

Abstract
The scaffold protein caspase recruitment domain-containing protein 11 (CARD11) is implicated in the regulation of inflammation and autoimmunity. The present study aimed to explore the role of CARD11 in the pathogenesis of rheumatoid arthritis (RA). Mice with collagen-induced arthritis (CIA) were treated with either CARD11-targeted interfering RNA (CARD11 siRNA) or control siRNA by intraperitoneal injection every 3 days after CIA establishment. The clinical score of arthritis was recorded every other day. Synovial inflammation and cartilage erosion were evaluated by histology and microcomputed tomography (micro-CT). Serum anti-type II collagen (anti-CII) antibodies and cytokines were measured by enzyme-linked immunosorbent assay (ELISA). The CARD11/Bcl10 formation and nuclear factor-kappa B (NF-κB) activation was assessed by immunoprecipitation and immunoblotting, and the percentage of T helper type 17 (Th17) cells was determined by flow cytometry. Systemic administration of CARD11 siRNA significantly reduced the clinical score of CIA severity. As indicated by the histology, joint inflammation and destruction were attenuated by CARD11 siRNA treatment. Micro-CT demonstrated less severe joint destruction in CARD11 siRNA-treated mice than in control mice. CARD11 siRNA treatment resulted in inhibition of CARD11/Bcl10 formation and the subsequent NF-κB activation. In addition, treatment with CARD11 siRNA resulted in a pronounced decrease in proinflammatory cytokines interleukin (IL)-1β, IL-6 and IL-17. Serum anti-CII antibody and the percentage of Th17 cells were also significantly reduced. CARD11 is involved in the pathogenesis of CIA by formation of the CARD11/Bcl10 complex and enhancement of the Th17 cell response. Targeting CARD11 provides a novel research direction in the development of therapeutic strategies for RA.
Keywords: CARD11, collagen-induced arthritis, small interfering RNA, Th17 cells
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that primarily targets the joints and cartilage. Patients with RA usually present as progressive disease with excessive inflammatory response which contributes greatly to joint destruction and deformity, leading to functional disability 1,2. The past decades have witnessed striking improvements in the management of RA. Treatment for RA has evolved from the strategy of providing symptomatic relief to a combination of different conventional and biological disease-modifying anti-rheumatic drugs (DMARDs) 3. Despite these advances and improved long-term outcomes, 30–40% of RA patients are still inadequately controlled 4. Thus, novel therapeutic approaches with greater potency and less toxicity to halt this disease are required.
Caspase recruitment domain-containing protein 11 (CARD11), also termed CARMA1, belongs to both the CARD and the CARMA families. CARD11 is an essential adaptor protein that activates the nuclear factor (NF)-κB signalling pathway 5. Upon stimulation, active CARD11 functions as a scaffold molecule that assembles Bcl10, which in turn recruits mucosa-associated lymphoid tissue lymphoma translocation 1 (MALT1) to form the CARMA1/Bcl10/MALT1 (CBM) signalosome; the CBM signalosome subsequently recruits downstream signal proteins to trigger the inhibitor of nuclear factor kappa-B kinase (IKK) complex/NF-κB and c-Jun N-terminal kinase (JNK) activation collaboratively 6,7. In the CBM signalosome, the scaffold protein CARD11 is crucial for the recruitment of downstream signal proteins 7. Although the NF-κB signalling pathway is central to the regulation of inflammation and immune response in RA 8, the functional role of CARD11 in RA remains unclear. In the present study, we aimed to investigate whether CARD11 is involved in the pathogenesis of RA using the collagen-induced arthritis (CIA) model.
Materials and methods
Mice
Specific pathogen-free male dilute, brown and non-Agouti (DBA)/1 mice were obtained from the Shanghai SLAC Laboratory Animal Company (Shanghai, China). The animals were maintained in a barrier animal facility with a 12-h light/dark cycle at the Laboratory Animal Center, Sun Yat-sen University. Mice in the control and treatment groups were matched for age and sex. All experimental procedures were approved by the Ethics Committee of the First Affiliated Hospital, Sun Yat-sen University and performed in accordance with the National Institute of Health (NIH) guidelines for the use of experimental animals.
Induction of CIA model
CIA was generated according to the following protocol 9: on day 0, 8-week-old male DBA1/J mice were immunized with 100 μl of type II bovine collagen (Chondrex, Redmond, WA, USA) emulsified with an equal volume of complete Freund's adjuvant containing Mycobacterium tuberculosis (Beijing Biolead, Beijing, China). A second vaccination was given on day 21 using the same dose of type II bovine collagen emulsified with an equal volume of incomplete Freund's adjuvant.
CARD11-targeted interfering RNA (CARD11siRNA) treatment
The administration of in-vivo siRNA was based on previous reports 10,11. In brief, 0·45 mg/kg CARD11 siRNA (Santa Cruz Biotechnology, Santa Crux, CA, USA) was reconstituted in RNAase-free water, mixed with an equal volume of siPORT™ Amine Transfection Agent (Life Technologies Corporation, Grand Island, NY, USA) and then administered to the mice via intraperitoneal injection. A control sequence (scrambled siRNA) was injected intraperitoneally into the control mice. Western blot, using the primary antibody against CARD11 (1:1000 dilution, CST), was performed to assess the silencing effects.
Both groups of mice (n = 8 per group) were randomized and treated every 3 days for four injections at 6, 9, 12 and 15 days after boosted immunization, as evident arthritis symptoms were present in all mice 6 days after boosted immunization. The animals were killed 24 h after the last injection (16 days after boosted immunization).
