

Abstract
The marcfortines are complex secondary metabolites that show potent anthelmintic activity and are characterized by the presence of a bicyclo[2.2.2]diazaoctane fused to a spirooxindole. Herein, we report the synthesis of two members of this family. The synthesis of marcfortine B utilizes a carboxylative TMM cycloaddition to establish the spirocyclic core, followed by an intramolecular Michael addition and oxidative radical cyclization to access the strained bicyclic ring system. In addition, the first asymmetric synthesis of (–)-marcfortine C is described. The key step involves a cyano-substituted TMM cycloaddition, which proceeds in nearly quantitative yield with high diastereo- and enantioselectivity. The resulting chiral center was used to establish all remaining stereocenters in the natural product.
INTRODUCTION
The marcfortines are a class of secondary metabolites first isolated by Polonsky and coworkers from Penicillium roqueforti, a fungus used in the production of blue cheese (Figure 1).1 Structurally, they are complex heptacyclic molecules with densely functionalized cores containing four quaternary centers. Characterized by the presence of a bicyclo[2.2.2]diazaoctane and a fused spirooxindole, they are related to several families of prenylated indole alkaloids, including paraherquamides,2,3,4 sclerotiamide,5 notoamides,6 brevianamides7 and versicolamide B.8 As with many of these compounds, the marcfortines possess potent antiparasitic and anthelmintic activity.9 Along with their derivatives, they have attracted attention as potential clinical candidates for the treatment of gastrointestinal nematodes in domestic livestock.10
Figure 1.

Marcfortines and selected related alkaloids.
The biosynthesis of these natural products has been the subject of several studies.11,12 They are all derived from tryptophan, one to two isoprene units, and a cyclic amino acid, typically a proline derivative. A distinguishing structural feature of the marcfortines, however, is the presence of a fused 6-membered ring derived from pipecolic acid. Stereochemically, the vast majority of these alkaloids share the same relative and absolute configurations, with the only known exceptions being brevianamides A and B, and versicolamide B. Interestingly, both enantiomers of notoamide B and versicolamide B are known, depending on whether the products were isolated from marine-derived Aspergillus sp. or the terrestrial organism Aspergillus versicolor NRRL 35600.6c
Given their biological activity and intriguing molecular architecture, the synthesis of these alkaloids has been widely explored. Williams and coworkers, in particular, have studied these compounds extensively, reporting asymmetric syntheses of paraherquamides A and B13 and versicolamide B,14 and racemic syntheses of brevianamide B,15 notoamide B,16 and marcfortine C.17 The reigning theme within this work is the use of a biomimetic intramolecular Diels-Alder reaction to construct the bicyclo[2.2.2]diazaoctane ring, while the spirooxindole moiety is typically prepared via oxidative rearragnement (Scheme 1a). We became interested in this class of compounds because their heavily substituted spirocyclic cyclopentane core could be effectively accessed by a palladium-catalyzed [3+2] cycloaddition of trimethylenemethane (TMM) with an isopropylidene oxindole (Scheme 1b). First disclosed by Trost and Chan in 1979,18 the TMM cycloaddition represents a powerful method for the construction of carbocycles.19 In 2006, the first general protocol for asymmetric TMM cycloaddition was developed by utilizing phosphoramidites as the chiral ligands.20 Subsequent efforts expanded the scope of this reaction to include the synthesis of carbo- and heterocycles.21,22 To date, however, the enantioselective methodology has not been successfully applied within the context of a total synthesis.23 In a previous paper, we reported an efficient approach to (±)-marfortine B using a carboxylative TMM cycloaddition.24 Herein, we disclose our full studies into this class of molecules, including an asymmetric synthesis of (–)-marcfortine C, utilizing an asymmetric cyano-TMM cycloaddition to establish the crucial C3 oxindole stereochemistry. Both syntheses effectively use this stereocenter as the sole control element to establish all remaining stereochemistry within the natural product.
Scheme 1.

Approaches to the spirooxindole core of marcfortines and related alkaloids.
RESULTS AND DISCUSSION
Total Synthesis of (±)-Marcfortine B
Our retrosynthetic approach to marcfortine B is depicted in Scheme 2. Late-stage installation of the dioxepin ring would be achieved by regioselective prenylation of the dihydroxyoxindole derived from 19.25 The initial strategy to generate the bicylic moiety in 19 involved a ring-closing alkylation using 20. However, we found that the required deprotonation α to the amide was not possible and the installed leaving group X (e.g., mesylate or tosylate) tended to quaternize the tertiary amine. Accordingly, we turned to the dehydro pipecolic acid derivative 21, which set the stage for ring closure by radical cyclization. The critical relative stereochemistry in 21 would be derived by a diastereoselective, intramolecular Michael addition between the carboxamide and α,β-unsaturated ester in 22, whereas the the pipecolic acid residue could be introduced from allylic alcohol 23. Slight functional group manipulation led us to spriocycle 24, which would be elegantly and efficiently prepared by carboxylative [3+2] cycloaddition26 between oxindole 25 and TMM donor 26.
Scheme 2.

Retrosynthetic analysis for the synthesis of marcfortine B.
In the forward direction, 25 was prepared from the known oxindole 2727 in two steps, and subsequently underwent a highly efficient cycloaddition with silyl donor 26 in the presence of 5 mol% palladium(II) acetate and 35 mol% triisopropyl phosphite (Scheme 3). After hydrolytic workup and alkylation, methyl ester 24 was isolated in excellent yield as a 1:1 mixture of diastereomers. The use of dimethyl sulfate/potassium carbonate over alternative conditions for methylation (e.g., DCC/methanol or DBU/CH3I) minimized any isomerization of the double bond and moreover provided higher yields than in the case of the optimized conditions utilizing either CH2N2 or TMSCHN2. For characterization purposes, the 1:1 mixture of diastereomers could easily be separated via recrystallization from petroleum ether. However, with regard to the further synthesis, 24 was used as a diastereomeric mixture since the allylic stereocenter is subsequently destroyed and, therefore, of no consequence. The exo-methylene was converted by epoxidation-elimination to allylic alcohol 23, and the pipecolic acid derivative was introduced by way of mesylation and SN2 coupling with 30, to provide an unstable secondary alcohol 28, which was immediately subjected to conditions for elimination to generate 22 in good overall yield.
Scheme 3.

Initial steps in the synthesis of marcfortine B.
Removal of the tert-butyl carbamate (Boc) protecting group for the Michael cyclization proved to be necessary to avoid decomposition. The deprotection was best achieved with tin tetrachloride in ethyl acetate (Scheme 4).28 Other more common protocols such as TFA, TMSI, TMSCl/phenol or thermolysis led to no product or only very poor yields. Michael addition was induced by treatment of the amide with either 2–3 equivalents of sodium hydride or potassium hexamethyldisilazide in THF. The crucial stereochemical outcome was analyzed by nOe studies. The unsubstituted amide 32 gave exclusively the desired diastereomer 35 in excellent yields, notably without any observed enhancement between the α-proton of the ester moiety (clearly identified as a singlet at δ 3.84 in the 1H NMR spectrum) and the aryl-proton of the oxindole residue (at δ 6.82). This is in sharp contrast to the N-methylated congener 33 (a forerunner for marcfortine A (1), which was prepared in analogous manner). Under all examined cyclization and quench conditions, the opposite diastereomer 37 was isolated exclusively, as evidenced by a 6% enhancement between the α-proton at δ 3.84 and the aryl-proton on δ 6.90. This can be rationalized by an internal protonation of the ester enolate in the case of the derivative with a free amide proton (34). In the case of the methyl substituted compound (36), external protonation from the least hindered face of the enolate led to the epimer at that stereocenter. At this point, only the synthesis of the desmethyl-congener marcfortine B (2) was further studied.
Scheme 4.

Michael addition and resultant diastereoselectivity.
Due to the poor solubility of the pentacycle 35, reprotection of the free amide nitrogen of the oxindole was necessary, where the best results were obtained using p-methoxybenzyl (PMB) chloride in acetone with potassium carbonate as base (Scheme 5). The choice of acetone compared to other solvents (e.g., DMF, DMSO or THF) proved to be crucial for high yields. Subsequent reduction of the methyl ester with DIBAL proceeded smoothly in 86% yield and gave the primary alcohol 21, which was converted into the required precursor 38 for the proposed radical cyclization. The radical cyclization required thorough optimization studies as the expected saturated cyclized product 19 was not obtained. Instead, the ring-closed compound 42, still bearing the same degree of unsaturation, was formed as the sole observable product (Table 1, entry 1). The use of alternative radical initiators or terminal reductants failed to affect the reaction outcome (entries 2–6). A potential explanation of this result is reaction of the secondary alkyl radical 39 with AIBN, rather than reaction with the tributylstannane. Accordingly, we found that superstoichiometric amounts of AIBN and catalytic amounts of tributylstannane were necessary for optimum yields (entries 7–9). The resulting nitrogen-centered radical 40 can then participate in a 1,4-hydrogen abstraction to generate alkyl radical 41, which undergoes fragmentation to the observed unsaturated product 42, an isobutyronitrile radical and a monoalkyl diazene. To our knowledge, this “oxidative” type of process under “reductive” conditions is unprecedented and results in a very useful alkene capable of further modification. For the purposes of our current synthesis, that modification is reduction, but useful analogs available by oxidation are also available and resemble other members of the family.
Scheme 5.

Elaboration of Michael adduct and reductive cyclization.
Table 1.
Optimization of the radical cyclization for the synthesis of 42.
| Entry | Conditions | % yield 42 |
|---|---|---|
| 1 | 1.5 eq. Bu3SnH slow add., 0.2 eq. AIBN, 1 mM, reflux benzene | 37 |
| 2 | Et3B, O2, Bu3SnH, benzene, 23°C | NR |
| 3 | 0.2 eq. AIBN, 1.6 eq. (TMS)3SiH, benzene reflux | 31 |
| 4 | 0.2 eq. AIBN slow add., 1.6 eq. (TMS)3SiH, benzene reflux | 41 |
| 5 | 1.2 eq. AIBN, 1.2 eq. Bu3SnH, benzene reflux | 24 |
| 6 | AIBN, (Bu3Sn)2, benzene reflux | Decomp. |
| 7 | 1.2 eq. AIBN slow add., 0.2 eq. Bu3SnH, 1 mM, reflux benzene, 46 µmol | 72–76 |
| 8 | 1.7 eq. AIBN slow add., 0.2 eq. Bu3SnH, 1 mM, reflux benzene, 0.3 mmol | 61 |
| 9 | 1.7 eq. AIBN slow add., 0.2 eq. Bu3SnH, 1 mM, reflux benzene, 3 mmol | 51 |
The double bond of 42 proved to be very resilient towards attempted hydrogenation. The starting material was further purified by stirring over Raney Nickel to remove any sulfur catalyst poisons from the previous step. Subsequently, the reduction was possible with Crabtree’s catalyst29 and worked best with a hydrogen pressure of 150 psi to obtain a fast and clean reaction (Scheme 6). Removal of the PMB protecting group in the next step required some optimization. Oxidative methods were not compatible with the electron-rich oxindole and led to complete decomposition within seconds. Protolytic conditions, involving reflux in trifluoroacetic acid with anisole as scavenger, cleaved the PMB group slowly but very cleanly. The resulting compound 43 was a crystalline solid whose structure and relative configuration could be unambiguously verified by x-ray crystal analysis (Figure 2). With all but the remaining dioxepin ring in place, the synthesis was completed using a known four-step protocol.25 The aryl methyl ethers were deprotected with boron tribromide, and selective monoprenylation provided intermediate 44. After epoxidation with m-chloroperoxybenzoic acid, a tin tetrachloride-mediated endo cyclization and subsequent dehydration afforded marcfortine B.
Scheme 6.

