

Abstract
Synthetic biology aims to rationally design and build synthetic circuits with desired quantitative properties, as well as provide tools to interrogate the structure of native control circuits. In both cases, the ability to program gene expression in a rapid and tunable fashion, with no off-target effects, can be useful. We have constructed yeast strains containing the ACT1 promoter upstream of a URA3 cassette followed by the ligand-binding domain of the human estrogen receptor and VP16. By transforming this strain with a linear PCR product containing a DNA binding domain and selecting against the presence of URA3, a constitutively expressed artificial transcription factor (ATF) can be generated by homologous recombination. ATFs engineered in this fashion can activate a unique target gene in the presence of inducer, thereby eliminating both the off-target activation and nonphysiological growth conditions found with commonly used conditional gene expression systems. A simple method for the rapid construction of GFP reporter plasmids that respond specifically to a native or artificial transcription factor of interest is also provided.
Keywords: Genetics, Issue 81, transcription, transcription factors, artificial transcription factors, zinc fingers, Zif268, synthetic biology
Introduction
Developing genetic switches in yeast is of great interest in both academic and industrial research. In the ideal case, genetic switches can be used to activate a particular gene (or set of genes) only when desired by the experimenter. A gene's coding sequence placed downstream of a synthetic promoter is not expressed in the absence of an inducing molecule: upon inducer addition the gene should be rapidly expressed. It has only recently been demonstrated that such a switch can be engineered in yeast, where in the absence of inducer, the strain displays a deletion phenotype, but in the presence of inducer, expression is activated in proportion to the level of inducer in all cells1,2. Historically, conditional expression systems in yeast have relied heavily upon nutrient-responsive DNA sequences, including the MET, PHO, and GAL promoters (whose activities are sensitive to extracellular levels of methionine, phosphate, and galactose, respectively). While conditional expression can be achieved, it comes at the cost of significant pleiotropic effects.
Hormone receptors such as the human estrogen receptors have proven to be effective switches in eukaryotes3,4. In the absence of hormone, a protein fused to a hormone receptor can be sequestered in the cytoplasm where it interacts with the Hsp90 chaperone complex. Upon binding the appropriate ligand, the receptor undergoes a conformational change, causing it to be released from the Hsp90 chaperone complex and revealing a nuclear localization signal. To observe the localization dynamics of a particular hormone receptor-containing protein, we created a C-terminal fusion of Gal4dbd.ER.VP16 (GEV, a synthetic transcription factor containing the Gal4p DNA binding domain, the ligand binding domain of the human estrogen receptor, and VP16) with GFP2. Nuclear localization was observed within 8 min following addition of saturating amounts of the inducer, ß-estradiol, to which wild type yeast is completely inert. By this time, the majority of cells have clearly visible active transcription sites for GEV target genes (assayed via fluorescence in situ hybridization) as well as fully mature mRNAs2,5. GEV target genes increased in expression >2-fold within 2.5 min2,6 (measured using microarrays), demonstrating that it takes <2.5 min for the chimeric activator to translocate to the nucleus, bind DNA, and activate transcription.
While several other on-switches have been applied in yeast that utilize various small molecules or drugs (including the classic doxycycline-based switches from Escherichia coli7,8 and an indigo-based switch9 from Arabidopsis thaliana), none have achieved the speed, specificity, or tightness of regulation exhibited by the hormone-based switches. It is worth mentioning that rapid off-switches have also been developed in yeast, and typically work by fusing a gene to a protease or ubiquitin ligase targeting sequence2,10,11,12. The ability to rapidly remove a target protein from the cell facilitates the study of essential genes as well as genes that are prone to genetic suppression when deleted10.
The methods described in this protocol are for building and characterizing synthetic on-switches in yeast. First, we show how to rapidly generate genomically-integrated fusion proteins consisting of a DNA binding domain of interest, the ligand binding domain of the human estrogen receptor, and VP16 (DBD-EV’s). Following our protocol, the fusion protein is expressed from an ACT1 promoter. We then show how to create a cognate reporter plasmid for the ATF and test its functionality using flow cytometry. Though we envision these reporter plasmids being used with new ATFs (Figure 1), they can be used as reporters for a native transcriptional activator as well. Finally, details for testing the effect of ATF activation on cell growth in 96-well plates and for engineering inducible genomic alleles are provided.
