

Abstract
The therapeutic armamentarium for autoimmune diseases of the central nervous system, specifically multiple sclerosis and neuromyelitis optica, is steadily increasing, with a large spectrum of immunomodulatory and immunosuppressive agents targeting different mechanisms of the immune system. However, increasingly efficacious treatment options also entail higher potential for severe adverse drug reactions. Especially in cases failing first-line treatment, thorough evaluation of the risk–benefit profile of treatment alternatives is necessary. This argues for the need of algorithms to identify patients more likely to benefit from a specific treatment. Moreover, paradigms to stratify the risk for severe adverse drug reactions need to be established. In addition to clinical/paraclinical measures, biomarkers may aid in individualized risk–benefit assessment. A recent example is the routine testing for anti-John Cunningham virus antibodies in natalizumab-treated multiple sclerosis patients to assess the risk for the development of progressive multi-focal leucoencephalopathy. Refined algorithms for individualized risk assessment may also facilitate early initiation of induction treatment schemes in patient groups with high disease activity rather than classical escalation concepts. In this review, we will discuss approaches for individiualized risk–benefit assessment both for newly introduced agents as well as medications with established side-effect profiles. In addition to clinical parameters, we will also focus on biomarkers that may assist in patient selection.
Other Articles published in this series
Paraneoplastic neurological syndromes. Clinical and Experimental Immunology 2014, 175: 336–48.
Disease-modifying therapy in multiple sclerosis and chronic inflammatory demyelinating polyradiculoneuropathy: common and divergent current and future strategies. Clinical and Experimental Immunology 2014, 175: 359–72.
Monoclonal antibodies in treatment of multiple sclerosis. Clinical and Experimental Immunology 2014, 175: 373–84.
CLIPPERS: chronic lymphocytic inflammation with pontine perivascular enhancement responsive to steroids. Review of an increasingly recognized entity within the spectrum of inflammatory central nervous system disorders. Clinical and Experimental Immunology 2014, 175: 385–96.
Requirement for safety monitoring for approved multiple sclerosis therapies: an overview. Clinical and Experimental Immunology 2014, 175: 397–407.
Myasthenia gravis: an update for the clinician. Clinical and Experimental Immunology 2014, 175: 408–18.
Cerebral vasculitis in adults: what are the steps in order to establish the diagnosis? Red flags and pitfalls. Clinical and Experimental Immunology 2014, 175: 419–24.
Multiple sclerosis treatment and infectious issues: update 2013. Clinical and Experimental Immunology 2014, 175: 425–38.
Diagnosis, pathogenesis and treatment of myositis: recent advances 2014, 175: 349–58.
Neuromyelitis optica: clinical features, immunopathogenesis and treatment 2014, 176: 149–64.
Keywords: monoclonal antibodies, multiple sclerosis, neuromyelitis optica, progressive multi-focal leukoencephalopathy
Introduction
Multiple sclerosis (MS) and neuromyelitis optica (NMO) are two distinct chronic progressive inflammatory diseases of the central nervous system (CNS) with different pathophysiology and epidemiology. Both are commonly associated with disability, impairment in quality of life, decreased work capacity and high socioeconomic burden 1–4.
The pathophysiology of MS is complex and highly heterogeneous with both inflammatory and neurodegenerative features 5, resulting in various phenotypes and disease courses.
In contrast, the discovery of aquaporin-4 immunoglobulin (Ig)G as an autoantibody with pathogenetic relevance for NMO 6,7 had a direct impact on therapeutic approaches.
As most immunotherapies in neuroimmunology have been studied in MS 8–22 and – to a lesser extent – in NMO 23–27, this review focuses on disease-modifying drugs (DMDs) for these autoimmune CNS entities. Treatment options for other neuroimmunological diseases of the central or peripheral nervous system and neuromuscular disorders such as neuro-sarcoidosis 28,29, myasthenia gravis 30 or chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) 31 have been reviewed in 32,33.
Whereas first-line agents used in MS such as interferons and glatirameracetate exhibit moderate efficacy, we have witnessed several decades of use with highly favourable safety profiles 34. In contrast, newer agents have surprised us with unexpected and sometimes even severe adverse drug reactions (SADR) or unanticipated high frequency of SADRs (Table 1) 35–37.
Table 1.
Drugs and reported (severe) adverse reactions.
| Drug | Reported adverse reactions |
|---|---|
| Alemtuzumab | Leucopenia 71, infusion reactions, infections (herpes viruses, tuberculosis), immune-thrombopenia, thyreoiditis, nephritis (Goodpasture syndrome), cancer (thyroid, colon, vulvar) 10–12 |
| Daclizumab | Elevated liver enzymes, cutaneous symptoms including severe dermatitis, infections 14,22 |
| Dimethylfumarate | Gastrointestinal symptoms, flushing, lymphopenia, proteinuria, pruritus 123,124 |
| Eculizumab | Meningococcal infection and sepsis 27, other infections, infusion reactions, pain (headache, arthralgia, myalgia, back, neck), leucopenia, thrombopenia, anaphylactic and cutaneous reactions, dizziness and vertigo, hypertension, oedema, gastrointestinal symptoms, paraesthesia 161 |
| Fingolimod | Elevated liver enzymes, gastrointestinal symptoms, viral infections (herpes viruses, influenza), other infections (upper respiratory tract), cardiac arrhythmia, macula oedema, dermal malignancy, lymphopenia, pain (head, back) 9,18, respiratory effects 105, haemophagocytic syndrome |
| Mitoxantrone | Elevated liver enzymes 162, gastrointestinal symptoms, transient alopecia 130,163, infections 145, neutro-/eosinophilia 164,165, systemic/cutaneous symptoms 164,165, teratogenicity, transient/permanent infertility 162, amenorrhoea 130,166, anaemia/leucopenia/thrombopenia 37,162, cardiotoxicity 36,143,150,153,155,167, TRAL 36,37,137,138,142,147–149,159,168 |
| Natalizumab | Elevated liver enzymes, gastrointestinal and cutaneous symptoms, pain (muscular, headache, arthralgia), dysmenorrhoea, infections, infusion reactions, hypersensitivity 169, neutralizing antibodies 170, PML 35,45,169, melanoma 171 |
| Rituximab/CD20-antibodies | Infusion reactions, hypotension, anaphylactic and cutaneous reactions, cytokine release, acute respiratory distress syndrome, cardiac events, infections (bacterial, viral) 102, PML 100 |
| Teriflunomide | Elevated liver enzymes, gastrointestinal symptoms, alopecia, neutro-/lymphopenia, infections (urinary tract, pyelonephritis, nasopharynx), pain (back, arthralgia), paraesthesia 116,117 |
| Tocilizumab | Elevated liver enzymes, increased cholesterol levels, gastrointestinal and cutaneous symptoms, headache, hypertension, infusion/injection site reactions, hypersensitivity, infections (respiratory tract, pneumonia), neutropenia, thrombopenia, neutralizing antibodies 172–174 |
PML = progressive multifocal leucoencephalopathy; TRAL = therapy-related acute leukaemia.
Due to the hypothesized selective mechanisms of action, fewer side effects were anticipated for different therapeutic monoclonal antibodies (mAB) coined initially as ‘magic bullets’ 38. Rare but occasionally fatal adverse drug reactions have evolved; however, their pathophysiology is still not well explained. Based on potential SADRs, approval for substances such as natalizumab (NAT), mitoxantrone (MX) and – at least in some countries – fingolimod (FTY) was restricted to patients refractory to first-line MS treatment options or with highly aggressive disease course; but labelling is different from the formal inclusion criteria of respective clinical trials. In addition, restriction to escalation therapy may carry the risk of omission bias, i.e. the decision not to treat patients with potential high benefit in order not to put them actively at risk for SADRs. In the face of newly introduced highly efficacious treatment options, strategies are thus needed that allow patient selection and counselling based on individualized safety and efficacy considerations. Selected patient groups at risk of rapidly developing high disability may particularly benefit from a ‘hit hard and (relatively) early’ treatment strategy.
Optimization of the benefit-to-risk ratio for individual substances can be achieved on multiple levels, including (a) patient selection according to clinical/paraclinical criteria, (b) optimization of treatment and monitoring protocols, (c) identification of patients at higher risk for SADRs and (d) the development of biomarkers for treatment response and/or risk profile (Fig. 1).
Figure 1.
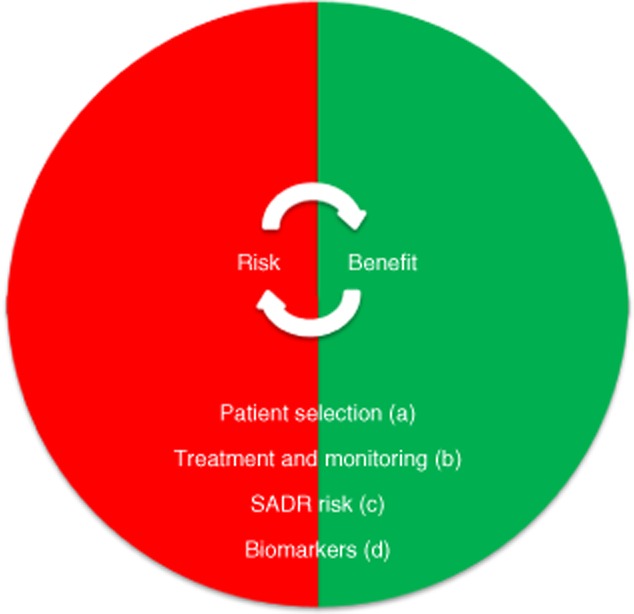
Factors of risk–benefit assessment.
In the following we will discuss these aspects, focusing on treatment of MS and NMO with mAbs (NAT, alemtuzumab, daclizumab and others), FTY, teriflunomide, dimethylfumarate (DMF) and MX.
Monoclonal antibodies
Natalizumab
Patient selection
The alpha-4-integrin-inhibitor natalizumab (Tysabri®) 39 was approved by the Food and Drug Administration (FDA) and European Medicines Agency (EMA) in 2005/06 for the treatment of highly active forms of the relapsing–remitting disease course (RRMS), but not chronic progressive forms [primary or secondary progressive MS (PPMS, SPMS)]. Efficacy in SPMS is under investigation in a Phase IIIb study, ASCEND in SPMS (A Clinical Study of the Efficacy of Natalizumab on Reducing Disability Progression in Subjects With SPMS; ClinicalTrials.gov NCT01416181). Therapeutic efficacy has also been reported in paediatric cohorts with high disease activity 40,41.
In NMO, the use of NAT should be avoided, as current data suggest negative effects on relapse rate and disease progression as well as severe astrocyte damage in spite of natalizumab treatment 42,43.
Treatment and monitoring
Monthly NAT administration is standard treatment. So far, there are only few data on the prolongation of infusion intervals 44. The REFINE trial (Exploratory Study of the Safety, Tolerability and Efficacy of Multiple Regimens of Natalizumab in Adult Subjects With Relapsing Multiple Sclerosis (MS); ClinicalTrials.gov NCT01405820) is investigating both different dosing schemes and application routes [intravenous (i.v.), subcutaneous (s.c.)]; thus far, this approach cannot be recommended outside clinical trials.