Completion of the synthesis of marcfortine B.
Figure 2.

X-ray crystallographic representation of oxindole 43.
Development of an Asymmetric Cycloaddition
Considering that the relative stereochemistry in marcfortine B was derived exclusively from the C3-oxindole stereocenter, we recognized the potential to access a member of this family by asymmetric synthesis, provided that an asymmetric cycloaddition could be developed. Following our initial disclosure of an asymmetric TMM cycloaddition utilizing phosphoramidite ligand L9,20 we investigated the potential for alkylidene oxindoles to undergo enantioselective cycloadditions with substituted TMM donors. While most of these donors were unsuccessful, including the carboxylative TMM donor 26, oxindoles proved to be excellent acceptors for the cyano donor (Table 2).21a First, the effect of the nitrogen substituent of the oxindole residue was examined. The free oxindole produced a myriad of different products (entry 1) and alkyl substituents such as methyl and PMB displayed either low reactivity or diastereoselectivity (entries 2 and 3). Electron withdrawing groups accelerated the reaction significantly and allowed later a reduction of the reaction temperature (entries 4–8). The methoxy carbonyl (Moc) group showed the best overall performance and was chosen for further optimization. Unfortunately, additives had no influence on the diastereoselectivity (entries 9–10), and halide ions seem to shutdown the reaction completely (entry 11).
Table 2.
Initial Optimization of the asymmetric cyano-TMM cycloaddition with oxindoles.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | R | Additive | T, °C | Yield (%) | dr (47:48) | 47, %ee | 48, %ee |
| 1 | H | – | 23 | NDb | – | – | – |
| 2 | Me | – | 23 | 91 | 1:1 | 91 | 53 |
| 3 | PMB | – | 23 | 60c | 1:1 | – | – |
| 4 | Ts | – | 23 | 50 | 1:2 | – | – |
| 5 | Boc | – | 23 | 95 | 1:1.1 | 74 | 93 |
| 6 | Ac | – | 23 | 98 | 1:2.2 | 85 | 89 |
| 7 | CO2Me | – | 23 | 94 | 1:2.3 | 72 | 73 |
| 8 | CO2Me | – | 0 | 93 | 1:3 | 80 | 97 |
| 9 | CO2Me | In(acac)3 | 0 | 98 | 1:3 | 82 | 98 |
| 10 | CO2Me | (n-Bu)3SnOAc | 0 | 96 | 1:3 | 81 | 96 |
| 11 | CO2Me | (n-C6H13)4NCl | 0 | 0 | – | – | – |
All reactions were performed at 0.2 M in toluene with 1.5 equivalents 45, 2.5 mol% Pd2(dba)3•CHCl3 and 10 mol% ligand for 12 hours. Yields were combined, isolated product; ee’s were determined by chiral HPLC.
Complex mixture.
Conversion.
A large number of ligands were then screened with this model substrate, with representative results shown in Table 3. Poor reactivity was observed with a variety of ligand classes, including phosphines and phosphites. On the other hand, reactivity was generally excellent using phosphoramidite ligands. In particular, the incorporation of bulky pyrrolidine derivatives, such as 2,5-bis(1-naphthyl) (L13) and 2,5-bis(2-naphthyl) (L14) proved critical for obtaining both high ee and dr. Interestingly, both configurations at the spirocyclic stereocenter could be obtained by using either L13 or L14, a remarkable stereochemical divergence considering that the ligands differ only by the relative orientation of the naphthyl substituents.21a
Table 3.
Optimization of the asymmetric cyano-TMM cycloaddition using oxindole 46 (R = Moc).
 |
All reactions were performed at 0.2 M in toluene with 1.5 equivalents 45, 2.5 mol% Pd2(dba)3•CHCl3 and 10 mol% ligand for 12 hours. Yields were combined, isolated product; ee’s were determined by chiral HPLC.
With these results in hand, we explored substituted oxindoles that would provide the requisite functionality to access the marcfortines and related natural products (Table 4). To our delight, we observed that the reaction was highly tolerant to oxindole substitution. As expected, both configurations at the spirocyclic stereocenter could be selectively prepared, where L13 in particular gave excellent yields, diastereo- and enantioselectivities. Accordingly, we targeted the asymmetric synthesis of marcfortine C, since it would demonstrate access to a member of this family with a different oxindole core.
Table 4.
Asymmetric TMM cycloaddition to generate precursors to spirocyclic oxindole natural products.
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | Substrate | Ligand | T, °C | Yield (%) | dr (50:51) | 50, %ee | 51, %ee |
| 1 |  |
L14 | −20 | 96 | 1.3:1 | >99 | 99 |
| 2 | L13 | 0 | 97 | 14:1 | 96 | 80 | |
| 3 |  |
L14 | −20 | 99 | 1:2.3 | 99 | >99 |
| 4 | L13 | 0 | 99 | 15:1 | 86 | 76 | |
All reactions were performed at 0.2 M in toluene with 1.5 equivalents 45, 2.5 mol% Pd2(dba)3•CHCl3 and 10 mol% ligand for 12 hours. Yields were combined, isolated product; ee’s were determined by chiral HPLC.
Initial Efforts Towards Marcfortine C
Using the synthesis of marcfortine B as a blueprint, we envisioned that the bicyclo[2.2.2]diazaoctane in marcfortine C could be synthesized by a radical cyclization of xanthate ester 52 (Scheme 7), which itself would derive from nitrile 53 by hydrolytic reduction. We anticipated that the requisite stereochemistry of nitrile 53 would be set by intramolecular Michael addition. In turn, 54 would derive from piperidine (±)−30 and spirooxindole 55, which could be accessed by asymmetric TMM cycloaddition.
Scheme 7.

Retrosynthetic strategy for the synthesis of marcfortine C.
The forward synthesis of oxindole 56 was commenced from commercially available 6-benzyloxindole (57) using a known procedure,30 involving Boc protection, benzyl ether hydrogenolysis and copper-catalyzed propargylation, to generate indole 58 in excellent overall yield (Scheme 8). Thermal Claisen rearrangement of 58 was known to proceed with loss of the Boc group; on the other hand, the platinum-catalyzed methodology originally developed by Sames31 permitted smooth formation of 59 in 82% yield. Treatment of this compound with LDA in the presence of triisopropyl borate led to the 2-indolylboronate, which was oxidized to yield oxindole 60 in 85% yield. At this stage, the Boc group was removed with trifluoroacetic acid and the acetone adduct 61 was prepared by treatment with N-methyl piperazine, which gave improved yields relative to the previously reported method involving HCl. The installation of the Moc protecting group was capricious and proceeded in moderate yield, but provided the key substrate 62. To further develop the TMM reaction, a series of phosphoramidite ligands were screened for the optimal selectivity (Table 5). Ligands L16 and L17 gave high enantioselectivities and good diastereoselectivities, whereas L18 gave high diastereoselectivity and good enantioselectivity. All diastereomers were inseparable at this stage. The TMM reaction generated two stereocenters (a and b), one of which (center b) would be eliminated later in the synthesis. Due to the ease of synthesis of ligand L11 compared to other ligands, and the relative high selectivity it offered, L11 was the ligand of choice for the synthetic study of our target natural product. Furthermore, as the scale of the TMM reaction increased, the loading of the catalyst could be lowered to 1 mol% Pd(0) and 2 mol% chiral ligand.
Scheme 8.

Initial synthesis of the spirocyclic oxindole.
Table 5.
Optimization of the asymmetric TMM cycloaddition using Moc-oxindole 62.
| entry | Pd2(dba)3•CHCl3 (mol%) |
Ligand (mol%) |
Yield (%) |
dr | ee (%) |
|---|---|---|---|---|---|
| 1 | 2.5 | L13 (10) | 99 | 15:1 | 86 |
| 2 | 2.5 | L11 (10) | 99 | 6:1 | 94 |
| 3 | 1.0 | L11 (4) | 100 | 6:1 | 94 |
| 4 | 0.5 | L11 (2) | 98 | 6:1 | 93 |
| 5 | 2.5 | L16 (10) | 100 | 8:1 | 96 |
| 6 | 2.5 | L17 (10) | 100 | 2:1 | 73 |
| 7 | 2.5 | L18 (10) | 100 | 4:1 | 70 |
Developing a simple, one-step protocol for the functionalization of the exocyclic double bond in TMM adduct 63 proved to be a significant challenge (Scheme 9). Both oxidation and bromination strategies led to reaction at the chromene, while attempts at allylic deprotonation-electrophilic capture gave recovered starting material or decomposition. Ultimately, rhodium-catalyzed hydroboration was successfully demonstrated, but this strategy then required a cumbersome series of additional steps to obtain the required allylic alcohol 69 (Scheme 10). During these experiments, the methyl carbonate was found to be an additional complicating factor and was removed using sodium methoxide. Thus, primary alcohol 66 was oxidized with Dess-Martin periodinane in the presence of 4Å molecular sieves. The resultant aldehyde was further oxidized by N-chlorosuccinimide in the presence of proline to yield an α-chloroaldehyde (67) that underwent elimination in the presence of DBU to the α,β-unsaturated aldehyde (68), which was finally reduced with sodium borohydride to provide allylic alcohol 69 in 44% yield over 5 steps.
Scheme 9.

Further elaboration plan for TMM adduct.
Scheme 10.

