

Abstract
We have developed a tissue-based model of the human trabecular meshwork (TM) using viable postmortem corneoscleral donor tissue. Two-photon microscopy is used to optically section and image deep in the tissue to analyze cells and extracellular matrix (ECM) within the original three-dimensional (3D) environment of the TM. Multimodal techniques, including autofluorescence (AF), second harmonic generation (SHG), intravital dye fluorescence, and epifluorescence, are combined to provide unique views of the tissue at the cellular and subcellular level. SHG and AF imaging are non-invasive tissue imaging techniques with potential for clinical application, which can be modeled in the system. We describe the following in the tissue-based model: analysis of live cellularity to determine tissue viability; characteristics of live cells based on intravital labeling; features and composition of the TM's structural ECM; localization of specific ECM proteins to regions such as basement membrane; in situ induction and expression of tissue markers characteristic of cultured TM cells relevant to glaucoma; analysis of TM actin and pharmacological effects; in situ visualization of TM, inner wall endothelium, and Schlemm's canal; and application of 3D reconstruction, modeling, and quantitative analysis to the TM. The human model represents a cost-effective use of valuable and scarce yet available human tissue that allows unique cell biology, pharmacology, and translational studies of the TM.
Introduction
Cell and extracellular matrix (ECM) interactions within the three-dimensional (3D) trabecular meshwork (TM) play important roles in modulating aqueous outflow resistance and intraocular pressure (IOP).1–3 Traditionally, TM biological interactions have been modeled in vitro,4 but cultured cells in a two-dimensional environment may not sufficiently replicate the true context and behavior of 3D tissue.5,6 While tissue cell biology may be studied by histological analysis, processing steps for conventional light and electron microscopy irretrievably alter tissue structure and cells, rendering them non-viable for further experimentation. Probing cell behavior in vivo is a further attractive alternative, but we are not yet able to resolve cellular or subcellular biological events in the TM of live animals or humans.
We have sought to develop a primary tissue model of the human TM in which interactions between cells, ECM, and fine structure can be directly resolved by two-photon microscopy (TPM) at the subcellular level within the tissue's preserved 3D environment. This provides a tissue-based platform for probing and simulating human TM cell biology within an environment mimicking the original tissue context.
TPM uses near-infrared laser that permits tissue and cellular imaging with less scatter, absorption and phototoxicity, and deeper penetration than is possible by conventional single photon microscopy.7–10 The resulting high-resolution deep tissue optical sectioning provides versatile options for analyzing whole live or fixed tissue without conventional histological sectioning. Two-photon excitation fluorescence (TPEF) imaging may utilize multiple modalities such as endogenous fluorescence [autofluorescence (AF)], direct labeled fluorescence using intravital dyes or transduced fluorescent proteins (ie, GFP), or indirect antibody-labeled epifluorescence. Non-excited fluorescence modalities such as second harmonic generation (SHG) may also be used.
We have applied TPM to probe and analyze TM cell biology within human tissue.11–15 We routinely image as deep as 100–200 μm in the TM. By this approach, we have performed tissue-based cell biological studies with the resolution and easy accessibility of in vitro methods. This has led to the development of a novel yet practical and authentic human TM cell imaging model that permits simulation of the in vivo TM. We are mindful that our approaches could provide fresh perspectives on the TM and lead to future clinical applications.
Human Postmortem Corneoscleral Tissue
We image recently postmortem corneoscleral tissue that is considered suitable for human therapeutic transplantation.11,12,14,15 An Appendix at the end of this article summarizes imaging methodology that we have applied to this tissue. We were drawn to the possibility of studying the TM in donor tissue, as eye banks consider this tissue to be of good enough quality for human transplantation for up to 2 weeks postmortem. Human TM cells have been grown from corneoscleral rim tissue and established in primary culture over long periods postmortem.16 Furthermore, experiments have shown that postmortem TM is viable in culture medium for 4 weeks17 and has been used in week-long organ culture perfusion studies.18
Following transplant surgery, we receive donor corneoscleral rims (Fig. 1A) stored in Optisol GS transport medium (Bausch & Lomb, Rochester, NY). The tissue is processed for imaging within a day of receipt. Schlemm's canal (SC) is readily identifiable when blood reflux is present, as this provides an excellent marker to localize the TM.11 We cut the corneoscleral rim into sector wedges before imaging (Fig. 1B). Our use of postmortem transplant tissue simply, inexpensively, and sustainably salvages good-quality but scarce human tissue for research.
FIG. 1.
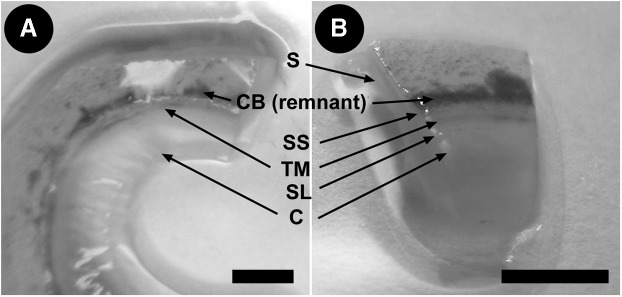
Human donor corneoscleral rims. (A) Angle structures in tissue. (B) Location of TM and Schlemm's canal (SC) in a corneoscleral wedge. S, sclera; CB, ciliary body; SS, scleral spur; TM, trabecular meshwork; SL, Schwalbe's line; C, cornea. Scale bar=3 mm.
We empirically assess each donor sample to confirm tissue quality. We have used fresh postmortem eyes obtained within 24–48 h postmortem as reference controls for viable tissue.14 AF screening for viability is coupled with labeling for vitality.11,12
Analysis of Live Cellularity and Viability
Viability screening of the tissue was assessed by AF (Fig. 2) and intravital dye analysis (Fig. 3). In viable tissue from donor rims received 48 h postmortem, TPEF shows linear autofluorescent beams and fibers with sharp branching and a lack of abnormal aggregates (Fig. 2A). Conversely, non-viable tissue shows indistinct, ragged, wavy, and tangled fibers with abnormal aggregates (Fig. 2B).
FIG. 2.
