

Abstract
Macrophages are critical components of the innate and adaptive immune responses, and they are the first line of defense against foreign invaders because of their powerful microbicidal activities. Macrophages are widely distributed throughout the body and are present in the lymphoid organs, liver, lungs, gastrointestinal tract, central nervous system, bone, and skin. Because of their repartition, they participate in a wide range of physiological and pathological processes. Macrophages are highly versatile cells that are able to recognize microenvironmental alterations and to maintain tissue homeostasis. Numerous pathogens have evolved mechanisms to use macrophages as Trojan horses to survive, replicate in, and infect both humans and animals and to propagate throughout the body. The recent explosion of interest in evolutionary, genetic, and biochemical aspects of host-pathogen interactions has renewed scientific attention regarding macrophages. Here, we describe a procedure to isolate and cultivate macrophages from murine bone marrow that will provide large numbers of macrophages for studying host-pathogen interactions as well as other processes.
Keywords: Immunology, Issue 81, biology (general), immunology, Life Sciences (General) macrophages, bone marrow, phagocytosis, phagosomes, lysosomes, endocytosis
Introduction
A significant aspect of macrophage function is their role in innate and adaptive immunity. Because of their capacity to phagocytose inert particles, bacteria or parasites, macrophages are a first line of defense against foreign invaders. Once internalized, microbes are degraded within phagolysosomes. Macrophages also send recruitment signals for and present antigens to other immune cells such as T lymphocytes. Macrophages are derived from monocytes. Monocytes arise in the bone marrow from myeloid stem cells and migrate to peripheral blood and various tissues where they differentiate into macrophages. It is estimated that a healthy adult mouse contains approximately 108 macrophages that are distributed throughout the body in various organs and tissues (Table 1)1,2. Macrophages display great phenotypic and functional diversity because of their ability to adapt to their microenvironment3,4. The most important macrophage property is their microbicidal activity, which is defined by their capacity to phagocytose microbes and destroy them. The phagocytic response is defined by the activation of complex signaling networks that are stimulated by microbial contact; thus, macrophages modulate gene expression appropriately in response to diverse stimuli. After phagocytosis, microbes are eliminated in a structure called the phagolysosome; however, many pathogenic microbes have developed strategies to subvert the microbicidal function of macrophages5. The diversity of subversion mechanisms that are utilized by different microbial species is a testament to the complexity of the phagocytic process6 and phagolysosome biogenesis. Infectious diseases are major human health problems, and numerous mechanisms and molecules participate in macrophage antimicrobial activities. Furthermore, the targets of the microbicidal properties that are hijacked by microbes remain unknown; thus, there is an explosion of interest in the evolutionary, genetic, and biochemical aspects of host-pathogen interactions that has renewed scientific attention regarding macrophages. Currently, the majority of the research in the field is done on macrophages cell lines, which differ from primary macrophages in the phagocytic activity, the cytokines production and the regulation of the oxidative burst. In addition, they are less suitable for microscopy. To investigate the interaction macrophages-pathogens it is recommended to use primary macrophages, such as Bone Marrow Derived Macrophages (BMDMs), which exhibit more physiological features. Moreover, it is possible to work on genetically modified BMDMs, because these macrophages might be isolated directly from transgenic mice and, with the availability of novel technologies such as lentiviral transfection, their gene expression profile can be altered by gene overexpression or RNA interference. Here, we describe a procedure to differentiate murine bone marrow into macrophages that will provide large numbers of macrophages in 7 days for various functional analyses such as proteomics7, transcriptomics8, intracellular trafficking studies9, dynamic studies10, genetic screens (RNAi) and drug screening11.
Protocol
Ethics Statement
The protocol for animal handling was approved by our institutional Animal Ethics Committee "Conseil Scientifique du Centre de Formation et de Recherche Experimental Médico-Chirurgical" (CFREMC, Project permit 10-300122013 to Eric Ghigo) from Aix-Marseille University in accord with the rules of Décret N° 87-848 of 10/19/1987. The experiments were performed at the Faculté de Médecine de la Timone (Experimentation permit number 13.385 to Eric Ghigo).
