101/gad.1596308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gerdes JM, Liu Y, Zaghloul NA, Leitch CC, Lawson SS, Kato M, Beachy PA, Beales PL, DeMartino GN, Fisher S, Badano JL, Katsanis N. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet. 2007;39:1350–60. doi: 10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- 35.Wigley WC, Fabunmi RP, Lee MG, Marino CR, Muallem S, DeMartino GN, Thomas PJ. Dynamic association of proteasomal machinery with the centrosome. J Cell Biol. 1999;145:481–90. doi: 10.1083/jcb.145.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi JW, Ahn SE, Rengaraj D, Seo HW, Lim W, Song G, Han JY. Matrix metalloproteinase 3 is a stromal marker for chicken ovarian cancer. Oncology letters. 2011;2:1047–51. doi: 10.3892/ol.2011.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao H, Yang Z, Wang X, Zhang X, Wang M, Wang Y, Mei Q, Wang Z. Triptolide inhibits ovarian cancer cell invasion by repression of matrix metalloproteinase 7 and 19 and upregulation of E-cadherin. Experimental & molecular medicine. 2012;44:633–41. doi: 10.3858/emm.2012.44.11.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakravarty D, Roy SS, Babu CR, Dandamudi R, Curiel TJ, Vivas-Mejia P, Lopez-Berestein G, Sood AK, Vadlamudi RK. Therapeutic targeting of PELP1 prevents ovarian cancer growth and metastasis. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:2250–9. doi: 10.1158/1078-0432.CCR-10-2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chou CH, Teng CM, Tzen KY, Chang YC, Chen JH, Cheng JC. MMP-9 from sublethally irradiated tumor promotes Lewis lung carcinoma cell invasiveness and pulmonary metastasis. Oncogene. 2012;31:458–68. doi: 10.1038/onc.2011.240. [DOI] [PubMed] [Google Scholar]
- 40.Naiki T, Asamoto M, Toyoda-Hokaiwado N, Naiki-Ito A, Tozawa K, Kohri K, Takahashi S, Shirai T. Organ specific Gst-pi expression of the metastatic androgen independent prostate cancer cells in nude mice. The Prostate. 2012;72:533–41. doi: 10.1002/pros.21455. [DOI] [PubMed] [Google Scholar]
- 41.Schmalfeldt B, Prechtel D, Harting K, Spathe K, Rutke S, Konik E, Fridman R, Berger U, Schmitt M, Kuhn W, Lengyel E. Increased expression of matrix metalloproteinases (MMP)-2, MMP-9, and the urokinase-type plasminogen activator is associated with progression from benign to advanced ovarian cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2001;7:2396–404. [PubMed] [Google Scholar]
- 42.Lengyel E, Schmalfeldt B, Konik E, Spathe K, Harting K, Fenn A, Berger U, Fridman R, Schmitt M, Prechtel D, Kuhn W. Expression of latent matrix metalloproteinase 9 (MMP-9) predicts survival in advanced ovarian cancer. Gynecol Oncol. 2001;82:291–8. doi: 10.1006/gyno.2001.6243. [DOI] [PubMed] [Google Scholar]
- 43.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 44.Lai SL, Chien AJ, Moon RT. Wnt/Fz signaling and the cytoskeleton: potential roles in tumorigenesis. Cell Res. 2009;19:532–45. doi: 10.1038/cr.2009.41. [DOI] [PubMed] [Google Scholar]
- 45.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–6. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 46.Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, Batlle E, Simon-Assmann P, Clevers H, Nathke IS, Clarke AR, Winton DJ. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18:1385–90. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, et al. The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell. 2002;111:241–50. doi: 10.1016/s0092-8674(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 48.Dufour A, Overall CM. Missing the target: matrix metalloproteinase antitargets in inflammation and cancer. Trends in pharmacological sciences. 2013;34:233–42. doi: 10.1016/j.tips.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 49.Amin N, Vincan E. The Wnt signaling pathways and cell adhesion. Front Biosci. 2012;17:784–804. doi: 10.2741/3957. [DOI] [PubMed] [Google Scholar]