Protocol
Figure 2 provides a schematic of protocol sections 1-3.
1. Creation of ATFs
Create primers for PCR amplifying DNA binding domain of interest. Homology to the ACT1 promoter and hormone receptor (as described in Figures 3 and 4) is added through PCR to facilitate correct recombination into the strain yMN15.
PCR amplify DBD of interest using a high fidelity polymerase.
Transform >1 µg of purified PCR product into yeast using the lithium acetate method13.
Following transformation, spin cells at top speed for 15 sec in microcentrifuge and remove transformation mix.
Resuspend in ~200 µl of sterile water and plate on YPD.
The next day, replica plate from YPD onto 5-FOA plates (5-FOA plates are made as described in the Cold Spring Harbor Yeast Manual)13.
Allow growth for 2-4 days, and restreak at least 5-10 colonies onto YPD. Perform a colony PCR using the primers in Table 1 and sequence to verify inclusion.
2. Creation of ATF Plasmid Reporter
Determine the binding site of the ATF (most likely to come from the literature).
Order desired binding region as oligos (or Ultramers, if needed). Order as top and bottom strands with correct overhangs for NotI & XbaI as shown in Table 2.
Dilute oligos to equimolar concentrations (100 µM).
Phosphorylate using T4 Polynucleotide Kinase and then anneal in a thermocycler. The reaction conditions are in Table 3.
Digest ~1-2 µg of pMN8 with NotI-HF & XbaI for 1 hr at 37 °C (reaction conditions in Table 4).
Gel purify the backbone band.
Dilute the purified backbone to 10 ng/µl.
Dilute the double-stranded, phosphorylated binding site to 5x the molar concentration of backbone per µl.
Using T4 DNA Ligase, ligate the binding sites into the digested backbone at room temperature for 30 min (Table 5).
For transformation, add half the ligation reaction to 50 µl of standard, chemically competent Escherichia coli such as XL1-Blue or DH5-Alpha.
Heat shock cells for 45 sec in a 42 °C water bath and then place on ice for 2 min.
Add 900 µl of SOC medium to transformation reaction.
Grow at 37 °C for 1 hr on a roller drum.
Spread 100-200 µl cells per LB+AMP (100 µg/ml) plate and incubate overnight.
Grow up E. coli in LB+AMP liquid and harvest plasmid DNA. Sequence verify new reporter plasmid from ligation colonies using the primer 5'-GTACCTTATATTGAATTTTC-3'.
Transform new reporter plasmid into ATF-containing yeast strain from Section 1 using lithium acetate transformation.
3. Quantify the Effect of ATF Induction on Gene Expression Using GFP Reporter
Preparing the samples:
Make 25 mM β-estradiol stock (10 ml of ethanol + 0.0681 g of β-estradiol). Keep refrigerated.
Grow ATF + Reporter-containing strain overnight in 5ml SC-URA.
Obtain nine 250 ml shake flasks and add 25 ml SC-URA to each.
Add 0 nM, 0.1 nM, 1 nM, 10 nM, 100 nM, 1 µM, or 10 µM β-estradiol. This is most easily done by making 10-fold serial dilutions of the 25 mM β-estradiol stock. For a final concentration of 10 µM β-estradiol, add 10 µl of the 25 mM β-estradiol stock to 25 ml of SC-URA.
Add 125 µl of overnight cultures to flasks (1:200 dilution).
For the 2 remaining flasks, inoculate with a constitutive GFP producing strain and a strain lacking GFP. These are needed for calibrating the flow cytometer.
Shake at 200 rpm at 30 °C for 12-18 hr. Note: The optimal incubation time is determined by identifying when GFP induction no longer changes following addition of β-estradiol.
Take 500 µl of each culture and spin down in separate 1.7 ml microcentrifuge tubes.
Aspirate SC-URA.
Wash once in 1 ml PBS+0.1% Tween-20 and aspirate.
Add 1 ml of PBS+0.1% Tween-20 and resuspend.
Add 1 ml of PBS+0.1% Tween-20 to Falcon tubes.
Add the resuspended cells to 5 ml tubes (final volume = 2 ml) (can be stored at 4 °C for several days).