Safety considerations and monitoring were profoundly influenced by the occurrence of progressive multi-focal leucoencephalopathy (PML). This is a relatively rare but potentially fatal (22%) opportunistic viral infection of the CNS which can result in severe disability in 40% of the patients 45. Epidemiological data on the frequency of NAT-associated PML has shown an increase of PML incidence after a treatment duration of 2 years (i.e. 24 infusions) 45. Thus, therapy continuation for more than 24 infusions requires updated documented informed consent 46 and re-evaluation of the individual risk–benefit ratio. In addition, adequate counselling of patients and relatives is crucial for the early recognition of symptoms and signs of possible PML, as neuropsychological symptoms may prevail initially.
Regular clinical monitoring and magnetic resonance imaging (MRI) are required to detect symptoms suggestive of PML or suspicious lesions 47. More frequent MRI scanning should be performed in high-risk patient groups (e.g. 3-monthly after a treatment duration of > 24 months).
SADR risk
Pathogenesis of PML – the most feared potential SADR of NAT – is multi-factorial, comprising cellular immunity of the host 48, reactivation of latent John Cunningham virus (JCV) infection or new infection combined with genetic variation of the virus. Both viral and host factors predisposing for PML development are under investigation. The differentiation between virulent and non-virulent JCV variants may be helpful, but relies on viraemia 49 and so far is not sufficiently validated.
Epidemiological risk factors for PML development are previous use of immunosuppressants, a positive anti-JCV antibody status and treatment duration 45,50–52. Hence, the estimated PML incidence ranges from ≤ 0·09/1000 to 11·1/1000 45. A total of 418 NAT-PML cases have been reported (as of November 2013 53).
PML must be suspected when new neurological symptoms occur in individuals on NAT therapy. In particular, neuropsychological symptoms and seizures are highly suspicious, whereas spinal or optic nerve symptoms are uncommon. Its diagnosis is based on clinical findings, MRI 47 and the detection of JCV DNA in cerebrospinal fluid (CSF) 35,54, although there are JCV DNA-negative NAT–PML reports 55,56. In uncertain cases, biopsy of suspicious lesions has to be discussed.
In the course of PML, immune reconstitution inflammatory syndrome (IRIS) can occur with a mean of about 1 month after NAT removal via plasma exchange 57. This inflammatory reaction directed against JCV can cause additional tissue damage with neurological deterioration after initial improvement after PML diagnosis. NAT and JCV elimination as well as control of IRIS evolution must be covered by PML treatment strategies which comprise plasma exchange, mefloquine, mirtazapine and corticosteroid pulses 35,58. However, due to relatively low patient numbers, none of these treatment options are evidence-based.
Although the outcome of NAT–PML seems to be better than HIV-associated PML 57, it is associated with disability 45,57. Seizures occur in more than 50% of patients 59 and are often linked to the appearance of IRIS, explaining the higher rate than in other PML cases; preventive anti-convulsive therapy may thus be beneficial 59.
Biomarkers
Routine anti-JCV antibody testing is established in clinical practice. However, false negative rates have to be considered for both first-and second-generation anti-JCV antibody testing. There is also a considerable proportion of seroconverters and – possibly linked to fluctuating antibody titres at the detection threshold – patients reverting from seropositive to seronegative 45,52,60,61.
The prevalence of anti-JCV antibodies differs in patient groups according to age and gender 52. Two studies reported antibody titres rather than mere serostatus. In one study, sera from patients who were diagnosed later with PML exhibited higher anti-JCV antibody reactivity than other seropositive patients 52. In five patients from whom sera prior to PML diagnosis were available, antibody titres increased 5–10 months before PML diagnosis 61. Methodological issues such as fluctuating serostatus around assay cut-points 52,61 and false negative rates 60 argue for a refinement of assay procedures with better reproducibility in low-antibody reactivity ranges. Thus, a second-generation enzyme-linked immunosorbent assay (ELISA) with a reported sensitivity of 98% 62 was introduced; however, so far an independent validation is lacking. Using this refined assay, the possible value of antibody reactivity for PML risk stratification was reported recently as abstract. Whereas increased immunoreactivity to JCV prior to PML would be biologically plausible, more data are needed to corroborate these initial findings.
Higher NAT plasma levels have been associated with lower body mass index and a supposedly higher risk for the development of PML, which needs to be further confirmed as a possible biomarker feasible for clinical routine 44.
Host factors promoting PML development include the determination of immunocompetence. It has been shown conclusively that both CD4+ and CD8+ T cells are important in the immune response to JCV and containment of PML 48,63. Investigation of the role of CD4+ T cells has demonstrated a lacking or even anti-inflammatory interleukin (IL)-10 response to JCV in a small number of PML patients 64. Intracellular adenosine triphosphate (ATP) levels as a functional parameter of T cell function were decreased in CD4+ T cells both after long-term NAT treatment and PML of different aetiology 65. However, this assay was confronted with pre-analytical difficulties, so far impeding application in larger validating studies or clinical routine, as shown by analysis of STRATA samples (Natalizumab Re-Initiation of Dosing; ClinicalTrials.gov NCT00297232) that could not confirm ATP decrease in five pre-PML samples 66. However, heterogeneous intervals of testing before PML onset may have influenced these results. It may be hypothesized that individual courses of ATP levels are more critical than absolute ATP level, and that a critical time-point of ATP decrease before PML onset has to be determined.
Recently, a lower proportion of L-selectin-expressing CD4+ T cells was associated with higher PML risk in NAT-treated MS patients (n = 8). Further validation as a potential biomarker for PML risk stratification is warranted 67. The determination of its biological plausibility remains unclear thus far, as it might express the general activation status of the peripheral immune system or a defective T cell response to JCV infection on different levels 67.
Lesions suggestive of PML may be detected on MRI before clinical PML manifestation 68, which argues for frequent MRI scanning in patients at high risk for PML evolution such as patients on long-term NAT treatment (> 24 infusions) or with previous immunosuppressive treatment. Characteristic PML lesions have been described as large, subcortical, grey-matter-sparing lesions appearing hyperintense on T2 and fluid-attenuated inversion recovery and hypointense on T1 scans; contrast enhancement may occur 47.
Alemtuzumab
Patient selection
The anti-CD52 mAb alemtuzumab (Lemtrada®) has been shown to be highly effective and is approved for active relapsing MS in Europe 10–12,69. Disease activity is defined as clinical or radiological deterioration 70. Mechanisms of action include depletion of CD52-expressing T/B lymphocytes, natural killer (NK) cells, dendritic cells and monocytes/macrophages with skewed repopulation leading to a reprogramming of the immune repertoire 71,72. Already in earlier studies, patients especially with an early relapsing disease course appeared to benefit most from alemtuzumab treatment, leading to the concept of a therapeutic window relatively early during the disease, when highly active immunotherapy may exert most profound effects 72. This was reflected in the inclusion criteria for the pivotal Phase III trials CARE-MS I and II (Comparison of Alemtuzumab and Rebif® Efficacy in Multiple Sclerosis, Studies One and Two). CARE-MS I included active relapsing, therapy-naive MS patients, whereas CARE-MS II focused on relapsing MS refractory to first-line therapy 10,12. Especially in terms of disease progression, the latter patient group appeared to benefit most. Whereas current EMA approval is relatively broad 70, careful patient selection is mandatory, as SADRs have been reported and thorough adherence to safety assessments is necessary. This is stressed by long-term data from the Phase II trial CAMMS223, with one additional SADR (Goodpasture syndrome), but also sustained reduction of disability accumulation and relapse rates compared to active comparator 73, revealing the dilemma of long-lasting efficacy versus potential SADRs.
Treatment and monitoring
Alemtuzumab is applied intravenously with a first treatment cycle of 12 mg over 5 days, followed by a second therapy cycle over 3 days after 12 months 10,12,69. Further cycles are not intended, but the question of when and how to continue DMD treatment after two cycles is unanswered. There is no class I evidence for different treatment protocols in this indication.
During and for 1 month after treatment, acyclovir (200 mg twice daily) has to be administered prophylactically.
Therapy surveillance with large treatment intervals, but necessarily close safety monitoring, will be a challenge in clinical practice 74 and emphasizes even more the importance of patient education, counselling and informed consent to assure adherence to safety measures. These include differential blood count, serum creatinine and urine analysis before first administration and monthly afterwards; regular testing of thyroid stimulating hormone (TSH) levels has to be performed before treatment initiation and every 3 months up to 4 years after the last administration 70.
SADR risk
Secondary antibody-mediated autoimmunity, even with fatal outcome, has been observed. This includes cases of autoimmune thrombocytopenia (1–3%), thyroiditis (16–30%) and nephritis due to glomerular basal membrane disease (single cases) (Table 1) 10–12,69. These SADRs may occur with late onset up to 4 years after treatment cessation 73, which highlights the need for adequate monitoring long after the actual infusion cycles (see above).
SADRs from oncological indications, e.g. myelodysplastic changes and tuberculous hepatitis 75,76, have thus far not been experienced in MS based on available long-term data from applications of CAMPATH-IH in the 1990s 77 or the Phase II trial CAMMS223 73.
Biomarkers
Pathogenesis of secondary autoimmune phenomena remains incompletely understood, but the skewed repopulation with an imbalance of B cells and regulatory T cells may partly account for these SADRs 78. The prognostic value of serum IL-21 as a risk marker for the development of secondary autoimmunity 79 was not confirmed. Hence, routine blood parameters and urinalysis remain critical regarding patient safety and early detection of SADRs.
Daclizumab
Patient selection
Daclizumab, used initially in transplant medicine, targets CD25, the alpha chain of the IL-2 receptor (IL-2Rα) 80,81. It is currently investigated on a Phase III level in RRMS after promising Phase II data. Daclizumab was investigated initially in combination with interferon (IFN)-beta 22. Meanwhile a modified formulation for s.c. monotherapy [daclizumab high-yield process (dac-HYP)] demonstrated clinical and paraclinical efficacy in a Phase II study in RRMS 14. Inclusion criteria required confirmed clinical or MRI disease activity 14. A paediatric study on seven patients showed some efficacy of daclizumab as second-line treatment; however, four children experienced further disease activity 82.
Treatment and monitoring
The ongoing dac-HYP Phase III trial DECIDE (Efficacy and Safety of Daclizumab High Yield Process Versus Interferon β 1a in Patients With Relapsing-Remitting Multiple Sclerosis; ClinicalTrials.gov NCT01064401) has left the 300-mg dosage in favour of a 150-mg subcutaneous dosage every 4 weeks.
SADR risk
The mode of action of daclizumab appears to be pleiotropic despite selective blockade of IL-2Rα: thus, expansion of regulatory CD56bright NK cells 80,83, reduction of proinflammatory signals 84 and interaction between T cells and antigen-presenting cells (APC) have been described 81.
To date, data on daclizumab show good tolerability and safety (Table 1) 14,22. However, the Safety and Efficacy Study of Daclizumab High Yield Process to Treat Relapsing-Remitting Multiple Sclerosis (SELECT) reports a fatal case after a series of events with initial possibly drug-related dermatitis 14. A single case report on secondary CNS vasculitis has recently been published and was evaluated as linked to daclizumab treatment 85. Long-term data and data from the Phase III trial are pending.