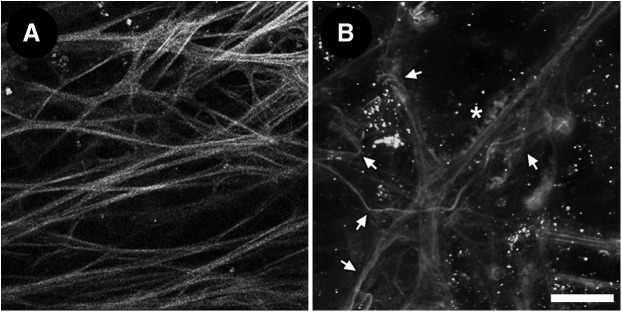
Autofluorescence clues of tissue viability. (A) Linear branching autofluorescent fibers without abnormal aggregates in viable tissue. (B) Indistinct, wavy and tangled autofluorescent fibers (arrows) with abnormal aggregates (asterisks) in non-viable tissue. Oval nuclei are labeled with Hoechst 33342. Scale bar=25 μm.
FIG. 3.

Live cellularity analysis by calcein AM (calcein; green) and propidium iodide (PI; red) co-labeling. (A) Freshly postmortem tissue: predominantly calcein-positive, PI-negative cellular labeling confirming viable tissue. (B) Viable postmortem tissue labeling similar to (A). (C) Dead tissue following exposure to Triton X-100 showing universally calcein-negative, PI-positive co-labeled cells among autofluorescent fibers. (D) Nonviable postmortem tissue showing similar co-labeling as (C). Scale bar=25 μm. Color images available online at www.liebertpub.com/jop
Intravital dye-labeled live cells can be visualized by TPEF within the intact TM.11,12,14 Intravital dyes are nontoxic, deeply penetrate tissues, and selectively stain cells and subcellular compartments. This allows live cells to be seen in association with the ECM within the original TM tissue environment. We co-label postmortem tissue with calcein AM (calcein), propidium iodide (PI), and Hoechst 33342 to qualitatively and quantitatively analyze live cellularity and tissue viability.
Tissue viability analysis is performed in serial optical sections in 8 μm steps, with viability calculated in 246×246×100 μm 3D z-stack volumes per tissue. Percent (%) live cellularity is calculated as the ratio of calcein-positive cells to total cells (live calcein-positive plus dead PI-positive cells).14 An arbitrary cut-off of 50% live cellularity is used to define tissue as viable. Tissues with less than 50% live cellularity do not meet our standards and are excluded from further analysis.
In our studies, freshly postmortem eyes within 24–48 h of death (Fig. 3A) and good quality donor corneoscleral tissue (Fig. 3B) had a co-labeling pattern of predominantly calcein-positive (live) and PI-negative (not dead) cells, indicating good live cellularity within the tissue.14 Conversely, universally calcein-negative (not alive) and PI-positive (dead) cells were seen in tissues exposed to Triton X-100 (0.2% nonionic surfactant), indicating non-viable tissue; we used this to represent dead controls (Fig. 3C). A similar, but less pronounced pattern of non-viable calcein and PI co-labeling was seen in screens of non-viable postmortem tissue (Fig. 3D).
In fresh sub-48 h postmortem tissues, live cellularity was 76%±10%. Two-thirds of standard postmortem donor tissue that we received met live cellularity criteria (>50% live cells) for viability. Mean (±SD) live cellularity in viable donor tissue was 71%±14% and in non-viable tissue, it was 11%±13%.14
This tissue-based live-dead analytical approach is not only useful for evaluating postmortem tissue viability in our model system, but it has other novel uses such as for studying reduced cellularity in primary open angle glaucoma and assessing potential drug toxicity during pharmacological screening.
Live Cellular Detail and Distribution
Various aspects of in situ live cell morphology and distribution are revealed by different intravital dyes.14 In our studies, CellTracker localized to the cytosol and perinuclear cellular regions (Fig. 4A). This dye allowed discernment of nuclei even in the absence of nuclear co-labeling (asterisk in Fig. 4A). Labeling by calcein, another cytosolic dye, revealed roundish bright cells in the TM with perinuclear fluorescence (Fig. 4B) and lamellipodia-like features (Fig. 4C). Unlabeled gaps between neighboring cells made it possible to identify and count individual cells.14 Tiny cytosolic signal voids of CellTracker-labeled cells (arrows in Fig. 4A) and calcein-labeled cells (arrows in Fig. 4B) likely represented unlabeled organelle compartments. We used Octadecyl rhodamine B chloride (R18) for cell membrane labeling (Fig. 4D). R18 labeling made it possible to see distinct membrane features such as filaments linking cells and complex lamellipodia-like structures. Extent and variation of cell shape and size was better appreciated by R18 than cytosolic labeling, although it was harder to distinguish the distinct boundaries of cells because the fluorescence signal from membranes of neighboring cells blended into one continuous label. This difficulty could be overcome by nuclear co-labeling to identify individual cells (Fig. 4D).
FIG. 4.
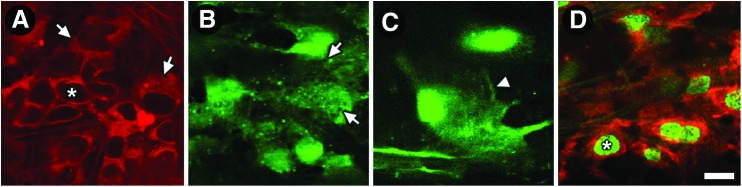
Live cell morphology by intravital imaging. (A) CellTracker (red) in the cytosol and perinuclear cellular regions. Asterisk: unlabeled nucleus. Arrows: tiny signal voids in cytosol likely represent unlabeled organelle compartments. (B) Calcein AM (green) in the cytosol of roundish cells. Arrows: tiny signal voids in cytosol are suggestive of organelle compartments. (C) Detail of calcein-labeled cell (green) showing lamellipodia-like configuration (arrowhead). (D) Octadecyl rhodamine B chloride (R18; red)-labeled cell membranes. Asterisk: Hoechst-labeled nucleus. Scale bar=10 μm. Color images available online at www.liebertpub.com/jop
Deep penetration of intravital dyes into the aqueous drainage tissue allowed for labeling of cells across all regions and depths in the TM. Live cells were identified with reference to neighboring cells, ECM, and fine tissue structures. In the uveal meshwork, cells labeled with Hoechst 33342 (nuclear label) were associated with autofluorescent trabecular beams (Fig. 5A). Adjacent and external to the uveal meshwork is the corneoscleral meshwork, where Hoechst-labeled nuclear labeling was seen among tissue plates, beams, and pores (Fig. 5B). In the juxtacanalicular meshwork (JCT), cells were densely organized amongst fine autofluorescent tissue fibers (Fig. 5C). In the corneal endothelium, adjacent to the corneoscleral meshwork, nuclei were organized in a regular mosaic pattern (Fig. 5D).11
FIG. 5.