Olefin functionalization, installment of pipecolic acid derivative and Michael cyclization.
With 69 in hand, chemoselective mesylation was possible to set up an SN2 displacement with (±)−30 that proceeded in good yield when two equivalents of the hydrobromide were used. Double mesylation of 70 was found to occur simultaneously both on the free oxindole and on the secondary alcohol. Interestingly, when this intermediate was treated with excess DBU, the desired elimination took place concurrently with deprotection of the mesyl group on the free oxindole to deliver our desired product 71. However, caution had to be taken in order to avoid excess methanesulfonyl chloride, which gave the undesired nitrile compound 73 after treatment with DBU. Michael cyclization of 71 could be effected with 2.5 equivalents of potassium hexamethyldisilazide to give the desired stereochemistry at both the spirocenter and the vicinal carbon, established by a 5% nOe between the N-H amide (observed as a broad singlet at δ 7.75 in the 1H NMR) and the proton at the carbon bearing the nitrile (a singlet at δ4.19). Unfortunately, efforts to optimize this reaction were unsuccessful; the product could be isolated in up to 64% yield, but the reaction remained capricious and there was formation of what was tentatively assigned as the diastereomeric product with the opposite configuration at the carbon bearing the nitrile. The products were separable by TLC and clearly distinguished by their 1H NMR spectra, where the minor diastereomer had a singlet at δ 3.49 ppm assigned to the α-nitrile proton. The relative orientation of these peaks is consistent with increased de-shielding by the amide functionality in the major epimer. We suspected that adventitious water might be affecting the reaction, and that the slightly increased basicity and less demanding steric effects of the cyano-substituted carbocycle might lead to competitive intermolecular protonation.
Moving forward with 72 in hand, the critical challenge became reducing the nitrile functionality. All attempts to reduce the fully unprotected substrate were unsuccessful (Table 6). Treatment with 2.5 equivalents of DIBAL at low temperature led to no reaction (entry 1), while a larger excess at room temperature caused decomposition. Raney nickel hydrogenation in the presence of boric acid and PPTS effected reduction of the enamine (entry 2). Schwartz’s reagent was also tested, but gave either no conversion or decomposition if the reaction was pushed (entry 3). Realizing that the free amides were inhibiting reaction, we explored in situ protection. After significant optimization, we achieved reduction by employing two equivalents of trimethylaluminum followed by treament with DIBAL. After hydrolytic workup, the resulting aldehyde was treated with sodium borohydride, however, only poor yields of alcohol 75 were obtained due to significant decomposition in the first reaction (entry 4).
Table 6.
Michael cyclization and attempted nitrile reduction.
 | ||
|---|---|---|
| Entry | Conditions | Result |
| 1 | DIBAL (2.5 equiv.), toluene, −78 °C | Recovered SM |
| 2 | H2, Raney Ni, B(OH)3, PPTS, IPA, H2O, 23 °C | Enamine reduction |
| 3 | Cp2Zr(H)Cl, THF, 23 °C | Recovered SM |
| 4 | 1) AlMe3 (2.0 equiv.), then DIBAL (2.5 equiv.) 2) NaBH4, DCM, MeOH, 0 °C | 22% yield 75 |
Second Generation Strategy Towards the Synthesis of Marcfortine C
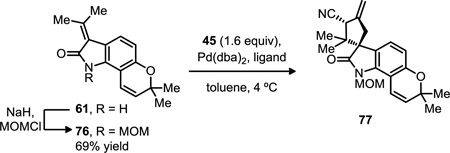
In light of the challenges in optimizing the nitrile reduction of 72, we considered that the reduction of a protected oxindole might be easier to achieve. We identified the TMM substrate 61 as an optimal entry point for this new protecting group, since 1) we anticipated it might simplify the functionalization of the exocyclic olefin that had been so problematic in our initial studies (see Scheme 10), and 2) we expected that such a change would streamline our overall synthesis by avoiding the need for multiple protection/deprotection steps. We initially attempted to install a PMB group on 61, but surprisingly we were unable to obtain any of the desired product. However, we could prepare Nmethoxymethyl (MOM) derivative 76 in good yield under standard alkylation conditions (Table 7). We next established that this oxindole was a competent acceptor in the TMM reaction. Surprisingly, both ligands L13 and L14 gave the same diastereomer as the major product in the reaction (Table 3, entries 1–2). While the dr was relatively poor with the latter, both gave the major product with excellent ee. Ligand L11 emerged as a possible alternative, since it furnished 77 in 89% yield, 7:1 dr and 86% ee under our standard conditions (entry 3). However, all attempts to use less than 5 mol% Pd catalyst with this ligand were unsuccessful (entry 4), including tests with either Pd(dba)2 or Pd2(dba)3•CHCl3, or reactions where catalyst/ligand charges were made sequentially. Since we preferred to achieve a higher catalyst turnover, we turned to azetidine ligand L10 and were pleased to observe excellent reactivity (entry 5). In contrast to our initial optimization efforts using an unsubstituted, Moc-protected oxindole (see Table 3), the diastereoselectivity using substrate 76 was high. Furthermore, catalyst loading could be reduced to 1 mol% Pd(dba)2, which gave 77 in 97% yield and 10:1 dr, with >99% ee for the major enantiomer (entry 6). The absolute configuration of cycloadduct 77 was assigned by analogy to our previous work,21a in which the use of (R,R,R)-L13 gave the R configuration at the spirocenter.
Table 7.
Preparation and TMM cycloaddition of N-MOM oxindole 76.
 | |||||
|---|---|---|---|---|---|
| Entry | Pd (mol%) |
Ligand (mol%) |
Yield (%) |
dr | ee (%) |
| 1 | 5 | L14 (10) | 99 | 2.6:1 | 97 |
| 2 | 5 | L13 (10) | 54 | 8.4:1 | 90 |
| 3 | 5 | L11 (10) | 89 | 7.0:1 | 86 |
| 4 | 1 | L11 (2) | 28 | ND | ND |
| 5 | 5 | L10 (10) | 99 | 9.7:1 | 96 |
| 6 | 1 | L10 (2) | 97 | 10.0:1 | 99 |
We also anticipated that the MOM protecting group might simplify the functionalization of the exocyclic olefin. In the studies using methyl carbonate 63, attempts to deprotonate generally returned unreacted starting material or decomposition via loss of the carbonate. Since the MOM group is more stable to base, we anticipated more flexibility in achieving the desired reaction. Accordingly, 77 was subjected to a variety of conditions for deprotonation (Table 8). Surprisingly, adding 1 equivalent of LDA at −78 °C led to only trace deprotonation (entry 1). Increasing to room temperature and 2 equivalents of either LDA, KHMDS or KOt-Bu gave complete deprotonation, but also substantial decomposition (entries 2–4). To separate the results of deprotonation from those of the electrophilic capture of the anion, these reactions were simply quenched with dilute acid to recover the isomerized product 78a. Despite nearly complete consumption of starting material, several unidentified products were observed and only poor yields of the desired products were recovered. Nonetheless, among these bases, KOt-Bu afforded the cleanest reaction. With these results in hand, we attempted to trap the allylic anion with molecular bromine, but obtained only 78a and unidentified by-products (entry 5).
Table 8.
One-step functionalization of the exocyclic olefin.
 | |||
|---|---|---|---|
| Entry | Conditions | Product | Result |
| 1 | LDA (1 equiv.), THF, −78 °C, then NaHSO4 | 78a (X=H) | >90% conv., 18% yield |
| 2 | LDA (2 equiv.), THF, 23 °C, then NaHSO4 | 78a | >90% conv., 18% yield |
| 3 | KHMDS (2.0 equiv.), THF, 23 °C, then NaHSO4 | 78a | 100% conv., <36% yield |
| 4 | KOt-Bu (2 equiv.), THF, 23 °C, then NaHSO4 | 78a | 100% conv., 58% yield |
| 5 | KOt-Bu (2 equiv.), THF, 23 °C, then Br2 | 78b (X=Br) | 100% conv, 0% yield |
| 6 | LiOt-Bu (1 equiv.), n-BuLi (1.5 equiv.), then CD3CO2D | 78c (X=D) | 85% yield |
| 7 | LiOt-Bu (1 equiv.), n-BuLi (1.5 equiv.), then (BzO)2 | 78d (X=OBn) | decomp. |
| 8 | LiOt-Bu (1 equiv.), n-BuLi (1.5 equiv.), then DMDO | 78e (X=OH) | trace product |
| 9 | LiOt-Bu (1 equiv.), n-BuLi (1.5 equiv.), then 79 | 78e | 64% yield |
| 10 | HOt-Bu (0.5 equiv.), then n-BuLi (1.1 equiv.), then 79 | 78e | 0% yield |
At this point, we became concerned that the tert-butoxide might be simply acting as a proton shuttle, catalyzing the formation of 78a rather than stoichiometrically generating the desired allylic anion (see below). Notably, 77 underwent no isomerization in the presence of n-BuLi and was stable at temperatures up to 0 °C. We reasoned that LiOt-Bu in the presence of an excess of n-BuLi would ensure stoichiometric formation of the allylic anion. Indeed, this strategy was successfully realized using 1 equivalent LiOt-Bu in the presence 1.5 equivalents of n-BuLi. After quenching with deuterated acetic acid, 78c was isolated in 85% yield with greater than 90% deuterium incorporation by 1H NMR (entry 5). Having developed an effective strategy to access the allylic anion of 77, we next explored potential oxidants. Benzoyl peroxide provided a complex mixture of products (entry 7). Dimethyl dioxirane (DMDO) gave trace formation of allylic alcohol 78e (entry 8), but the main product appeared to be an acetone adduct. The best results were obtained using Davis oxaziridine 79, which gave 78e in good yield (entry 9). We also confirmed our suspicions that LiOt-Bu could simply isomerize the exocyclic olefin rather than stoichiometrically deprotonate. When an excess of n-BuLi was added to a solution containing cycloadduct 77 and catalytic t-BuOH, only the isomerized by-product 78a was obtained after quenching with Davis reagent (entry 10).
In practice, it was easier to perform both the TMM reaction and subsequent oxidation based on isopropylidene oxindole 76 (Scheme 11). We also found we could reduce the equivalents of all reagents in the second step. At gram scale, we obtained 60% overall yield of allylic alcohol 82e in 89% ee. Accordingly, this two-step sequence effectively set the stereochemistry at the spirocenter, which we anticipated would allow us to establish all the remaining stereocenters in (–)-marcfortine C by analogy to our previous work. The drop in ee confirms that the cycloadduct 77 was diastereomeric at the spirocenter and not at the carbon bearing the nitrile. We could now readily introduce the required piperidine functionality by SN2 alkylation and eliminate the secondary alcohol to obtain 80 in 84% yield over 3 steps. Notably, while the N-H compound was highly sensitive to excess methanesulfonyl chloride during this elimination (see Scheme 10), the N-MOM species tolerated excess reagent and no nitrile formation was observed. Finally, primary carboxamide 80 underwent intramolecular Michael addition in the presence of one equivalent LiOt-Bu to provide 81 in 71% yield. The stereochemistry of 81 was consistent with previous work and was established by nOe. Irradiation of the proton geminal to the nitrile showed a strong 8.3% enhancement at the N-H amide, and no enhancement with any aromatic protons.
Scheme 11.