Biomarkers
Putative surrogate markers, especially CD56bright NK status, have been described for daclizumab-treatment responders 84. In addition, they have been suggested for risk evaluation 85.
Other mAbs
Several other mAbs are being investigated in clinical programmes or used on an off-label basis for otherwise treatment-refractory neuroimmunological disease.
The chimeric anti-CD20 mAb rituximab (MabThera®) is approved for haematological indications. In several countries, rituximab is recommended as first-line treatment for NMO, although not approved for this indication. For the malignant NMO disease course refractory to other treatment options, use of the IL-6-receptor mAb tocilizumab (RoActemra®, approved for rheumatoid arthritis) or the terminal complement inhibitor eculizumab (Soliris®, approved for paroxysmal nocturnal haemoglobinuria) has been reported.
Patient selection
Especially for substances used on an off-label basis, patient selection is based on single-case decisions, sometimes supported by preclinical experimental data.
Beneficial outcomes in smaller studies were reported for the anti-CD20 mAb rituximab in different neurological autoimmune conditions such as RRMS 8,15, NMO 86–88, myasthenia gravis 30,89 and multi-focal motor neuropathy 90,91. In PPMS, only a subgroup of younger patients with focal inflammatory activity on cranial MRI appeared to have some benefit from rituximab treatment. There are some data on rituximab use in paediatric populations with different neuroimmunological conditions 92–94.
Treatment with the IL-6 receptor mAb tocilizumab was efficacious in single cases of NMO refractory to rituximab 23,95 and other neuroimmunological conditions 96–98.
Inhibition of the complement system via eculizumab has been tested in a small number of NMO patients with positive results. As mostly feared from treatment of paroxysmal nocturnal haemoglobinuria and atypical haemolytic uraemic syndrome, it was associated with one case of meningococcal sepsis from a total of 14 patients 27.
These concepts will have to be confirmed in larger prospective trials to evaluate efficacy and safety in neurological patient cohorts.
Treatment
Although formally off-label in each of the neuroimmunological disorders, rituximab is recommended as the first-line DMD for treatment of NMO in respective guidelines with two suggested regimens (haematological protocol 375 mg/m2 body surface area weekly over 4 weeks versus 2 × 1 g) 46,99.
SADR risk
Adverse effects reported mainly from other indications are given in Table 1.
Rituximab-associated PML cases are described in rheumatoid arthritis, systemic lupus erythematosus and haematological populations, with combined rituximab and immunosuppressants 100,101. However, the risk appears to be considerably lower than with NAT–PML in MS 101. Due to the high frequency of infusion-related adverse events 102, newer anti-CD20 mAb have been studied on a Phase II level, the humanized ocrelizumab 17 and human ofatumumab 21. Results of further studies are pending. The clinical development of ocrelizumab in rheumatoid arthritis has been abandoned due to adverse effects. Eculizumab treatment has raised the special concern of meningococcal infections 27.
Biomarkers
Data on specific biomarkers for most of the agents described are widely lacking.
Repopulation of B cells via detection of CD19+ and CD20+ cells is sometimes used to determine reinfusion intervals for rituximab treatment, as it may be correlated with disease activity 103.
Fingolimod
Patient selection
FTY entails peripheral immunomodulatory effects and direct interactions within the CNS resulting from modulation of sphingosin-phosphate receptors (S1PR) 104. Approval of Gilenya® for treatment of RRMS differs substantially between FDA and EMA 105,106, reflecting divergent evaluations of its risk–benefit profile. Whereas, in the United States, FTY is approved as first-line therapy, in the European Union it is considered second-line therapy predominantly after a failure of IFN-beta or glatirameracetate. This approach is supported, at least in part, by subgroup analyses of the TRANSFORMS (TRial Assessing injectable interferoN vS FTY720 Oral in RrMS) study, especially for patients with high disease activity on IFN-beta therapy 107. Ongoing studies investigate the use of FTY in PPMS (ClinicalTrials.gov NCT00731692), in paediatric MS (ClinicalTrials.gov NCT01892722) and in CIDP (ClinicalTrials.gov NCT01625182). Siponimod, a specific modulator of S1PR subtypes 1 and 5, 108 is being evaluated in a trial in SPMS patients (ClinicalTrials.gov NCT01665144).
Specific risk populations comprise patients with predisposing conditions for the development of macula oedema such as diabetes mellitus and (recurrent) uveitis. Patients with pre-existing cardiac arrhythmia, negative dromo-and chronotropic co-medication and pre-existing pulmonary disease should be evaluated closely. In addition, assessment of varizella zoster (VZV) immune status is mandatory 106.
Treatment and monitoring
FTY is administered orally as a 0·5-mg capsule once daily. Before treatment initiation, laboratory investigations including differential blood count, liver enzymes, pregnancy test and VZV status have to be performed. VZV-IgG-negative patients should be vaccinated. Electrocardiography (ECG) and continuous ECG monitoring are recommended during first-dose administration and selectively afterwards. Ophthalmological and dermatological screening are recommended as routine pretreatment investigation, most importantly in risk populations (see Patient selection). Routine laboratory testing, especially for lymphopenia, is required at close intervals; dermatological, opthalmological and pneumological check-up should be implied in bigger, but regular, intervals or by clinical indication 106. Because FTY can moderately raise blood pressure, especially in hypertensive patients, blood pressure measurements should be performed regularly.
SADR risk
The described safety measures account for the potential adverse effects of FTY such as pronounced lymphopenia (0·2–1·0%) and cardiac arrhythmia, including symptomatic bradycardia (0·5–2·4%) and atrio-ventricular block (0·2–0·7%), macular oedema (0·5–1%), herpes infections (2·1–5·5%) and skin malignancies (0–0·7%) 9. Optical coherence tomography (OCT) may help to detect increased macular volume that seems to occur frequently under FTY treatment; however, macular oedema is a rare condition 109,110.
Two deaths were reported due to herpes virus infections: a primary VZV infection and a herpes-simplex encephalitis 9. A PML case is being discussed 111, but thus far has not been fully elucidated.
In the post-marketing setting, mainly cardiac events have been reported thus far and have led to extended cardiovascular safety monitoring 112. Recently, the marketing authorization holder published two fatal cases of haemophagocytic syndrome (HPS) associated with a 9-and 15-month treatment period with FTY. HPS is triggered typically by (viral) infections such as Epstein–Barr virus, as in the cases described. It results in a severe disturbance of the immune system and multi-organ involvement including fever, lymphadenopathy, organomegaly, cytopenia, liver failure and various neurological symptoms. Early diagnosis and treatment of both the triggering condition and the overwhelming immune response via immunosuppressive means are crucial to reduce mortality of HPS.
Biomarkers
The described safety set-up implies several putative biomarkers, although not evaluated formally thus far in terms of prediction of response or determination of SADR development. However, evaluation of lymphocyte counts may serve not only as a necessary safety measurement, but also as a therapy adherence marker.
Subclinical impairment of VZV and Epstein–Barr-virus reactivity have been found recently 113.
Teriflunomide
Patient selection
Teriflunomide (Aubagio®) is the active metabolite of leflunomide, a disease-modifying anti-rheumatic drug (DMARD). It is an inhibitor of the dihydroorotate dehydrogenase and interacts with de-novo pyrimidine synthesis 114.
Although the pivotal trial included 8·6% of SPMS patients 115,116, it has been approved by the FDA and EMA for RRMS. Specific contraindications for teriflunomide include severe hepatic or renal disorders and hypoproteinaemia (due to high plasma protein-binding) 117. As experimental data hint at teratogenic potential, FDA prescription guidelines emphasize the restriction of teriflunomide during pregnancy 118. It may be hypothesized that teriflunomide treatment may be especially beneficial with co-existing neuroimmunological and rheumatic disorders. Due to the long half-life of the drug and pronounced enterohepatic recirculation, teriflunomide might be an option in patients having difficulties with adherence to treatment schedules, but may be used more cautiously in patients with an impending wish for children.
Treatment and monitoring
Oral teriflunomide is administered once daily, 7 or 14 mg (FDA approval), or 14 mg (EMA approval) 116. Due to moderate elevation of blood pressure, regular blood pressure controls before and on treatment as well as the exclusion of severe infections before initiation are recommended. Interactions with warfarin [decrease of international normalized ratio (INR)] need to be controlled with frequent INR monitoring. There are no data with regard to marcumar, which is used more commonly in European countries. Adjunctive teriflunomide treatment with IFN-beta or glatirameracetate has been evaluated in several trials – Phase II trials showed a favourable safety profile and positive MRI outcomes 119 (and ClinicalTrials.gov NCT00475865), the results of extensions and other studies are pending. Regarding long drug half-life, drug washout after discontinuation can be accelerated via cholestyramine or activated charcoal powder 117, which is relevant in cases of unplanned pregnancy, newly acquired co-morbidities or rapid switch to other immune medications.
SADR risk
Long-term safety data on teriflunomide are being followed-up in extensions of Phases II and III trials (ClinicalTrials.gov NCT00228163, NCT00803049) 120. Experience on SADRs has been widely favourable, but includes the rare occurrence of potentially fatal infections and tuberculosis (Table 1). Whereas severe liver injury was not reported in the clinical development programme of teriflunomide, few cases were reported with leflunomide. Thus, risk assessment for teriflunomide is conservative, with extrapolation from post-marketing experience with leflunomide of more than 2·1 million patient years.
Biomarkers
Plasma levels of teriflunomide can be measured that might be useful in special situations such as pregnancy in order to monitor the rapid elimination procedure 117.
Ongoing or projected studies are investigating the influence of teriflunomide on brain pathology by use of MRI (ClinicalTrials.gov NCT01881191) and the role of lymphocyte subsets as biomarkers for teriflunomide therapy (ClinicalTrials.gov NCT01863888).
Dimethylfumarate
Patient selection
Dimethylfumarate (DMF) is described to have differential modes of action, including anti-inflammatory [e.g. enhanced T helper type 2 (Th2) response, T cell apoptosis] and potentially neuroprotective aspects [modulation of the nuclear (erythroid-derived 2)-related factor (Nrf2) pathway, anti-oxidative effects] 121,122. Two Phase III trials have shown efficacy of DMF in RRMS 123,124.
Due to possible gastrointestinal side effects, application of DMF in patients with severe gastrointestinal disorders such as peptic ulcers should be assessed cautiously. Whereas DMF (Tecfidera®) is approved in the United States, as of October 2013 marketing in the European Union has not yet begun.
Treatment and monitoring
DMF is an oral compound administered twice daily at a dose of 240 mg. The administration of 720 mg per day has not shown higher efficacy than the 480 mg daily dose 123,124. In order to improve the tolerability of DMF, dose titration is recommended. Lymphopenia will presumably be addressed in safety monitoring schedules in European treatment guidelines. This has not been accounted for in US prescription guidelines.