Autofluorescence (AF) and Hoechst-labeled nuclei in trabecular meshwork and corneal endothelium. (A) Nuclear association with trabecular beams in uveal meshwork. Inset: software-assisted measurement to determine width of trabecular beams. (B) Nuclei among coarser beams and pores in corneoscleral meshwork. (C) Dense nuclear distribution among AF fibers in juxtacannalicular meshwork. A number of smaller nuclei with high-intensity fluorescence labeling are seen in the adjacent inner wall endothelium of Schlemm's canal (arrows). (D) Regular mosaic of corneal endothelial nuclei with minimal/no associated AF. Scale bar=25 μm.
Nuclear density increased from inner to outer TM.14 In TPEF optical sectioning, 69.8±13.8 nuclei were present in the uveal meshwork within the 246×246 μm imaging fields; 103±26.1 nuclei in the corneoscleral meshwork within the same imaging field dimensions; and 98.2±29.8 nuclei in the JCT.14
The potential for tissue-based live cellular analysis is further enhanced by fluorescent protein transduction of TM cells to label subcellular structures such as actin that will permit exploration of live cell dynamics and drug effects in the viable TM.19
TM Autofluorescence
The TM has an intricate mesh-like structure formed by a complex network of branching autofluorescent beams that varies with depth in the tissue and can be visualized by TPEF.11 From an internal approach, the mesh-like TM borders ciliary muscle, which has wavy autofluorescent fibers running perpendicular to the circumferential orientation of TM and SC, and cornea. The cornea has minimal AF (Fig. 5D).
AF imaging reveals a uveal meshwork of slender branching beams that are separated by large gaps (Fig. 5A). Adjacent and external to the uveal meshwork is the corneoscleral meshwork that is organized as coarser autofluorescent beams, which appear as plate-like structures with pores that diminish in size toward the more external JCT (Fig. 5B). Figure 5A (inset) shows how software-assisted measurement may be used to determine the width of trabecular beams based on AF imaging. We have previously reported the mean diameter of gaps in the uveal meshwork as greater than 40 μm. In the corneoscleral meshwork, pores vary from less than 10 μm to more than 20 μm.12 In the JCT, arrays of fine autofluorescent fibers form a cribriform network lacking pores (Fig. 5C).12
There are subtle differences in AF features of the anterior TM (overlying sclera, adjacent to cornea) and posterior TM (overlying SC, adjacent to ciliary muscle).11 The anterior and posterior TM have similar uveal and corneoscleral regions, but they differ in their more external portions. The posterior TM has a JCT adjacent to SC, but the anterior TM transitions to an organization resembling denser and more homogenous sclera crisscrossed by randomly aligned fine autofluorescent fibers.11 Thus it is possible to non-invasively examine the fine structure of the TM in situ and in great detail by AF imaging.
Tissue Localization of SHG and AF
SHG has been used to characterize collagen in various tissues,8 including the human eye.9,11,12,20,21 SHG is a polarization wave phenomenon produced by nonlinear optical recombination of photons that yields a signal half the wavelength of the incident light.7,22,23 SHG permits direct imaging of uniquely organized structures such as collagen that are anisotropic, non-centrosymmetric and possess large hyperpolarizabilities.7 SHG detection requires a narrow band-pass filter with full width half maximum centered at a wavelength half that of the incident light; however, a wider, short wavelength filter could also be used as an alternative.8,24
In trabecular beams, fine, high-intensity autofluorescent fibers are aligned with beam axes. Brighter fibers also appear to coil around beams, forming discrete beam protuberances at 5 μm intervals.11 These intensely autofluorescent fibers are distinguishable from a background of dimmer and more homogenous AF within the uveal and corneoscleral beams.
We hypothesized that different protein fluorophores contribute to the heterogeneity in TM AF and tested the possibility that collagen and elastin are sources of this heterogeneity,11,12 as may be predicted by foregoing descriptions of human TM.25–30 We compared the tissue distribution of high- and lower-intensity AF signals, SHG (for collagen), and eosin dye fluorescence (for elastin).
Our analysis showed qualitatively and quantitatively that low-intensity AF (Fig. 6A) and SHG-positive regions (Fig. 6B) in the uveal and corneoscleral meshwork co-localized. Conversely, high-intensity AF (Fig. 6A) co-localized with SHG signal voids (Fig. 6B). Separately, SHG signal voids (Fig. 6C) co-localized with eosin-positive fibers (Fig. 6D), corresponding to fibers that were positively labeled with anti-tropoelastin antibody (Fig. 6E). Thus the source of high-intensity AF in trabecular beams is elastin.31–34 Conversely, lower-intensity AF in beams originates from collagen, agreeing with prior understanding that beam cores consist of elastin encased in collagen. Hence AF imaging resolves both structure and ECM protein composition in the TM.15,25–30,35 This has implications for potential non-invasive clinical imaging to probe ECM proteins in disease states. For example, collagen and elastin aberrations have been associated with glaucoma and IOP abnormality.36–39
FIG. 6.
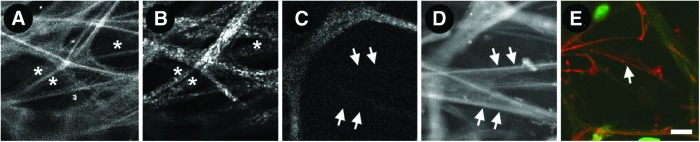
Imaging of autofluorescence second harmonic generation (SHG) of collagen, and eosin-labeled fluorescence of elastin in human trabecular meshwork. (A) Low-intensity (asterisks) and high-intensity AF signals (bright fibers); (B) corresponding SHG-positive (asterisks; for collagen) and SHG-negative (linear void between SHG-positive signal indicated by asterisks) signals; and (C) SHG-signal voids (arrows). (D) Eosin-positive regions (arrows; for elastin) that co-localized with SHG signal voids of (C). (E) These fine fibers positively label with anti-tropoelastin antibody (red, arrow). Scale bar=10 μm. Color images available online at www.liebertpub.com/jop
Schlemm's Canal
We locate SC within postmortem primary tissue by identifying a broad circumferential AF signal void in serial optical sectioning just beyond the final cellular or nuclear layer of the TM. This final cellular layer represents the region of the JCT-SC endothelial inner wall complex (Fig. 7A–C). In our studies, high-intensity nuclear fluorescence labeling was seen in this region, likely reflecting the high density of cells here.11 No AF was present in the SC lumen; instead, a signal void was seen (Fig. 7A–C).