Elaboration of the N-MOM TMM cycloadduct.
With compound 81 in hand, we sought to effect the crucial nitrile reduction (Table 9). As we had observed previously with the N-H oxindole, Cp2Zr(H)Cl was unreactive when a slight excess of the reagent was used (entry 1), while a large excess provided complete decomposition. The nitrile was also resistant to borohydride reductants such as K-selectride (entry 2). However, when substrate 81 was treated with an excess of DIBAL, some conversion to the aldehyde was observed, albeit poor and with decomposition (entry 3). On the other hand, the use of the n-BuLi/DIBAL-ate complex failed to afford any of the desired aldehyde, instead providing unidentified by-products along with unreacted starting material (entry 4). Improved results were obtained by employing trimethylaluminum as an in situ protecting group for the secondary amide, which provided the primary alcohol 83 in up to 29% yield over two steps (entry 5). The modest yield was due largely to poor selectivity in the first reduction, since an unidentified by-product was formed along with the desired aldehyde in roughly equimolar amounts. Performing this reaction in either toluene or dichloromethane had little effect on the overall yield. In order to increase the electrophilicity of the nitrile, we also attempted to add BF3•OEt2, but observed poor conversion (entry 6), and a similar result was observed when triethylborane was used as the protecting agent (entry 7). However, the use of triethylaluminum was significantly more successful (entry 8). While the reaction was more sluggish under these conditions, we were pleased to observe that the aldehyde product was formed cleanly with a small amount of unreacted starting material. In practice, these two components were challenging to separate, however, treatment of the mixture with excess of sodium borohydride effectively reduced the aldehyde and allowed recovery of any unreacted starting material. Under these conditions, we were able to isolate the primary alcohol 83 in 58% based on recovered starting material (brsm).
Table 9.
Optimization of the hydrolytic nitrile reduction.
| Entry | Conditions | Result (Stage 1) | [H-] source | Yield (83, 2 steps) |
|---|---|---|---|---|
| 1 | Cp2Zr(H)Cl (2.0 equiv.), THF, 23 °C | trace conv. | – | – |
| 2 | K-selectride (2.0 equiv.), THF, 0 °C | trace conv. | – | – |
| 3 | DIBAL (2.3 equiv.), DCM, −78 °C | complex mixture | – | – |
| 4 | DIBAL-n-BuLi (2.4 equiv.), THF, DCM, 0 °C | SM + decomposition | – | – |
| 5 | AlMe3 (1.0 eq.), DCM, 0 °C, then DIBAL (2.0 equiv.), −78 °C | – | DIBAL | 29% (41% brsm) |
| 6 | AlMe3 (1.0 equiv.), DCM, −78 °C then BF3•OEt2 (1.0 equiv.), DIBAL (2.5 equiv.), −78 °C | trace conv. | – | – |
| 7 | BEt3 (1.0 equiv.), DCM, 0 °C then DIBAL (2.5 equiv.), −78 °C | trace conv. | – | – |
| 8 | Et3Al (1.0 equiv.), DCM, 0 °C then DIBAL (2.5 equiv.), −78 °C | 85% mass recovery | NaBH4 | 46% (58% brsm) |
With primary alcohol 83 in hand, we were able to complete the synthesis of marcfortine C (Scheme 12). Xanthate ester formation proceeded in 75% yield using LiOt-Bu as the base to give 84. Surprisingly, the initial attempts to employ 84 in the radical cyclization, using the procedure developed for marcfortine B, were unsuccessful and led only to decomposition. Reasoning that the MOM protecting group was contributing to undesired side-reactions, we tried adding an excess of N,O-bis(trimethylsilyl)acetamide (BSA). To our delight, the bicyclo[2.2.2]diazaoctane 85 was generated in approximately 40% yield, and doubling the equivalents of AIBN improved the yield to 54%. To the best of our knowledge, the use of BSA in such radical cyclizations is unprecedented. In a number of reactions wherein the substrates and/or products may be subject to decomposition due to the adventitious presence of acid or moisture, addition of BSA has proven to be an innocuous acid and moisture trap. We suspect it plays a similar role here. On the other hand, we cannot rule out a direct role in the radical process such as by turning over the tin catalyst by silylating the methylthio fragment. As was observed earlier (see Scheme 5), the cyclization reaction proceeds in an oxidative fashion to provide the unsaturated product. Thus, we next had to effect a chemoselective reduction of this newly formed olefin in the presence of the chromene moiety. Notably, both olefins are part of a fused 6-membered heterocycle and possess a vicinal tertiary carbon. Initial tests using either Wilkinson’s or Crabtree’s catalysts were highly chemoselective, and the latter proceeded in nearly quantitative yield. Finally, deprotection was achieved with aqueous HCl in DME to provide (–)-marcfortine C in 74% yield. The absolute configuration was confirmed by comparing the observed optical rotation ([a]D −68.0 (c 1.10, CHCl3)) with the reported value1b ([a]D −64.4 (c 1.10, CHCl3)), and all spectral properties of the synthetic material were consistent with the literature reports.32
Scheme 12.

