

Abstract
Toll-like receptors (TLRs) recognize pathogen-associated molecular patterns and results in innate immune system activation that results in elicitation of the adaptive immune response. One crucial modulator of the adaptive immune response is CD40. However, whether these molecules influence each other's expression and functions is not known. Therefore, we examined the effects of TLRs on CD40 expression on macrophages, the host cell for the protozoan parasite L eishmania major. While polyinosinic-polycytidylic acid [poly (I:C)], a TLR-3 ligand, lipopolysaccharide (LPS), a TLR-4 ligand, imiquimod, a TLR-7/8 ligand and cytosine–phosphate–guanosine (CpG), a TLR-9 ligand, were shown to enhance CD40 expression, CD40 stimulation enhanced only TLR-9 expression. Therefore, we tested the synergism between CD40 and CpG in anti-leishmanial immune response. In L eishmania-infected macrophages, CpG was found to reduce CD40-induced extracellular stress-regulated kinase (ERK)1/2 activation; with the exception of interleukin (IL)-10, these ligands had differential effects on CD40-induced IL-1α, IL-6 and IL-12 production. CpG significantly enhanced the anti-leishmanial function of CD40 with differential effects on IL-4, IL-10 and interferon (IFN)-γ production in susceptible BALB/c mice. Thus, we report the first systematic study on CD40–TLR cross-talk that regulated the experimental L . major infection.
Keywords: CD40, Leishmania, macrophages, Toll-like receptor
Introduction
Toll-like receptors (TLRs) recognize various molecular patterns on pathogens and activate the cells of the innate immune system 1–3, such as macrophages and dendritic cells, both of which are antigen-presenting cells. It is reported that activation of the innate immune system by the ligands of TLR results in the activation of the adaptive immune system, particularly the T cells 4,5. The TLR-expressing antigen-presenting cells provide not only the major histocompatibility complex (MHC)–peptide complex for recognition by the antigen-specific T cell receptor but also the co-stimulatory signals through co-stimulatory ligand–receptor interaction, such as CD80–CD28 and CD40–CD40–ligand interactions for optimal T cell activation 6–8. It is reported that in the absence of CD40 or CD40-L, infections with Leishmania major or L. amazonensis exacerbate due to deficient T cell response 9–11. We have also shown that CD40 exerts strong anti-leishmanial activity 12–14. Thus, once it is known that TLRs activate the adaptive immune system and that CD40 is a crucial component in the anti-leishmanial T cell responses, it remains to be examined whether TLRs and CD40 modulate each other's expressions and Leishmania major infection.
TLRs recognize different ligands with restricted specificity. It is shown that TLR-2 heterodimerizes with TLR-1 or TLR-6 and recognizes triacylated or diacylated lipopeptides, respectively 15. TLR-3, TLR-4 and TLR-5 recognize polyinosinic-polycytidylic acid [poly (I:C)], lipolysaccharide (LPS) complex and flagellin, respectively, whereas TLR-7/-8, TLR-9 and TLR-11 are shown to recognize imiquimod, cytosine–phosphate–guanosine (CpG) and profilin, respectively 16–19. Thus, each of these ligands is used to activate cells through a specific TLR. Once these ligands bind to their specific TLRs, signals induce the production of proinflammatory cytokines such as interleukin (IL)-1α, IL-6 and IL-12 20, which serve as an index of cellular activation. Another measure of activation is the control of intracellular pathogens such as L. major. It has been reported that several TLRs play important roles in the control of Leishmania infections 21. As CD40 is also shown to exert anti-leishmanial functions in macrophages 12–14, it remains to be assessed whether the TLR ligands can alter the anti-leishmanial effects of CD40 in macrophages and in susceptible hosts such as BALB/c mice.
Based on these arguments, we examined whether TLRs and CD40 modulated each other's expressions in thioglycolate-elicited macrophages and whether TLRs could modulate the CD40-induced phosphorylation of p38 mitogen-activated protein kinase (MAPK), extracellular stress-regulated kinase (ERK)-1/2 and nuclear factor (NF)-κB and the CD40-induced release of IL-1α, IL-6, IL-10 and IL-12 from macrophages. Finally, we tested whether the TLR ligands with distinct effects on CD40-induced functions could modulate the L. major infection in BALB/c mice. We observed that most TLRs enhanced CD40 expression, but the effects of poly (I:C), LPS, imiquimod and CpG were most prominent. Conversely, CD40 altered the expression of only TLR-9. These ligands had differential effects on CD40 signalling, cytokine induction and parasite load. Corroborating these observations, these ligands, CpG in particular, were found to exert significant anti-leishmanial effects, accompanied by differential effects on cytokine production in BALB/c mice. Thus, we report the CD40–TLR cross-talk for the first time, to our knowledge.