1. Material and Culture Media Preparation
Sterilize two forceps, two scissors, two surgical blades, a mortar and a pestle.
Obtain complete DMEM containing 10% fetal calf serum (FCS), 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin .
Dilute 10x PBS in sterile distillated water to obtain 1x PBS.
Obtain ice-cold 1x PBS, ice-cold complete DMEM, complete DMEM warmed to 37 °C.
2. Preparation of L929 Cell Supernatants
Grow L929 cells to confluence (twenty 175 cm2 flasks) in complete DMEM at 37 °C, 5% CO2.
Note: Granulocyte-macrophage colony stimulating factor (GM-CSF) is required to induce hematopoietic cell differentiation into macrophages12. L929 cells produce GM-CSF.
At confluence, replace culture media with fresh complete DMEM. Transfer flasks to 32 °C, 5% CO2 for 10 days.
Collect, pool and centrifuge the supernatants at 750 x g for 10 min. Discard cell pellets.
Store supernatants in 15 ml tubes and store at -20 °C.
3. Bone Marrow-derived Macrophage (BMDM) Preparation
- Sacrifice 1 mouse by cervical dislocation.
- Use sterile surgical blades throughout the experiment. Disinfect the skin with 70% alcohol. Make an incision at the top of each hind leg and pull the skin down towards the foot to expose the muscle.
- Cut off the hind legs, remove the skin with sterile scissors and sterile forceps. Place legs within a sterile Petri dish (35/10 mm) containing sterile, ice-cold 1x PBS (5 ml).
- Remove the flesh and muscles that are adhering to the bones with sterile scissors and forceps.
Transfer the bones into a new, sterile Petri dish (35/10 mm) containing ice-cold, sterile 1x PBS (5 ml). Wash the bones two times with 5 ml ice-cold, sterile 1x PBS.
Transfer the bone into a sterile mortar containing 5 ml ice-cold, sterile 1x PBS.
Cut the tibia from the femur at the joint with sterile scissors. Smash the bones gently in a sterile mortar containing 5 ml ice-cold, sterile 1x PBS using a pestle.
Collect the supernatant in ice-cold 15 ml tubes. Repeat this step 3x.
Filter through a 70 μm Nylon cell strainer to remove solid fragments. Centrifuge the filtrate at 450 x g for 10 min at 4 °C.
Gently discard the supernatant. Dissociate the pellet in 10 ml red blood cell lysis buffer for 30 sec. Add 20 ml ice-cold, complete DMEM.
Note: The aim of this step is to remove contaminating red blood cells; thus, this step must be performed within 2 min to avoid hematopoietic cell alteration by the red blood cell lysis buffer.
Centrifuge at 450 x g for 10 min at 4 °C. Gently discard the supernatant. Dissociate the pellet in 20 ml complete DMEM that has been warmed to 37 °C.
Transfer the dissociated cells into 2 Petri dishes (100/20 mm). Incubate them for 4 hr at 37 °C.
Collect the supernatants in 50 ml tubes at room temperature. Discard the dishes that contain the resident macrophages.
Note: The objective of this step is to eliminate the resident bone marrow macrophages by their ability to adhere to culture-treated plastic. These resident macrophages may be used in other experiments.
Centrifuge the collected supernatants at 450 x g for 10 min at 4 °C. Discard the supernatant.
Dissociate gently the pellet in 10 ml complete DMEM containing 15% L929 cell supernatant. Filter the cells through a 40 μm Nylon cell strainer.
Recover the filtrate. Add the collected filtrate (10 ml) to 140 ml complete DMEM that has been supplemented with 15% L929 cell media.
Distribute 10 ml of the cell suspension per Petri dish (15 Petri dishes, 100/20 mm). Incubate the cells at 37 °C, 5% CO2.
Grow cells for 3 days
Add 10 ml complete DMEM that has been supplemented with 15% L929. Incubate cells for 4 additional days.
Note: Monitor cell growth periodically with an inverted microscope (steps 3.14-3.16). Adherent macrophages will be observed after 3 days of culture.