Completion of the synthesis of marcfortine C.
CONCLUSION
In summary, we have described an efficient approach to spirocyclic oxindole alkaloids such as the marcfortines. The synthesis of marcfortine B was accomplished using a carboxylative TMM cycloaddition as a key step, whereas the synthesis of marcfortine C utilized an enantioselective cyano-substituted TMM cycloaddition. These cycloadditions allowed for a rapid synthesis of the spirooxindole moiety in nearly quantitative yield, with excellent enantio- and diastereoselectivity in the case of marcfortine C. Both syntheses took advantage of an intramolecular Michael addition and an unusual oxidative radical cyclization to build the bicyclo[2.2.2]diazaoctane core. The radical cyclization potentially allows for late-stage structural diversification in order to access more biologically active analogues. In addition, we presented a novel method for the rapid functionalization of the exocyclic olefin by oxidation of an allylic anion with an oxaziridine, and a triethylaluminum-protected reduction of a nitrile in the presence of multiple amide derivatives. Our approach allowed for the synthesis of marcfortine B in 25 steps and 1.7% overall yield, and marcfortine C in 19 steps and 2.6% overall yield.
EXPERIMENTAL DETAILS
1'-tert-butyl 3-methyl 6',7'-dimethoxy-2,2-dimethyl-4-methylene-2'-oxospiro[cyclopentane-1,3'-indoline]-1',3-dicarboxylate (24)
A solution of palladium acetate (892 mg, 3.96 mmol) in 500 mL dry toluene at room temperature was treated with triisopropyl phosphite (6.82 mL, 27.6 mmol). After 15 min N-(tert-butyloxycarbonyl)-3- isopropylidene-6,7-dimethoxy-indoline-2-one (25) (26.3 g, 78.9 mmol) and methyl 3-(trimethylsilyl)-2-(trimethylsilylmethyl)-allyl carbonate (26) (43.6 g, 157.8 mmol) were added and the reaction mixture was heated to 110 °C for 1.5 h (preheated oil bath). After cooling to ambient temperature, the reaction mixture was concentrated in vacuo, and the resulting residue was chromatographed on silica gel (CH2Cl2/MeOH, 20:1 to 10:1) to afford the cycloadduct as a 1:1 mixture of the two diastereomers as faint red oil, which was used directly in the next step. IR (NaCl, film): 2981, 2942, 1759, 1742, 1624, 1503, 1462, 1371, 1261, 1150 cm−1. 1H NMR (500 MHz, CDCl3): δ 6.87 (d, J = 8.3 Hz, 1H), 6.87 (d, J = 8.3 Hz, 1H), 6.64 (d, J = 8.3 Hz, 1H), 6.63 (d, J = 8.3 Hz, 1H), 5.35-5.20 (m, 4H), 4.28 (bs, 1H), 3.88 (s, 3H), 3.87 (s, 3H), 3.83 (s, 3H), 3.83 (s, 3H), 3.72 (s, br., 1H), 3.10 (d, J = 16.4 Hz, 1H), 2.91 (d, J = 17.6 Hz, 1H), 2.79 (d, J = 17.6 Hz, 1H), 2.72 (d, J = 16.4 Hz, 1H), 1.60 (s, 9H), 1.59 (s, 9H), 1.14 (s, 3H), 1.09 (s, 3H), 1.03 (s, 3H) ppm. 13C NMR (75 MHz, CDCl3) δ: 178.5, 177.9, 176.9, 176.1, 153.5, 153.3, 149.2, 144.2, 143.3, 134.5, 134.4, 133.7, 132.6, 124.4, 120.8, 120.5, 119.1, 113.1, 111.8, 107.1, 106.6, 85.1, 84.8, 60.4, 60.3, 60.1, 60.0, 56.8, 56.0, 50.0, 49.1, 40.9, 39.4, 27.5, 27.4 ppm. HRMS (EI): Calc’d for C23H29NO7 + [M]+: 431.1944. Found: 431.1939.
A solution of the above cycloadduct in 1.5 L acetone was treated with 14.2 g (103 mmol) K2CO3 and 10.6 mL (112 mmol) dimethyl sulfate. The reaction mixture was stirred at 40 °C for 2 h, cooled to r.t. and concentrated in vacuo. The residue was redissolved in 600 mL water and extracted into CH2Cl2 (3× 600 mL). The combined organic layers were dried (MgSO4) and evaporated. The residue was purified via chromatography on silica gel (petrol ether/EtOAc 4:1) to give 32.8 g (93% over 2 steps) of 24 as a 1:1 mixture of the two diastereomers. IR (NaCl, film): 2981.1, 2947.0, 1759.2, 1740.2, 1622.8, 1503.0, 1462.0, 1370.6, 1261.2, 1234.9, 1194.9, 1056.2, 1117.4, 1022.1 cm−1. 1H NMR (500 MHz, CDCl3, trans-isomer): d 6.87 (d, J = 8.4 Hz, 1H), 6.64 (d, J = 8.4 Hz, 1H), 5.24-5.22 (m, 1H), 5.17-5.15 (m, 1H), 4.24 (s, br., 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.74 (s, 3H), 2.93-2.86 (m, 1H), 2.80 (d, J = 18.1 Hz, 1H), 1.60 (s, 9H), 1.02 (s, 3H), 0.98 (s, 3H) ppm. 1H-NMR (500 MHz, CDCl3, cis-isomer): d 6.84 (d, J = 8.3 Hz, 1H), 6.60 (d, J = 8.3 Hz, 1H), 5.34-5.30 (m, 1H), 5.20-5.17 (m, 1H), 3.87 (s, 3H), 3.83 (s, 3H), 3.75 (s, 3H), 3.70-3.68 (m, 1H), 3.33-3.28 (m, 1H), 2.53 (d, J = 16.7 Hz, 1H), 1.61 (s, 9H), 1.13 (s, 3H), 0.79 (s, 3H) ppm. 13C-NMR (125 MHz, CDCl3, trans-isomer): d 178.6, 172.6, 153.5, 149.3, 144.0, 134.5, 133.8, 120.8, 120.8, 111.3, 106.6, 84.8, 60.4, 60.0, 57.1, 56.0, 51.5, 49.0, 39.5, 27.6, 21.9, 21.6 ppm. 13C-NMR (125 MHz, CDCl3, cis-isomer): d 175.5, 172.1, 153.1, 149.5, 144.6, 134.5, 132.4, 126.0, 118.4, 112.6, 106.9, 84.7, 59.6, 58.8, 58.7, 56.0, 51.7, 50.3, 40.3, 27.5, 23.0, 21.1 ppm. HRMS (EI): Calc’d for C24H31NO7+ [M]+: 445.2101. Found: 445.2103.
Allylic alcohol (23)
A solution of methyl esters 24 (12.13 g, 27.23 mmol) in 750 mL dry CH2Cl2 was treated with m-CPBA (13.43 g, 54.45 mmol, 70%, 2 equiv.) at 0 °C. The reaction mixture was allowed to warm to room temperature and stirred overnight. The reaction was quenched with sat. aq. Na2SO3 (400 mL). After vigorous stirring for 30 min, the aqueous layer was separated and extracted with CH2Cl2 (3× 300 mL). The combined organic phases were dried (MgSO4) and concentrated in vacuo. Chromatography on silica gel (petroleum ether/EtOAc 2:1) yielded the diastereomeric epoxides as a complex mixture (11.27 g, 24.42 mmol, 90%). To an ice-cold solution of the epoxides (1.09 g, 2.37 mmol) in 5 mL of dry THF was added DBU (0.54 mL, 3.55 mmol). The mixture was stirred at ambient temperature for 1 day, then diluted with diethyl ether, washed with 1N KHSO4, sat. NaHCO3, brine, dried (MgSO4) and evaporated. Chromatography of the residue on silica gel (petroleum ether/EtOAc 1:1) yielded 550 mg, (50%) of 23 as colourless foam. The recovered starting material (483 mg, 44%) was subjected to DBU again to give additional allylic alcohol 23 (233 mg, 22%). The total yield after two iterations was 783 mg (72%). IR (NaCl, film): 3515.8, 2979.7, 2940.4, 1763.8, 1736.6, 1709.4, 1623.4, 1503.1, 1462.4, 1369.5, 1268.3, 1215.8, 1148.2 cm−1. 1H NMR (500 MHz, CDCl3): δ 6.94 (d, J = 8.3 Hz, 1H), 6.62 (d, J = 8.3 Hz, 1H), 4.60-4.46 (m, 2H), 3.87 (s, 3H), 3.83 (s, 3H), 3.77 (s, 3H), 3.06 (d, J = 17.9 Hz, 1H), 2.93 (s, br., 1H), 2.86 (d, J = 17.9 Hz, 1H), 1.59 (s, 9H), 1.22 (s, 3H), 1.09 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ 177.3, 166.0, 155.3, 153.3, 149.4, 134.5, 134.4, 133.2, 122.7, 119.9, 106.9, 84.7, 60.4, 60.3, 59.9, 56.0, 53.6, 51.5, 42.3, 27.5, 24.2, 22.1 ppm. Anal: Calc’d. for C24H31NO8: C 62.46, H 6.77, N 3.04. Found: C 62.70, H 6.77, N 3.04.
α,β-Unsaturated amide (22)
To an ice-cold solution of 23 (10.8 g, 23.5 mmol) and triethylamine (3.7 mL, 25.9 mmol) in 200 mL dry CH2Cl2 was added methanesulfonylchloride (2.00 mL, 2.96 g, 25.9 mmol). The mixture was stirred for 10 min at 0 °C and then diluted with 600 mL Et2O, washed with sat. NaHCO3 and brine, dried (MgSO4) and evaporated. The residue was taken up in 30 mL dry DMSO and added to a solution of 10.53 g (47 mmol) piperidine hydrobromide 30 and 14.0 mL (98 mmol) triethylamine in 120 mL dry DMSO. The mixture was stirred at room temperature overnight, diluted with 800 mL EtOAc, washed with water and brine, dried (MgSO4) and evaporated. Column chromatography on silica gel (petroleum ether/EtOAc 1:1 to CH2Cl2/MeOH 20:1) gave the unstable tertiary amine which was immediately dissolved in 150 mL dry CH2Cl2 and cooled to 0 °C. 4.0 mL (28.2 mmol) triethylamine and 2.00 mL (2.96 g, 25.9 mmol) methanesulfonylchloride were added. After 10 min, 19 mL DBU were added, the reaction was brought back to room temperature and stirred for 2 h. The mixture was extracted with 600 mL EtOAc, washed with water and brine, dried (MgSO4) and evaporated. The residue was chromatographed on silica gel (CH2Cl2/MeOH 20:1) yielding 11.28 g (84% over 3 steps) of the α,β-unsaturated amide 22 as pale yellow foam. mp 81–83 °C. IR (KBr): 3458, 3354, 2934, 1761, 1713, 1681, 1624, 1502, 1462, 1435, 1369, 1267, 1209, 1148, 1114, 1063, 1020, 916, 845, 798, 732 cm−1. 1H NMR (500 MHz, CDCl3): δ 6.96 (d, J = 8.5 Hz, 1H), 6.65 (d, J = 8.5 Hz, 1H), 6.56 (br, 1H), 6.04 (t, J = 3.1 Hz, 1H), 5.28 (br, 1H), 4.01 (d, J = 16.2 Hz, 1H), 3.91 (s, 3H), 3.85 (s, 3H), 3.77 (s, 3H), 3.66 (d, J = 16.2 Hz, 1H), 3.15 (d, J = 17.0 Hz, 1H), 3.07-3.02 (m, 1H), 2.94-2.89 (m, 1H), 2.82 (d, J = 17.0 Hz, 1H), 2.19-2.14 (m, 2H), 1.73-1.67 (m, 2H), 1.61 (s, 9H), 1.24 (s, 3H), 1.14 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3): δ 177.6, 168.6, 166.1, 153.6, 149.6, 142.3, 136.7, 134.8, 133.5, 122.6, 120.2, 118.0, 107.1, 85.0, 60.6, 60.0, 56.2, 54.2, 51.8, 51.6, 47.6, 42.6, 27.8, 24.6, 23.4, 22.1, 17.3 ppm. HRMS (EI): Calc’d for C30H39N3O8 + [M]+: 569.2737. Found: 569.2738.
Michael Adduct (35)