Clinical scoring in CIA
The evaluation of inflammation was conducted as described previously 12. The total clinical score was defined as the sum of digit inflammation score (0–2 per animal) and paw swelling score (0–3 per paw), resulting in a maximum combined clinical score of 14 per animal. Digit inflammation scores were as follows: score 0 = no inflamed digit; score 0·5 = one to five digits inflamed; score 1 = six to 10 digits inflamed; score 1·5 = 11–15 digits inflamed and score 2 = 16 or more digits inflamed. Paw swelling (paw thickness increase compared to day 0 before CIA induction) was assessed as follows: score 0 ≤ 30% increase; score 1 > 30% increase; score 2 > 50% increase and score 3 > 80% increase.
Microcomputed tomography (micro-CT) analysis
Live mice were subjected to the micro-CT analyses under anaesthesia before being killed at the end of this study. Micro-CT imaging of joints was performed using a Siemens Inveon CT/PET Multimodality system.
Histological analysis
At the end of the experiment, mice were killed and hind limbs were fixed in 4% paraformaldehyde, decalcified in ethylenediamine tetraacetic acid (EDTA) solution for 21 days and then embedded in paraffin. Serial ankle joint sections (5 μm) were stained with haematoxylin and eosin according to standard protocols for morphological analysis. Safranin O staining was performed to estimate the proteoglycan content in the cartilage using the established procedure 12.
Histological evaluation in CIA
Synovial inflammation and cartilage erosion were evaluated and scored 12. Synovial inflammation was scored on a scale of 0–4, where 0 = no inflammation; 1 = slight thickening of the lining layer or some infiltrating cells in the sublining layer; 2 = slight thickening of the lining layer plus some infiltrating cells in the sublining layer; 3 = thickening of the lining layer, influx of cells in the sublining layer and the presence of cells in the synovial space; and 4 = synovium highly infiltrated with many inflammatory cells. Cartilage erosion was determined by the depletion of cartilage proteoglycan (loss of Safranin O staining) and scored on a scale of 0–4, where 0 = no destruction; 1 = minimal erosion limited to single spots; 2 = slight to moderate erosion in a limited area; 3 = more extensive areas of erosion; and 4 = general destruction.
Measurements of cytokines
Sera were obtained from anaesthetized animals by cardiac puncture at the end of the experiment under anaesthesia. Joints of mice were snap-frozen in liquid nitrogen and homogenized mechanically in cell lysis buffer (Cell Signaling Technology, Danvers, MA, USA) and homogenized followed by centrifugation at 15 000 g. The extracted protein and serum were subjected to enzyme-linked immunosorbent assay (ELISA) for detection of interleukin (IL)-1β, IL-6 and IL-17 (R&D Systems, Minneapolis, MN, USA), according to the manufacturer's protocols.
Measurement of anti-type II collagen (anti-CII) antibody
At termination of the study, serum anti-CII IgG antibody was determined using an ELISA as described previously 13. In brief, the ELISA plates (Corning, Inc., Corning, NY, USA) were coated overnight at 4°C with 1 mg/ml (0·1 ml/well) bovine type II collagen in carbonate/bicarbonate buffer. Then, the plates were washed three times with phosphate-buffered saline containing 0·05% Tween 20 (PBST) and blocked with 5% skimmed milk powder. Serum samples were diluted 50-fold in milk diluents, followed by threefold serial dilutions. Fifty μl of diluted sample was added per well in duplicate, the plates were incubated for 2 h at room temperature and then washed with PBST. Horseradish peroxidase (HRP)-conjugated goat anti-mouse immunoglobulin (Ig)G, IgG1 and IgG2a (Santa Cruz Technology, Santa Cruz, CA, USA) were added to each well and incubated for 2 h at room temperature. The plates were washed before the addition of 100 ml/well tetramethylbenzidine substrate (Sigma-Aldrich, St Louis, MO, USA). Reaction was stopped with 1 M H2SO4. Absorbance was read at 450 nm.
Immunoprecipitation and immunoblotting
Total or nuclear proteins from the splenocytes were isolated, respectively. Protein concentrations were determined by bicinchoninic acid assay (BCA) Protein Assay (Pierce Thermoscientific, Rockford, IL, USA). Immunoprecipitation and immunoblotting were performed to assess the CARD11/Bcl10 complex formation, as described previously 14. In brief, 300 μg of extracted protein were mixed with 1 μg/sample of the appropriate antibody, and samples were incubated overnight at 4°C. Following incubation, 10 μl of 50% protein G slurry was added, and the samples were incubated further for 1 h. Subsequently, the Tris-NaCl-Tween (TNT) buffer was used to wash the precipitates extensively. The resulting immunopurified proteins were used for immunoblotting experiments.
For the immunoblotting analysis, either the immunopurified protein complexes or, as indicated, protein extract was loaded onto a sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) 10% gel and transferred onto a nitrocellulose membrane. After blockade with 5% skimmed milk powder in TBST (Tris-buffered saline with 0·1% Tween 20), membranes were incubated with the following primary antibodies: mouse anti-CARD11 (Cell Signaling Technology), anti-Bcl10 (Cell Signaling Technology), anti-NF-κB p65 (Santa Cruz Technology) and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Santa Cruz Technology) or anti-fibrillarin (Cell Signaling Technology), respectively, overnight at 4°C. Protein visualization on the membrane was achieved using enhanced chemiluminescence (ECL) (Cell Signaling Technology).
Statistical analysis
Data are presented as the mean ± standard deviation (s.d.). Statistical analyses were processed by spss version 16·0 software. Statistical comparisons were conducted using Student's t-test or the Mann-Whitney U-test. P < 0·05 was considered statistically significant.