Optional: sonicate cells lightly to breakup any clumped cells. Flow cytometry analysis: Overview: The fluorescence of 10,000-100,000 cells is typically analyzed per sample. Data can be processed either in FlowJo or using custom scripts in MATLAB or R. Make sure to calibrate voltage of the GFP channel using GFP positive and negative controls. Once the FCS files are exported, a script such as the one in Figure 6 along with the function fca_readfcs (http://www.mathworks.com/matlabcentral/fileexchange/9608-fcs-data-reader) can be used to process the data.
- Perform flow cytometry.
- Use forward scatter (FSC) and side scatter (SSC) to identify yeast cells.
- Set the flow rate to ~3,000 cells/sec.
- Measure GFP (FITC channel).
- Once data are collected, export .FCS files
Load MATLAB.
Move the .FCS files, fcs_readfcs.m, and a script similar to that from Figure 6 to the same folder.
Change the present working directory to the folder containing the data and scripts.
Run the script from Figure 6 (note: the legend has been manually entered to produce what's seen in Figure 7).
4. Quantify the Effect of ATF Induction on Cell Growth
From frozen stocks, streak out both (1) an ATF-containing strain and (2) an ATF-lacking strain (such as yMN15) onto separate YPD plates.
After 2 days of growth, inoculate separate culture tubes containing 5 ml of YPD liquid with each strain and grow overnight at 30 °C.
Perform 10-fold serial dilutions of a 0.25 mM β-estradiol stock solution up to 10,000-fold (0.025 µM β-estradiol) in 100% ethanol.
Add 40 µl of overnight cultures to 10 ml of YPD liquid (1-250 dilution).
For each strain, add 96 µl of diluted cells to 18 wells of a 96-well plate.
Add 4 µl of β-estradiol to the cells. An essential point is to make sure each well has the same amount of total ethanol. (Alternatively, a more concentrated stock of β-estradiol can be used and subsequently diluted to the point the effect of ethanol on growth is not measurable).
Perform in triplicate. Three of the wells for each strain should be ethanol-only controls.
Cover the 96-well plate in gas permeable membrane.
Monitor growth (OD600) in plate reader.
Quantify the growth rates using any number of standard methods such as finding the maximum slope. Note: We've had good success with the open source software package grofit14, which runs in the R programming environment. It is worth mentioning that while growth rates computed from 96-well plate assays compared to shake-flasks (25 ml cultures) are not necessarily the same (due to both differences in physiology and in instrumentation), we've observed that the relative growth rates are similar (though this may not always be the case).
5. Create Inducible Genomic Alleles
Obtain pMN10, a noncentromeric plasmid with the ATF-responsive promoter from pMN8 fused to KanMX (in the opposite orientation).
Restriction digest the vector backbone and gel extract as in Section 2.
Anneal, phosphorylate, and ligate oligos containing appropriate binding sites as described in Section 2.
Sequence verify. This plasmid will now serve as a DNA template for creating the inducible allele.
Obtain the yeast strain expressing the corresponding ATF of interest.
PCR amplify the KanMX-Promoter cassette (~2 kb) using primers from Table 6.
Transform the PCR product into the ATF-expressing strain using the lithium acetate method.
Plate on YPD and replica plate onto YPD+ G418 the following day.
Confirm appropriate insertion of the promoter using the forward primer (5'-CTGCAGCGAGGAGCCGTAAT-3') and a reverse primer internal to the gene whose promoter has been replaced. The reverse primer is typically designed to be between 200-300 bp downstream of the first ATG15. The final PCR product is ~1.2 kb.
Representative Results
Figure 5 shows induction of GFP by Z3EV, Z4EV, or GEV over time. Note the increase in GFP production in response to the ATFs containing nonyeast DNA binding domains. We recently speculated that this results from these factors achieving single-gene specificity in the yeast genome1. GEV induction alone leads to activation/repression of hundreds of genes, whereas Z3EV and Z4EV only activate expression of a target gene placed downstream of a synthetic promoter containing appropriate binding sites (5'-GCGTGGGCG-3' and 5'-GCGGCGGAGGAG-3', respectively)1. Figure 7 demonstrates the tight regulation of the system (no inducer results in no detectable GFP) and that all cells get induced in response to inducer The ability of Z3EV to induce GFP across a range of β-estradiol concentrations is shown in Figure 8.