Materials and methods
Reagents
Antibodies specific for p-p38 MAPK, total p38 MAPK, pERK-1/2, total ERK-1/2 and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), p-NF-κB, total NF-κB (Cell Signaling Technology, Danvers, MA), anti-cytokine antibodies [IL-1α, IL-6, IL-12, IL-10, interferon (IFN)-γ and IL-4] and standard cytokines for enzyme-linked immunosorbent assay (ELISA), anti-CD40 antibody [no-azide/low endotoxin (NA/LE); clone 3/23], anti-CD11b, anti-CD16/32, anti-CD40 antibody for fluorescence-activated cell sorter (FACS) analyses, anti-CD3 and anti-CD28 antibodies (BD Biosciences, San Diego, CA, USA) were procured. TLR ligands – Pam3CSK4, peptidoglycan (PGN), poly (I:C), LPS, flagellin, fibroblast-stimulating lipopeptide (FSL), imiquimod and CpG from (Invivogen, San Diego, CA, USA) and Profilin (Alexis, San Diego, CA, USA) – were procured, as mentioned. TLR-9 antibody and myeloid differentiation-associated gene 88 (MyD88) inhibitory and control peptides were from Imgenex (San Diego, CA, USA). IL-4 and granulocyte–macrophage colony-stimulating factor (GM-CSF) were procured from BD Biosciences.
Mice, parasites and infection
Susceptible BALB/c mice were procured from Jackson Laboratories (Bar Harbour, ME, USA). Mice were bred, maintained and monitored by resident veterinarians in the National Centre for Cell Science's experimental animal facility. The animal use protocol was approved by the Institutional Animal Care and Use Committee (IACUC) and the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), the regulatory authorities for animal experimentation in the country.
The L. major strain (MHOM/Su73/5ASKH) was maintained in vitro in RPMI-1640, 10% fetal calf serum (FCS); for maintenance of virulence, the parasite was passaged regularly through BALB/c mice by subcutaneous infection of the stationary-phase promastigotes (2 × 106/mouse).
The BALB/c-derived, thioglycolate-elicited macrophages were harvested and plated in vitro. In some experiments macrophages were infected with L. major promastigotes at a 1:10 ratio for 12 h, followed by washing of the extracellular parasites, cultured with or without TLR ligand or anti-CD40 antibody for 60 h and Giemsa-stained, followed by enumeration of amastigotes per 100 macrophages.
Generation of bone marrow-derived dendritic cells (BMDC)
DC were generated from bone marrow progenitor cells. BALB/c-derived femoral cells were cultured in DC culture media (advanced RPMI medium, 10% FCS, 100 U/ml penicillin, 100 μg/ml streptomycin, 50 μM 2-mercaptoethanol, 10 ng/ml IL-4 and 20 ng/ml GM-CSF) in a 24-well plate (1 × 106/ml/well). Culture medium was replaced with fresh medium on alternate days till day 6. BMDC were dislodged for further experiments; 2·5 × 106 cells were plated in six-well plates and infected with L. major promastigotes at a 1:10 ratio, 64 h cells were stimulated with TLR ligands for a further 8 h (total 72 h), then total RNA was extracted and reverse transcription–polymerase chain reaction (RT–PCR) was performed 22.
Reverse transcriptase PCR and real-time PCR
BALB/c-derived 2·5 × 106 peritoneal macrophages, uninfected or 64 h L. major-infected, were treated with or without TLR ligands alone or in combination with anti-CD40 antibody for further 8 h (total 72 h of infection). RNA was extracted using TRI-reagent (Sigma-Aldrich, St Louis, MO, USA); cDNA was synthesized and amplified using gene-specific primers, as described previously 23. Each sample was amplified for mouse glyceraldehyde 3-phosphate dehydrogenase (GAPDH) to ensure equal cDNA input. Amplified PCR products were analysed in 1·2% agarose gel. The primers were: GAPDH, forward 5′-GAG CCA AAC GGG TCA TCA TC-3′, reverse 5′-CCT GCT TCA CCA CCT TCT TG-3′; IL-12 p40 forward 5′-CAC GCC TGA AGA AGA TGA CA-3′, reverse 5′-GAC AGA GAC GCC ATT CCA CA-3′; inducible nitric oxide synthase (iNOS), forward 5′-CAG AGG ACC CAG AGA CAA GC-3′, reverse 5′-AAG ACC AGA GGC AGC ACA TC- 3′; and CD40, forward 5′-TCC CTG CCC AGT CGG CTT CT-3′, reverse 5′-CTG TCT TGG CTC ATC TCA AA-3′.