Results
CARD11 silencing reduced severity and radiographic damage in CIA mice
Mice received treatment starting at 6 days after boosted immunization. The clinical score of CIA mice was recorded every 2 days until the end of the study. Animals with targeted silencing of CARD11 showed a marked reduction in disease severity after two injections (day 10 after boosted immunization) compared to controls, determined by clinical disease severity score (Fig. 1a). Consistently, micro-CT imaging showed that CARD11 siRNA treatment reduced the joint destruction significantly compared with the control siRNA-treated animals (Fig. 1b).
Figure 1.
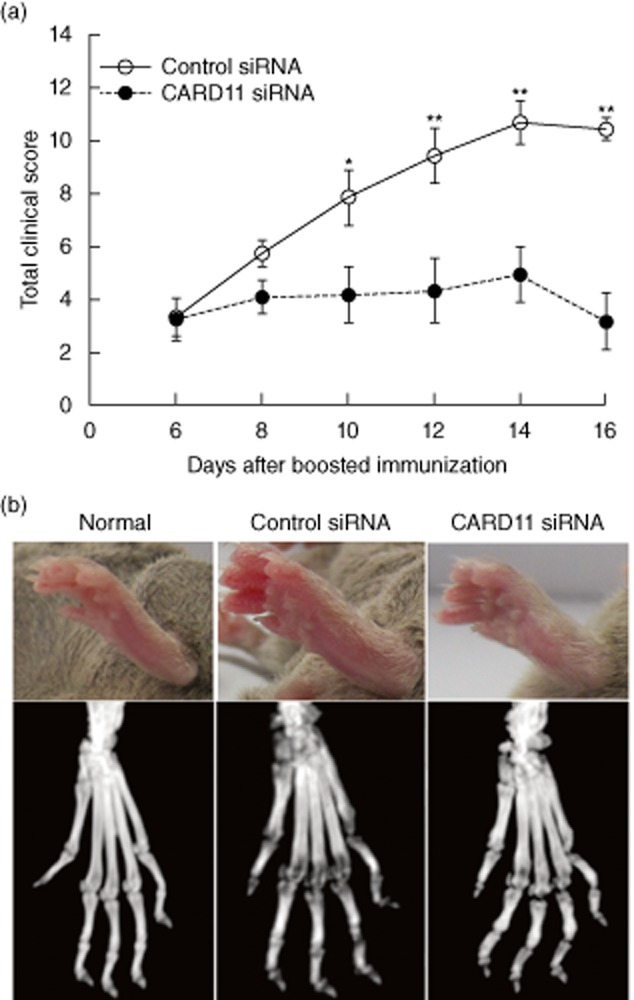
Caspase recruitment domain-containing protein 11 (CARD11) silencing reduced the severity of collagen-induced arthritis (CIA). (a) Clinical scoring in CIA. (b) Microcomputed tomography (micro-CT) analysis of live mice under anaesthesia using a Siemens Inveon CT/PET Multimodality system. Data are mean ± standard deviation (s.d.). *P < 0·05; **P < 0·01 versus control siRNA-treated group; n = 8 mice per group.
CARD11 silencing attenuated synovial inflammation and cartilage erosion in CIA mice
After CIA induction, control mice developed severe paw swelling and joint inflammation with massive inflammatory infiltration, synovial proliferation and pannus formation, whereas CARD11siRNA-treated mice showed less infiltration of leucocytes and synovial hyperplasia (Fig. 2a). In addition, as reflected by the Safranin O staining, cartilage erosion in the CIA mice was inhibited significantly by CARD11 siRNA treatment (Fig. 2b). The results were confirmed by subsequent semi-quantitative analysis of the histological score (Fig. 2c). These results showed that CARD11 silencing reduced the synovial inflammation and cartilage erosion.
Figure 2.
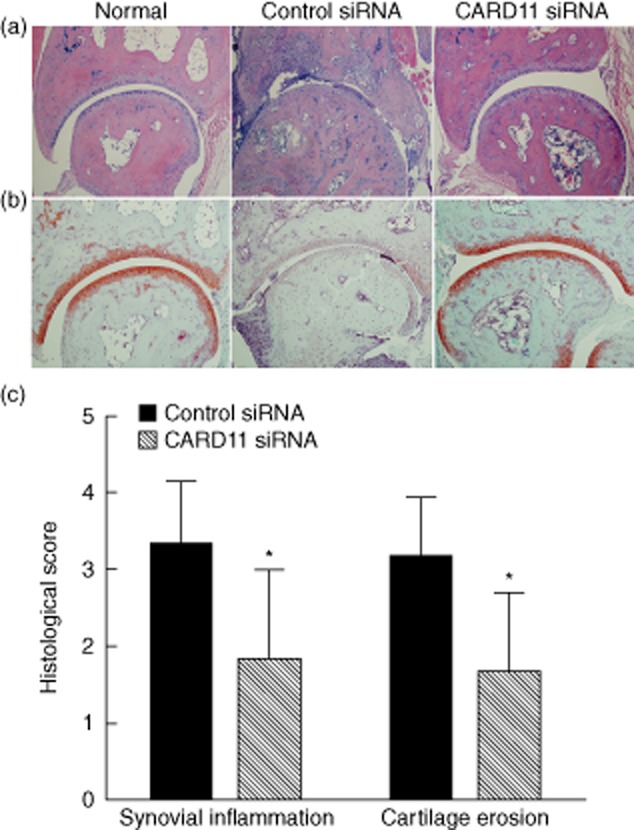
Caspase recruitment domain-containing protein 11 (CARD11) silencing suppressed joint destruction in collagen-induced arthritis (CIA). (a) Representative haematoxylin and eosin (H&E) staining images (×10). (b) Representative Safranin-O staining images (×10). (c) Semi-quanitative score. Data are mean ± standard deviation (s.d.). *P < 0·05 versus control siRNA-treated group; n = eight mice per group.