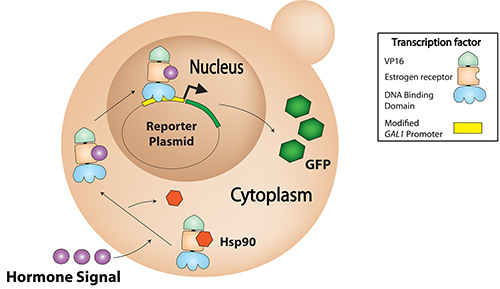 Figure 1.Artificial transcription factors interact with Hsp90 in the absence of β-estradiol. When β-estradiol binds to the ATF, Hsp90 dissociates, allowing the ATF to enter the nucleus and activate expression from the plasmid reporter.
Figure 1.Artificial transcription factors interact with Hsp90 in the absence of β-estradiol. When β-estradiol binds to the ATF, Hsp90 dissociates, allowing the ATF to enter the nucleus and activate expression from the plasmid reporter.
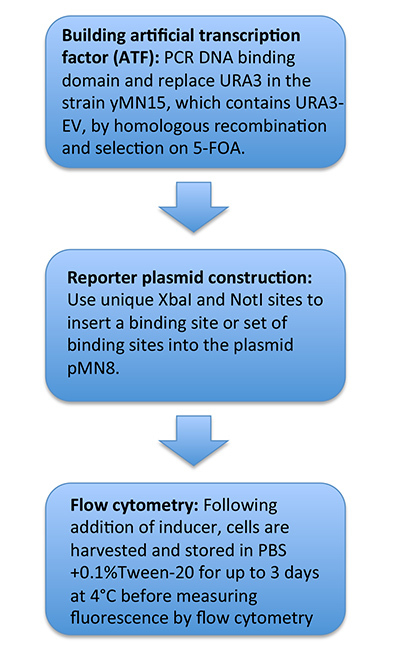 Figure 2.
Schematic of protocol for engineering strains that contain artificial transcription factors and building/testing cognate GFP reporters.
Figure 2.
Schematic of protocol for engineering strains that contain artificial transcription factors and building/testing cognate GFP reporters.
 Figure 3.
The ACT1pr-URA3-EV sequence in the strain yMN15 at the LEU2 locus.
Figure 3.
The ACT1pr-URA3-EV sequence in the strain yMN15 at the LEU2 locus.
 Figure 4.
Design of PCR primers for amplifying a DNA binding domain of interest with additional sequence homology to generate a new fusion protein when successfully transformed into yMN15.
Figure 4.
Design of PCR primers for amplifying a DNA binding domain of interest with additional sequence homology to generate a new fusion protein when successfully transformed into yMN15.
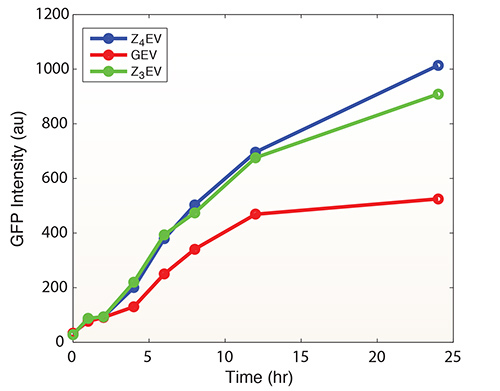 Figure 5.
GFP induction by three different ATF-promoter pairs in response to 1 µM β-estradiol
.
Figure 5.
GFP induction by three different ATF-promoter pairs in response to 1 µM β-estradiol
.
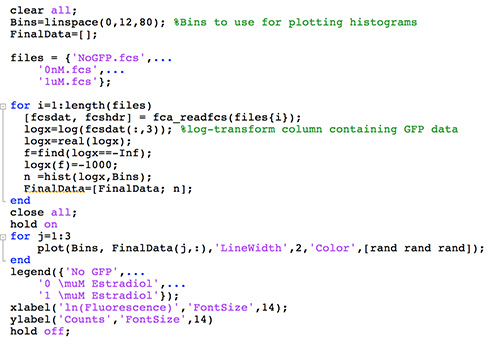 Figure 6.A MATLAB script for processing flow cytometry data. This script was adapted from http://openwetware.org/wiki/McClean:_Matlab_Code_for_Analyzing_FACS_Data.