To a 0 °C cold solution of amide 32 (140 mg, 0.3 mmol, azeotropically dried twice with toluene) in 6 mL dry THF was added a KHMDS in THF (1.2 mL, 2.2 equivalents, freshly prepared, 0.55 M). After 1 h at 0 °C, the cooling bath was removed and the mixture stirred for 15 h at room temperature. The reaction was quenched with 0.1 mL water and poured in 60 mL CH2Cl2/MeOH 10:1, dried (MgSO4) and evaporated. The residue was washed with methanol (3 × 2 mL) and acetone (2 × 1mL) to give pure 35 (140 mg, quant.) as colorless amorphous powder. mp 250 °C (dec.). IR (NaCl, film): 3257, 3142, 2951, 2921, 2852, 1731, 1698, 1662, 1633, 1615, 1505, 1456, 1392, 1345, 1269, 1259, 1224, 1196, 1171, 1135, 1109, 1087, 796 cm−1. 1H NMR (500 MHz, CDCl3): δ 8.17 (s, 1H), 7.31 (s, 1H), 6.82 (d, J = 8.4 Hz, 1H), 6.53 (d, J = 8.4 Hz, 1H), 6.01 (dd, J = 4.8, J = 3.7 Hz, 1H), 3.89 (s, 3H), 3.87 (s, 3H), 3.84 (s, 1H), 3.61 (s, 3H), 3.27 (d, J = 11.7 Hz, 1H), 3.03-2.98 (m, 1H), 2.94 (d, J = 11.2 Hz, 1H), 2.73 (td, J = 10.7, J = 2.8 Hz, 1H), 2.36 (d, J = 15.1 Hz, 1H), 2.21- 2.12 (m, 3H), 1.88-1.79 (m, 2H), 1.34 (s, 3H), 0.88 (s, 3H) ppm. 13C NMR (125 MHz, DMSO, 50 °C): δ 182.5, 170.8, 158.8, 152.3, 135.9, 135.5, 132.4, 121.2, 120.3, 106.0, 105.0, 61.1, 60.6, 60.1, 59.6, 55.6, 55.1, 50.8, 48.6, 47.8, 44.6, 22.7, 21.5, 20.7, 20.5 ppm. Anal: Calc’d for C25H31N3O6: C, 63.95; H, 6.65; N, 8.95. Found: C, 63.97; H, 6.47; N, 8.83.
Unsaturated Bicyclo[2.2.2]diazaoctane (42)
A solution of xanthate ester 38 (200 mg, 0.307 mmol) in 350 mL dry benzene was degassed for 15 min with argon. 8 µL (30 µmol) tributylstannane and 60 mg (0.368 mmol) AIBN were added and the mixture was immersed in a preheated oil bath. When the solvent began to reflux, a mixture of 8 µL (30 µmol) tributylstannane and 24 mg (0.150 mmol) AIBN in 50 mL benzene was added slowly over 1.5 h. The reaction was cooled after 2 h and evaporated. The residue was purified by chromatography on silica gel (CH2Cl2/acetone 10:1 to CH2Cl2/MeOH 20:1) yielding 101 mg (61%) of the cyclized compound 42 as colourless glass. IR (NaCl, film): 3324, 2933, 2836, 1691, 1673, 1616, 1513, 1500, 1462, 1378, 1344, 1266, 1247, 1176, 1118, 1104, 1036, 982, 901, 809, 733 cm−1. 1H NMR (500 MHz, CDCl3): δ 7.25 (d, J = 8.8 Hz, 2H), 6.87 (d, J = 8.2 Hz, 1H), 6.80 (d, J = 8.8 Hz, 2H), 6.60 (br, 1H), 5.53 (d, J = 8.2 Hz, 1H), 5.96 (ddd, J = 10.0 Hz, J = 4.7 Hz, J = 2.6 Hz, 1H), 5.76 (ddd, J = 10.0 Hz, J = 2.4 Hz, J = 1.6 Hz, 1H), 5.07 (d, J = 14.9 Hz, 1H), 4.98 (d, J = 14.9 Hz, 1H), 3.86 (s, 3H), 3.76 (s, 3H), 3.71 (br d, J = 10.6 Hz, 1H), 3.60 (s, 3H), 3.25 (ddd, J = 10.5 Hz, J = 10.4 Hz, J = 1.6 Hz, 1H), 2.72 (dd, J = 8.4 Hz, J = 3.6 Hz, 2H), 2.64 (dd, J = 10.8 Hz, J = 1.7 Hz, 1H), 2.42-2.32 (m, 1H), 2.22 (d, J = 15.2 Hz, 1H), 2.11-2.04 (m, 1H), 1.98 (d, J = 15.2 Hz, 1H), 1.85-1.68 (m, 2H), 1.07 (s, 3H), 0.70 (s, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ: 181.6, 175.5, 158.6, 153.5, 135.5, 133.8, 129.8, 128.7, 128.6, 127.6, 127.5, 122.8, 120.5, 113.7, 113.7, 113.6, 104.8, 61.7, 61.6, 61.3, 61.1, 60.8, 55.8, 55.1, 54.1, 49.2, 46.7, 44.6, 40.8, 29.8, 26.4, 24.0, 20.7 ppm. HRMS (EI): Calc’d for C32H37N3O5 + [M]+: 543.2733. Found: 543.2727.
Marcfortine B (2)
Freshly purified m-CPBA (25.6 mg, 0.15 mmol) was dissolved in 1 mL chloroform and cooled to 0 °C. Alkene 44 (20.0 mg, 43 µmol) was dissolved in 1 mL CHCl3 (0.5 mL additional CHCl3 to rinse) and added to the mCPBA solution. The mixture was slowly warmed to room temperature overnight. 1 mL of 10% aq. NaHSO3 was added to reduce the formed N-oxide and the mixture was stirred for 30 min vigorously. The solution was extracted 5 times with 7 mL EtOAc. The combined organic phases were washed twice with 3 mL sat. aq. NaHCO3. The aqueous washes were back extracted twice with 7 mL EtOAc. The combined organic phases were washed with 5 mL brine, dried over Na2SO4 and concentrated in vacuo and used immediately for the next step. Neat tin tetrachloride (60 µL, 512 µmol) was added dropwise to 6 mL dry dioxane. When the turbid solution cleared up, a solution of the crude epoxides in 1.5 mL dry THF was added and the reaction mixture was stirred for 20 min. The reaction was quenched with 10 mL 10% aq. KF and extracted twice with 50 mL EtOAc. The combined organic layers were washed with brine, dried (Na2SO4) and evaporated. The residue was filtered over silica gel (CH2Cl2/MeOH 20:1) and used directly for the next step. A solution of 90 mg (222 µmol) methyltriphenoxyphosphonium iodide (thoroughly washed with dry ether before use) in 1 mL freshly destilled dry DMPU was added to the above cyclised compound. The mixture was stirred under exclusion of light for 24 h at room temperature. After quenching the reaction with 1 mL methanol, 50 mL EtOAc were added and the solution washed with 10 mL sat. aq. Na2S2O3, 3 times with 10 mL water and 10 mL brine. The organic phase was dried (Na2SO4), evaporated and purified by silica gel chromatography (CH2Cl2 to CH2Cl2/MeOH 20:1). The fractions containing 1 were purified further by preparative TLC (CH2Cl2/MeOH: 25:1) and finally crystallized from EtOAc/CH2Cl2 (5:1) to yield 8.4 mg (42% over 3 steps) of synthetic marcfortine B. mp 220 °C (dec.). IR (NaCl, film): 3232, 1702, 1670, 1633, 1592, 1503, 1467, 1382, 1366, 1331, 1289, 1233, 1200, 1146, 1120, 1049 cm−1. 1H NMR (500 MHz, DMSO): δ 10.54 (s, 1H), 8.51 (s, 1H), 6.98 (d, J = 8.1 Hz, 1H), 6.60 (d, J = 8.1 Hz, 1H), 6.35 (d, J = 7.6 Hz, 1H), 4.97 (d, J = 7.6 Hz, 1H), 3.61 (d, J = 11.3 Hz, 1H), 2.81 (t, J = 10.5 Hz, 1H), 2.54 (d, J = 11.7 Hz, 1H), 2.28 (d, J = 14.9 Hz, 1H), 2.23-2.12 (m, 2H), 1.90 (d, J = 12.7 Hz, 1H), 1.85 (d, J = 14.9 Hz, 1H), 1.72-1.60 (m, 1H), 1.54 (dd, J = 12.4, 11.3 Hz, 1H), 1.48-1.39 (m, 3H), 1.36 (s, 3H), 1.34 (s, 3H), 1.23-1.13 (m, 1H), 0.96 (s, 3H), 0.69 (s, 3H) ppm. 13C NMR (125 MHz, DMSO): δ 182.2, 173.9, 145.6, 138.9, 135.0, 133.5, 125.8, 120.9, 116.1, 115.1, 79.2, 61.9, 61.0, 59.8, 59.7, 53.3, 45.3, 38.3, 31.0, 30.7, 29.5, 29.1, 25.4, 24.1, 20.5, 20.3 ppm.
1-(methoxymethyl)-7,7-dimethyl-3-(propan-2-ylidene)-3,7-dihydropyrano[2,3-g]indol-2(1H)-one (76)
To a well-stirred suspension of sodium hydride (970 mg, 60% in mineral oil, 24.3 mmol) in THF (24 mL) was added a suspension of N-H oxindole 61 (4.16 g, 16.3 mmol) in THF/DMF (1:1, 24 mL) dropwise over 5–10 minutes. The vial initially containing oxindole was washed with THF/DMF (1:1, 6 mL), and the wash was added by cannula to the sodium suspension. After 5 minutes, chloromethyl methyl ether (1.83 mL, 24.1 mmol) was added dropwise over 3–4 minutes. The resultant yellow suspension was stirred at room temperature for 4 hours before it was quenched with 1 M NaHSO4 (120 mL) and extracted with ethyl acetate (3 × 120 mL). The organic layer was washed with brine (120 mL), dried over MgSO4, concentrated and purified by chromatography (7% ethyl acetate in hexanes) to yield the product as a pale yellow solid (3.39 g, 69% yield), mp 112–116 °C (recrystallized from ethyl acetate in hexanes). 1H NMR (400 MHz, CDCl3): δ 7.34 (d, J = 8.3 Hz, 1H), 7.00 (d, J = 9.9 Hz, 1H), 6.56 (d, J = 8.3 Hz, 1H), 5.70 (d, J = 9.90 Hz, 1H), 5.22 (s, 2H), 3.42 (s, 3H), 2.59 (s, 3H), 2.34 (s, 3H), 1.44 (s, 6H). 13C NMR (100 MHz, CDCl3): δ 169.9, 153.7, 152.9, 137.2, 131.2, 124.1, 121.9, 118.5, 117.9, 110.6, 107.2, 75.4, 72.1, 56.3, 27.7, 25.7, 23.8. IR (thin film): 2980, 1704, 1633, 1601, 1447 cm−1. HRMS: calcd for (M+Na+) C18H21NO3Na 322.1414; found 322.1405.
(1R,3R)-1'-(methoxymethyl)-2,2,7',7'-tetramethyl-4-methylene-2'-oxo-2',7'-dihydro-1'H-spiro[cyclopentane-1,3'-pyrano[2,3-g]indole]-3-carbonitrile (77)
To an argon-purged vial of oxindole 76 (299 mg, 1.0 mmol), ligand L10 (10.5 mg, 0.02 mmol) and Pd(dba)2 (5.8 mg, 0.01 mmol) was added toluene (1.7 mL) and the solution was stirred for 2–3 minutes at room temperature before cooling to 4 °C. 1-Cyano-2-((trimethylsilyl)methyl)allyl acetate (340 mg, 355 µL, 1.6 mmol) was added and the reaction was stirred for 18 hours at 4 °C, then purified directly by flash chromatography (7% ethyl acetate in hexanes) to yield the product as a white foam (365.4 mg, 97% yield; dr 10:1 as detected by 1H NMR resonances assigned to the methyl groups at δ 0.92 (major) and δ 0.82 (minor) ppm; >99% ee and 92% ee, respectively). 1H NMR (major diastereomer only, 400 MHz, CDCl3): δ 6.98 (d, J = 7.8 Hz, 1H), 6.93 (d, J = 9.8 Hz, 1H), 6.57 (d, J = 7.8 Hz, 1H), 5.72 (d, J = 9.8 Hz, 1H), 5.48 (s, 1H), 5.25-5.21 (m, 2H), 5.02 (d, J = 11.7 Hz, 1H), 4.59 (s, 1H), 3.41 (s, 3H), 2.80 (s, 2H), 1.47 (s, 3H), 1.41 (s, 3H), 1.24 (s, 3H), 0.97 (s, 3H). 13C NMR (major diastereomer only, 100 MHz, CDCl3): δ 182.5, 154.4, 142.4, 139.1, 131.6, 125.6, 119.8, 119.0, 117.9, 111.6, 111.2, 107.9, 75.5, 72.5, 58.0, 56.8, 48.4, 45.9, 39.1, 28.2, 27.4, 22.3, 21.4. IR (thin film): 2974, 2230, 1713, 1635, 1608, 1461 cm−1. [α]24D = −67.0 (c 1.15, CHCl3). Chiral HPLC: Chiralpak OD-H, 0.8 mL/min, 1% i-PrOH in heptane, λ = 254 nm; MAJOR: tR, major = 14.7 min, tR, minor = 17.5 min; MINOR: tR, minor = 21.8 min, tR, major = 29.3 min. HRMS: calcd for (M+Na+) C23H26N2O3Na 401.1836; found 401.1820.