4. Harvesting BMDMs
Remove the supernatant. Wash BMDMs 2 times with complete DMEM
Add 5 ml complete DMEM that has been warmed to 37 °C. Detach BMDMs by gently scraping with a rubber policeman.
Collect BMDMs in 50 ml tubes and centrifuge at 450 x g for 10 min. Gently dissociate cell pellets in 20 ml complete DMEM.
Count BMDMs in the presence of trypan blue (no more than 10% mortality should be observed).
Note: Generally, approximately 6-7.5 x 107 macrophages are obtained from 15 Petri dishes (100/20 mm) of macrophages. There are approximately 4-5 x 106 macrophages/Petri dish (100/20 mm).
Prepare BMDMs as required for the experiment. After 16 hr in complete media at 37 °C, 5% CO2, macrophages will again adhere to the support.
5. Storing BMDMs
Collect isolated BMDMs (step 4.3). Centrifuge at 450 x g for 10 min. Note: BMDMs may be frozen in liquid nitrogen.
Resuspend cell pellets in freezing media consisting of 10% DMSO and 90% FCS at a final concentration of 4 x 106 cells/ml and pipette 1 ml into each ampoule.
Freeze the cells at a cooling rate of 1 °C/min. After 24 hr, transfer the ampoule to a liquid nitrogen container for long-term storage.
Representative Results
The aim of this method was to easily obtain large numbers of macrophages in few days. The bone marrow cell preparation is illustrated in Figure 1. Bones from the hind leg were collected and smashed in a mortar. Once the resident macrophages were removed from the bone marrow cell preparation, bone marrow cells were incubated with GM-CSF (Day 0). After 3 days, the cells, which were round and nonadherent before culture, start to differentiate into macrophages and adhere (Figure 1A). Cytometry analysis using F4/80 and HLA-DR markers revealed that cells expressed both markers thus proving that they are differentiated into macrophages (Figure 1B). After 7 days, a BMDM monolayer was obtained with 6-7.5 x 107 BMDM/mouse. Macrophages can be used in different types of experiments (Figure 2). The functional activities of the BMDMs may be determined in different ways. For example, BMDM phagocytosis was monitored by latex bead internalization. The number of beads per cell increased over time (Figure 2A). BMDM endocytic potential (Figure 2B), bacterial pathogen interactions (Figure 2C) and signal transduction pathways (Figure 2D) were also assessed.
| Location | Cell types |
| Bone | Osteoclast |
| Bone marrow | Pro-monocyte, macrophage |
| Central nervous system | Microglial cell |
| Lamina propria | Resident macrophage |
| Liver | Kupffer cell |
| Lung | Alveolar macrophage |
| Peripheral blood | Monocyte |
| Peritoneal cavity | Peritoneal macrophage |
| Skin | Langerhans cell |
| Spleen | Resident macrophage |
| Thymus | Resident macrophage |
Table 1. Macrophage sources in tissues and fluids.
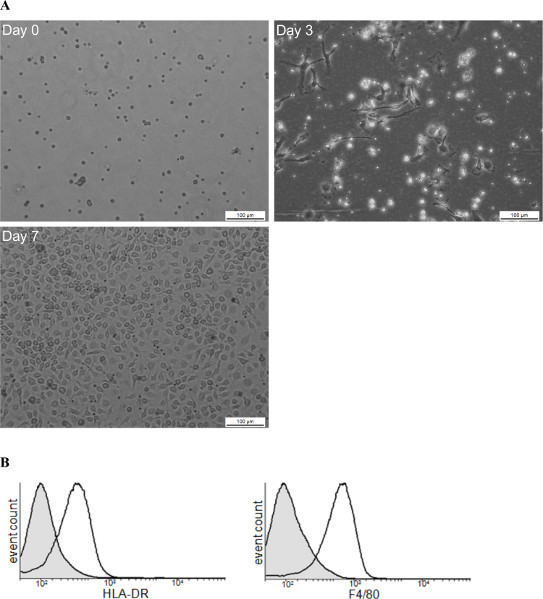 Figure 1. Schematic view of BMDM production. (A) This figure illustrates the steps of BMDM differentiation from progenitor differentiation (day 0) to mature macrophages (day7). (B) FACS analysis illustrates the purity of isolated cells at day 7 using as macrophages markers HLA-DR and F4/80. Click here to view larger figure.