FIG. 7.

Schlemm's canal (SC) and juxtacanalicular meshwork (JCT) interface. Orthogonal reconstruction (A) and tangential views (B–D) of interface region of JCT/inner wall endothelium next to SC lumen. (A) Orthogonal view through JCT/inner wall region, next to SC signal void (between dotted lines) region (b: JCT/inner wall interface; c: inner wall nuclei; d: SC lumen). (B) Fine autofluorescent fibers (arrows) are seen among Hoechst-labeled nuclei in the JCT region. Region of SC endothelium should have nuclei only without autofluorescent fibers (darker background). (C) SC signal void in a more external optical section just beyond JCT. (D) Autofluorescence signal from nylon marker (asterisk) in SC lumen showing course of SC without exogenous labeling. Arrowheads: Hoechst-labeled nuclei. Scale bar=25 μm.
Placing an autofluorescent nylon marker in the lumen of SC allows the course of the canal to be visualized by TPEF without exogenous labeling (Fig. 7D). In experiments, SC may be cannulated for a short distance to enable verification of downstream SC, which continues as a signal void beyond the autofluorescent suture tip.
Visualizing the JCT and SC inner wall endothelium side by side could provide meaningful possibilities for studying biological interactions within this physiologically high resistance region.
Characteristic Tissue and Cellular Markers
We use a multimodal approach combining indirect epifluorescence, intravital dye fluorescence, and AF imaging to characterize cells and ECM in the TM. In our studies, induction and expression of α-smooth muscle actin (α-SMA) (Fig. 8A; red) and myocilin (Fig. 8B; red) by transforming growth factor-β1 (TGF-β1) and dexamethasone respectively—both important to glaucoma—were localized in situ.12 Characteristic TM ECM markers such as fibronectin (Fig. 8C; red) and collagen type IV (Fig. 9A) were also localized in the tissue.12
FIG. 8.
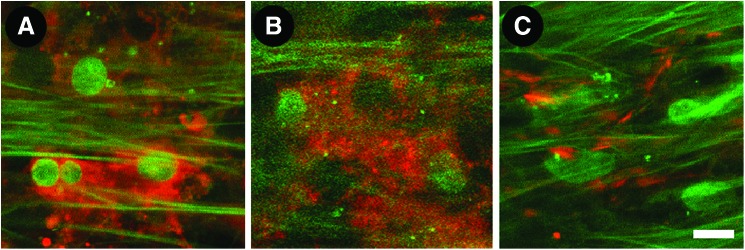
Induction of both intra-and extracellular markers in the trabecular meshwork. (A) α-smooth muscle actin (red) is seen intracellularly after induction by transforming growth factor-β1. (B) Myocilin (red), also an intracellular marker, is observed after induction by dexamethasone; (C) Fibronectin (red), a surface marker, is in cell-cell borders. Scale bar=10 μm. Color images available online at www.liebertpub.com/jop
FIG. 9.
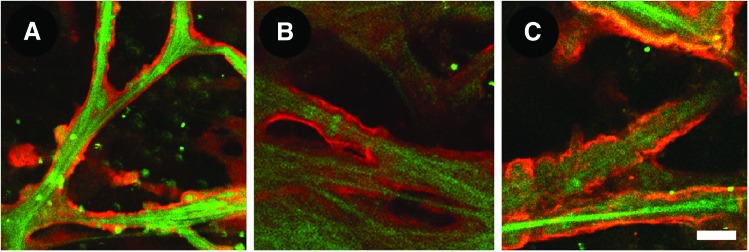
Extracellular matrix (ECM) proteins in basement membrane. ECM proteins that were not evident by autofluorescence and second harmonic generation imaging were visualized by two-photon excitation fluorescence after whole tissue immunolabeling. These ECM proteins, which included (A) Collagen type IV (red), (B) Laminin (red) and (C) Heparan sulfate (red) were observed in the basement membrane surrounding trabecular beams (green AF). Scale bar=10 μm. Color images available online at www.liebertpub.com/jop
We were able to resolve associations between tissue architecture, ECM proteins, and cells by TPEF in situ. Our findings agreed with cell culture, electron microscopy, and immunohistochemistry findings.26,29,40–52 We found that collagen type IV was associated with autofluorescent structures in a region which was consistent with basement membrane.26,29,40–42 Our myocilin findings based on tissue induction by dexamethasone agreed with prior reports of the protein's localization to intracellular compartments43–49,53 and extracellular association with long-spacing collagens and sheath material surrounding elastin.40,43 α-SMA, which is induced by TGF-β1 in situ, was seen expressed intracellularly.54
TPEF identification of markers characteristic of cultured primary TM cells and pertinent to glaucoma indicates that it is possible to study biological interactions involving these important proteins within the TM.
Extracellular Matrix
AF and SHG imaging may be used to visualize certain structural ECM proteins such as elastin and collagen. Other ECM proteins not seen by AF or SHG may be visualized by whole tissue immunolabeling and epifluorescence imaging. In our studies, collagen type IV labeling was seen in the basement membrane region surrounding trabecular beams (Fig. 9A). Localization of collagen type IV was similar to that of other basement membrane-associated ECM proteins such as laminin (Fig. 9B) and heparan sulfate54 (Fig. 9C). Localization of these ECM proteins was different from that of fibronectin54 (Fig. 8C), which was seen at cell surface and border regions. Thus, specific localization of ECM proteins within the TM is possible by TPEF.
Actin and Pharmacological Analysis
The state of the TM actin cytoskeleton influences outflow resistance and IOP,55 but specific actin dynamics pertinent to IOP modulation remain to be better understood. If it were possible to resolve actin and actin dynamics in situ, authentic perspectives into tissue function, glaucoma pathogenesis, and therapeutic targets may be gained.