Real-time PCR was performed using the cDNA sample, as prepared above, in a 10-μl reaction mixture containing 10 ng cDNA, 2 ng forward primer, 2 ng reverse primer and 2× iQ SYBR Green Supermix (5 μl; Bio-Rad; Hercules, CA, USA) in thin-walled 0·2-ml strip tubes (Axygen, Union City, CA, USA). Quantitative real-time PCR was performed on an Eppendorf realplex4 Mastercycler under the following conditions: 95°C for 2 min, 40 cycles of 95°C for 1 min, 60°C for 30 s and 72°C for 35 s. Reactions were performed in duplicate. Relative quantitation was performed by normalizing the ΔCt value of the target gene with the ΔCt value of GADPH, and relative fold changes were expressed using the comparative threshold (ΔΔCt) method compared with untreated controls. The sequences for the gene-specific primers used were: TLR-3, forward 5′-TCC TTG CGT TGC GAA GTG AA-3′, reverse: 5′-TTG GGC GTT GTT CAA GAG GA-3′; TLR-7, forward 5′-TGC AAC TGT GAT GCT GTG TGG T-3′, reverse 5′-TTT GAC CTT TGT GTG CTC CTG G-3′; TLR-9, forward 5′-ACT GAG CAC CCC TGC TTC TA-3′, reverse 5′-AGA TTA GTC AGC GGC AGG AA-3′; CD40, forward 5′-AGG AAC GAG TCA GAC TAA TGT-3′, reverse 5′-GGA TCT TGC CGT CGA GC-3′; and GAPDH, forward 5′-ATG GAC TGT GGT CAT GAG CC-3′ and reverse 5′-ATT GTC AGC AAT GCA TCC TG-3′.
Flow cytometry
BALB/c-derived peritoneal macrophages, uninfected or 48 h L. major-infected, were treated with or without TLR ligands. Twenty-four h after stimulation, cells were scraped out and incubated with anti-CD16/32 for 30 min. After washing with FACS buffer (PBS with 2% FCS), cells were incubated with phycoerythrin (PE)-conjugated monoclonal anti-CD40 antibody for 1 h, washed twice with FACS buffer 12,24 and acquired on a FACS CyAn™ ADP Analyzer flow cytometer and analysed by Summit software (Beckman Coulter, Brea, CA, USA).
Preparation of cell lysates and Western blotting
BALB/c-derived thioglycolate-elicited macrophages were treated with or without TLR ligands and anti-CD40 antibody. Cells were washed twice with chilled PBS and lysed in cell lysis buffer [20 mM Tris (pH 7·4), 150 mM NaCl, 1% Nonidet P-40, 10% glycerol, 2 mM ethylenediamine tetraacetic acid (EDTA), protease inhibitor mixture (Roche Applied Science, Mannheim, Germany) and phosphatase inhibitor cocktail (Pierce, Rockford, IL, USA)]. Lysates were centrifuged (12 000 g for 30 min) and protein was quantified by a bicinchoninic acid (BCA) kit (Pierce) and an equal amount of protein was resolved on sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE). Resolved proteins were immunoblotted and the immunoreactive bands were visualized with luminol reagent (Santa Cruz Biotechnology), as described earlier 25. Densitometric analyses of bands were performed using Quantity One 4·6·1 (basic) software (Bio-Rad).
Cytokine ELISA
Culture supernatants from macrophages – uninfected or infected with L. major for 12 h and treated with various TLR ligands and/or anti-CD40 antibody (4 μg/ml) stimulation for 48 h – or from popliteal lymph node cells from L. major-infected and TLR ligand- or anti-CD40-treated were assayed for IL-1α, IL-6, IL-10 and IL-12 (p70), IL-4 or IFN-γ, as shown 23.
Treatment of mice with poly (I:C), imiquimod, CpG and anti-CD40 antibody
Susceptible BALB/c mice were infected subcutaneously into the hind leg with 2 × 106 L. major stationary phase promastigotes. Mice were treated with ligands for TLR-3 [poly (I:C)]; 10 μg/ml), TLR-7 (imiquimod; 10 μg/ml), TLR-9 (CpG ODN; 10 μg/ml) or with CpG ODN control subcutaneously into the same footpad infected with Leishmania and/or with anti-CD40 antibody [intraperitoneally (i.p.), 50 μg/mouse; clone 3/23, a gift from Dr G. Klaus, London, UK] from the third day, starting from infection, alternately for 3 days. Disease severity was assessed by measurement of footpad thickness using a digital micrometer (Mitutoyo, Kawasaki, Kanagawa, Japan); mice were sacrificed after 5 weeks of infection and parasite load was enumerated from the popliteal lymph node cells. Lymph node cells from various mice in a group were pooled for assaying the cytokine response. Seven mice were kept in each group.
Statistical analyses
Student's t-tests were performed to evaluate the significance between the mean values of the control and the treated groups or between two treated groups. The fiducial limit was fixed at 0·03, such that P-values less than 0·05 are significant. All experiments were performed in biological duplicates and repeated at least thrice.