Figure 6.A MATLAB script for processing flow cytometry data. This script was adapted from http://openwetware.org/wiki/McClean:_Matlab_Code_for_Analyzing_FACS_Data.
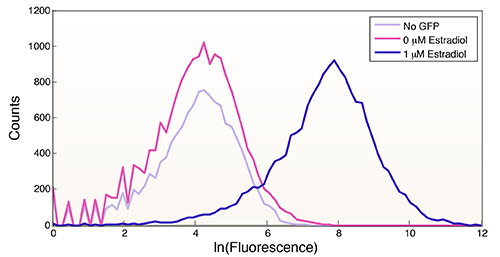 Figure 7.Induction of GFP by Z3EV in response to 0 µM β-estradiol and 1 µM β-estradiol following 18 hr of induction. Fluorescence was also measured from cells lacking GFP to illustrate the tight regulation of the Z3EV system. This figure was generated using the code in Figure 6.
Figure 7.Induction of GFP by Z3EV in response to 0 µM β-estradiol and 1 µM β-estradiol following 18 hr of induction. Fluorescence was also measured from cells lacking GFP to illustrate the tight regulation of the Z3EV system. This figure was generated using the code in Figure 6.
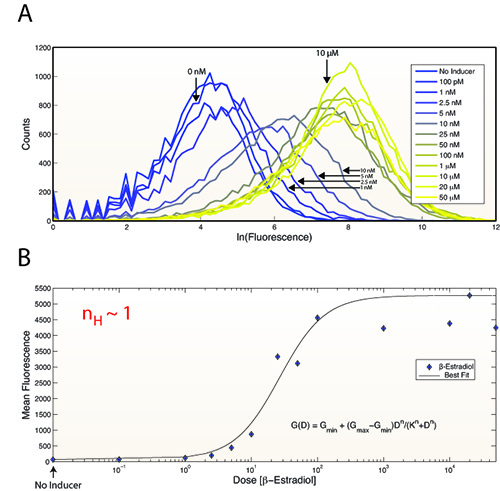 Figure 8.The relationship between inducer concentration and expression output is graded with a Hill coefficient of ~1. (A) Distributions of GFP expression as a function of β-estradiol concentration. (B) The mean fluorescence of the distributions from (A). The data were fit to the function G(D) = Gmin + (Gmax−Gmin)Dn/(Kn+Dn) where D is the β-estradiol dose, Gmin and Gmax are the minimum and maximum GFP values, respectively, n is the Hill coefficient, and K is the β-estradiol dose that yields ½ (Gmax + Gmin).
Figure 8.The relationship between inducer concentration and expression output is graded with a Hill coefficient of ~1. (A) Distributions of GFP expression as a function of β-estradiol concentration. (B) The mean fluorescence of the distributions from (A). The data were fit to the function G(D) = Gmin + (Gmax−Gmin)Dn/(Kn+Dn) where D is the β-estradiol dose, Gmin and Gmax are the minimum and maximum GFP values, respectively, n is the Hill coefficient, and K is the β-estradiol dose that yields ½ (Gmax + Gmin).
| Oligonucleotide Sequence | Name |
| 5’-TTGAAACCA AACTCGCCTCT-3’ | DBD Check Forward |
| 5’-tccagagac ttcagggtgct-3’ | DBD Check Reverse |
Table 1. Primers for confirming correct fusion of DBD of interest to the Estrogen Receptor. The forward primer is homologous the ACT1 promoter and the reverse primer is homologous to the estrogen receptor. For Typical DBDs (~300 bp), the PCR product is <1.5 kb.
| Oligonucleotide Sequence | Name |
| 5'-ggccgc…DBD binging site... t-3' | Top oligo |
| 5’-ctaga…DNA binding site reverse…gc-3’ | Bottom oligo |
| 5'-gccgcGTGGGCGTGCGTGGGCGGG CGTGGGCGTGCGTGGGCGGGCGTG GGCGTGCGTGGGCGt-3' | Top oligo + 6x Zif268 Binding Sites |
| 5’-ctagaCGCCCACGCACGCCCACGCC CGCCCACGCACGCCCACGCCCGCC CACGCACGCCCACgc-3’ | Bottom oligo + 6x Zif268 Binding Sites |
Table 2. Structure of primers for creating dsDNA containing binding sites of interest. To create the Z3EV-responsive promoter, the oligonucleotides Top oligo + 6x Zif268 Binding Sites and Bottom oligo + 6x Zif268 Binding were used. Sequences for creating the correct DNA overhangs are in lowercase. The Zif268 consensus binding site, GCGTGGGCG, is underlined in the top oligo.