CARD11 silencing reduced levels of proinflammatory cytokines in CIA mice
Proinflammatory cytokines are key players in the development and progression of RA. We found that CARD11 siRNA administration resulted in a significant reduction in the levels of serum IL-1β, IL-6 and IL-17, as demonstrated by ELISA, in comparison with control siRNA treatment (Fig. 3a). Consistently, the levels of IL-1β, IL-6 and IL-17 in homogenized joints were also down-regulated significantly after CARD11 silencing (Fig. 3b). These results indicate that CARD11 blockade may have therapeutic effects on CIA via inhibiting the production of proinflammatory cytokines in this model.
Figure 3.
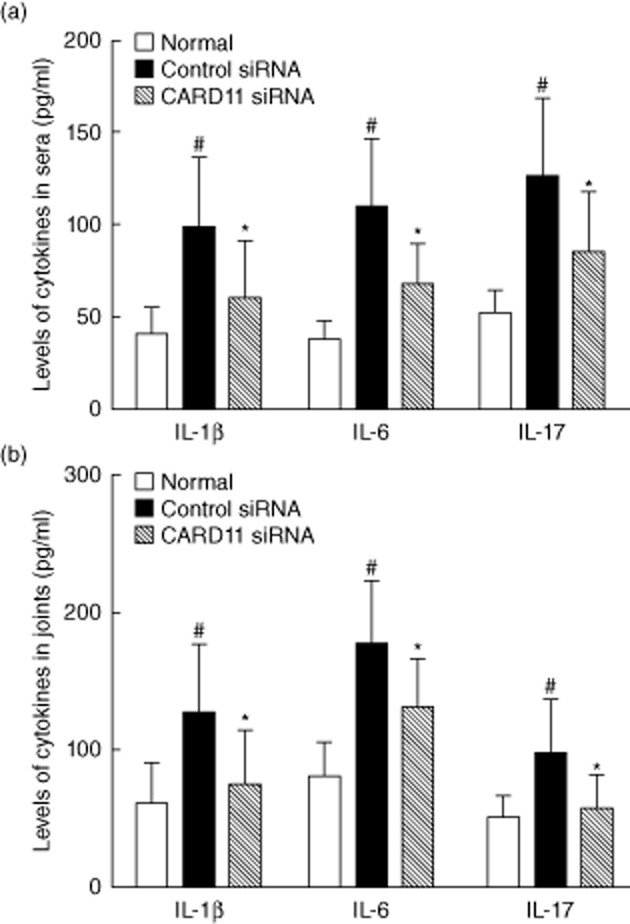
Caspase recruitment domain-containing protein 11 (CARD11) silencing inhibited the production of proinflammatory cytokines in collagen-induced arthritis (CIA). (a) Cytokine levels in sera. (b) Cytokine levels in joints. Data are mean ± standard deviation (s.d.). #P < 0·01 versus normal control; *P < 0·05 versus control siRNA-treated group; n = eight mice per group.
CARD11 silencing inhibited CARD11/Bcl10 assembly and NF-κB activation in the splenocytes in CIA mice
First, Western blot analysis of the splenocytes confirmed the silencing effect of CARD11 siRNA, showing that more than 80% of CARD11 protein expression was inhibited (Fig. 4a,b).
Figure 4.
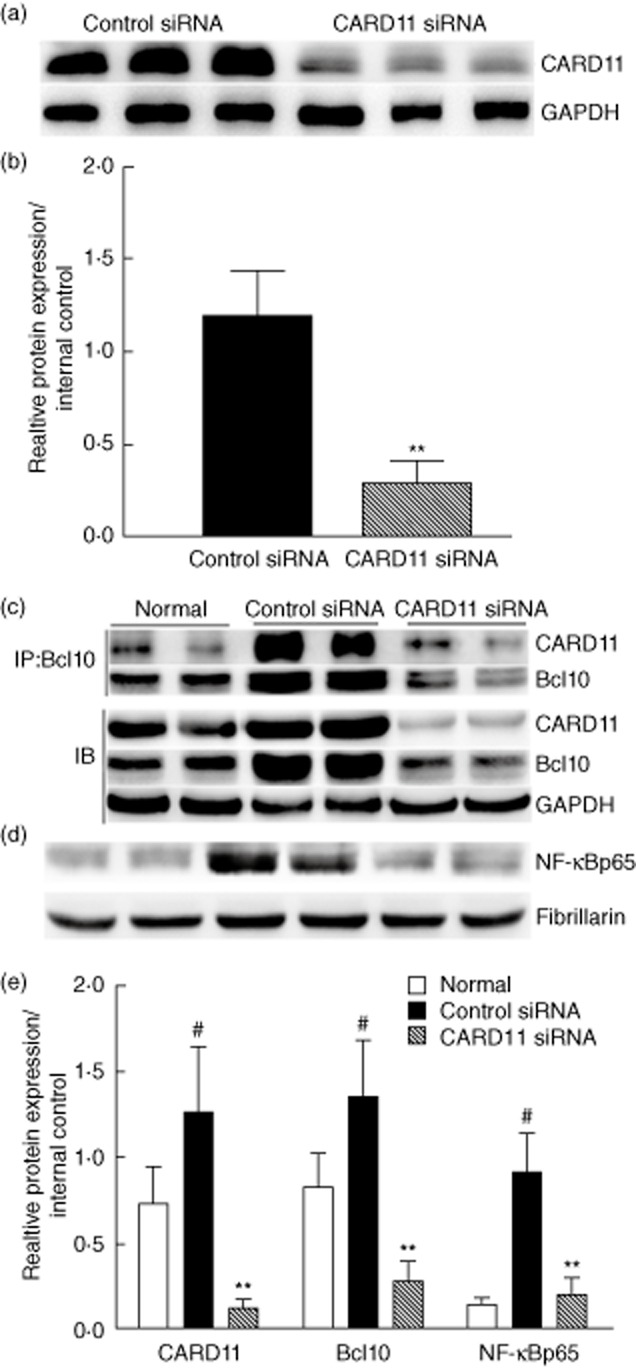
Caspase recruitment domain-containing protein 11 (CARD11) silencing inhibited CARD11/Bcl110 assembly and nuclear factor (NF)-κB activation. (a,b) CARD11 protein was reduced by 80% by siRNA treatment, as measured by Western blotting. (c) Bcl10 was immunoprecipitated (IP) from cell lysates. The resulting immunopurified protein complexes were subjected to immunoblot analyses (IB) with the indicated antibodies. To determine the cellular levels of the CARD11/Bcl110 complex, control immunoblot analyses were performed with the indicated antibodies. (d) CARD11 silencing inhibited NF-κB activation. (e) Analysis of band intensity of Western blot. Values are the mean ± standard deviation (s.d.). #P < 0·01 versus normal control; **P < 0·01 versus control siRNA-treated group; n = eight mice per group.