SADR risk
Similar to other agents discussed in this review, for fumaric acid esters there is also experience on the safety profile from other indications than MS. Fumaderm®, a mixture containing DMF as well as other different monoethyl fumarate salts, has been approved for the treatment of psoriasis since the early 1990s, and dermatological experience suggests a favourable safety profile with more than 185 000 patient years. However, PML cases have been reported recently during psoriasis treatment with fumaric esters 125–128, although confounding factors were identified in these cases. Two cases had experienced long-lasting lymphopenia without treatment adaption, as recommended 126,127; the other cases had a history of sarcoidosis, cancer, previous mAb (efalizumab) and immunosuppressive (methotrexate) treatment 128. Tecfidera®, also with differences regarding galenics, is approved for MS. Thus far, no signal for opportunistic infections such as PML have been reported from the clinical programme or the short post-marketing interval (US) with Tecfidera®.
Biomarkers
The regular assessment of leuco-and lymphocyte counts is sensible and may serve treatment surveillance. At 1 year of treatment, leuco-and lymphocyte counts decreased by 10–12% and 28–32% (mean), respectively; 4–5% of patients experienced total lymphocyte counts below 0·5 × 109 per litre 123,124.
As in other DMD treatments, regular MRI under DMF therapy will be reasonable for both therapy monitoring and determining effectiveness.
Mitoxantrone
Patient selection
Mitoxantrone (MX, Ralenova®/Novantrone®) has been approved for the treatment of secondary progressive and progressive relapsing MS following two placebo-controlled trials 19,129 and two studies comparing MX or MX in combination with methylprednisolone (MP) to MP alone 130,131. Data on MX in primary progressive MS (PPMS) is discouraging 132–134, but has gained relevance in NMO treatment 24,25. Although not formally approved, MX has been used in children with aggressive forms of MS 135.
Treatment and monitoring
Different treatment protocols may be an influencing factor for SADR development, especially in terms of therapy-related acute leukaemia (TRAL) 136. Whereas an intravenous infusion every 3 months according to the placebo-controlled, double-blind, randomised, multicentre, phase III trial of mitoxantrone in secondary progressive multiple sclerosis (MIMS) protocol 129, including dose adaption according to leucocyte nadir, is used widely in Germany, dose regimens differ substantially and may not include regular dose adaption 137,138.
Additional differences may comprise pre-or co-treatments 37. Thus, MP co-treatment has been shown to increase intracellular MX dosage in vitro 139, and may thus increase cellular toxicity.
Treatment de-escalation should be considered after 1 year of clinical and paraclinical stability of disease to minimize the risk of at least partially dose-dependent SADRs (e.g. cardiotoxicity). Haematological monitoring should include regular examination of blood counts up to 5 years after cessation of MX 140–142. Cardiac ultrasound and electrocardiography (ECG) should be performed accordingly, as late-onset cardiotoxicity is described 143.
Thorough monitoring and vigilance is especially relevant for TRAL, as secondary leukaemia is potentially curable if diagnosed early and treated adequately 144, but is associated with potentially fatal complications 145–147 if overlooked.
SADR risk
Discussions about SADR incidence, especially TRAL and cardiotoxicity 36,37,137,138,142,148–152, have led to reassessment of the proper risk–benefit profile of MX.
TRAL incidences vary from 0·07% 149 to 2·82% 138 and are subject to methodological difficulties (e.g. reporting bias especially for meta-analyses 36,149 and largely lacking prospective data). Interestingly, there seem to be regional differences of TRAL incidence with similar German and French estimates 37,142, but higher Italian and Spanish rates 137,138.
Estimates of the incidence of cardiotoxicity are complicated by different definitions of an adverse cardiac event [reporting of clinical events versus paraclinical abnormalities in ECG, transthoracal echocardiography (TTE) 153 and radionuclide ventriculography 143,150,154]. Subclinical decrease of left ventricular ejection fraction (LVEF) in TTE may be a dose-dependent effect 153; however, this has not been confirmed by a study with 14% incidence of LVEF decrease in radionuclide ventriculography without dose-dependency 150. Data on recovery and prognosis of cardiac events are inconsistent 143,150,151,153,155,156.
Biomarkers
Clinical and paraclinical parameters for the prediction of MX response have been established 157.
SADR development might be associated with pronounced or lasting leucopenia before TRAL onset 37 and increased brain natriuretic peptide (BNP) in subclinical myocardial injury 158.
In addition to treatment-related factors, genetic factors (genes involved in detoxification: CYP3A4; cellular drug efflux: ABCB1, ABCG2; DNA repair: BRCA2, XRCC5) may influence susceptibility for SADRs 139,155,159. Pharmacogenetic approaches may help early identification of patients at higher risk for side effects or even individualized treatment schemes.
Discussion and conclusions
The growing spectrum of treatment options for neuroimmunological diseases confronts us with complex risk–benefit considerations and treatment decisions. Whereas established first-line DMDs such as interferon-beta formulations and glatirameracetate are generally safe, newly emerging DMDs with higher efficacy often carry a higher potential of adverse effects with thorough therapy monitoring requirements. Long monitoring intervals, even after cessation of therapy, also pose new challenges for adherence to respective protocols.
If not in the clinical trial setting (FTY, alemtuzumab), post-marketing experience (NAT) has revealed relevant or even completely new safety issues not anticipated previously. We have to keep in mind that, for example, in order to identify an event occurring at a frequency of 0·1% as an adverse drug reaction, a sample size of 50 000 would be necessary to observe a twofold increased adverse event rate in comparison to a control group 160. This emphasizes the need for thorough post-marketing surveillance, Phase IV trials and drug registries to enhance patient safety. Such studies would also be valuable as validation studies for putative biomarkers.
Principles for optimization of the benefit-to-risk ratio comprise thorough patient selection according to distinct clinical criteria, proper treatment intervals, dosage and duration, the evaluation of (individual) risk profiles for SADRs and the investigation and validation of biomarkers for risk stratification and treatment benefit. The transfer of these principles into clinical practice is difficult, and has thus far been only partially achieved for the substances described.
Treatment decisions may be based not only on ‘classic’ first-and second-line dichotomy and parallel concepts [‘hit hard and (relatively) early'] may be beneficial in distinct patient groups. Current guidelines tend to emphasize individual factors and contraindications to alleviate treatment decisions.
Safety monitoring of patients begins before treatment initiation and outlasts the actual active treatment period as, for many SADRs, late-onset cases have been reported.
For all treatment options discussed, routine laboratory investigations of liver and renal function, thorough assessment of existing severe infections or immunosuppression for any cause is relevant to allow safe treatment initiation, just as important as the assessment of pregnancy and information of (especially female) patients in terms of reproductive issues.
Regular safety assessments help in the early detection of severe side effects or their prodromal signs and symptoms. Clinical vigilance and education of patients for signs and symptoms of SADRs is key for improving the safety of modern DMD therapy, as early accurate treatment of SADRs is crucial and of prognostic relevance. Early interdisciplinary co-operation is necessary, as SADRs for many agents are described not only in the neurological field (PML, neuropathy, CNS infection), but also in dermatological, ophthalmological and internal medicine. Counselling of patients may also include gynaecological and/or andrological advice.
Biomarkers for SADR prediction and pharmacogenetic approaches for different agents will have to be validated in larger patient cohorts and may alleviate therapeutic decisions in the future.
Acknowledgments
This work was supported by the German Bundesministerium für Bildung und Forschung (BMBF), German competence Network Multiple Sclerosis (KKNMS), 01GI0914.
Disclosures
A. S. has received personal compensation for activities with Novartis, Sanofi and Almirall Hermal GmbH. R. G. has received personal compensation for activities with Bayer Healthcare, Biogen Idec and Teva Neuroscience and in an editorial capacity from Therapeutic Advances in Neurological Disorders, and also received patent payments from Biogen Idec and research support from Bayer Healthcare, Biogen Idec, Merck Serono, Teva Neuroscience, Novartis and from the German Ministry for Education and Research [BMBF, ‘German Competence Network Multiple Sclerosis’ (KKNMS), CONTROL MS, 01GI0914]. A. C. has received personal compensation for activities with Almirall Hermal GmbH, Bayer Schering, Biogen Idec, Merck Serono, Novartis and Teva Neuroscience, research support from Bayer Schering, Biogen Idec, Merck Serono and Novartis and research grants from the German Ministry for Education and Research [BMBF, ‘German Competence Network Multiple Sclerosis’ (KKNMS), CONTROL MS, 01GI0914].
References
- 1.Thompson AJ, Toosy AT, Ciccarelli O. Pharmacological management of symptoms in multiple sclerosis: current approaches and future directions. Lancet Neurol. 2010;9:1182–1199. doi: 10.1016/S1474-4422(10)70249-0. [DOI] [PubMed] [Google Scholar]
- 2.Jacob A, McKeon A, Nakashima I, et al. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2013;84:922–930. doi: 10.1136/jnnp-2012-302310. [DOI] [PubMed] [Google Scholar]
- 3.Flensner G, Landtblom AM, Soderhamn O, Ek AC. Work capacity and health-related quality of life among individuals with multiple sclerosis reduced by fatigue: a cross-sectional study. BMC Public Health. 2013;13:224. doi: 10.1186/1471-2458-13-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chanson JB, Zephir H, Collongues N, et al. Evaluation of health-related quality of life, fatigue and depression in neuromyelitis optica. Eur J Neurol. 2011;18:836–841. doi: 10.1111/j.1468-1331.2010.03252.x. [DOI] [PubMed] [Google Scholar]
- 5.Hafler DA, Slavik JM, Anderson DE, O'Connor KC, De Jager P, Baecher-Allan C. Multiple sclerosis. Immunol Rev. 2005;204:208–231. doi: 10.1111/j.0105-2896.2005.00240.x. [DOI] [PubMed] [Google Scholar]
- 6.Hinson SR, Pittock SJ, Lucchinetti CF, et al. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69:2221–2231. doi: 10.1212/01.WNL.0000289761.64862.ce. [DOI] [PubMed] [Google Scholar]
- 7.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet. 2004;364:2106–2112. doi: 10.1016/S0140-6736(04)17551-X. [DOI] [PubMed] [Google Scholar]
- 8.Bar-Or A, Calabresi PA, Arnold D, et al. Rituximab in relapsing–remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol. 2008;63:395–400. doi: 10.1002/ana.21363. [DOI] [PubMed] [Google Scholar]
- 9.Cohen JA, Barkhof F, Comi G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 10.Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing–remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet. 2012;380:1819–1828. doi: 10.1016/S0140-6736(12)61769-3. [DOI] [PubMed] [Google Scholar]
- 11.Coles AJ, Fox E, Vladic A, et al. Alemtuzumab versus interferon beta-1a in early relapsing–remitting multiple sclerosis: post-hoc and subset analyses of clinical efficacy outcomes. Lancet Neurol. 2011;10:338–348. doi: 10.1016/S1474-4422(11)70020-5. [DOI] [PubMed] [Google Scholar]
- 12.Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet. 2012;380:1829–1839. doi: 10.1016/S0140-6736(12)61768-1. [DOI] [PubMed] [Google Scholar]
- 13.Edan G, Comi G, Le Page E, Leray E, Rocca MA, Filippi M. Mitoxantrone prior to interferon beta-1b in aggressive relapsing multiple sclerosis: a 3-year randomised trial. J Neurol Neurosurg Psychiatry. 2011;82:1344–1350. doi: 10.1136/jnnp.2010.229724. [DOI] [PubMed] [Google Scholar]
- 14.Gold R, Giovannoni G, Selmaj K, et al. Daclizumab high-yield process in relapsing–remitting multiple sclerosis (SELECT): a randomised, double-blind, placebo-controlled trial. Lancet. 2013;381:2167–2175. doi: 10.1016/S0140-6736(12)62190-4. [DOI] [PubMed] [Google Scholar]
- 15.Hauser SL, Waubant E, Arnold DL, et al. B-cell depletion with rituximab in relapsing–remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 16.Kappos L, Bates D, Edan G, et al. Natalizumab treatment for multiple sclerosis: updated recommendations for patient selection and monitoring. Lancet Neurol. 2011;10:745–758. doi: 10.1016/S1474-4422(11)70149-1. [DOI] [PubMed] [Google Scholar]
- 17.Kappos L, Li D, Calabresi PA, et al. Ocrelizumab in relapsing–remitting multiple sclerosis: a phase 2, randomised, placebo-controlled, multicentre trial. Lancet. 2011;378:1779–1787. doi: 10.1016/S0140-6736(11)61649-8. [DOI] [PubMed] [Google Scholar]
- 18.Kappos L, Radue EW, O'Connor P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 19.Millefiorini E, Gasperini C, Pozzilli C, et al. Randomized placebo-controlled trial of mitoxantrone in relapsing–remitting multiple sclerosis: 24-month clinical and MRI outcome. J Neurol. 1997;244:153–159. doi: 10.1007/s004150050066. [DOI] [PubMed] [Google Scholar]
- 20.Rose JW, Burns JB, Bjorklund J, Klein J, Watt HE, Carlson NG. Daclizumab phase II trial in relapsing and remitting multiple sclerosis: MRI and clinical results. Neurology. 2007;69:785–789. doi: 10.1212/01.wnl.0000267662.41734.1f. [DOI] [PubMed] [Google Scholar]
- 21.Soelberg Sorensen P, Drulovic J, Havrdova E, Lisby S, Graff O, Shackelford S. 2010. Magnetic resonance imaging (MRI) efficacy of ofatumumab in relapsing–remitting multiple sclerosis (RRMS) = 24-week results of a phase II study. ECTRIMS; October 13–16,; Gothenburg, Sweden P136 2010.