| Reagent | Amount |
| T4 Polynucleotide Kinase Buffer | 5 µl |
| T4 Polynucleotide Kinase (@10,000 units/ml) | 1 µl |
| Top oligo (100 µM) | 1.5 µl |
| Bottom oligo (100 µM) | 1.5 µl |
| Water | 41 µl |
Table 3. Phosphorylate and anneal primers in a thermocycler using 50 µl of reaction described above. There are two thermocycler steps. Step 1: 37 °C 30 min (Phosphorylate), and step 2: 95 °C 30 sec (step down 1 °C every 30 sec until at 4 °C; Anneal).
| Reagent | Amount |
| XbaI | 1 µl |
| NotI-HF | 1 µl |
| Plasmid DNA | 5 µl (1-2 µg) |
| 10x Cut Smart Buffer | 5 µl |
| Water | 38 µl |
Table 4. Restriction digest of plasmid pMN8 with XbaI and NotI.
| Reagent | Amount |
| T4 Ligase Buffer | 2 µl |
| T4 Ligase | 0.2 µl |
| Backbone @ 10 ng/µl | 1 µl |
| Binding Sequences@ 5x molar concentration/µl | 1 µl |
| Water | 15.8 µl |
Table 5. Ligate binding sites into plasmid DNA.
| Primer | Description |
| ~40 bp upstream of ATG + CGCACTTAACTTCGCATCTG-3’ | Forward primer for amplifying KanMX-Promoter |
| 5’- reverse complement(ATG + ~37bp) + TATAGTTTTTTCTCCTTGACG-3’ | Reverse primer for amplifying KanMX-Promoter |
| 5’-caatttgtctgctcaagaaaataaa ttaaatacaaataaaCGCAC TTAACTTCGCATCTG-3’ | Forward primer for making GCN4 promoter swap. |
| 5’-tggatttaaagcaaataaacttgg ctgatattcggacatTATAGTT TTTTCTCCTTGACG-3’ | Reverse primer for making GCN4 promoter swap. |
Table 6. PCR amplify KanMX-Promoter for making titratible allele.
Discussion
The hormone-based switches described here have myriad applications, from studying native cell biology to testing the ability of a DNA binding domain of interest to target a DNA sequence in vivo. The system has many desirable features, including a graded response to inducer, fast action, and tight regulation (i.e. no measurable leakiness, see Figure 7). However, there is still room for improvement and expansion.
Recent work in synthetic biology has emphasized multiplexing synthetic systems and expanding the repertoire of well-characterized, programmable DNA parts16. Independent, conditional expression of distinct target genes requires both multiple inducers and DNA binding domains that lack overlapping specificity. The availability of engineered and naturally occurring receptors provides a large set of parts with which to develop new artificial transcription factors. Expanding the repertoire of mutually orthogonal switches has been greatly aided by directed evolution approaches17,18. Current efforts to reprogram the ligand-binding specificity of our artificial transcription factors will be presented in the future.
Previously, the phenotypic consequences of gene deletion/overexpression have been extensively studied in yeast19,20. However, it has not been straightforward to study the effect of expression on different phenotypes over a range of expression levels. A primary feature of the system presented herein is its graded nature (Figure 8). While often assumed, the relationship between gene expression and phenotypes of interest may not necessarily be monotonic. Artificial transcription factors such as Z3EV and Z4EV will make the task of assaying the phenotypic responses to changes in gene expression straightforward.
Finally, the protocols discussed in this manuscript are for engineering and characterizing artificial transcription factors that respond to β-estradiol; however, an important alternative to chemically responsive domains is the use of light-responsive domains (such as the PhyB/Pif3, LOV, and BLUF), whose activities are reversible without having to perturb the culture growth medium21-23. Light-based systems facilitate spatiotemporal control of gene expression, protein-protein interactions, ion transport, and enzymatic activity, which have already proven powerful21-26.