Results
TLRs enhance CD40 expression on macrophages
Because TLRs are reported to activate an adaptive immune response 4,5, wherein CD40 plays a crucial role 6–8, we tested the effects of known TLR ligands on CD40 expression by RT–PCR, real-time PCR and FACS. We observed that all TLR ligands except profilin, a TLR-11 ligand, enhanced CD40 expression in a dose-dependent manner in both uninfected and L. major-infected macrophages (Fig. 1a). In addition, these TLR ligands also enhanced iNOS and IL-12, known for nitric oxide (NO) generation and host-protective T helper type 1 (Th1) cell differentiation, respectively 26,27, in macrophages (Fig. 1). We confirmed the findings by real-time PCR; of all the TLR ligands, poly (I:C), LPS, imiquimod and CpG were found to be highly active (Fig. 1b). While confirming these observations using FACS, which checked CD40 expression on the macrophage surface, it was observed that poly (I:C), imiquimod and CpG were among the most active TLR ligands in enhancing CD40 expressions (Fig. 1c). We also observed that poly (I:C), imiquimod and CpG also enhanced CD40 and IL-12 expression in BMDC (Fig. 1d). These results demonstrated that all intracellular TLRs – TLR-3, TLR-7/-8 and TLR-9 – significantly enhanced CD40 expression on both uninfected and L. major-infected macrophages.
Figure 1.

Toll-like receptor (TLR) ligands induce expression of CD40, inducible nitric oxide synthase (iNOS) and interleukin (IL)-12. Thioglycolate-elicited peritoneal mouse macrophages were harvested and plated. (a) Mouse macrophages were infected with either Leishmania major promastigoes (IM) at a 1:10 macrophage to parasite ratio or left uninfected (UIM), as described in Materials and methods, and were treated with the indicated doses of the TLR ligands: Pam3CSK4, peptidoglycan (PGN), polyinosinic-polycytidylic acid [poly( I:C)], lipopolysaccharide (LPS), flagellin (FLG), fibroblast-stimulating lipopeptide (FSL), imiquimod (IMQ), cytosine–phosphate–guanosine (CpG) and Profilin for 8 h, followed by RNA extraction and reverse transcriptase polymerase chain reaction (PCR) to assess the expression of CD40, iNOS and IL-12 (left panel). (b,c) uninfected (UIM) and infected (IM) mouse macrophages were treated with the TLR ligands: Pam3CSK4 (50 ng/ml), PGN (5 μg/ml), poly (I:C) (10 μg/ml), LPS (50 ng/ml), FLG (50 ng/ml), fibroblast-stimulating lipopeptide (FSL) (50 ng/ml), imiquimod (IMQ; 2 μg/ml)), CpG (0·12 μM) and Profilin (250 ng/ml) for 8 h and 24 h, to check the expression of CD40 by real-time PCR (upper right panel) and by fluorescence activated cell sorter (FACS) (middle panel), respectively. Expression of CD40 was analysed by FACS in uninfected (medium) and infected (infection) macrophages (lower panel). (d) Bone marrow-derived dendritic cells (BMDC) were treated with poly (I:C) (10 μg/ml), imiquimod (2 μg/ml) and CpG (0·12 μM) for 8 h followed by RNA extraction and reverse transcriptase PCR was performed to assess the expression of CD40 and IL-12p40. UIDC = uninfected BMDC; IDC = infected BMDC. Error bars shown are the mean ± standard error (SE) from three experiments; *P < 0·05.
CD40 and TLRs cross-talk
Because CD40 activates macrophages to kill Leishmania amastigotes 12–14, it is possible that CD40 stimulation may alter the expression of TLRs. Therefore, we assessed the expression of all TLRs following CD40 stimulation in uninfected and L. major-infected macrophages. We observed that compared to the untreated control macrophages, only TLR-9 expression was enhanced significantly in the CD40-stimulated macrophages (Fig. 2a). The heightened expression of TLR-9 in CD40-stimulated macrophages was confirmed by Western blot (Fig. 2a). Furthermore, inhibition of TLR-2 signalling by the MyD88 inhibitory peptide but not the control peptide impaired CD40 expression (Fig. 2b).
Figure 2.