- 22.Wynn D, Kaufman M, Montalban X, et al. Daclizumab in active relapsing multiple sclerosis (CHOICE study): a phase 2, randomised, double-blind, placebo-controlled, add-on trial with interferon beta. Lancet Neurol. 2010;9:381–390. doi: 10.1016/S1474-4422(10)70033-8. [DOI] [PubMed] [Google Scholar]
- 23.Ayzenberg I, Kleiter I, Schroder A, et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol. 2013;70:394–397. doi: 10.1001/jamaneurol.2013.1246. [DOI] [PubMed] [Google Scholar]
- 24.Cabre P, Olindo S, Marignier R, Jeannin S, Merle H, Smadja D. Efficacy of mitoxantrone in neuromyelitis optica spectrum: clinical and neuroradiological study. J Neurol Neurosurg Psychiatry. 2013;84:511–516. doi: 10.1136/jnnp-2012-303121. [DOI] [PubMed] [Google Scholar]
- 25.Kim SH, Kim W, Park MS, Sohn EH, Li XF, Kim HJ. Efficacy and safety of mitoxantrone in patients with highly relapsing neuromyelitis optica. Arch Neurol. 2011;68:473–479. doi: 10.1001/archneurol.2010.322. [DOI] [PubMed] [Google Scholar]
- 26.Lehmann-Horn K, Schleich E, Hertzenberg D, et al. Anti-CD20 B-cell depletion enhances monocyte reactivity in neuroimmunological disorders. J Neuroinflammation. 2011;8:146. doi: 10.1186/1742-2094-8-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pittock SJ, Lennon VA, McKeon A, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol. 2013;12:554–562. doi: 10.1016/S1474-4422(13)70076-0. [DOI] [PubMed] [Google Scholar]
- 28.O'Reilly MW, Sexton DJ, Dennedy MC, et al. Radiological remission and recovery of thirst appreciation after infliximab therapy in adipsic diabetes insipidus secondary to neurosarcoidosis. QJM. 2013 doi: 10.1093/qjmed/hct023. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 29.Segal BM. Neurosarcoidosis: diagnostic approaches and therapeutic strategies. Curr Opin Neurol. 2013;26:307–313. doi: 10.1097/WCO.0b013e3283608459. [DOI] [PubMed] [Google Scholar]
- 30.Keung B, Robeson KR, Dicapua DB, et al. Long-term benefit of rituximab in MuSK autoantibody myasthenia gravis patients. J Neurol Neurosurg Psychiatry. 2013;84:1407–1409. doi: 10.1136/jnnp-2012-303664. [DOI] [PubMed] [Google Scholar]
- 31.Markvardsen LH, Debost JC, Harbo T, et al. Subcutaneous immunoglobulin in responders to intravenous therapy with chronic inflammatory demyelinating polyradiculoneuropathy. Eur J Neurol. 2013;20:836–842. doi: 10.1111/ene.12080. [DOI] [PubMed] [Google Scholar]
- 32.Yoon MS, Chan A, Gold R. Standard and escalating treatment of chronic inflammatory demyelinating polyradiculoneuropathy. Ther Adv Neurol Disord. 2011;4:193–200. doi: 10.1177/1756285611405564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahsen von N, Chan A. Therapeutic monitoring of immunotherapies in autoimmune diseases. Curr Pharm Des. 2012;18:4550–4555. doi: 10.2174/138161212802502152. [DOI] [PubMed] [Google Scholar]
- 34.Goodin DS, Ebers GC, Cutter G, et al. Cause of death in MS: long-term follow-up of a randomised cohort, 21 years after the start of the pivotal IFNbeta-1b study. BMJ Open. 2012;2:e001972. doi: 10.1136/bmjopen-2012-001972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hellwig K, Gold R. Progressive multifocal leukoencephalopathy and natalizumab. J Neurol. 2011;258:1920–1928. doi: 10.1007/s00415-011-6116-8. [DOI] [PubMed] [Google Scholar]
- 36.Marriott JJ, Miyasaki JM, Gronseth G, O'Connor PW. Evidence report: the efficacy and safety of mitoxantrone (Novantrone) in the treatment of multiple sclerosis: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 2010;74:1463–1470. doi: 10.1212/WNL.0b013e3181dc1ae0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stroet A, Hemmelmann C, Starck M, et al. Incidence of therapy-related acute leukaemia in mitoxantrone-treated multiple sclerosis patients in Germany. Ther Adv Neurol Disord. 2012;5:75–79. doi: 10.1177/1756285611433318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Correale P, Cusi MG, Tagliaferri P. Immunomodulatory properties of anticancer monoclonal antibodies: is the ‘magic bullet’ still a reliable paradigm? Immunotherapy. 2011;3:1–4. doi: 10.2217/imt.10.92. [DOI] [PubMed] [Google Scholar]
- 39.O'Connor P. Natalizumab and the role of alpha 4-integrin antagonism in the treatment of multiple sclerosis. Expert Opin Biol Ther. 2007;7:123–136. doi: 10.1517/14712598.7.1.123. [DOI] [PubMed] [Google Scholar]
- 40.Kornek B, Aboul-Enein F, Rostasy K, et al. Natalizumab therapy for highly active pediatric multiple sclerosis. JAMA Neurol. 2013;70:469–475. doi: 10.1001/jamaneurol.2013.923. [DOI] [PubMed] [Google Scholar]
- 41.Arnal-Garcia C, Garcia-Montero MR, Malaga I, et al. Natalizumab use in pediatric patients with relapsing–remitting multiple sclerosis. Eur J Paediatr Neurol. 2013;17:50–54. doi: 10.1016/j.ejpn.2012.09.004. [DOI] [PubMed] [Google Scholar]
- 42.Kleiter I, Hellwig K, Berthele A, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol. 2012;69:239–245. doi: 10.1001/archneurol.2011.216. [DOI] [PubMed] [Google Scholar]
- 43.Barnett MH, Prineas JW, Buckland ME, Parratt JD, Pollard JD. Massive astrocyte destruction in neuromyelitis optica despite natalizumab therapy. Mult Scler. 2012;18:108–112. doi: 10.1177/1352458511421185. [DOI] [PubMed] [Google Scholar]
- 44.Foley J. Natalizumab related PML: an evolving risk stratification paradigm. Neurology. 2013;80(Meeting Abstracts 1):S30.002. [Google Scholar]
- 45.Bloomgren G, Richman S, Hotermans C, et al. Risk of natalizumab-associated progressive multifocal leukoencephalopathy. N Engl J Med. 2012;366:1870–1880. doi: 10.1056/NEJMoa1107829. [DOI] [PubMed] [Google Scholar]
- 46.Gold R, Hanschke S, Hemmer B, Wiendl H. 2012. DGN/KKNMS Leitlinie zur Diagnose und Therapie der MS – online version, Stand: 09.08.2012 Available at: http://www.kompetenznetz-multiplesklerose.de/images/stories/PDF_Dateien/Leitlinie/dgn-kknms_ms-ll_20120809_frei.pdf (accessed 11 August 2013)
- 47.Yousry TA, Pelletier D, Cadavid D, et al. Magnetic resonance imaging pattern in natalizumab-associated progressive multifocal leukoencephalopathy. Ann Neurol. 2012;72:779–787. doi: 10.1002/ana.23676. [DOI] [PubMed] [Google Scholar]
- 48.Aly L, Yousef S, Schippling S, et al. Central role of JC virus-specific CD4+ lymphocytes in progressive multi-focal leucoencephalopathy-immune reconstitution inflammatory syndrome. Brain. 2011;134:2687–2702. doi: 10.1093/brain/awr206. [DOI] [PubMed] [Google Scholar]
- 49.Ryschkewitsch CF, Jensen PN, Major EO. Multiplex qPCR assay for ultra sensitive detection of JCV DNA with simultaneous identification of genotypes that discriminates non-virulent from virulent variants. J Clin Virol. 2013;57:243–248. doi: 10.1016/j.jcv.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bozic C, Richman S, Plavina T, et al. Anti-John Cunnigham virus antibody prevalence in multiple sclerosis patients: baseline results of STRATIFY-1. Ann Neurol. 2011;70:713–721. doi: 10.1002/ana.22606. [DOI] [PubMed] [Google Scholar]
- 51.Gorelik L, Lerner M, Bixler S, et al. Anti-JC virus antibodies: implications for PML risk stratification. Ann Neurol. 2010;68:295–303. doi: 10.1002/ana.22128. [DOI] [PubMed] [Google Scholar]
- 52.Trampe AK, Hemmelmann C, Stroet A, et al. Anti-JC virus antibodies in a large German natalizumab-treated multiple sclerosis cohort. Neurology. 2012;78:1736–1742. doi: 10.1212/WNL.0b013e3182583022. [DOI] [PubMed] [Google Scholar]
- 53.BiogenIdec. 2013. Tysabri Available at: http://tysabri.de/index.php?inhalt=tysabri.sicherheitsmanagement (accessed 17 December 2013)
- 54.Havla J, Berthele A, Kumpfel T, et al. Co-occurrence of two cases of progressive multifocal leukoencephalopathy in a natalizumab ‘infusion group’. Mult Scler. 2013;19:1213–1215. doi: 10.1177/1352458512466165. [DOI] [PubMed] [Google Scholar]
- 55.Kuhle J, Gosert R, Buhler R, et al. Management and outcome of CSF-JC virus PCR-negative PML in a natalizumab-treated patient with MS. Neurology. 2011;77:2010–2016. doi: 10.1212/WNL.0b013e31823b9b27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mazda ME, Brosch JR, Wiens AL, et al. A case of natalizumab-associated progressive multifocal leukoencephalopathy with repeated negative CSF JCV testing. Int J Neurosci. 2013;123:353–357. doi: 10.3109/00207454.2012.760561. [DOI] [PubMed] [Google Scholar]
- 57.Dahlhaus S, Hoepner R, Chan A, et al. Disease course and outcome of 15 monocentrically treated natalizumab-associated progressive multifocal leukoencephalopathy patients. J Neurol Neurosurg Psychiatry. 2013;84:1068–1074. doi: 10.1136/jnnp-2013-304897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hellwig K, Kleiter I, Gold R. Immune reconstitution inflammatory syndrome in natalizumab-associated PML. Neurology. 2012;78:371. doi: 10.1212/01.wnl.0000411451.56205.c3. author reply. [DOI] [PubMed] [Google Scholar]