The actin cytoskeleton is organized as a 3D network across adjacent cells and autofluorescent beams in the TM. Our in situ studies showed filamentous actin (F-actin) in prominent cortical distributions near cell borders and in punctate form in perinuclear regions (Fig. 10A). The same observations are made in live cells in the tissue transduced with a fluorescent protein labeling actin. In addition, the actin network is seen wrapped around and aligned with trabecular beams.19 Our observations of F-actin in the human TM agree with observations by electron microscopy, immunohistochemistry, indirect fluorescence, and confocal microscopy describing actin filaments that are concentrated in perinuclear and peripheral regions of TM cells.56–59,60
FIG. 10.
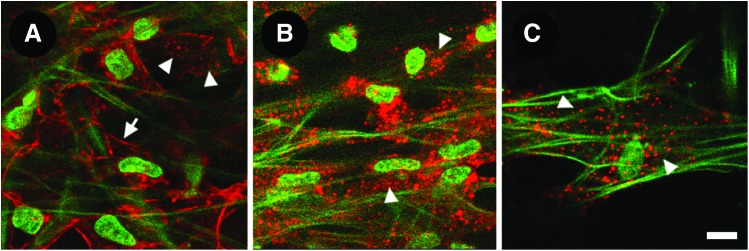
Filamentous actin (F-actin) in the trabecular meshwork. (A) F-actin (red) was prominent in a cortical distribution near cell borders (arrow) and in a perinuclear punctate distribution (arrowheads). (B, C) After Latruculin-A exposure, cortical F-actin was reduced leaving only perinuclear punctate actin (arrowheads). Autofluorescence signals were unchanged. Scale bar=10 μm. Color images available online at www.liebertpub.com/jop
1 μm latrunculin-A (Lat-A) caused actin reorganization leading to reduced cortical actin, but retained perinuclear punctate F-actin, as observed by TPEF in situ (arrowheads in Fig. 10B, C). Trabecular autofluorescent beam morphology was unchanged by Lat-A.13 Tissue-based quantitative analysis of the effect of Lat-A on actin and cell viability is being addressed in ongoing studies.
3D Reconstruction and Modeling
We perform 3D reconstructions of serial fine-cut optical sections to qualitatively and quantitatively analyze structural, cellular, and subcellular features of the TM.61 Specific features may be extracted, recombined, and modeled to analyze associations in 3D space. Three-dimensional models employing isosurface mapping may be combined with reconstructions to perform protein mapping and co-localization analysis in tissues (Fig. 11). We have used such analysis to quantify live cellularity in 3D space (Fig. 11A), perform tissue-based actin reconstructions (Fig. 11B), and map different structural ECM protein associations with AF signals within trabecular beams (Fig. 11C, D). These techniques provide novel ways to explore cell biology and glaucoma pathogenesis within human tissue.
FIG. 11.

Three-dimensional (3D) reconstruction and analysis of human trabecular meshwork (TM). (A) 3D reconstruction and analysis of live/dead co-labeling by calcein AM (green) and propidium iodide (PI; red) in TM. Tangential view (246×246 μm) is shown, with uveal meshwork seen on the upper face. Left half of image: Labeled cells are predominantly alive [calcein-positive (green) and PI-negative labeling (red) is scant]. Right half of image: spot mapping of individual cells during which different spheres are assigned to live (green) and dead (red) cells to allow automated live/dead cell counting. (B) Multimodal imaging combining F-actin epifluorescence (red filaments; left half of image) and structural matrix autofluorescence (AF); green fibers; right half of image). Actin filaments form an interconnected network among autofluorescent trabecular beams. (C) Molecular dissection of structural matrix by multimodal imaging and “isosurface” modeling. Model shows collagen (second harmonic generation (SHG); purple) encasing elastic fibers (AF; green) in the uveal meshwork. Upper third: 3D reconstruction of original SHG and AF signals; middle third: isosurface mapping segregating AF and SHG; lower third: SHG isosurface map after subtraction of AF signal to show space occupied by elastin in beam (arrow). (D) Mapping of proteins corresponding to components of AF signal. Left third: 3D reconstruction of original AF signal; middle third: isosurface model segregating high- and lower-intensity AF signals; right third: co-localization of eosin fluorescence (elastin) with high-intensity AF but not lower-intensity AF. Color images available online at www.liebertpub.com/jop
Conclusion and Future Developments
Application of TPM in our human TM model has particular benefits for tissue-based cell biology, pharmacological and translational studies. The tissue-based model closely mimics the cell, ECM, and 3D organization of the conventional drainage pathway, permitting authentic insights into the tissue's systems biology. The model goes beyond merely recapitulating tissue and cellular morphology. Rather, cells within the tissue environment can be probed and responses can be observed and quantified. Characteristic cellular responses in the model system involving dexamethasone and TGF-β1 induction mimic observations in human primary cell culture, immunohistochemistry, and electron microscopy.12 It will be a technical advance if biological interactions can be examined directly, live, and quantitatively within the human TM. This will provide a platform for exploring cell and molecular dynamics, screening drugs, and elucidating drug-induced mechanisms in a way that is directly relevant to humans.
The model does not replace in vitro, ex vivo, and in vivo systems already in use. Rather, it applies alternative technological approaches and provides perspectives of a different kind. Our use of postmortem transplant tissue simply and inexpensively salvages good-quality human tissue for research that is otherwise scarce and costly. Potentially, this is achievable in a way that is sustainable and accessible to many researchers.
Our initial efforts have centered on characterizing the tissue model by TPM. Our findings agree with those of different TM models and methods. They are also relevant to applying clinical TPM imaging to the human eye, as is already being explored in other organ systems such as skin and gastrointestinal tract. Anticipating issues such as motion artifact in clinical imaging, we have started exploring techniques to correct for potential artifacts during such imaging.61 For now, our goal is to use the model system for tissue-based drug development and as a platform to translate findings in live animal models62,63 to humans.