(R)-4-(hydroxymethyl)-1'-(methoxymethyl)-2,2,7',7'-tetramethyl-2'-oxo-2',7'-dihydro-1'H-spiro[cyclopent[3]ene-1,3'-pyrano[2,3-g]indole]-3-carbonitrile (78e)
i) To an argon-purged vial of oxindole 76 (1.0 g, 3.34 mmol), ligand L10 (35.0 mg, 0.067 mmol) and Pd(dba)2 (19.2 mg, 0.033 mmol) was added toluene (8 mL) and the solution was stirred for 2–3 minutes at room temperature before cooling to 4 °C. 1-Cyano-2-((trimethylsilyl)methyl)allyl acetate (1.06 g, 5.0 mmol) was added and the reaction was stirred for 18 hours at 4 °C, then purified directly by flash chromatography (7% ethyl acetate in hexanes) to yield the product as a white foam (approximately 1.3 g). ii) This foam was dissolved in dry THF (24 mL) and cooled to −78 °C. To this flask was then added 7.8 mL of a 0.30 M LiOt-Bu/0.60 M n-BuLi solution (2.34 mmol and 4.7 mmol, respectively) in THF. The resultant yellow solution was stirred at −78 °C for 45 minutes, and then a −78 °C solution of oxaziridine (1.83 g, 7.0 mmol) in THF (8 mL) was added as quickly as possible using a thick cannula. After 30 minutes at −78 °C, the reaction was warmed to room temperature over 30 minutes, quenched with 1 M NaHSO4 (50 mL) and extracted with ethyl acetate (100 mL). The organic phase was washed with sat. NaHCO3 (2 × 50 mL) and brine (50 mL), dried over MgSO4, concentrated and purified by flash chromatography (40% ethyl acetate in hexanes) to yield the product as a faint yellow foam (785 mg, 60% yield over 2 steps, 89% ee). The product is sometimes contaminated by a small amount of benzenesulfonamide, which can be removed by washing an ethyl acetate solution with 2–3 portions of 1 M NaOH. 1H NMR (400 MHz, CDCl3): δ 7.01 (d, J = 8.3 Hz, 1H), 6.92 (d, J = 10.2 Hz, 1H), 6.55 (d, J = 8.3 Hz, 1H), 5.72 (d, J = 10.2 Hz, 1H), 5.26 (d, J = 11.0 Hz, 1H), 5.00 (d, J = 11.0 Hz, 1H), 4.52 (d, J = 14.9 Hz, 1H), 4.45 (d, J = 14.9 Hz, 1H), 3.39 (s, 3H), 2.99 (d, J = 18.1 Hz, 1H), 2.88 (d, J = 18.1 Hz, 1H), 1.48 (s, 3H), 1.40 (s, 3H), 1.19 (s, 3H), 1.12 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 180.9, 158.9, 154.3, 138.5, 131.6, 124.9, 121.2, 117.9, 117.6, 115.0, 111.3, 107.9, 75.5, 72.5, 60.6, 58.7, 56.6, 53.0, 41.8, 28.2, 27.2, 25.1, 22.6. IR (thin film): 3425 (br), 2969, 2933, 2216, 1722, 1609, 1461 cm−1. [α]23D = −41.0 (c 0.88, CHCl3, 89% ee). Chiral HPLC: Chiralpak IA, 0.8 mL/min, 5% i-PrOH in heptane, λ = 254 nm; tR, minor = 16.6 min, tR, major = 20.2 min. HRMS: calcd for (M+Na+) C23H26N2O4Na 417.1785; found 417.1779.
Michael Adduct (81)
A solution of unsaturated nitrile 80 (376 mg, 0.75 mmol, concentrated thrice from toluene (4 mL) in THF (24 mL) was cooled to 0 °C. Lithium tert-butoxide solution (1.82 mL, 0.41 M in 4.5:1 THF/hexanes, 0.75 mmol) was added by syringe pump over 60 minutes, and the resulting solution was stirred at 0 °C for an addition 2 hours. It was then concentrated and purified by flash chromatography (70:1 dichloromethane/methanol) to yield the product as a glassy, colorless film (267 mg, 71% yield). 1H NMR (400 MHz, CDCl3): δ 7.07 (bs, 1H), 6.93 (d, J = 7.9 Hz, 1H), 6.90 (d, J = 9.9 Hz, 1H), 6.57 (d, J = 7.9 Hz, 1H), 6.05 (t, J = 3.9 Hz, 1H), 5.73 (d, J = 9.9 Hz, 1H), 5.23 (d, J = 11.4 Hz, 1H), 5.03 (d, J = 11.4 Hz, 1H), 3.91 (s, 1H), 3.41 (s, 3H), 3.23 (d, J = 11.5 Hz, 1H), 3.15-3.08 (m, 2H), 3.00-2.94 (m, 1H), 2.48 (d, J = 15.1 Hz, 1H), 2.24-2.17 (m, 2H), 2.12 (d, J = 15.1 Hz, 1H), 2.02-1.81 (m, 2H), 1.47 (s, 3H), 1.41 (s, 3H), 1.32 (s, 3H), 0.96 (s, 3H). 13C NMR (125 MHz, CD2Cl2): δ 183.2, 160.9, 154.9, 139.4, 136.5, 132.1, 125.7, 118.8, 117.9, 117.8, 111.4, 109.3, 108.4, 75.8, 72.9, 60.7, 59.5, 58.7, 56.9, 51.4, 50.5, 49.3, 47.0, 28.0, 27.2, 23.0, 22.7, 21.8. IR (thin film): 3361, 2933, 1702, 1674, 1629, 1451 cm−1. [α]25D = −9.0 (c 0.94, CH2Cl2, 89% ee). HRMS: calcd for (M+H+) C29H35N4O4 503.2653; found 503.2647.
Primary alcohol (83)
1) To a solution of nitrile 81 (210 mg, 0.42 mmol, dried from 3 × 4 mL benzene) in dichloromethane (10.5 mL) at 0 °C was added triethylaluminum (0.42 mL, 1 M in heptane, 0..42 mmol) and the pale pink solution was stirred for 90 minutes at 0 °C. It was then cooled to −78 °C and DIBAL (1.04 mL, 1.2 M in toluene, 1.04 mmol) was added. The solution was stirred for 60 minutes at −78 °C and quenched by the addition of methanol (125 µL). The cooling bath was removed, sat. aq. Rochelle’s salt (10 mL) was added, and the mixture was stirred for 1 hour before it was extracted with ethyl acetate (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over MgSO4, concentrated and purified by chromatography (2% to 3% methanol in dichloromethane) to yield a mixture of the nitrile and the desired aldehyde that was carried on directly to the next step. 1H NMR (aldehyde only, 600 MHz, CDCl3): δ 9.85 (d, J = 4.0 Hz, 1H), 7.08 (bs, 1H), 6.92 (d, J = 7.6 Hz, 1H), 6.89 (d, J = 8.2 Hz, 1H), 6.55 (d, J = 8.2 Hz, 1H), 6.08 (t, J = 4.3 Hz, 1H), 5.71 (d, J = 7.6 Hz, 1H), 5.23 (d, J = 11.0 Hz, 1H), 5.01 (d, J = 11.0 Hz, 1H), 3.62 (d, J = 4.0 Hz, 1H), 3.41 (s, 3H), 3.28-3.09 (m, 4H), 2.45 (d, J = 14.8 Hz, 1H), 2.23-2.16 (m, 2H), 2.07 (d, J = 14.8 Hz, 1H), 1.99-1.83 (m, 2H), 1.47 (s, 3H), 1.41 (s, 3H), 1.30 (s, 3H), 0.85 (s, 3H).
2) The above mixture was dissolved in dichloromethane and methanol (30 mL, 4:1) and cooled to 0 °C. Sodium borohydride (79.0 mg, 2.09 mmol) was added and the reaction was stirred for 4 hours at 0 °C. It was then carefully quenched by the addition of 1 M NaHSO4 (2 mL) and extracted with ethyl acetate (2 × 100 mL). The combined organic layers were dried over MgSO4, concentrated and purified by chromatography (2.0% to 3.5% methanol in dichloromethane) to yield recovered starting material (44 mg) followed by the desired product (98 mg, 46% yield, 58% yield brsm) as a white film. 1H NMR (400 MHz, CDCl3): δ 7.60 (bs, 1H), 6.93 (d, J = 9.9 Hz, 1H), 6.81 (d, J = 8.1 Hz, 1H), 6.53 (d, J = 8.1 Hz, 1H), 6.27 (t, J = 4.3 Hz, 1H), 5.71 (d, J = 9.9 Hz, 1H), 5.26 (d, J = 10.3 Hz, 1H), 5.01 (d, J = 10.3 Hz, 1H), 4.19 (d, J = 10.5 Hz, 1H), 3.87 (t, J = 11.5 Hz, 1H), 3.68- 3.60 (m, 1H), 3.42 (s, 3H), 3.28 (d, J = 12.0 Hz, 1H), 3.25-3.19 (m, 1H), 3.08 (dd, J = 3.4, 10.5 Hz, 1H), 2.94 (d, J = 11.5 Hz, 1H), 2.77 (dt, J = 3.7, 11.3 Hz, 1H), 2.27-2.20 (m, 3H), 2.04 (d, J = 14.6 Hz, 1H), 1.97-1.89 (m, 2H), 1.47 (s, 3H), 1.40 (s, 3H), 0.90 (s, 3H), 0.85 (s, 3H). 13C NMR (100 MHz, C6D6): δ 183.4, 160.3, 154.4, 139.8, 137.4, 130.9, 125.7, 120.4, 118.5, 110.8, 110.7, 107.8, 75.1, 72.4, 61.2, 60.1, 59.7, 58.8, 57.7, 56.4, 50.8, 48.3, 48.1, 28.1, 27.0, 23.7, 22.8, 22.2, 20.6. IR (thin film): 3353, 2971, 2934, 1703, 1668, 1626 cm−1. [α]24D = −14.8 (c 0.96, CH2Cl2, 89% ee). HRMS: calcd for (M+H+) C29H38N3O5 508.2806; found 508.2802.
Unsaturated bicyclo[2.2.2]diazaoctane (85)
In a round-bottom flask equipped with a reflux condenser, a solution of xanthate ester 84 (178 mg, 0.3 mmol) in dry benzene (300 mL) was sparged with argon for 15 minutes. A solution of AIBN (117.3 mg, 0.71 mmol), n-Bu3SnH (8.1 µL, 0.03 mmol) and N,O-bis(trimethylsilyl)acetamide (0.73 mL, 3.0 mmol) in benzene (5 mL) was then added and the flask was immersed in a preheated 90 °C oil bath. Once reflux was observed, a solution of AIBN (49.3 mg, 0.3 mmol) and n-Bu3SnH (8.1 µL, 0.03 mmol) in benzene (25 mL) was added by syringe pump over 90 minutes. After an additional 30 minutes at reflux, the reaction was cooled to room temperature, concentrated, and held under high vacuum (~100–200 mtorr) in order to remove excess BSA. It was then purified by flash chromatography (4% methanol in dichloromethane) to yield the product as a clear, colorless film (79 mg, 54% yield). 1H NMR (400 MHz, CDCl3): δ 6.95 (d, J = 8.0 Hz, 1H), 6.94 (d, J = 9.9 Hz, 1H), 6.93 (bs, 1H), 6.55 (d, J = 8.0 Hz, 1H), 5.99-5.94 (m, 1H), 5.74-5.70 (m, 1H), 5.70 (d, J = 9.9 Hz, 1H), 5.27 (d, J = 11.2 Hz, 1H), 4.95 (d, J = 11.2 Hz, 1H), 3.72 (d, J = 10.9 Hz, 1H), 3.39 (s, 3H), 3.22-3.15 (m, 1H), 2.75-2.70 (m, 2H), 2.65 (d, J = 10.9 Hz, 1H), 2.43-2.33 (m, 1H), 2.25 (d, J = 14.6 Hz, 1H), 2.08 (bd, J = 17.7 Hz, 1H), 1.99 (d, J = 14.6 Hz, 1H), 1.89-1.76 (m, 2H), 1.48 (s, 3H), 1.40 (s, 3H), 1.08 (s, 3H), 0.81 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 183.6, 175.9, 154.0, 139.2, 131.4, 128.1, 127.8, 125.5, 121.7, 118.1, 110.8, 107.7, 75.5, 72.8, 61.9, 61.9, 61.5, 61.1, 56.7, 54.7, 49.6, 46.9, 41.0, 30.2, 28.3, 27.2, 26.8, 24.7, 21.0. IR (thin film): 2971, 2928, 1713, 1674, 1636, 1461 cm−1. [α]24D = −71.4 (c 1.04, CHCl3). HRMS: calcd for (M+H+) C29H26N3O4 490.2700; found 490.2702.
Marcfortine C (10)