- 59.Hoepner R, Dahlhaus S, Kollar S, et al. Prophylactic antiepileptic treatment reduces seizure frequency in natalizumab-associated progressive multifocal leukoencephalopathy. Ther Adv Neurol Disord. 7:3–6. doi: 10.1177/1756285613503515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Berger JR, Houff SA, Gurwell J, Vega N, Miller CS, Danaher RJ. JC virus antibody status underestimates infection rates. Ann Neurol. 2013;74:84–90. doi: 10.1002/ana.23893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Warnke C, Ramanujam R, Plavina T, et al. Changes to anti-JCV antibody levels in a Swedish national MS cohort. J Neurol Neurosurg Psychiatry. 2013;84:1199–1205. doi: 10.1136/jnnp-2012-304332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee P, Plavina T, Castro A, et al. A second-generation ELISA (STRATIFY JCV DxSelect) for detection of JC virus antibodies in human serum and plasma to support progressive multifocal leukoencephalopathy risk stratification. J Clin Virol. 2013;57:141–146. doi: 10.1016/j.jcv.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 63.Koralnik IJ, Du Pasquier RA, Kuroda MJ, et al. Association of prolonged survival in HLA-A2+ progressive multifocal leukoencephalopathy patients with a CTL response specific for a commonly recognized JC virus epitope. J Immunol. 2002;168:499–504. doi: 10.4049/jimmunol.168.1.499. [DOI] [PubMed] [Google Scholar]
- 64.Perkins MR, Ryschkewitsch C, Liebner JC, et al. Changes in JC virus-specific T cell responses during natalizumab treatment and in natalizumab-associated progressive multifocal leukoencephalopathy. PLOS Pathog. 2012;8:e1003014. doi: 10.1371/journal.ppat.1003014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haghikia A, Perrech M, Pula B, et al. Functional energetics of CD4+-cellular immunity in monoclonal antibody-associated progressive multifocal leukoencephalopathy in autoimmune disorders. PLOS ONE. 2011;6:e18506. doi: 10.1371/journal.pone.0018506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Goelz S, Polman C, Rudick R, et al. ImmuKnow® (Cylex) does not appear to be useful for PML risk stratification with natalizumab treatment. Mult Scler. 2011;17:S507–S524. P1118. [Google Scholar]
- 67.Schwab N, Schneider-Hohendorf T, Posevitz V, et al. L-Selectin is a possible biomarker for individual PML risk in natalizumab-treated MS patients. Neurology. 2013;81:865–871. doi: 10.1212/WNL.0b013e3182a351fb. [DOI] [PubMed] [Google Scholar]
- 68.Linda H, Heijne von A. Presymptomatic diagnosis with MRI and adequate treatment ameliorate the outcome after natalizumab-associated progressive multifocal leukoencephalopathy. Front Neurol. 2013;4:11. doi: 10.3389/fneur.2013.00011. doi: 10.3389/fneur.2013.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coles AJ, Compston DA, Selmaj KW, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med. 2008;359:1786–1801. doi: 10.1056/NEJMoa0802670. [DOI] [PubMed] [Google Scholar]
- 70.European Medicines Agency (EMA) 2013. Summary of Product Characteristics – Lemtrada Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003718/WC500150521.pdf (accessed 28 September 2013)
- 71.Klotz L, Meuth SG, Wiendl H. Immune mechanisms of new therapeutic strategies in multiple sclerosis-A focus on alemtuzumab. Clin Immunol. 2012;142:25–30. doi: 10.1016/j.clim.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 72.Coles AJ, Cox A, Le Page E, et al. The window of therapeutic opportunity in multiple sclerosis: evidence from monoclonal antibody therapy. J Neurol. 2006;253:98–108. doi: 10.1007/s00415-005-0934-5. [DOI] [PubMed] [Google Scholar]
- 73.Coles AJ, Fox E, Vladic A, et al. Alemtuzumab more effective than interferon beta-1a at 5-year follow-up of CAMMS223 clinical trial. Neurology. 2012;78:1069–1078. doi: 10.1212/WNL.0b013e31824e8ee7. [DOI] [PubMed] [Google Scholar]
- 74.Ontaneda D, Cohen JA. The benefits and risks of alemtuzumab in multiple sclerosis. Expert Rev Clin Immunol. 2013;9:189–191. doi: 10.1586/eci.13.1. [DOI] [PubMed] [Google Scholar]
- 75.Braester A, Akria L, Suriu C, Sonkin V. Alemtuzumab and myelodysplasia: a story of love and hate (possible alemtuzumab-induced myelodysplastic changes) Acta Haematol. 2012;129:185–186. doi: 10.1159/000345254. [DOI] [PubMed] [Google Scholar]
- 76.Bosch W, Poowanawittayakom N, Chaikriangkrai K, et al. Tuberculous hepatitis in renal transplant recipients following alemtuzumab induction therapy. Transpl Infect Dis. 2013;15:E33–39. doi: 10.1111/tid.12048. [DOI] [PubMed] [Google Scholar]
- 77.Moreau T, Coles A, Wing M, et al. CAMPATH-IH in multiple sclerosis. Mult Scler. 1996;1:357–365. doi: 10.1177/135245859600100616. [DOI] [PubMed] [Google Scholar]
- 78.Costelloe L, Jones J, Coles A. Secondary autoimmune diseases following alemtuzumab therapy for multiple sclerosis. Expert Rev Neurother. 2012;12:335–341. doi: 10.1586/ern.12.5. [DOI] [PubMed] [Google Scholar]
- 79.Jones JL, Phuah CL, Cox AL, et al. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H) J Clin Invest. 2009;119:2052–2061. doi: 10.1172/JCI37878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bielekova B, Catalfamo M, Reichert-Scrivner S, et al. Regulatory CD56(bright) natural killer cells mediate immunomodulatory effects of IL-2Ralpha-targeted therapy (daclizumab) in multiple sclerosis. Proc Natl Acad Sci U S A. 2006;103:5941–5946. doi: 10.1073/pnas.0601335103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wuest SC, Edwan JH, Martin JF, et al. A role for interleukin-2 trans-presentation in dendritic cell-mediated T cell activation in humans, as revealed by daclizumab therapy. Nat Med. 2011;17:604–609. doi: 10.1038/nm.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gorman MP, Tillema JM, Ciliax AM, Guttmann CR, Chitnis T. Daclizumab use in patients with pediatric multiple sclerosis. Arch Neurol. 2012;69:78–81. doi: 10.1001/archneurol.2011.581. [DOI] [PubMed] [Google Scholar]
- 83.Sheridan JP, Zhang Y, Riester K, et al. Intermediate-affinity interleukin-2 receptor expression predicts CD56(bright) natural killer cell expansion after daclizumab treatment in the CHOICE study of patients with multiple sclerosis. Mult Scler. 2011;17:1441–1448. doi: 10.1177/1352458511414755. [DOI] [PubMed] [Google Scholar]
- 84.Perry JS, Han S, Xu Q, et al. Inhibition of LTi cell development by CD25 blockade is associated with decreased intrathecal inflammation in multiple sclerosis. Sci Transl Med. 2012;4:145ra106. doi: 10.1126/scitranslmed.3004140. doi: 10.1126/scitranslmed.3004140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ohayon J, Oh U, Richert N, et al. CNS vasculitis in a patient with MS on daclizumab monotherapy. Neurology. 2013;80:453–457. doi: 10.1212/WNL.0b013e31827f0f42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim SH, Huh SY, Lee SJ, Joung A, Kim HJ. A 5-year follow-up of rituximab treatment in patients with neuromyelitis optica spectrum disorder. JAMA Neurol. 2013;70:1110–1117. doi: 10.1001/jamaneurol.2013.3071. [DOI] [PubMed] [Google Scholar]
- 87.Cree BA, Lamb S, Morgan K, Chen A, Waubant E, Genain C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology. 2005;64:1270–1272. doi: 10.1212/01.WNL.0000159399.81861.D5. [DOI] [PubMed] [Google Scholar]
- 88.Jacob A, Weinshenker BG, Violich I, et al. Treatment of neuromyelitis optica with rituximab: retrospective analysis of 25 patients. Arch Neurol. 2008;65:1443–1448. doi: 10.1001/archneur.65.11.noc80069. [DOI] [PubMed] [Google Scholar]
- 89.Collongues N, Casez O, Lacour A, et al. Rituximab in refractory and non-refractory myasthenia: a retrospective multicenter study. Muscle Nerve. 2012;46:687–691. doi: 10.1002/mus.23412. [DOI] [PubMed] [Google Scholar]
- 90.Chaudhry V, Cornblath DR. An open-label trial of rituximab (Rituxan(R)) in multifocal motor neuropathy. J Peripher Nerv Syst. 2010;15:196–201. doi: 10.1111/j.1529-8027.2010.00270.x. [DOI] [PubMed] [Google Scholar]
- 91.Stieglbauer K, Topakian R, Hinterberger G, Aichner FT. Beneficial effect of rituximab monotherapy in multifocal motor neuropathy. Neuromuscul Disord. 2009;19:473–475. doi: 10.1016/j.nmd.2009.04.013. [DOI] [PubMed] [Google Scholar]
- 92.Leen WG, Weemaes CM, Verbeek MM, Willemsen MA, Rotteveel JJ. Rituximab and intravenous immunoglobulins for relapsing postinfectious opsoclonus–myoclonus syndrome. Pediatr Neurol. 2008;39:213–217. doi: 10.1016/j.pediatrneurol.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 93.Koul R, Al Futaisi A, Abdwani R. Rituximab in severe seronegative juvenile myasthenia gravis: review of the literature. Pediatr Neurol. 2012;47:209–212. doi: 10.1016/j.pediatrneurol.2012.05.017. [DOI] [PubMed] [Google Scholar]
- 94.Pranzatelli MR, Tate ED, Travelstead AL, et al. Rituximab (anti-CD20) adjunctive therapy for opsoclonus–myoclonus syndrome. J Pediatr Hematol Oncol. 2006;28:585–593. doi: 10.1097/01.mph.0000212991.64435.f0. [DOI] [PubMed] [Google Scholar]
- 95.Kieseier BC, Stuve O, Dehmel T, et al. Disease amelioration with tocilizumab in a treatment-resistant patient with neuromyelitis optica: implication for cellular immune responses. JAMA Neurol. 2013;70:390–393. doi: 10.1001/jamaneurol.2013.668. [DOI] [PubMed] [Google Scholar]
- 96.Urbaniak P, Hasler P, Kretzschmar S. Refractory neuro-Behçet treated by tocilizumab: a case report. Clin Exp Rheumatol. 2012;30:S73–75. [PubMed] [Google Scholar]