Appendix: Imaging Methodology
All experimental imaging methods reported here have previously been described.11–15,24,25 We captured images with a Leica SP5 microscope (Leica Microsystems, Heidelberg, Germany) coupled to a Chameleon Ultra-II multiphoton laser (Coherent, Santa Clara, CA). We typically use 850 nm excitation, pulsed, and focused through inverted 20X/0.7NA or 63X/1.3NA objectives. Two-photon excitation fluorescence (TPEF) emission is passed through green (525/50 nm) or red (635/90 nm) filters (Chroma, Bellows Falls, VT) onto a non-descanned photomultiplier tube detector (Hamamatsu, Bridgewater, NJ). Collagen second harmonic generation signals are collected through a shorter wavelength narrow band-pass filter (425/10 nm; Chroma).
Images were collected as multiple channel z-stacks using 512×512 or 1024×1024 pixel frames, a 16-bit grayscale resolution, and 16×line averaging using 1–8 μm step sizes.11–15 We have used LAS AF and AF Lite 2.2.1 (Leica Microsystems), Volocity 5.4.1 (PerkinElmer, Waltham, MA), Imaris 7.3.0 (Bitplane, Zurich, Switzerland), Image J (NIH, Bethesda, MD), and Photoshop CS5 (Adobe, San Jose, CA) for software-assisted image analysis. Isosurface modeling was conducted in Imaris.11,12,14,15
The following techniques assisted TPEF visualization: (a) 6-0 black nylon suture (Ethilon black monofilament, Ethicon, Somerville, NJ) is autofluorescent and when inserted into Schlemm's canal, it aids canal visualization with emission collected between 500 and 550 nm.11 (b) For intravital dye viability analysis, wedges were co-incubated with 0.33 μM calcein-AM (calcein; a cytosolic dye of living cells) and 1 μg/mL propidium iodide (PI; a nuclear dye only labeling necrotic and apoptotic cells with compromised plasma membrane; Life Technologies, Carlsbad, CA) for 30 min at 37°C immediately prior to imaging.14 (c) Other intravital dyes used included: CellTracker Red CMTPX (cytosolic label), Hoechst 33342 (chromosomal DNA), and octadecyl rhodamine B chloride (R18; plasma membrane; all from Life Technologies, St. Louis, MO).14 (d) Antibody labeling for immunofluorescence analysis12,24 was performed by first fixing tissue with 4% paraformaldehyde (JT Baker) in PBS for 30 min at room temperature or overnight at 4°C, washed with PBS, permeabilized with 5% TX-100/PBS for 2 h at 4°C, blocked with 1% BSA/PBS for 30 min at room temperature, and then incubated with antibodies in 0.1% BSA/PBS and 1 μg/mL Hoechst 33342 overnight at 4°C. Antibodies used were as follows: rabbit anti-laminin primary anitibody (pAb), rabbit anti-α-smooth muscle actin (α-SMA) pAb, rabbit anti-myocilin pAb (Abcam, San Francisco, CA), rabbit anti-elastin pAb (Elastin Products Company, Owensville, MO), mouse anti-type I collagen (clone 23IIC3), anti-type III collagen and anti-type IV collagen mAb (Millipore, Billerica, MA), and mouse anti-heparan sulfate mAb (10E4; US Biologicals, Swampscott, MA).12,24 Controls were mouse or rabbit IgG (Santa Cruz, CA). Alexa 568-conjugated secondary antibodies and Alexa 568-conjugated phalloidin were purchased from Life Technologies (Carlsbad, CA).13 All antibodies except anti-myocilin were used at 1:100 dilution. Anti-myocilin and phalloidin were used at 1:50 dilution. Some tissues were incubated with 200 nM dexamethasone (EMD Chemicals) for 3 days and the media was changed every 48 h or with 50 ng/mL TGF-β1 (Abcam) for 48 h with media changed at 24 h; up-regulated myocilin and α-SMA, respectively12; or Latrunculin-A (Cayman Chemicals, Ann Arbor, MI) prior to antibody or phalloidin labeling.12,13 Elastin was labeled with Eosin Y (Lerner Labs, Pittsburgh, PA).15
Acknowledgments
This study was supported by National Institutes of Health, Bethesda, MD, Grants EY020863 (J.C.H.T.), EY03040 (Doheny Vision Research Institute Imaging Core), 1S10RR024754 (USC Multiphoton Core); Kirchgessner Foundation Research Grant (J.C.H.T.); American Glaucoma Society Mentoring for Physician Scientists Award and Young Clinician Scientist Award (J.C.H.T.); Career Development Award from Research to Prevent Blindness (J.C.H.T.); and an unrestricted grant from the Research to Prevent Blindness, Inc., New York, New York.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Fuchshofer R., and Tamm E.R.Modulation of extracellular matrix turnover in the trabecular meshwork. Exp. Eye Res. 88:683–688, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Rhee D.J., Haddadin R.I., Kang M.H., and Oh D.J.Matricellular proteins in the trabecular meshwork. Exp. Eye Res. 88:694–703, 2009 [DOI] [PubMed] [Google Scholar]
- 3.Keller K.E., Aga M., Bradley J.M., Kelley M.J., and Acott T.S.Extracellular matrix turnover and outflow resistance. Exp. Eye Res. 88:676–682, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polansky J.R., Wood I.S., Maglio M.T., and Alvarado J.A.Trabecular meshwork cell culture in glaucoma research: evaluation of biological activity and structural properties of human trabecular cells in vitro. Ophthalmology. 91:580–595, 1984 [DOI] [PubMed] [Google Scholar]
- 5.Cukierman E., Pankov R., Stevens D.R., and Yamada K.M.Taking cell-matrix adhesions to the third dimension. Science. 294:1708–1712, 2001 [DOI] [PubMed] [Google Scholar]
- 6.Beebe D.C.The use of cell lines to “model” ocular tissues: cautionary tales. Invest. Ophthalmol. Vis. Sci. 54:5720, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Zipfel W.R., Williams R.M., and Webb W.W.Nonlinear magic: multiphoton microscopy in the biosciences. Nat. Biotechnol. 21:1369–1377, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Schenke-Layland K.Non-invasive multiphoton imaging of extracellular matrix structures. J. Biophotonics. 1:451–462, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aptel F., Olivier N., Deniset-Besseau A., et al. Multimodal nonlinear imaging of the human cornea. Invest. Ophthalmol. Vis. Sci. 51:2459–2465, 2010 [DOI] [PubMed] [Google Scholar]