Figure 1. Schematic view of BMDM production. (A) This figure illustrates the steps of BMDM differentiation from progenitor differentiation (day 0) to mature macrophages (day7). (B) FACS analysis illustrates the purity of isolated cells at day 7 using as macrophages markers HLA-DR and F4/80. Click here to view larger figure.
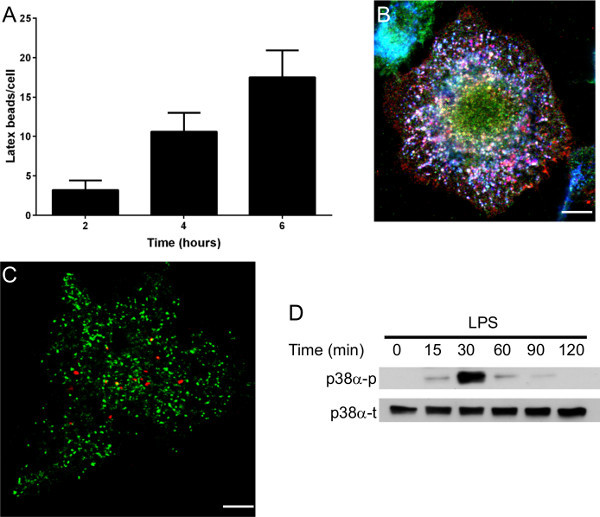 Figure 2. Examples of experiments that are possible with BMDMs. (A) Phagocytosis assay. BMDMs were incubated for different periods of time with latex beads, and bead uptake was observed by microscopy. (B) Intracellular Coxiella burnetii lipopolysaccharide (LPS) localization in BMDMs. LPS (in red) was localized to a compartment that harbors lysosomal-associated membrane protein-1 (LAMP-1) (in blue) but not cathepsin D (in green). This experiment demonstrates that LPS is present in late endosomes that are unable to fuse with lysosomes. (C) BMDMs infected with Tropheryma whipplei (in red) was not localized in compartment that harbors cathepsin D (in green). (D) Representative immunoblot demonstrating total (t) and phosphorylated (p) p38α MAP Kinase levels in Escherichia coli LPS-stimulated BMDMs (1 μg/ml). Scale bars represent 5 μm.
Figure 2. Examples of experiments that are possible with BMDMs. (A) Phagocytosis assay. BMDMs were incubated for different periods of time with latex beads, and bead uptake was observed by microscopy. (B) Intracellular Coxiella burnetii lipopolysaccharide (LPS) localization in BMDMs. LPS (in red) was localized to a compartment that harbors lysosomal-associated membrane protein-1 (LAMP-1) (in blue) but not cathepsin D (in green). This experiment demonstrates that LPS is present in late endosomes that are unable to fuse with lysosomes. (C) BMDMs infected with Tropheryma whipplei (in red) was not localized in compartment that harbors cathepsin D (in green). (D) Representative immunoblot demonstrating total (t) and phosphorylated (p) p38α MAP Kinase levels in Escherichia coli LPS-stimulated BMDMs (1 μg/ml). Scale bars represent 5 μm.
Discussion
The protocol described herein details a method to produce large numbers of BMDMs. BMDM are primary cells and have the biological function and properties of macrophages differentiated from monocytes because there are mature, in contrast to macrophage cell lines, which are immature. BMDMs may be used for genetic screening (RNAi), drug screening, functional studies, host-pathogen interaction studies and many other areas of investigation.
The procedure presented herein is very simple and does not require any specialized equipment; therefore, it can be easily performed in a laboratory or classroom. This method requires some practice and manual dexterity. It is also possible to store the BMDMs in liquid nitrogen, thus facilitating future experiments.
There are several critical steps for successful BMDM culture: 1) maintaining a sterile, healthy culture; 2) bone marrow extraction needs to be efficient; 3) L929 supernatant quality is critical; 4) do not expose cells to the red blood cell lysis buffer for more than 2 min to avoid hematopoietic cell damage.