- 97.Abisror N, Mekinian A, Lavigne C, Vandenhende MA, Soussan M, Fain O. Tocilizumab in refractory Takayasu arteritis: a case series and updated literature review. Autoimmun Rev. 2013;12:1143–1149. doi: 10.1016/j.autrev.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 98.Krogias C, Hoepner R, Muller A, Schneider-Gold C, Schroder A, Gold R. Successful treatment of anti-Caspr2 syndrome by interleukin 6 receptor blockade through tocilizumab. JAMA Neurol. 2013;70:1056–1059. doi: 10.1001/jamaneurol.2013.143. [DOI] [PubMed] [Google Scholar]
- 99.Sellner J, Boggild M, Clanet M, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019–1032. doi: 10.1111/j.1468-1331.2010.03066.x. [DOI] [PubMed] [Google Scholar]
- 100.Food and Drug Administration (FDA) 2009. Safety Information – Rituximab Availale at: http://www.fda.gov/downloads/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/UCM187792.pdf (accessed 11 August 2013)
- 101.Carson KR, Focosi D, Major EO, et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol. 2009;10:816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 102.Kosmidis ML, Dalakas MC. Practical considerations on the use of rituximab in autoimmune neurological disorders. Ther Adv Neurol Disord. 2010;3:93–105. doi: 10.1177/1756285609356135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pellkofer HL, Krumbholz M, Berthele A, et al. Long-term follow-up of patients with neuromyelitis optica after repeated therapy with rituximab. Neurology. 2011;76:1310–1315. doi: 10.1212/WNL.0b013e3182152881. [DOI] [PubMed] [Google Scholar]
- 104.Horga A, Montalban X. FTY720 (fingolimod) for relapsing multiple sclerosis. Expert Rev Neurother. 2008;8:699–714. doi: 10.1586/14737175.8.5.699. [DOI] [PubMed] [Google Scholar]
- 105.Food and Drug Administration (FDA) 2010. Approved labelling text – Gilenya Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022527s000lbl.pdf (accessed 11 August 2013) [DOI] [PubMed]
- 106.European Medicines Agency (EMA) 2012. Summary of Product Characteristics – Gilenya Available at: http://www.ema.europa.eu/docs/en_GB/document_library/Other/2012/04/WC500125687.pdf (accessed 11 August 2013)
- 107.Cohen JA, Barkhof F, Comi G, et al. Fingolimod versus intramuscular interferon in patient subgroups from TRANSFORMS. J Neurol. 2013;260:2023–2032. doi: 10.1007/s00415-013-6932-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gergely P, Nuesslein-Hildesheim B, Guerini D, et al. The selective sphingosine 1-phosphate receptor modulator BAF312 redirects lymphocyte distribution and has species-specific effects on heart rate. Br J Pharmacol. 2012;167:1035–1047. doi: 10.1111/j.1476-5381.2012.02061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zarbin MA, Jampol LM, Jager RD, et al. Ophthalmic evaluations in clinical studies of fingolimod (FTY720) in multiple sclerosis. Ophthalmology. 2013;120:1432–1439. doi: 10.1016/j.ophtha.2012.12.040. [DOI] [PubMed] [Google Scholar]
- 110.Nolan R, Gelfand JM, Green AJ. Fingolimod treatment in multiple sclerosis leads to increased macular volume. Neurology. 2013;80:139–144. doi: 10.1212/WNL.0b013e31827b9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Reuters. Suspected PML on Gilenya Available at: http://www.reuters.com/article/2013/07/30/novartis-gilenya-idUSL6N0G02A520130730 (accessed 18 August 2013)
- 112.European Medicines Agency (EMA) 2012. European Medicines Agency Gives New Advice to Better Manage Risk of Adverse Effects on the Heart with Gilenya Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2012/04/news_detail_001498.jsp&mid=WC0b01ac058004d5c1 (accessed 11 August 2013)
- 113.Ricklin ME, Lorscheider J, Waschbisch A, et al. T-cell response against varicella-zoster virus in fingolimod-treated MS patients. Neurology. 2013;81:174–181. doi: 10.1212/WNL.0b013e31829a3311. [DOI] [PubMed] [Google Scholar]
- 114.Balague C, Pont M, Prats N, Godessart N. Profiling of dihydroorotate dehydrogenase, p38 and JAK inhibitors in the rat adjuvant-induced arthritis model: a translational study. Br J Pharmacol. 2012;166:1320–1332. doi: 10.1111/j.1476-5381.2012.01836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Miller AE, O'Connor P, Wolinsky JS, et al. Pre-specified subgroup analyses of a placebo-controlled phase III trial (TEMSO) of oral teriflunomide in relapsing multiple sclerosis. Mult Scler. 2012;18:1625–1632. doi: 10.1177/1352458512450354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.O'Connor P, Wolinsky JS, Confavreux C, et al. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011;365:1293–1303. doi: 10.1056/NEJMoa1014656. [DOI] [PubMed] [Google Scholar]
- 117.Sanofi/Genzyme. 2012. Aubagio Available at: http://products.sanofi.us/aubagio/aubagio.pdf (accessed 19 August 2013)
- 118.Food and Drug Administration (FDA) 2012. Approved labelling text – Aubagio Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202992s000lbl.pdf (accessed 27 October 2013)
- 119.Freedman MS, Wolinsky JS, Wamil B, et al. Teriflunomide added to interferon-beta in relapsing multiple sclerosis: a randomized phase II trial. Neurology. 2012;78:1877–1885. doi: 10.1212/WNL.0b013e318258f7d4. [DOI] [PubMed] [Google Scholar]
- 120.Confavreux C, Li DK, Freedman MS, et al. Long-term follow-up of a phase 2 study of oral teriflunomide in relapsing multiple sclerosis: safety and efficacy results up to 8·5 years. Mult Scler. 2012;18:1278–1289. doi: 10.1177/1352458512436594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Linker RA, Lee DH, Ryan S, et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain. 2011;134:678–692. doi: 10.1093/brain/awq386. [DOI] [PubMed] [Google Scholar]
- 122.Papadopoulou A, D'Souza M, Kappos L, Yaldizli O. Dimethyl fumarate for multiple sclerosis. Expert Opin Investig Drugs. 2010;19:1603–1612. doi: 10.1517/13543784.2010.534778. [DOI] [PubMed] [Google Scholar]
- 123.Gold R, Kappos L, Arnold DL, et al. Placebo-controlled phase 3 study of oral BG-12 for relapsing multiple sclerosis. N Engl J Med. 2012;367:1098–1107. doi: 10.1056/NEJMoa1114287. [DOI] [PubMed] [Google Scholar]
- 124.Fox RJ, Miller DH, Phillips JT, et al. Placebo-controlled phase 3 study of oral BG-12 or glatiramer in multiple sclerosis. N Engl J Med. 2012;367:1087–1097. doi: 10.1056/NEJMoa1206328. [DOI] [PubMed] [Google Scholar]
- 125.Schwab N, Ulzheimer JC, Fox RJ, et al. Fatal PML associated with efalizumab therapy: insights into integrin alphaLbeta2 in JC virus control. Neurology. 2012;78:458–467. doi: 10.1212/WNL.0b013e3182478d4b. discussion 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ermis U, Weis J, Schulz JB. PML in a patient treated with fumaric acid. N Engl J Med. 2013;368:1657–1658. doi: 10.1056/NEJMc1211805. [DOI] [PubMed] [Google Scholar]
- 127.Oosten van BW, Killestein J, Barkhof F, Polman CH, Wattjes MP. PML in a patient treated with dimethyl fumarate from a compounding pharmacy. N Engl J Med. 2013;368:1658–1659. doi: 10.1056/NEJMc1215357. [DOI] [PubMed] [Google Scholar]
- 128.Sweetser MT, Dawson KT, Bozic C. Manufacturer's response to case reports of PML. N Engl J Med. 2013;368:1659–1661. doi: 10.1056/NEJMc1300283. [DOI] [PubMed] [Google Scholar]
- 129.Hartung HP, Gonsette R, Konig N, et al. Mitoxantrone in progressive multiple sclerosis: a placebo-controlled, double-blind, randomised, multicentre trial. Lancet. 2002;360:2018–2025. doi: 10.1016/S0140-6736(02)12023-X. [DOI] [PubMed] [Google Scholar]
- 130.Edan G, Miller D, Clanet M, et al. Therapeutic effect of mitoxantrone combined with methylprednisolone in multiple sclerosis: a randomised multicentre study of active disease using MRI and clinical criteria. J Neurol Neurosurg Psychiatry. 1997;62:112–118. doi: 10.1136/jnnp.62.2.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wyngaert van de FA, Beguin C, D'Hooghe MB, et al. A double-blind clinical trial of mitoxantrone versus methylprednisolone in relapsing, secondary progressive multiple sclerosis. Acta Neurol Belg. 2001;101:210–216. [PubMed] [Google Scholar]
- 132.Cotte S, Stroet A, Ahsen van N, et al. Lack of therapeutic effect of mitoxantrone in primary progressive multiple sclerosis independent of pharmacogenetic factors: multicentre, retrospective analysis. Neurology. 2011;76:A613. P07.198. [Google Scholar]
- 133.Leary SM, Thompson AJ. Primary progressive multiple sclerosis: current and future treatment options. CNS Drugs. 2005;19:369–376. doi: 10.2165/00023210-200519050-00001. [DOI] [PubMed] [Google Scholar]
- 134.Pelfrey CM, Cotleur AC, Zamor N, Lee JC, Fox RJ. Immunological studies of mitoxantrone in primary progressive MS. J Neuroimmunol. 2006;175:192–199. doi: 10.1016/j.jneuroim.2006.03.003. [DOI] [PubMed] [Google Scholar]
- 135.Kornek B, Bernert G, Rostasy K, et al. Long-term follow-up of pediatric patients treated with mitoxantrone for multiple sclerosis. Neuropediatrics. 2011;42:7–12. doi: 10.1055/s-0031-1275345. [DOI] [PubMed] [Google Scholar]
- 136.Stroet A, Gold R, Chan A. Acute myeloid leukemia in Italian patients with multiple sclerosis treated with mitoxantrone. Neurology. 2012;78:933. doi: 10.1212/01.wnl.0000413366.14351.54. author reply 4. [DOI] [PubMed] [Google Scholar]
- 137.Martinelli V, Cocco E, Capra R, et al. Acute myeloid leukemia in Italian patients with multiple sclerosis treated with mitoxantrone. Neurology. 2011;77:1887–1895. doi: 10.1212/WNL.0b013e318238ee00. [DOI] [PubMed] [Google Scholar]