- 10.Johnson A.W., Ammar D.A., and Kahook M.Y.Two-photon imaging of the mouse eye. Invest. Ophthalmol. Vis. Sci. 52:4098–4105, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tan J.C., Gonzalez J.M., Jr., Hamm-Alvarez S., and Song J.In situ autofluorescence visualization of human trabecular meshwork structure. Invest. Ophthalmol. Vis. Sci. 53:2080–2088, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez J.M., Jr., Heur M., and Tan J.C.Two-photon immunofluorescence characterization of the trabecular meshwork in situ. Invest. Ophthalmol. Vis. Sci. 53:3395–3404, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez J.M. Jr., et al. Association for research in vision and ophthalmology. Invest. Ophthalmol. Vis. Sci. E-Abstract 2746, 2012 [Google Scholar]
- 14.Gonzalez J.M., Jr., Hamm-Alvarez S., and Tan J.C.Analyzing live cellularity in the human trabecular meshwork. Invest. Ophthalmol. Vis. Sci. 54:1039–1047, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang A., Gonzalez J.M., Jr., Le P.V., Heur M., and Tan J.C.Sources of structural autofluorescence in the human trabecular meshwork. Invest. Ophthalmol. Vis. Sci. 54:4813–4820, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhee D.J., Tamm E.R., and Russell P.Donor corneoscleral buttons: a new source of trabecular meshwork for research. Exp. Eye Res. 77:749–756, 2003 [DOI] [PubMed] [Google Scholar]
- 17.Johnson D.H., and Tschumper R.C.The effect of organ culture on human trabecular meshwork. Exp. Eye Res. 49:113–127, 1989 [DOI] [PubMed] [Google Scholar]
- 18.Gottanka J., Chan D., Eichhorn M., Lütjen-Drecoll E., and Ethier C.R.Effects of TGF-beta2 in perfused human eyes. Invest. Ophthalmol. Vis. Sci. 45:153–158, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez J.M., Jr., Hsu H.Y., and Tan J.C.Observing live actin in the human trabecular meshwork. Clin. Exp. Ophthalmol. 2013. DOI: 10.1111/ceo.12255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ammar D.A., Lei T.C., Gibson E.A., and Kahook M.Y.Two-photon imaging of the trabecular meshwork. Mol. Vis. 16:935–944, 2010 [PMC free article] [PubMed] [Google Scholar]
- 21.Ammar D.A., Lei T.C., Masihzadeh O., Gibson E.A., and Kahook M.Y.Trans-scleral imaging of the human trabecular meshwork by two-photon microscopy. Mol. Vis. 17:583–590, 2011 [PMC free article] [PubMed] [Google Scholar]
- 22.Franken P.A., Hill A.E., Peters C.W., and Weinreich G.Generation of optical harmonics. Phys. Rev. Lett. 7:118–119, 1961 [Google Scholar]
- 23.Denk W., Strickler J.H., and Webb W.W.Two-photon laser scanning fluorescence microscopy. Science. 248: 73–76, 1990 [DOI] [PubMed] [Google Scholar]
- 24.Williams R.M., Zipfel W.R., and Webb W.W.Interpreting second-harmonic generation images of collagen I fibrils. Biophys. J. 88:1377–1386, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lütjen-Drecoll E., Shimizu T., Rohrbach M., and Rohen J.W.Quantitative analysis of ‘plaque material’ in the inner- and outer wall of Schlemm's canal in normal and glaucomatous eyes. Exp. Eye Res. 42:443–445, 1986 [DOI] [PubMed] [Google Scholar]
- 26.Hann C.R., Springett M.J., Wang X., and Johnson D.H.Ultrastructural localization of collagen IV, fibronectin, and laminin in the trabecular meshwork of normal and glaucomatous eyes. Ophthalmic Res. 33:314–324, 2001 [DOI] [PubMed] [Google Scholar]
- 27.Gong H.Y., Trinkaus-Randall V., and Freddo T.F.Ultrastructural immunocytochemical localization of elastin in normal human trabecular meshwork. Curr. Eye Res. 8:1071–1082, 1989 [DOI] [PubMed] [Google Scholar]
- 28.Rohen J.W.Why is intraocular pressure elevated in chronic simple galucoma? Anatomical considerations. Ophthalmology. 90:758–765, 1983 [DOI] [PubMed] [Google Scholar]
- 29.Murphy C.G., Yun A.J., Newsome D.A., and Alvarado J.A.Localization of extracellular proteins of the human trabecular meshwork by indirect immunofluorescence. Am. J. Ophthalmol. 104:33–43, 1987 [DOI] [PubMed] [Google Scholar]
- 30.Marshall G.E., Konstas A.G., and Lee W.R.Immunogold ultrastructural localization of collagens in the aged human outflow system. Ophthalmology. 98:692–700, 1991 [DOI] [PubMed] [Google Scholar]
- 31.Goldstein D.J.The fluorescence of elastic fibers stained with eosin and excited by visible light. Histochem. J. 1:187–198, 1969 [DOI] [PubMed] [Google Scholar]
- 32.Huntington H.Autofluorescence of eosinophilic substances. Arch. Pathol. Lab. Med. 110:93, 1986 [PubMed] [Google Scholar]
- 33.Deeb S., Nesr K.H., Mahdy E., Badawey M., and Badei M.Autofluorescence of routinely hematoxylin and eosin-stained sections without exogenous markers. Afr. J. Biotechnol. 7:504–507, 2008 [Google Scholar]
- 34.Heo Y.S., and Song H.J.Characterizing cutaneous elastic fibers by eosin fluorescence detected by fluorescence microscopy. Ann. Dermatol. 23:44–52, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson M.‘What controls aqueous humour outflow resistance?’ Exp. Eye Res. 82:545–557, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dai Y., Lindsey J.D., Duong-Polk X., Nguyen D., Hofer A., and Weinreb R.N.Outflow facility in mice with a targeted type I collagen mutation. Invest. Ophthalmol. Vis. Sci. 50:5749–5753, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steinhart M.R., Cone F.E., Nguyen C., et al. Mice with an induced mutation in collagen 8A2 develop larger eyes and are resistant to retinal ganglion cell damage in an experimental glaucoma model. Mol. Vis. 18:1093–1106, 2012 [PMC free article] [PubMed] [Google Scholar]