- 50.Kurayoshi M, Oue N, Yamamoto H, Kishida M, Inoue A, Asahara T, Yasui W, Kikuchi A. Expression of Wnt-5a is correlated with aggressiveness of gastric cancer by stimulating cell migration and invasion. Cancer Res. 2006;66:10439–48. doi: 10.1158/0008-5472.CAN-06-2359. [DOI] [PubMed] [Google Scholar]
- 51.Matsumoto S, Fumoto K, Okamoto T, Kaibuchi K, Kikuchi A. Binding of APC and dishevelled mediates Wnt5a-regulated focal adhesion dynamics in migrating cells. EMBO J. 2010;29:1192–204. doi: 10.1038/emboj.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent JM. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell. 2002;1:279–88. doi: 10.1016/s1535-6108(02)00045-4. [DOI] [PubMed] [Google Scholar]
- 53.Badiglian Filho L, Oshima CT, De Oliveira Lima F, De Oliveira Costa H, De Sousa Damiao R, Gomes TS, Goncalves WJ. Canonical and noncanonical Wnt pathway: a comparison among normal ovary, benign ovarian tumor and ovarian cancer. Oncol Rep. 2009;21:313–20. [PubMed] [Google Scholar]
- 54.Huang CL, Liu D, Nakano J, Ishikawa S, Kontani K, Yokomise H, Ueno M. Wnt5a expression is associated with the tumor proliferation and the stromal vascular endothelial growth factor--an expression in non-small-cell lung cancer. J Clin Oncol. 2005;23:8765–73. doi: 10.1200/JCO.2005.02.2871. [DOI] [PubMed] [Google Scholar]
- 55.Pukrop T, Klemm F, Hagemann T, Gradl D, Schulz M, Siemes S, Trumper L, Binder C. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc Natl Acad Sci U S A. 2006;103:5454–9. doi: 10.1073/pnas.0509703103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanaki H, Yamamoto H, Sakane H, Matsumoto S, Ohdan H, Sato A, Kikuchi A. An anti-Wnt5a antibody suppresses metastasis of gastric cancer cells in vivo by inhibiting receptor-mediated endocytosis. Mol Cancer Ther. 2012;11:298–307. doi: 10.1158/1535-7163.MCT-11-0682. [DOI] [PubMed] [Google Scholar]
- 57.Dwyer MA, Joseph JD, Wade HE, Eaton ML, Kunder RS, Kazmin D, Chang CY, McDonnell DP. WNT11 expression is induced by estrogen-related receptor alpha and beta-catenin and acts in an autocrine manner to increase cancer cell migration. Cancer Res. 2010;70:9298–308. doi: 10.1158/0008-5472.CAN-10-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Uysal-Onganer P, Kawano Y, Caro M, Walker MM, Diez S, Darrington RS, Waxman J, Kypta RM. Wnt-11 promotes neuroendocrine-like differentiation, survival and migration of prostate cancer cells. Mol Cancer. 2010;9:55. doi: 10.1186/1476-4598-9-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mochmann LH, Bock J, Ortiz-Tanchez J, Schlee C, Bohne A, Neumann K, Hofmann WK, Thiel E, Baldus CD. Genome-wide screen reveals WNT11, a non-canonical WNT gene, as a direct target of ETS transcription factor ERG. Oncogene. 2011;30:2044–56. doi: 10.1038/onc.2010.582. [DOI] [PubMed] [Google Scholar]
- 60.Dubeau L. The cell of origin of ovarian epithelial tumours. The lancet oncology. 2008;9:1191–7. doi: 10.1016/S1470-2045(08)70308-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boyer A, Goff AK, Boerboom D. WNT signaling in ovarian follicle biology and tumorigenesis. Trends in endocrinology and metabolism: TEM. 2010;21:25–32. doi: 10.1016/j.tem.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 62.Rask K, Nilsson A, Brannstrom M, Carlsson P, Hellberg P, Janson PO, Hedin L, Sundfeldt K. Wnt-signalling pathway in ovarian epithelial tumours: increased expression of beta-catenin and GSK3beta. British journal of cancer. 2003;89:1298–304. doi: 10.1038/sj.bjc.6601265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gatcliffe TA, Monk BJ, Planutis K, Holcombe RF. Wnt signaling in ovarian tumorigenesis. International journal of gynecological cancer: official journal of the International Gynecological Cancer Society. 2008;18:954–62. doi: 10.1111/j.1525-1438.2007.01127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.