Then, we measured whether or not CARD11 siRNA administration suppressed the formation of CARD11/Bcl10 complex by immunoprecipitation. As shown in Fig. 4c, the formation of CARD11/Bcl10 complex in the splenocytes was inhibited significantly by CARD11 siRNA treatment compared to control mice. Furthermore, the expression of NF-κBp65 in the nuclear extracts (indicative of NF-κB activation) was also down-regulated significantly in the splenocytes in CIA treated with CARD11 siRNA in comparison with the control siRNA-treated animals (Fig. 4d). These findings suggest that CARD11 blockade suppressed CARD11/Bcl10 assembly and subsequent NF-κB activation in CIA mice.
CARD11 silencing decreased serum anti-CII antibody in CIA mice
We also determined the impact of CARD11 silencing on the production of autoantibody at the end of the study. The serum levels of total anti-CII-IgG, anti-CII IgG1 and anti-CII IgG2a antibodies were elevated dramatically in the control siRNA-treated mice compared with those in normal controls. In contrast, CARD11 siRNA-treated animals showed a significant reduction in the serum total anti-CII IgG, anti-CII IgG1 and anti-CII IgG2a antibodies (Fig. 5a).
Figure 5.
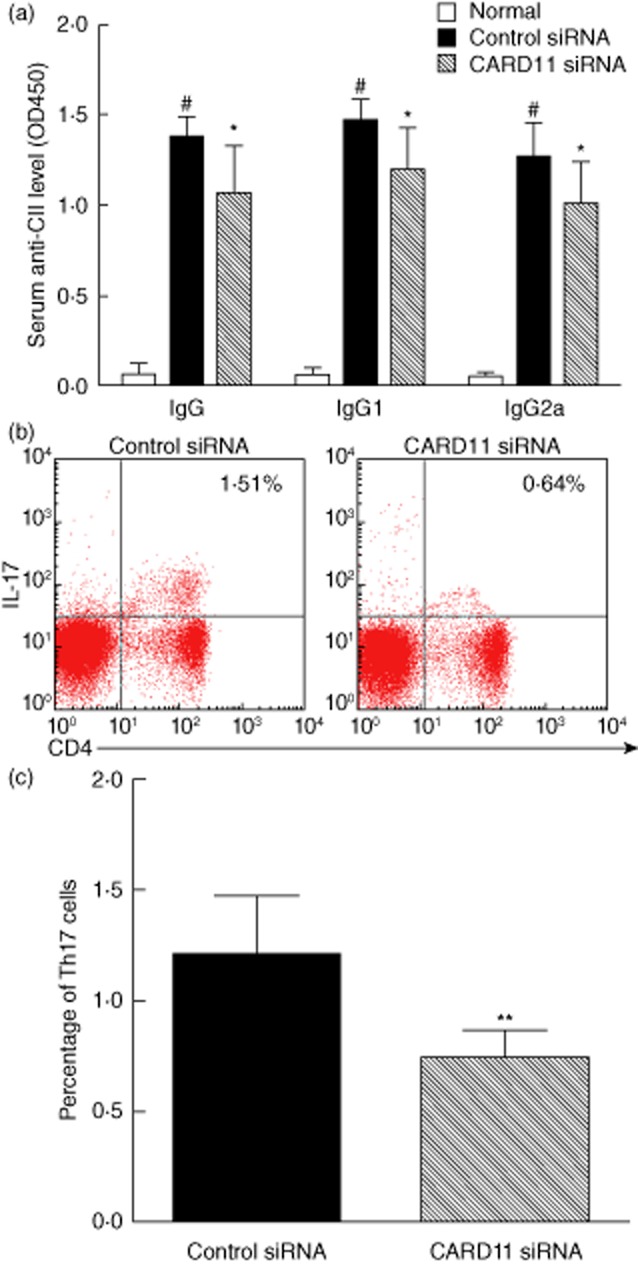
Caspase recruitment domain-containing protein 11 (CARD11) silencing reduced anti-CII antibodies and T helper type 17 (Th17) response in collagen-induced arthritis (CIA). (a) Serum level of total anti-CII-immunoglobulin (Ig)G, anti-CII IgG1 and anti-CII IgG2a antibodies. (b) Representive images of CD4+interleukin (IL)17+ Th17 cells by flow cytometry. (c) The percentage of Th17 cells in the splenocytes of CIA mice. Values are mean ± standard deviation (s.d.). #P < 0·01 versus normal control; *P < 0·05; **P < 0·01 versus control siRNA-treated group; n = eight mice per group.