- 38.Vahedi K., and Alamowitch S.Clinical spectrum of type IV collagen (COL4A1) mutations: a novel genetic multisystem disease. Curr. Opin. Neurol. 24:63–68, 2011 [DOI] [PubMed] [Google Scholar]
- 39.Thorleifsson G., Magnusson K.P., Sulem P., et al. Common sequence variants in the LOXL1 gene confers susceptibility to exfoliation glaucoma. Science. 317:1397–1400, 2007 [DOI] [PubMed] [Google Scholar]
- 40.Ueda J., and Yue B.Y.Distribution of myocilin and extracellular matrix components in the corneoscleral meshwork of human eyes. Invest. Ophthalmol. Vis. Sci. 44:4772–4779, 2003 [DOI] [PubMed] [Google Scholar]
- 41.Tawara A., Tou N., Kubota T., Harada Y., and Yokota K.Immunohistochemical evaluation of the extracellular matrix in trabecular meshwork in steroid-induced glaucoma. Graefes Arch. Clin. Exp. Ophthalmol. 246:1021–1028, 2008 [DOI] [PubMed] [Google Scholar]
- 42.Fuchshofer R., Welge-Lüssen U., Lütjen-Drecoll E., and Birke M.Biochemical and morphological analysis of basement membrane component expression in corneoscleral and cribriform human trabecular meshwork cells. Invest. Ophthalmol. Vis. Sci. 47:794–801, 2006 [DOI] [PubMed] [Google Scholar]
- 43.Rao P.V., Allingham R.R., and Epstein D.L.TIGR/myocilin in human aqueous humor. Exp. Eye Res. 71:637–641, 2000 [DOI] [PubMed] [Google Scholar]
- 44.Russell P., Tamm E.R., Grehn F.J., Picht G., and Johnson M.The presence and properties of myocilin in the aqueous humor. Invest. Ophthalmol. Vis. Sci. 42:983–986, 2001 [PubMed] [Google Scholar]
- 45.Ezzat M.K., Howell K.G., Bahler C.K., et al. Characterization of monoclonal antibodies against the glaucoma-associated protein myocilin. Exp. Eye Res. 87:376–384, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sakai H., Shen X., Koga T., et al. Mitochondrial association of myocilin, product of a glaucoma gene, in human trabecular meshwork cells. J. Cell. Physiol. 213:775–784, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Wentz-Hunter K., Ueda J., Shimizu N., and Yue B.Y.Myocilin is associated with mitochondria in human trabecular meshwork cells. J. Cell. Physiol. 190:46–53, 2002 [DOI] [PubMed] [Google Scholar]
- 48.Fautsch M.P., Vrabel A.M., and Johnson D.H.The identification of myocilin-associated proteins in the human trabecular meshwork. Exp. Eye Res. 82:1046–1052, 2006 [DOI] [PubMed] [Google Scholar]
- 49.Sohn S., Hur W., Joe M.K., et al. Expression of wild-type and truncated myocilins in trabecular meshwork cells: their subcellular localizations and cytotoxicities. Invest. Ophthalmol. Vis. Sci. 43:3680–3685, 2002 [PubMed] [Google Scholar]
- 50.Stamer W.D., Perkumas K.M., Hoffman E.A., Roberts B.C., Epstein D.L., and McKay B.S.Coiled-coil targeting of myocilin to intracellular membranes. Exp. Eye Res. 83:1386–1395, 2006 [DOI] [PubMed] [Google Scholar]
- 51.Rohen J.W., Futa R., and Lütjen-Drecoll E.The fine structure of the cribriform meshwork in normal and glaucomatous eyes as seen in tangential sections. Invest. Ophthalmol. Vis. Sci. 21:574–585, 1981 [PubMed] [Google Scholar]
- 52.Streeten B.W.Elastic fibers and microfibrils in the eyes. In: Lütjen-Drecoll E., ed. Basic Aspects of Glaucoma Research: v. 3. Stuttgart: Schattauer GmbH; 1993; p. 67 [Google Scholar]
- 53.Ueda J., Wentz-Hunter K.K., Cheng E.L., Fukuchi T., Abe H., and Yue B.Y.Ultrastructural localization of myocilin in human trabecular meshwork cells and tissues. J. Histochem. Cytochem. 48:1321–1330, 2000 [DOI] [PubMed] [Google Scholar]
- 54.de Kater A.W., Shahsafaei A., and Epstein D.L.Localization of smooth muscle and nonmuscle actin isoforms in the human aqueous outflow pathway. Invest. Ophthalmol. Vis. Sci. 33:424–429, 1992 [PubMed] [Google Scholar]
- 55.Kaufman P.L.Enhancing trabecular outflow by disrupting the actin cytoskeleton, increasing uveoscleral outflow with prostaglandins, and understanding the pathophysiology of presbyopia interrogating Mother Nature: asking why, asking how, recognizing the signs, following the trail. Exp. Eye Res. 86:3–17, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weinreb R.N., and Ryder M.I.In situ localization of cytoskeletal elements in the human trabecular meshwork and cornea. Invest. Ophthalmol. Vis. Sci. 31:1839–1847, 1990 [PubMed] [Google Scholar]
- 57.Ethier C.R., Read A.T., and Chan D.Biomechanics of Schlemm's canal endothelial cells: influence on F-actin architecture. Biophys. J. 87:2828–2837, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Read A.T., Chan D.W., and Ethier C.R.Actin structure in the outflow tract of normal and glaucomatous eyes. Exp. Eye Res. 82:974–985, 2006 [DOI] [PubMed] [Google Scholar]
- 59.Stamer W.D., Read A.T., Sumida G.M., and Ethier C.R.Sphingosine-1-phosphate effects on the inner wall of Schlemm's canal and outflow facility in perfused human eyes. Exp. Eye Res. 89:980–988, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grierson I., and Rahi A.H.Microfilaments in the cells of the human trabecular meshwork. Br. J. Ophthalmol. 63:3–8, 1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Le P.V., et al. Association for research in vision and ophthalmology. Invest. Ophthalmol. Vis. Sci. E-Abstract 3268, 2012 [Google Scholar]
- 62.Tan J.C., et al. Association for research in vision and ophthalmology. Invest. Ophthalmol. Vis. Sci. E-Abstract 3553, 2013 [Google Scholar]
- 63.Gonzalez J.M. Jr., et al. Association for research in vision and ophthalmology. Invest. Ophthalmol. Vis. Sci. E-Abstract 3550, 2013 [Google Scholar]