CD40 and Toll-like receptors (TLRs) cross-talk. Thioglycolate-elicited mouse macrophages were infected with Leishmania major (IM) or left uninfected (UIM) for 72 h (a–c). (a) UIM and IM were treated with the indicated doses of anti-CD40 antibody for 8 h and RNA extraction was performed to check the expression of TLR-3, TLR-7 and TLR-9 by real-time polymerase chain reaction (PCR). Mouse macrophages treated with anti-CD40 (6 μg/ml) for the indicated time-point and cells were lysed to check the expression of TLR-9 by Western blotting (upper right subpanel). TLR-9 expression in UIM and IM were checked by macrophages treated with anti-CD40 (6 μg/ml) for 24 h, followed by cell lysis and immunoblotting. (b) Mouse macrophages were pretreated with myeloid differentiation-associated gene 88 (MyD88) inhibitory peptide (150 μg/ml) for 1 h, followed by treatment with Pam3CSK4 (50 ng/ml), peptidoglycan (PGN) (5 μg/ml) and fibroblast-stimulating lipopeptide (FSL) (50 ng/ml) for 8 h and RNA extraction to check the expression of CD40. (c) UIM and IM cells were treated with or without polyinosinic-polycytidylic acid poly-(I:C)] (10 μg/ml), imiquimod (2 μg/ml), cytosine–phosphate–guanosine (CpG) (0·12 μM) and anti-CD40 antibody (4 μg/ml) for 15 min; cells were lysed and the activation of phospho-p65, phospho-p38 and phospho-extracellular stress-regulated kinase (ERK)1/2 mitogen-activated protein kinases (MAPKs) were observed by Western blotting. Ratio of p-ERK : p-p38 and p-65 were calculated from the densitometric values obtained from the quantity one program (lower panel). (d) Uninfected (UIM) and infected (IM) mouse macrophages were treated with poly (I:C) (10 μg/ml), imiquimod (2 μg/ml), CpG (0·12 μM) and/or anti-CD40 antibody (4 μg/ml) for 48 h; culture supernatants were collected to assess interleukin (IL)-1α, IL-6, IL-10 and IL-12 production by enzyme-linked immunosorbent assay (ELISA). The data shown are representative of one experiment, which was performed thrice in duplicate. The error bars shown represent mean ± standard error; *P < 0·05.
To test whether the ligands for TLRs could modulate the CD40-induced ERK-1/2 and p38 MAPK phosphorylation, both uninfected and L. major-infected macrophages were treated with CD40 or respective TLR ligands or both, followed by Western blotting. It was observed that TLR-9 decreased ERK-1/2 phosphorylation in L. major-infected macrophages, compared to uninfected macrophages (Fig. 2c). We did not observe any differential changes in NF-κB phosphorylation in response to the indicated TLR ligands alone or CD40 alone, or the CD40+TLR ligand combination (Fig. 2c).
As described earlier 28, cytokines also play important roles in anti-leishmanial functions. As TLRs can induce different proinflammatory cytokines, they have been implicated in host-protective anti-leishmanial functions 25. However, whether TLRs enhance the cytokine induction by CD40 is not known. Therefore, in order to test whether TLRs enhance CD40-induced cytokine production, we treated uninfected and L. major-infected macrophages with TLR ligands alone or in combination with anti-CD40 and assessed cytokine production, as indicated. TLR-3 induces IL-1α in combination with anti-CD40. It was observed that there were no differences between the three TLR ligands in inducing IL-10 production (Fig. 2d). However, IL-6 production was significantly less in CpG+CD40-treated macrophages and IL-12p70 production was significantly higher in macrophages treated with imiquimod+anti-CD40 or CpG+anti-CD40 antibody (Fig. 2d). Taken together, these data imply that poly (I:C), imiquimod and CpG might exert significant host-protective anti-leishmanial effects.
Anti-leishmanial function of CD40 is enhanced by TLRs
As both CD40 and TLRs are shown to exert anti-leishmanial effects 9–14,25, we tested if combinations of a TLR ligand with CD40 exerted a better anti-leishmanial effect. First, BALB/c-derived thioglycolate-elicited macrophages were infected with L. major, as described in Materials and methods, followed by the indicated treatments from 24 h after the infection. The amastigotes in macrophages were enumerated 72 h after infection. It was observed that the anti-leishmanial function of CD40 was enhanced significantly by poly (I:C), imiquimod and CpG (Fig. 3a).
Figure 3.
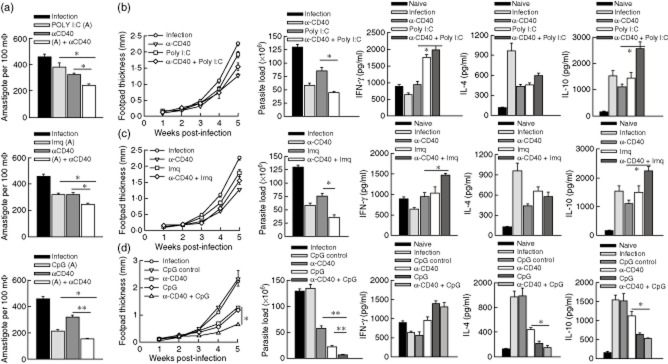
Anti-leishmanial function of CD40 is enhanced by Toll-like receptor (TLR) ligands. (a) Mouse macrophages were infected with Leishmania major for 12 h, extracellular parasites were washed out and after 12 h incubation cells were treated with polyinosinic-polycytidylic acid [poly (I:C)] (10 μg/ml), imiquimod (2 μg/ml), cytosine–phosphate–guanosine (CpG) (0·12 μm) and/or anti-CD40 antibody (4 μg/ml) for 60 h. Cells were washed with 1 × phosphate-buffered saline (PBS) and fixed with chilled methanol, stained with Giemsa for enumeration of amastigotes per 100 macrophages under a light microscope. (b–d) The indicated groups of mice, comprising seven mice in each group, injected subcutaneously with L. major (2 × 106/mouse) into one of the hind leg footpads or left uninfected as a control. Mice were treated with poly (I:C) (30 μg/mouse), imiquimod (10 μg/mouse) or CpG (10 μg/mouse) subcutaneously into the same infected footpad and/or intraperitoneally with anti-CD40 antibody (50 μg/mouse) starting from the third day of infection for 3 alternate days. Footpad thickness was assessed weekly until the mice were sacrificed 5 weeks after the infection. Parasite loads in the popliteal lymph node cells were enumerated. Lymph node cells from the mice in each group were pooled, stimulated with anti-CD3 (0·5 μg/ml) and anti-CD28 (2·0 μg/ml) for 48 h, and the culture supernatants were assessed for interleukin (IL)-4, IL-10 and interferon (IFN)-γ by (ELISA). The data shown are from one representative experiment that was repeated twice. The error bars shown represent mean ± standard error; *P < 0·05; **P ≤ 0·001.