CARD11 silencing reduced T helper type 17 (Th17) response in CIA mice
Furthermore, we analysed the percentage of Th17 cells in splenocytes by flow cytometry. CARD11 siRNA-treated mice showed a markedly reduced ratio of Th17 cells (CD4+IL-17+) in splenocytes in comparison with control siRNA-treated mice (P < 0·05) (Fig. 5b,c). These results were consistent with the above results, demonstrating markedly reduced levels of serum and intra-articular IL-17.
Discussion
Small RNA interference is now established as an important biological strategy for silencing genes both in vitro and in vivo 15,16. Using in-vivo silencing, we present evidence that knock-down of CARD11 by systemic administration of CARD11 siRNA reduced disease severity and synovial inflammation significantly in mice with established CIA. In addition, CARD11 siRNA treatment also attenuated joint destruction. Further investigation showed that the therapeutic effects of CARD11 blockade were mediated via inhibiting CARD11/Bcl10 assembly and Th17 response.
Several lines of evidence have suggested that CARD11 and its related signalling molecules are involved in inflammation and autoimmune responses 17–20. Mice deficient in CARD11 gene do not develop inflammation in a murine model of asthma due to a defect in the activation of naive T cells and NF-κB 17. Subsequent data have demonstrated a critical role for CARD11 in effector and memory T cell responses 18. CARD11 also plays an essential role in T cell differentiation, and adoptive transfer of bone marrow cells expressing constitutively active CARD11 resulted in lung inflammation 19. A recent report has indicated that CARD11 knock-out mice are resistant to experimental autoimmune encephalomyelitis 20. However, it remains unknown whether CARD11 plays crucial role in the development of RA. In the present study, we found that CARD11 blockade attenuated joint inflammation and destruction in mouse CIA, suggesting the involvement of the CARDA11 signalling pathway in the pathogenesis of RA.
It is well accepted that NF-κB activation and proinflammatory cytokines contribute to inflammation and development of RA 8,21. NF-κB activation occurs in cultured synovial fibroblasts and synovial tissues from RA patients. Animal models of RA have also confirmed the pivotal role of NF-κB in the development and progression of RA 22. Of interest, intra-articular or systemic blockade of NF-κB signalling is effective in the treatment of arthritis in animal models of RA 23,24. Genetic analysis also reveals the association of several NF-κB-related genes with RA pathogenesis 25. Our results suggested that in-vivo inhibition of CARD11 (upstream of NF-κB activation) suppressed the activation of NF-κB, which conferred benefit on CIA.
In addition to its direct role in RA, NF-κB is also crucial for the expression of multiple proinflammatory genes in the microenvironment of the arthritic joints, including IL-1, IL-6 and IL-17, that exert their influences both on osteoclast differentiation and osteoblasts, thereby contributing to progressive joint destruction in RA 21. Overexpression of IL-1β or deficiency of the soluble IL-1 receptor antagonist (IL-1Ra) in murine RA models resulted in the development of arthritis with associated bone and cartilage destruction 26,27. Mice deficient in IL-6 were protected against bone destruction in an antigen-induced arthritis model 28. Notably, tocilizumab (a recombinant humanized anti-IL-6 receptor monoclonal antibody) has been shown to be effective for RA patients in several clinical trials 29. IL-17 production by activated peripheral blood mononuclear cells (PBMC) from RA patients was blocked by NF-κB inhibition, suggesting that the NF-κB pathway is involved in the overproduction of IL-17 in RA 30. IL-17, as the signature cytokine of Th17 cells, can enhance the production of IL-1β and IL-6 31. Importantly, targeting IL-17 has demonstrated therapeutic effects in animal models of inflammatory and autoimmune diseases, including RA 32. Our data showed that silencing of CARD11 in vivo suppressed the production of IL-1β, IL-6 and IL-17. These results suggest that CARD11 may promote the development of RA via NF-κB activation and expression of proinflammatory cytokines.
Humoral and cellular autoimmunity play indispensable roles in the pathogenesis of RA. Human anti-CII antibody has been reported to be associated with increased joint destruction in patients with RA 33. We also investigated the effects of CARD11 blockade on the production of autoantibody. CARD11 siRNA treatment suppressed production of total anti-CII-IgG, anti-CII-IgG1 and anti-CII-IgG2a antibodies. In support of our findings, a recent study by Jeelall et al. demonstrated that CARD11 was involved in autoantibody secretion in vivo 34. Additionally, the present study demonstrated that CARD11 inhibition reduced the Th17 response. Th17 cells are pathogenic in several autoimmune and inflammatory diseases, including RA 35. CARD11 also critically links NF-κB signalling activation to completion of Th17 differentiation 20,32. Our findings suggest that CARD11 signalling promotes aberrant responses of B cells and Th17 cells, resulting in humoral and cellular autoimmunity.
Of note, CARD11 is a key regulator in T cell/B cell co-operation and the normal immune response. Impaired CARD11 function might affect T cell/B cell co-operation and result in immunodeficiency 36,37. Under disease conditions, overactivation of CARD11 may contribute to abundance of inflammatory cytokines in a wide range of autoimmune and inflammatory diseases 17,38, and targeting CARD11 may be a potential therapeutic strategy for these disorders 39. However, one should bear in mind that the concerns of severe infection be addressed when a key signalling pathway is targeted, as in the case of tofacitinib, a JAK kinase inhibitor used for treatment of RA, which was shown to lead to a significant increase in the serious infection rate compared with controls 40. Our results showed that serum levels of cytokines were reduced significantly after CARD11 siRNA treatment, but were still higher than those in the normal controls (Fig. 4). In addition, NF-κB activation in the siRNA-treated group was comparable to that of normal mice, suggesting that profound immunosuppression was less likely to occur.