- 138.Pascual AM, Tellez N, Bosca I, et al. Revision of the risk of secondary leukaemia after mitoxantrone in multiple sclerosis populations is required. Mult Scler. 2009;15:1303–1310. doi: 10.1177/1352458509107015. [DOI] [PubMed] [Google Scholar]
- 139.Cotte S, Ahsen von N, Kruse N, et al. ABC-transporter gene-polymorphisms are potential pharmacogenetic markers for mitoxantrone response in multiple sclerosis. Brain. 2009;132:2517–2530. doi: 10.1093/brain/awp164. [DOI] [PubMed] [Google Scholar]
- 140.Food and Drug Administration (FDA) 2008. Mitoxantrone Hydrochloride (marketed as Novantrone and generics) – Healthcare Professional Sheet Available at: http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm126445.htm (accessed 11 August 2013)
- 141.Lee GR, Foerster J, Lukons J, Paraskevar F, Greer JP, Rodgers GM. Philadelphia, PA, USA: Lippincott Williams & Wilkins; [Google Scholar]
- 142.Le Page E, Leray E, Edan G. Long-term safety profile of mitoxantrone in a French cohort of 802 multiple sclerosis patients: a 5-year prospective study. Mult Scler. 2011;17:867–875. doi: 10.1177/1352458511398371. [DOI] [PubMed] [Google Scholar]
- 143.Goffette S, Pesch van V, Vanoverschelde JL, Morandini E, Sindic CJ. Severe delayed heart failure in three multiple sclerosis patients previously treated with mitoxantrone. J Neurol. 2005;252:1217–1222. doi: 10.1007/s00415-005-0839-3. [DOI] [PubMed] [Google Scholar]
- 144.Lo-Coco F, Avvisati G, Vignetti M, et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111–121. doi: 10.1056/NEJMoa1300874. [DOI] [PubMed] [Google Scholar]
- 145.Ammatuna E, Montesinos P, Hasan SK, et al. Presenting features and treatment outcome of acute promyelocytic leukemia arising after multiple sclerosis. Haematologica. 2011;96:621–625. doi: 10.3324/haematol.2010.036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Taube F, Stolzel F, Thiede C, Ehninger G, Laniado M, Schaich M. Increased incidence of central nervous system hemorrhages in patients with secondary acute promyelocytic leukemia after treatment of multiple sclerosis with mitoxantrone? Haematologica. 2011;96:e31. doi: 10.3324/haematol.2011.045583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Meyer C, Ansorge N, Siglienti I, et al. [Mitoxantrone-related acute leukemia by multiple sclerosis. Case report and practical approach by unclear cytopenia] Nervenarzt. 2010;81:1483–1489. doi: 10.1007/s00115-010-3041-5. [DOI] [PubMed] [Google Scholar]
- 148.Ellis R, Boggild M. Therapy-related acute leukaemia with mitoxantrone: what is the risk and can we minimise it? Mult Scler. 2009;15:505–508. doi: 10.1177/1352458508100967. [DOI] [PubMed] [Google Scholar]
- 149.Ghalie RG, Mauch E, Edan G, et al. A study of therapy-related acute leukaemia after mitoxantrone therapy for multiple sclerosis. Mult Scler. 2002;8:441–445. doi: 10.1191/1352458502ms836oa. [DOI] [PubMed] [Google Scholar]
- 150.Kingwell E, Koch M, Leung B, et al. Cardiotoxicity and other adverse events associated with mitoxantrone treatment for MS. Neurology. 2010;74:1822–1826. doi: 10.1212/WNL.0b013e3181e0f7e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mulroy E, Joyce E, Scott J, et al. Long-term risk of leukaemia or cardiomyopathy after mitoxantrone therapy for multiple sclerosis. Eur Neurol. 2012;67:45–47. doi: 10.1159/000334101. [DOI] [PubMed] [Google Scholar]
- 152.Paul F, Dorr J, Wurfel J, Vogel HP, Zipp F. Early mitoxantrone-induced cardiotoxicity in secondary progressive multiple sclerosis. BMJ Case Rep. 2009 doi: 10.1136/bcr.06.2009.2004. pii: bcr06.2009.2004. doi: 10.1136/bcr.06.2009.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Franzen D, Haus A, Hellmich M. Serial assessment of cardiac function during and following mitoxantrone infusion in 30 consecutive patients with multiple sclerosis. Mult Scler Int. 2010;2010:351045. doi: 10.1155/2010/351045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bernitsas E, Wei W, Mikol DD. Suppression of mitoxantrone cardiotoxicity in multiple sclerosis patients by dexrazoxane. Ann Neurol. 2006;59:206–209. doi: 10.1002/ana.20747. [DOI] [PubMed] [Google Scholar]
- 155.Dorr J, Bitsch A, Schmailzl KJ, et al. Severe cardiac failure in a patient with multiple sclerosis following low-dose mitoxantrone treatment. Neurology. 2009;73:991–993. doi: 10.1212/WNL.0b013e3181b878f6. [DOI] [PubMed] [Google Scholar]
- 156.Killestein J, Meer van der ML, Regelink JC, Huijgens PC, Polman CH. Severe cardiac failure in a patient with multiple sclerosis following low-dose mitoxantrone treatment. Neurology. 2010;74:934. doi: 10.1212/WNL.0b013e3181d2b7de. author reply. [DOI] [PubMed] [Google Scholar]
- 157.Debouverie M, Vandenberghe N, Morrissey SP, et al. Predictive parameters of mitoxantrone effectiveness in the treatment of multiple sclerosis. Mult Scler. 2004;10:407–412. doi: 10.1191/1352458504ms1066oa. [DOI] [PubMed] [Google Scholar]
- 158.Luchowski P, Mitosek-Szewczyk K, Bartosik-Psujek H, et al. B-type natriuretic peptide as a marker of subclinical heart injury during mitoxantrone therapy in MS patients = preliminary study. Clin Neurol Neurosurg. 2009;111:676–678. doi: 10.1016/j.clineuro.2009.06.007. [DOI] [PubMed] [Google Scholar]
- 159.Hasan SK, Buttari F, Ottone T, et al. Risk of acute promyelocytic leukemia in multiple sclerosis: coding variants of DNA repair genes. Neurology. 2011;76:1059–1065. doi: 10.1212/WNL.0b013e318211c3c8. [DOI] [PubMed] [Google Scholar]
- 160.Berlin JA, Glasser SC, Ellenberg SS. Adverse event detection in drug development: recommendations and obligations beyond phase 3. Am J Public Health. 2008;98:1366–1371. doi: 10.2105/AJPH.2007.124537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.European Medicines Agency (EMA) 2013. Summary of Product Characteristics – Soliris Available at: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000791/WC500054208.pdf (accessed 18 August 2013)
- 162.Anonymous. 2009. Summary of Product Characteristics Mitoxantrone/Ralenova Available at: http://www.medapharma.de/extras/download/gebrauchsinfo.pdf?pdfIdx=214 (accessed 11 August 2013)
- 163.Martinelli V, Radaelli M, Straffi L, Rodegher M, Comi G. Mitoxantrone: benefits and risks in multiple sclerosis patients. Neurol Sci. 2009;30(Suppl. 2):S167–170. doi: 10.1007/s10072-009-0142-7. [DOI] [PubMed] [Google Scholar]
- 164.Caruso A, Vecchio R, Patti F, Neri S. Drug rash with eosinophilia and systemic signs syndrome in a patient with multiple sclerosis. Clin Ther. 2009;31:580–584. doi: 10.1016/j.clinthera.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 165.Kumpfel T, Gerdes LA, Flaig M, Hohlfeld R, Wollenberg A. Drug-induced Sweet's syndrome after mitoxantrone therapy in a patient with multiple sclerosis. Mult Scler. 2011;17:495–497. doi: 10.1177/1352458510390069. [DOI] [PubMed] [Google Scholar]
- 166.Cocco E, Sardu C, Gallo P, et al. Frequency and risk factors of mitoxantrone-induced amenorrhea in multiple sclerosis: the FEMIMS study. Mult Scler. 2008;14:1225–1233. doi: 10.1177/1352458508094882. [DOI] [PubMed] [Google Scholar]
- 167.Strotmann JM, Spindler M, Weilbach FX, Gold R, Ertl G, Voelker W. Myocardial function in patients with multiple sclerosis treated with low-dose mitoxantrone. Am J Cardiol. 2002;89:1222–1225. doi: 10.1016/s0002-9149(02)02312-3. [DOI] [PubMed] [Google Scholar]
- 168.Brassat D, Recher C, Waubant E, et al. Therapy-related acute myeloblastic leukemia after mitoxantrone treatment in a patient with MS. Neurology. 2002;59:954–955. doi: 10.1212/wnl.59.6.954. [DOI] [PubMed] [Google Scholar]
- 169.Food and Drug Administration (FDA) 2013. Prescribing Information – Tysabri Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125104s813lbl.pdf (accessed 11 August 2013)
- 170.Oliver B, Fernandez O, Orpez T, et al. Kinetics and incidence of anti-natalizumab antibodies in multiple sclerosis patients on treatment for 18 months. Mult Scler. 2011;17:368–371. doi: 10.1177/1352458510385508. [DOI] [PubMed] [Google Scholar]
- 171.Mullen JT, Vartanian TK, Atkins MB. Melanoma complicating treatment with natalizumab for multiple sclerosis. N Engl J Med. 2008;358:647–648. doi: 10.1056/NEJMc0706103. [DOI] [PubMed] [Google Scholar]
- 172.Burmester GR, Rubbert-Roth A, Cantagrel A, et al. A randomised, double-blind, parallel-group study of the safety and efficacy of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional disease-modifying antirheumatic drugs in patients with moderate to severe rheumatoid arthritis (SUMMACTA study) Ann Rheum Dis. 2014;73:69–74. doi: 10.1136/annrheumdis-2013-203523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Smolen JS, Beaulieu A, Rubbert-Roth A, et al. Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet. 2008;371:987–997. doi: 10.1016/S0140-6736(08)60453-5. [DOI] [PubMed] [Google Scholar]
- 174.Genovese MC, McKay JD, Nasonov EL, et al. Interleukin-6 receptor inhibition with tocilizumab reduces disease activity in rheumatoid arthritis with inadequate response to disease-modifying antirheumatic drugs: the tocilizumab in combination with traditional disease-modifying antirheumatic drug therapy study. Arthritis Rheum. 2008;58:2968–2980. doi: 10.1002/art.23940. [DOI] [PubMed] [Google Scholar]