Next, BALB/c mice were infected with L. major promastigotes and were treated with the respective TLR ligand and anti-CD40 antibody from the third day of infection on 3 alternate days. Mice were sacrificed 5 weeks after the L. major infection and the parasite load in the draining lymph node was enumerated 12. It was observed that, in combination with the anti-CD40 antibody, CpG was the most active anti-leishmanial TLR ligand, followed by imiquimod and poly (I:C) (Fig. 3b–d). The CpG+anti-CD40 antibody-mediated protection was accompanied by differential IFN-γ, IL-4 and IL-10 production (Fig. 3d). Therefore, these results suggest that CpG, along with the agonistic anti-CD40 antibody, can be a significant anti-leishmanial therapeutic agent.
Discussion
Leishmania infection begins with its interaction between the parasite-expressed lipophosphoglycan (LPG) and the host cell-expressed TLR-2 29. Previous reports, including ours, show that TLR-2 may not necessarily be host-protective, as proposed for all TLR anti-pathogen functions 29,30. However, there are nine other TLRs that are implicated differentially in infections with different Leishmania species 25. It is proposed that activation of TLRs leads to activation of adaptive immune responses, primarily maturation of antigen-presenting cells and T cell activation 4,5. CD40 plays a crucial role in adaptive immune responses to Leishmania infection 9–11. Therefore, we examined whether TLR ligands and CD40 modulated each other's expression and whether TLR ligands could effectively enhance the anti-leishmanial functions of CD40.
In this study we show, for the first time, the effect of different TLR ligands on CD40 expression and vice versa. Our observations demonstrate clearly that CD40 expression in L. major-infected macrophages is enhanced by poly (I:C), imiquimod and CpG, all of which are DNA- or RNA-recognizing intracellular TLRs. The observations suggest that the pathogen-derived nucleic acids may play important roles in enhancing the expression of CD40 in the host cell. As LPG–TLR-2 interaction suppresses TLR-9 expression as an immune evasion strategy, the up-regulation of CD40 by intracellular TLR can be a host-protective strategy. L. major infection deviates CD40 signalling through various signalling intermediates, particularly tumour necrosis factor (TNF) receptor-associated factor (TRAF)-6 and MAP kinases, but not its expression 12–14,23,25. Both TRAF-6 and MAPK are also used for TLR signalling, so it is possible that the parasite may modulate the use of the available signalling intermediates between these two pathways and that by triggering the TLR-2 signal during its initial interaction with the macrophages, Leishmania interferes with CD40 signalling either by competing for TRAFs or other signalling intermediates, including transcription factors or by modulating their availability to CD40. Therefore, a balance between TLR-2 signalling and TLR-3, TLR-7/-8 and TLR-9 signalling may decide the outcome of Leishmania infection.
Conversely, CD40 enhanced the expression of only TLR-9, which recognizes the CpG motifs in DNA. This observation implies that CpG may trigger such host-protective functions which may synergize the host-protective functions of CD40. Indeed, both CD40 and CpG induce IL-12, a cytokine shown to have a strong host-protective anti-leishmanial effect by promoting Th1 differentiation 31. CD40 induces iNOS expression in macrophages and IL-12-induced Th1 cells secrete IFN-γ, which also enhances iNOS expression in macrophages 32. Therefore, it is possible that CpG and CD40 positively regulate each other's expression to enhance host-protective anti-leishmanial effects. Corroborating this proposition, the combination of anti-CD40 antibody and CpG provided the best anti-leishmanial effect both in macrophages and in susceptible BALB/c mice.