It is possible to do some modification of the protocol but there are in limiting number. 1) DMEM can be replaced by RPMI 1640, but BMDM production is less efficient; 2) commercial M-CSF (500-1000 UI/ml) can be used instead of L929 supernatant, but it is expensive and M-CSF has to be added every 2 days to cells during the differentiation process; 3) alternatively bone marrow can be extruded from bone using a syringe equipped with 25 G needle instead of smashing the bones in a sterile mortar using a pestle.
Disclosures
There are no declared conflicts of interest.
Acknowledgments
This work was supported by the CNRS (PICS 2012-2014 to E.G.) and by a grant from Regione Campania (L.R. n.5, 28.03.2002 to Giovanna Mottola). Filippo Conti is a fellow of the Scientific Cooperation Foundation ''Infectiopole Sud.'' Nicola Boucherit is a fellow of the French Ministry for Research and Technology. The funding sources had no role in the study design, data collection, data analysis, decision to publish, or manuscript preparation.
References
- Rutherford MS, Witsell A, Schook LB. Mechanisms generating functionally heterogeneous macrophages: chaos revisited. J. Leukocyte Biol. 1993;53:602–618. doi: 10.1002/jlb.53.5.602. [DOI] [PubMed] [Google Scholar]
- Van Furth R. In: Inflammation: Basic Principles and Clinical Correlates. Gallin JI, Goldstein IM, Snyderman R, editors. Vol. 112. Raven Press; 1992. pp. 325–336. [Google Scholar]
- Adams D, Halmiton T. In: Inflammation: Basic Principles and Clinical Correlates. Gallin JI, Goldstein IM, Snyderman R, editors. Vol. 112. Raven Press; 1992. pp. 325–336. [Google Scholar]
- Morris L, Graham CF, Gordon S. Macrophages in haemopoietic and other tissues of the developing mouse detected by the monoclonal antibody F4/80. Development. 1991;112:517–526. doi: 10.1242/dev.112.2.517. [DOI] [PubMed] [Google Scholar]
- Haas A. The phagosome: compartment with a license to kill. Traffic. 2007;8:311–330. doi: 10.1111/j.1600-0854.2006.00531.x. [DOI] [PubMed] [Google Scholar]
- Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu. Rev. Immunol. 2002;20:825–852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- Castagna A, Polati R, Bossi AM, Girelli D. Monocyte/macrophage proteomics: recent findings and biomedical applications. Expert Rev. Proteomics. 2012;9:201–215. doi: 10.1586/epr.12.11. [DOI] [PubMed] [Google Scholar]
- Benoit M, Desnues B, Mege JL. Macrophage polarization in bacterial infections. J. Immunol. 2008;181:3733–3739. doi: 10.4049/jimmunol.181.6.3733. [DOI] [PubMed] [Google Scholar]
- Barry AO, et al. Impaired stimulation of p38alpha-MAPK/Vps41-HOPS by LPS from pathogenic Coxiella burnetii prevents trafficking to microbicidal phagolysosomes. Cell Host Microbe. 2012;12:751–763. doi: 10.1016/j.chom.2012.10.015. [DOI] [PubMed] [Google Scholar]
- Henry RM, Hoppe AD, Joshi N, Swanson JA. The uniformity of phagosome maturation in macrophages. J. Cell Biol. 2004;164:185–194. doi: 10.1083/jcb.200307080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaramurthy V, et al. Integration of Chemical and RNAi Multiparametric Profiles Identifies Triggers of Intracellular Mycobacterial Killing. Cell Host Microbe. 2013;13:129–142. doi: 10.1016/j.chom.2013.01.008. [DOI] [PubMed] [Google Scholar]
- Boltz-Nitulescu G, et al. Differentiation of rat bone marrow cells into macrophages under the influence of mouse L929 cell supernatant. J. Leukocyte Biol. 1987;41:83–91. doi: 10.1002/jlb.41.1.83. [DOI] [PubMed] [Google Scholar]