Taken together, silencing of CARD11 in vivo inhibited the development of experimental autoimmune arthritis. The therapeutic effects were mediated by inhibition of CARD11/Bcl10 assembly and subsequent NF-κB activation, Th17 cell response and autoantibody production, indicating that CARD11 is involved in the pathogenesis of CIA by formation of the CARD11/Bcl10 complex and enhancement of humoral and cellular autoimmunity. Targeting of the CARD11 pathway provides a novel research direction in the development of therapeutic strategies for RA.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81273278), the PhD Program Foundation of Ministry of Education of China (20120171110064), Guangdong Natural Science Foundation (S2012010008780 and S2011010004578), Guangzhou Science and Technology Planning Program (2012J4100085) and Sun Yat-sen Innovative Talents Cultivation Program for Doctoral Graduate Students (Jijun Zhao).
Disclosure
The authors declare no conflicts of interest.
References
- 1.Welsing PM, van Gestel AM, Swinkels HL, Kiemeney LA, van Riel PL. The relationship between disease activity, joint destruction, and functional capacity over the course of rheumatoid arthritis. Arthritis Rheum. 2001;44:2009–2017. doi: 10.1002/1529-0131(200109)44:9<2009::AID-ART349>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 2.Kwok SK, Cho ML, Park MK, et al. Interleukin-21 promotes osteoclastogenesis in humans with rheumatoid arthritis and in mice with collagen-induced arthritis. Arthritis Rheum. 2012;64:740–751. doi: 10.1002/art.33390. [DOI] [PubMed] [Google Scholar]
- 3.Upchurch KS, Kay J. Evolution of treatment for rheumatoid arthritis. Rheumatology (Oxf) 2012;51(Suppl. 6):vi28–36. doi: 10.1093/rheumatology/kes278. [DOI] [PubMed] [Google Scholar]
- 4.Yazici Y. Treatment of rheumatoid arthritis: we are getting there. Lancet. 2009;374:178–180. doi: 10.1016/S0140-6736(09)60792-3. [DOI] [PubMed] [Google Scholar]
- 5.Bertin J, Wang L, Guo Y, et al. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J Biol Chem. 2001;276:11877–11882. doi: 10.1074/jbc.M010512200. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y, Song R, Gao Y, et al. Protein kinase C-delta negatively regulates T cell receptor-induced NF-kappaB activation by inhibiting the assembly of CARMA1 signalosome. J Biol Chem. 2012;287:20081–20087. doi: 10.1074/jbc.M111.335463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blonska M, Lin X. CARMA1-mediated NF-kappaB and JNK activation in lymphocytes. Immunol Rev. 2009;228:199–211. doi: 10.1111/j.1600-065X.2008.00749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Loo G, Beyaert R. Negative regulation of NF-kappaB and its involvement in rheumatoid arthritis. Arthritis Res Ther. 2011;13:221. doi: 10.1186/ar3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simelyte E, Rosengren S, Boyle DL, Corr M, Green DR, Firestein GS. Regulation of arthritis by p53: critical role of adaptive immunity. Arthritis Rheum. 2005;52:1876–1884. doi: 10.1002/art.21099. [DOI] [PubMed] [Google Scholar]
- 10.Zhao J, Wang H, Dai C, et al. P2X7 blockade attenuates lupus nephritis by inhibiting NLRP3/ASC/caspase-1 activation. Arthritis Rheum. 2013;65:3176–3185. doi: 10.1002/art.38174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mezzaroma E, Toldo S, Farkas D, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci USA. 2011;108:19725–19730. doi: 10.1073/pnas.1108586108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Camps M, Ruckle T, Ji H, et al. Blockade of PI3Kgamma suppresses joint inflammation and damage in mouse models of rheumatoid arthritis. Nat Med. 2005;11:936–943. doi: 10.1038/nm1284. [DOI] [PubMed] [Google Scholar]
- 13.Suchard SJ, Stetsko DK, Davis PM, et al. An LFA-1 (alphaLbeta2) small-molecule antagonist reduces inflammation and joint destruction in murine models of arthritis. J Immunol. 2010;184:3917–3926. doi: 10.4049/jimmunol.0901095. [DOI] [PubMed] [Google Scholar]
- 14.Palkowitsch L, Marienfeld U, Brunner C, Eitelhuber A, Krappmann D, Marienfeld RB. The Ca2+-dependent phosphatase calcineurin controls the formation of the Carma1-Bcl10-Malt1 complex during T cell receptor-induced NF-kappaB activation. J Biol Chem. 2011;286:7522–7534. doi: 10.1074/jbc.M110.155895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Musacchio T, Torchilin VP. siRNA delivery: from basics to therapeutic applications. Front Biosci (Landmark Ed) 2013;18:58–79. doi: 10.2741/4087. [DOI] [PubMed] [Google Scholar]
- 16.Courties G, Baron M, Presumey J, et al. Cytosolic phospholipase A2alpha gene silencing in the myeloid lineage alters development of Th1 responses and reduces disease severity in collagen-induced arthritis. Arthritis Rheum. 2011;63:681–690. doi: 10.1002/art.30174. [DOI] [PubMed] [Google Scholar]
- 17.Medoff BD, Seed B, Jackobek R, et al. CARMA1 is critical for the development of allergic airway inflammation in a murine model of asthma. J Immunol. 2006;176:7272–7277. doi: 10.4049/jimmunol.176.12.7272. [DOI] [PubMed] [Google Scholar]