The host-protective function of CD40 and CpG was accompanied by low IL-10 and IL-4 secretion. Because IL-12 promotes Th1 cells 31 and may reciprocally reduce the disease-exacerbating Th2 cells, as reflected in low IL-4 and low IL-10 production, this observation corroborates well with the high IL-12 production by CpG-treated macrophages. Another subset of CD4+ T cells that may alter L. major infection is transforming growth factor (TGF)-β-dependent regulatory T (Treg) cells. It is also possible that TLR ligands may alter TGF-β production, altering T-reg cells; these require separate studies, however. None the less, the data reported here refine the rationale for anti-leishmanial immunotherapy by a combination of agents targeting the innate immune system and the adaptive immune system. To the best of our knowledge, this is the first report on CD40–TLR cross-talk and how the data derived from such a study can be used to rationalize a novel anti-leishmanial therapeutic strategy.
Acknowledgments
H. S. C., M. K. J. and S. P. P. are supported by fellowships from the Indian Council of Medical Research, New Delhi. S. K. S. is supported by University Grant Commission, New Delhi. The work was assisted financially by the Grant BT/PR/3288/BRB/10/966/2011 from the Department of Biotechnology, New Delhi.
Disclosure
The authors declare that they have no conflicts of interest.
Author contributions
H. S. C., S. P P., D. S., K. L., M. K. J. and S. K. S. performed the experiments; B. S. designed the experiments and prepared the manuscript.
References
- 1.Bauer S, Müller T, Hamm S. Pattern recognition by Toll-like receptors. Adv Exp Med Biol. 2009;653:15–34. doi: 10.1007/978-1-4419-0901-5_2. [DOI] [PubMed] [Google Scholar]
- 2.Martinez J, Huang X, Yang Y. Direct TLR2 signaling is critical for NK cell activation and function in response to vaccinia viral infection. PLOS Pathog. 2010;6:e1000811. doi: 10.1371/journal.ppat.1000811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janeway CA, Jr, Medzhitov R. Lipoproteins take their toll on the host. Curr Biol. 1999;9:R879–882. doi: 10.1016/s0960-9822(00)80073-1. [DOI] [PubMed] [Google Scholar]
- 4.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 5.Pasare C, Medzhitov R. Control of B-cell responses by Toll-like receptors. Nature. 2005;438:364–368. doi: 10.1038/nature04267. [DOI] [PubMed] [Google Scholar]
- 6.Schwartz RH. T-lymphocyte recognition of antigen in association with gene products of the major histocompatibility complex. Annu Rev Immunol. 1985;3:237–261. doi: 10.1146/annurev.iy.03.040185.001321. [DOI] [PubMed] [Google Scholar]
- 7.Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol. 1993;11:191–212. doi: 10.1146/annurev.iy.11.040193.001203. [DOI] [PubMed] [Google Scholar]
- 8.Banchereau J, Bazan F, Blanchard D, et al. The CD40 antigen and its ligand. Annu Rev Immunol. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 9.Campbell KA, Ovendale PJ, Kennedy MK, et al. CD40 ligand is required for protective cell-mediated immunity to Leishmania major. Immunity. 1996;4:283–289. doi: 10.1016/s1074-7613(00)80436-7. [DOI] [PubMed] [Google Scholar]
- 10.Kamanaka M, Yu P, Yasui T, et al. Protective role of CD40 in Leishmania major infection at two distinct phases of cell-mediated immunity. Immunity. 1996;4:275–281. doi: 10.1016/s1074-7613(00)80435-5. [DOI] [PubMed] [Google Scholar]
- 11.Soong L, Xu JC, Grewal IS, et al. Disruption of CD40–CD40 ligand interactions results in an enhanced susceptibility to Leishmania amazonensis infection. Immunity. 1996;4:263–273. doi: 10.1016/s1074-7613(00)80434-3. [DOI] [PubMed] [Google Scholar]
- 12.Awasthi A, Mathur R, Khan A, et al. CD40 signaling is impaired in L. major-infected macrophages and is rescued by a p38MAPK activator establishing a host-protective memory T cell response. J Exp Med. 2003;197:1037–1043. doi: 10.1084/jem.20022033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mathur RK, Awasthi A, Wadhone P, Ramanamurthy B, Saha B. Reciprocal CD40 signals through p38MAPK and ERK-1/2 induce counteracting immune responses. Nat Med. 2004;10:540–544. doi: 10.1038/nm1045. [DOI] [PubMed] [Google Scholar]
- 14.Rub A, Dey R, Jadhav M, et al. Cholesterol depletion associated with Leishmania major infection alters macrophage CD40 signalosome composition and effector function. Nat Immunol. 2009;10:273–280. doi: 10.1038/ni.1705. [DOI] [PubMed] [Google Scholar]