To a solution of N-MOM oxindole in dimethoxyethane (3.7 mL) was added conc. HCl (0.7 mL) and the biphasic mixture was stirred for 5 hours at 45 °C. The reaction was then cooled to room temperature, quenched with sat. NaHCO3 (5 mL) and extracted with ethyl acetate (10 mL). The organic layer was washed with NaHCO3 (5 mL), water (5 mL) and brine (5 mL), dried over MgSO4, concentrated and purified by chromatography (8% methanol in dichloromethane) to yield the product as a white solid (10.9 mg, 74% yield), mp >250 °C (lit. mp 264–267 °C)1b. Spectral properties matched known characterization.1b,17a 1H NMR (600 MHz, CDCl3): δ 9.02 (bs, 1H), 6.89 (d, J = 8.2 Hz, 1H), 6.47 (bs, 1H), 6.44 (d, J = 8.2 Hz, 1H), 6.39 (d, J = 9.8 Hz, 1H), 5.81 (d, J = 9.8 Hz, 1H), 3.70 (d, J = 11.7 Hz, 1H), 3.08 (t, J = 10.7 Hz, 1H), 2.68 (br d, J = 10.5 Hz, 1H), 2.48-2.42 (m, 2H), 2.15 (d, J = 15.1 Hz, 1H), 2.14 (br d, J = 13.0 Hz, 1H), 1.96 (d, J = 15.1 Hz, 1H), 1.55–1.85 (m, 7H), 1.46 (s, 3H), 1.44 (s, 3H), 1.09 (s, 3H), 0.83 (s, 3H). 13C NMR (125 MHz, CDCl3): δ 185.2, 177.2, 153.4, 137.8, 131.6, 125.9, 121.4, 116.5, 110.0, 105.8, 76.7, 62.8, 61.3, 61.1, 61.0, 54.9, 54.5, 46.7, 40.3, 31.3, 30.9, 28.3, 28.2, 25.7, 24.1, 20.9, 20.8. IR (thin film): 3251 (br), 2931, 1698, 1644 cm−1. [α]23D = −68.0 (c 1.10, CHCl3, 89% ee) (lit. [α]22D = −64.4 (c 1.10, CHCl3)). HRMS: calcd for (M+H+) C27H34N3O3 448.2600; found 448.2588.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the NSF and the NIH (GM 033049) for their generous support of our programs. Additional financial support was provided by the W.S. Johnson Graduate Fellowship (D.A.B.) and a Feodor-Lynen fellowship of the Alexander von Humboldt Foundation (N.C.). Palladium salts were a generous gift from Johnson Matthey.
Footnotes
ASSOCIATED CONTENT
Experimental details and spectral data for all unknown compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Polonsky J, Merrien M-A, Prange T, Pascard C. J. Chem. Soc., Chem. Commun. 1980:601. [Google Scholar]; (b) Prange T, Buillion M-A, Vuilhorgne M, Pascard C, Polonsky J. Tetrahedron Lett. 1981;22:1977. [Google Scholar]
- 2.(a) Yamazaki M, Okuyama E, Kobayashi M, Inoue H. Tetrahedron Lett. 1981;22:135. [Google Scholar]; (b) Ondeyka JG, Goegelman RT, Schaeffer JM, Kelemen L, Zitano L. J. Antibiot. 1990;43:1375. doi: 10.7164/antibiotics.43.1375. [DOI] [PubMed] [Google Scholar]; (c) Liesch JM, Wichmann CF. J. Antibiot. 1990;43:1380. doi: 10.7164/antibiotics.43.1380. [DOI] [PubMed] [Google Scholar]; (d) Blanchflower SE, Banks RM, Everett JR, Manger BR, Reading C. J. Antibiot. 1991;44:492. doi: 10.7164/antibiotics.44.492. [DOI] [PubMed] [Google Scholar]; (e) Lopez-Gresa MP, Gonzalez MC, Ciavatta L, Ayala I, Moya P, Primo J. J. Agric. Food Chem. 2006;54:2921. doi: 10.1021/jf0530998. [DOI] [PubMed] [Google Scholar]
- 3.For paraherquamide derivatives VM55595, VM55596 and VM55597, see Blanchflower SE, Banks RM, Everett JR, Reading C. J. Antibiot. 1993;46:1355. doi: 10.7164/antibiotics.46.1355.
- 4.For paraherquamide derivatives SB103105 and SB200437, see Banks RM, Blanchflower SE, Everett JR, Manger BR, Reading C. J. Antibiot. 1997;50:840. doi: 10.7164/antibiotics.50.840.
- 5.Whyte AC, Gloer JB, Wicklow DT, Dowd PF. J. Nat. Prod. 1996;59:1093. doi: 10.1021/np960607m. [DOI] [PubMed] [Google Scholar]
- 6.(a) Kato H, Yoshida T, Tokue T, Nojiri Y, Hirota H, Ohta T, Williams RM, Tsukamoto S. Angew. Chem., Int. Ed. 2007;46:2254. doi: 10.1002/anie.200604381. [DOI] [PubMed] [Google Scholar]; (b) Tsukamoto S, Kato H, Samizo M, Nojiri Y, Onuki H, Hirota H, Ohta T. J. Nat. Prod. 2008;71:2064. doi: 10.1021/np800471y. [DOI] [PubMed] [Google Scholar]; (c) Tsukamoto S, Kawabata T, Kato H, Greshock TJ, Hota H, Ohta T, Williams RM. Org. Lett. 2009;11:1297. doi: 10.1021/ol900071c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Birch AJ, Wright JJ. J. Chem. Soc., Chem. Commun. 1969:644. [Google Scholar]; (b) Birch AJ, Wright JJ. Tetrahedron. 1970;26:2329. doi: 10.1016/s0040-4020(01)92812-1. [DOI] [PubMed] [Google Scholar]; (c) Birch AJ, Russell RA. Tetrahedron. 1972;28:2999. [Google Scholar]; (d) Coetzer J. Acta Crystallogr. 1974;B30:2254. [Google Scholar]
- 8. Greshock TJ, Grubbs AW, Jiao P, Wicklow DT, Gloer JB, Williams RM. Angew. Chem., Int. Ed. 2008;47:3573. doi: 10.1002/anie.200800106. (b) Ref 6c
- 9.Lee BH. Stud. Nat. Prod. Chem. 2003;28:331. and references therein. [Google Scholar]
- 10.For full reference, see Supporting Information: Lee BH, et al. Curr. Top. Med. Chem. 2002;2:779. doi: 10.2174/1568026023393705.
- 11.For the biosynthesis of marcfortine A, see Kuo MS, Wiley VH, Cialdella JI, Yurek DA, Whaley HA, Marshall VP. J. Antibiot. 1996;49:1006. doi: 10.7164/antibiotics.49.1006. For studies on the biosynthesis of the paraherquamides, see Summer K, Williams RM. Tetrahedron. 2009;65:3246. doi: 10.1016/j.tet.2008.08.102.
- 12.For recent reviews, see Williams RM, Cox RJ. Acc. Chem. Res. 2003;36:127. doi: 10.1021/ar020229e. Williams RM, Stocking EM, Sanz-Cervera JF. Top. Curr. Chem. 2000;209:98.
- 13.(a) Williams RM, Cao J, Tsujishima H, Cox RJ. J. Am. Chem. Soc. 2003;125:12172. doi: 10.1021/ja036713+. [DOI] [PubMed] [Google Scholar]; (b) Cushing TD, Sanz-Cervera JF, Williams RM. J. Am. Chem. Soc. 1996;118:557. [Google Scholar]
- 14.Stocking EM, Sanz-Cervera JF, Williams RM. Angew. Chem. Int. Ed. 2001;40:1296. doi: 10.1002/1521-3773(20010401)40:7<1296::aid-anie1296>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 15.(a) Williams RM, Sanz-Cervera JF, Sancenon F, Marco JA, Halligan K. J. Am. Chem. Soc. 1998;120:1090. doi: 10.1016/s0968-0896(98)00102-3. [DOI] [PubMed] [Google Scholar]; (b) Williams RM, Sanz-Cervera JF, Sancenon F, Marco JA, Halligan K. Bioorg. Med. Chem. 1998;6:1233. doi: 10.1016/s0968-0896(98)00102-3. [DOI] [PubMed] [Google Scholar]
- 16.Greshock TJ, Grubbs AW, Tsukamoto T, Williams RM. Angew. Chem. Int. Ed. 2007;46:2262. doi: 10.1002/anie.200604378. [DOI] [PubMed] [Google Scholar]
- 17.(a) Greshock TJ, Grubbs AW, Williams RM. Tetrahedron. 2007;63:6124. doi: 10.1016/j.tet.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Greshock TJ, Williams RM. Org. Let. 2007;9:4255. doi: 10.1021/ol701845t. [DOI] [PubMed] [Google Scholar]
- 18.(a) Trost BM, Chan DMT. J. Am. Chem. Soc. 1979;101:6429. [Google Scholar]; (b) Trost BM, Chan DMT. J. Am. Chem. Soc. 1979;101:6432. [Google Scholar]
- 19.Chan DMT. Recent Advances in Palladium-Catalyzed Cycloadditions Involving Trimethylenemethane and its Analogs. In: Kobayashi S, Jorgensen KA, editors. Cycloaddition Reactions in Organic Synthesis. Weinheim, Germany: Wiley-VCH; 2002. pp. 57–83. [Google Scholar]
- 20.Trost BM, Stambuli JP, Silverman SM, Schworer U. J. Am. Chem. Soc. 2006;128:13328. doi: 10.1021/ja0640750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.For the asymmetric synthesis of carbocycles by TMM cycloaddition, see: Trost BM, Cramer N, Silverman SM. J. Am. Chem. Soc. 2007;129:12396. doi: 10.1021/ja075335w. Trost BM, McDougall PJ, Hartmann O, Wathen PT. J. Am. Chem. Soc. 2008;130:14960. doi: 10.1021/ja806979b. Trost BM, Silverman SM, Stambuli JP. J. Am. Chem. Soc. 2011;133:19483. doi: 10.1021/ja207550t. Trost BM, Bringley DA, Seng PS. Org. Lett. 2012;14:234. doi: 10.1021/ol2030179. Trost BM, Lam TM. J. Am. Chem. Soc. 2012;134:11319. doi: 10.1021/ja305717r. Trost BM, Maruniak A. Angew. Chem. Int. Ed. 2013;52:6262. doi: 10.1002/anie.201300275.
- 22.For the asymmetric synthesis of heterocycles by TMM cycloaddition, see: Trost BM, Silverman SM, Stambuli JP. J. Am. Chem. Soc. 2007;129:12398. doi: 10.1021/ja0753389. Trost BM, Silverman SM. J. Am. Chem. Soc. 2010;132:8238. doi: 10.1021/ja102102d. Trost BM, Bringley DA, Silverman SM. J. Am. Chem. Soc. 2011;133:7664. doi: 10.1021/ja201181g. Trost BM, Silverman SM. J. Am. Chem. Soc. 2012;134:4941. doi: 10.1021/ja210981a. Trost BM, Lam TM, Herbage MA. J. Am. Chem. Soc. 2013;135:2459. doi: 10.1021/ja312351s. Trost BM, Bringley DA. Angew. Chem. Int. Ed. 2013;52:4466. doi: 10.1002/anie.201300616.
- 23.For selected examples in racemic syntheses, see: Trost BM, Renaut P. J. Am. Chem. Soc. 1982;104:6668. Trost BM, Lynch J, Renaut P, Steinman DH. J. Am. Chem. Soc. 1986;108:284. Ikeda M, Okano M, Kosaka K, Kido M, Ishibashi H. Chem. Pharm. Bull. 1993;41:276. Trost BM, Parquette JR. J. Am. Chem. Soc. 1994;59:7568. Trost BM, Higuchi RI. J. Am. Chem. Soc. 1996;118:10094. Jacobsen MF, Moses JE, Adlington RM, Baldwin JE. Tetrahedron. 2006;62:1672.
- 24.Trost BM, Cramer N, Bernsmann H. J. Am. Chem. Soc. 2007;129:3086. doi: 10.1021/ja070142u. [DOI] [PubMed] [Google Scholar]
- 25.Williams RM, Cushing TD. Tetrahedron Lett. 1990;31:6325–6328. [Google Scholar]
- 26.(a) Trost BM, Mignani SM, Nanninga TN. J. Am. Chem. Soc. 1988;110:1602. doi: 10.1021/ja00279a070. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Mignani SM, Nanninga TN. J. Am. Chem. Soc. 1986;108:6051. doi: 10.1021/ja00279a070. [DOI] [PubMed] [Google Scholar]
- 27.Savall BM, McWhorter WW, Walker EA. J. Org. Chem. 1996;61:8696. [Google Scholar]
- 28.Frank R, Schutkowsky M. J. Chem. Soc., Chem. Commun. 1996:2509. [Google Scholar]
- 29.Crabtree R. Acc. Chem. Res. 1979;12:331. [Google Scholar]
- 30.Artman GD, III, Grubbs AW, Williams RM. J. Am. Chem. Soc. 2007;129:6336. doi: 10.1021/ja070259i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pastine SJ, Youn SW, Sames D. Tetrahedron. 2003;59:8859. [Google Scholar]
- 32.See Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.