- 18.Ramadas RA, Roche MI, Moon JJ, Ludwig T, Xavier RJ, Medoff BD. CARMA1 is necessary for optimal T cell responses in a murine model of allergic asthma. J Immunol. 2011;187:6197–6207. doi: 10.4049/jimmunol.1101348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blonska M, Joo D, Zweidler-McKay PA, Zhao Q, Lin X. CARMA1 controls Th2 cell-specific cytokine expression through regulating JunB and GATA3 transcription factors. J Immunol. 2012;188:3160–3168. doi: 10.4049/jimmunol.1102943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Molinero LL, Cubre A, Mora-Solano C, Wang Y, Alegre MLT. cell receptor/CARMA1/NF-kappaB signaling controls T-helper (Th) 17 differentiation. Proc Natl Acad Sci USA. 2012;109:18529–18534. doi: 10.1073/pnas.1204557109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karmakar S, Kay J, Gravallese EM. Bone damage in rheumatoid arthritis: mechanistic insights and approaches to prevention. Rheum Dis Clin North Am. 2010;36:385–404. doi: 10.1016/j.rdc.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roman-Blas JA, Jimenez SA. Targeting NF-kappaB: a promising molecular therapy in inflammatory arthritis. Int Rev Immunol. 2008;27:351–374. doi: 10.1080/08830180802295740. [DOI] [PubMed] [Google Scholar]
- 23.Min SY, Yan M, Du Y, et al. Intra-articular nuclear factor-kappaB blockade ameliorates collagen-induced arthritis in mice by eliciting regulatory T cells and macrophages. Clin Exp Immunol. 2013;172:217–227. doi: 10.1111/cei.12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okamoto H, Yoshio T, Kaneko H, Yamanaka H. Inhibition of NF-kappaB signaling by fasudil as a potential therapeutic strategy for rheumatoid arthritis. Arthritis Rheum. 2010;62:82–92. doi: 10.1002/art.25063. [DOI] [PubMed] [Google Scholar]
- 25.Criswell LA. Gene discovery in rheumatoid arthritis highlights the CD40/NF-kappaB signaling pathway in disease pathogenesis. Immunol Rev. 2010;233:55–61. doi: 10.1111/j.0105-2896.2009.00862.x. [DOI] [PubMed] [Google Scholar]
- 26.Ghivizzani SC, Kang R, Georgescu HI, et al. Constitutive intra-articular expression of human IL-1 beta following gene transfer to rabbit synovium produces all major pathologies of human rheumatoid arthritis. J Immunol. 1997;159:3604–3612. [PubMed] [Google Scholar]
- 27.Horai R, Saijo S, Tanioka H, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191:313–320. doi: 10.1084/jem.191.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ohshima S, Saeki Y, Mima T, et al. Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci USA. 1998;95:8222–8226. doi: 10.1073/pnas.95.14.8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ash Z, Emery P. The role of tocilizumab in the management of rheumatoid arthritis. Expert Opin Biol Ther. 2012;12:1277–1289. doi: 10.1517/14712598.2012.707178. [DOI] [PubMed] [Google Scholar]
- 30.Kim KW, Cho ML, Park MK, et al. Increased interleukin-17 production via a phosphoinositide 3-kinase/Akt and nuclear factor kappaB-dependent pathway in patients with rheumatoid arthritis. Arthritis Res Ther. 2005;7:R139–148. doi: 10.1186/ar1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chabaud M, Fossiez F, Taupin JL, Miossec P. Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol. 1998;161:409–414. [PubMed] [Google Scholar]
- 32.Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. 2012;11:763–776. doi: 10.1038/nrd3794. [DOI] [PubMed] [Google Scholar]
- 33.Mullazehi M, Wick MC, Klareskog L, van Vollenhoven R, Ronnelid J. Anti-type II collagen antibodies are associated with early radiographic destruction in rheumatoid arthritis. Arthritis Res Ther. 2012;14:R100. doi: 10.1186/ar3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jeelall YS, Wang JQ, Law HD, et al. Human lymphoma mutations reveal CARD11 as the switch between self-antigen-induced B cell death or proliferation and autoantibody production. J Exp Med. 2012;209:1907–1917. doi: 10.1084/jem.20112744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol. 2012;181:8–18. doi: 10.1016/j.ajpath.2012.03.044. [DOI] [PubMed] [Google Scholar]
- 36.Stepensky P, Keller B, Buchta M, et al. Deficiency of caspase recruitment domain family, member 11 (CARD11), causes profound combined immunodeficiency in human subjects. J Allergy Clin Immunol. 2013;131:477–485. doi: 10.1016/j.jaci.2012.11.050. [DOI] [PubMed] [Google Scholar]
- 37.Greil J, Rausch T, Giese T, et al. Whole-exome sequencing links caspase recruitment domain 11 (CARD11) inactivation to severe combined immunodeficiency. J Allergy Clin Immunol. 2013;131:1376–1833. doi: 10.1016/j.jaci.2013.02.012. [DOI] [PubMed] [Google Scholar]
- 38.Rajasekaran K, Kumar P, Schuldt KM, et al. Signaling by Fyn-ADAP via the Carma1-Bcl-10-MAP3K7 signalosome exclusively regulates inflammatory cytokine production in NK cells. Nat Immunol. 2013;14:1127–1136. doi: 10.1038/ni.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roche MI, Ramadas RA, Medoff BD. The role of CARMA1 in T cells. Crit Rev Immunol. 2013;33:219–243. doi: 10.1615/critrevimmunol.2013007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van Vollenhoven RF, Fleischmann R, Cohen S, et al. Tofacitinib or adalimumab versus placebo in rheumatoid arthritis. N Engl J Med. 2012;367:508–519. doi: 10.1056/NEJMoa1112072. [DOI] [PubMed] [Google Scholar]