- 15.Qiu Y, Ding Y, Zou L, et al. Divergent roles of amino acid residues inside and outside the BB loop affect human Toll-like receptor (TLR)2/2, TLR2/1 and TLR2/6 responsiveness. PLOS One. 2013;8:e61508. doi: 10.1371/journal.pone.0061508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uematsu S, Akira S. Toll-Like receptors (TLRs) and their ligands. Handb Exp Pharmacol. 2008;183:1–20. doi: 10.1007/978-3-540-72167-3_1. [DOI] [PubMed] [Google Scholar]
- 17.Hornung V, Barchet W, Schlee M, Hartmann G. RNA recognition via TLR7 and TLR8. Handb Exp Pharmacol. 2008;183:71–86. doi: 10.1007/978-3-540-72167-3_4. [DOI] [PubMed] [Google Scholar]
- 18.Jin MS, Lee JO. Structures of the Toll-like receptor family and its ligand complexes. Immunity. 2008;29:182–191. doi: 10.1016/j.immuni.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Smith KD, Ozinsky A. Toll-like receptor-5 and the innate immune response to bacterial flagellin. Curr Top Microbiol Immunol. 2002;270:93–108. doi: 10.1007/978-3-642-59430-4_6. [DOI] [PubMed] [Google Scholar]
- 20.Zeytun A, Chaudhary A, Pardington P, Cary R, Gupta G. Induction of cytokines and chemokines by Toll-like receptor signaling: strategies for control of inflammation. Crit Rev Immunol. 2010;30:53–67. doi: 10.1615/critrevimmunol.v30.i1.40. [DOI] [PubMed] [Google Scholar]
- 21.Tuon FF, Amato VS, Bacha HA, Almusawi T, Duarte MI, Amato Neto V. Toll-like receptors and leishmaniasis. Infect Immun. 2008;76:866–872. doi: 10.1128/IAI.01090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murugaiyan G, Agrawal R, Mishra GC, Mitra D, Saha B. Functional dichotomy in CD40 reciprocally regulates effector T cell functions. J Immunol. 2006;177:6642–6649. doi: 10.4049/jimmunol.177.10.6642. [DOI] [PubMed] [Google Scholar]
- 23.Sudan R, Srivastava N, Pandey SP, Majumdar S, Saha B. Reciprocal regulation of protein kinase C isoforms results in differential cellular responsiveness. J Immunol. 2012;188:2328–2337. doi: 10.4049/jimmunol.1101678. [DOI] [PubMed] [Google Scholar]
- 24.Martin S, Agarwal R, Murugaiyan G, Saha B. CD40 expression levels modulate regulatory T cells in Leishmania donovani infection. J Immunol. 2010;185:551–559. doi: 10.4049/jimmunol.0902206. [DOI] [PubMed] [Google Scholar]
- 25.Srivastava N, Sudan R, Saha B. CD40-modulated dual-specificity phosphatases MAPK phosphatase (MKP)-1 and MKP-3 reciprocally regulate L. major infection. J Immunol. 2011;186:5863–5872. doi: 10.4049/jimmunol.1003957. [DOI] [PubMed] [Google Scholar]
- 26.Liew FY, Wei XQ, Proudfoot L. Cytokines and nitric oxide as effector molecules against parasitic infections. Phil Trans R Soc Lond B Biol Sci. 1997;352:1311–1315. doi: 10.1098/rstb.1997.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan SH, Perussia B, Gupta JW, et al. Induction of interferon gamma production by natural killer cell stimulatory factor: characterization of the responder cells and synergy with other inducers. J Exp Med. 1991;173:869–879. doi: 10.1084/jem.173.4.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Launois P, Tacchini-Cottier F, Parra-Lopez C, Louis JA. Cytokines in parasitic diseases: the example of cutaneous leishmaniasis. Int Rev Immunol. 1998;17:157–180. doi: 10.3109/08830189809084491. [DOI] [PubMed] [Google Scholar]
- 29.Srivastava S, Pandey SP, Jha MK, Chandel HS, Saha B. Leishmania expressed lipophosphoglycan interacts with Toll-like receptor (TLR)-2 to decrease TLR-9 expression and reduce anti-leishmanial responses. Clin Exp Immunol. 2013;172:403–409. doi: 10.1111/cei.12074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vargas-Inchaustegui DA, Tai W, Xin L, Hogg AE, Corry DB, Soong L. Distinct roles for MyD88 and Toll-like receptor 2 during Leishmania braziliensis infection in mice. Infect Immun. 2009;77:2948–2956. doi: 10.1128/IAI.00154-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Afonso LC, Scharton TM, Vieira LQ, Wysocka M, Trinchieri G, Scott P. The adjuvant effect of interleukin-12 in a vaccine against Leishmania major. Science. 1994;263:235–237. doi: 10.1126/science.7904381. [DOI] [PubMed] [Google Scholar]
- 32.Bogdan C, Röllinghoff M, Diefenbach A. The role of nitric oxide in innate immunity. Immunol Rev. 2000;173:17–26. doi: 10.1034/j.1600-065x.2000.917307.x. [DOI] [PubMed] [Google Scholar]