In conclusion, we have presented methods for the rapid construction of inducible, artificial transcription factors in yeast. This protocol provides a template for their construction in conjunction with cognate GFP reporters, as well as methods for analyzing the reporter data using flow cytometry and assaying the effect of ATF activation on growth.
Disclosures
McIsaac, Noyes, and Botstein (with others) have submitted part of this system as a Patent Disclosure.
Acknowledgments
R.S.M. acknowledges funding from the NSF Graduate Research Fellowship. M.B.N. acknowledges funding from a Lewis-Sigler Fellowship and the endowed gift of Peter Lewis. D.B. acknowledges funding from the NIH (GM046406) the National Institute of General Medical Sciences Center for Quantitative Biology (GM071508). We acknowledge Christina DeCoste for assistance with flow cytometry experiments.
References
- McIsaac RS, et al. Synthetic gene expression perturbation systems with rapid, tunable, single-gene specificity in yeast. Nucleic acids res. 2013;41:e57. doi: 10.1093/nar/gks1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIsaac RS, et al. Fast-acting and nearly gratuitous induction of gene expression and protein depletion in Saccharomyces cerevisiae. Mol. Biol. cell. 2011;22:4447–4459. doi: 10.1091/mbc.E11-05-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louvion JF, Havaux-Copf B, Picard D. Fusion of GAL4-VP16 to a steroid-binding domain provides a tool for gratuitous induction of galactose-responsive genes in yeast. Gene. 1993;131:129–134. doi: 10.1016/0378-1119(93)90681-r. [DOI] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic acids res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIsaac RS, et al. Visualization and Analysis of mRNA Molecules Using Fluorescence In Situ Hybridization in Saccharomyces cerevisiae. J. Vis. Exp. 2013. p. e50382. [DOI] [PMC free article] [PubMed]
- McIsaac RS, Petti AA, Bussemaker HJ, Botstein D. Perturbation-based analysis and modeling of combinatorial regulation in the yeast sulfur assimilation pathway. Mol. Biol. cell. 2012;23:2993–3007. doi: 10.1091/mbc.E12-03-0232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belli G, Gari E, Piedrafita L, Aldea M, Herrero E. An activator/repressor dual system allows tight tetracycline-regulated gene expression in budding yeast. Nucleic acids res. 1998;26:942–947. doi: 10.1093/nar/26.4.942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron U, Bujard H. Tet repressor-based system for regulated gene expression in eukaryotic cells: principles and advances. Meth. Enzymol. 2000;327:401–421. doi: 10.1016/s0076-6879(00)27292-3. [DOI] [PubMed] [Google Scholar]
- Kodama S, Okada K, Akimoto K, Inui H, Ohkawa H. Recombinant aryl hydrocarbon receptors for bioassay of aryl hydrocarbon receptor ligands in transgenic tobacco plants. Plant Biotechnol. J. 2009;7:119–128. doi: 10.1111/j.1467-7652.2008.00378.x. [DOI] [PubMed] [Google Scholar]
- Hickman MJ, et al. Coordinated regulation of sulfur and phospholipid metabolism reflects the importance of methylation in the growth of yeast. Mol. Biol. cell. 2011;22:4192–4204. doi: 10.1091/mbc.E11-05-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taxis C, Stier G, Spadaccini R, Knop M. Efficient protein depletion by genetically controlled deprotection of a dormant N-degron. Mol. Syst. Biol. 2009;5:267. doi: 10.1038/msb.2009.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. methods. 2009;6:917–922. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Burke DJ, Amberg DC, Strathern JN. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. 2005.
- Kahm M, Hasenbrink G, Lichtenberg-Frate H, Ludwig J, Kschischo M. grofit: Fitting Biological Growth Curves with R. J. Stat. Softw. 2010;33:1–21. [Google Scholar]
- Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- Khalil AS, et al. A synthetic biology framework for programming eukaryotic transcription functions. Cell. 2012;150:647–658. doi: 10.1016/j.cell.2012.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chockalingam K, Chen Z, Katzenellenbogen JA, Zhao H. Directed evolution of specific receptor-ligand pairs for use in the creation of gene switches. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5691–5696. doi: 10.1073/pnas.0409206102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan MJ, Chockalingam K, Lai KC, Zhao H. Directed evolution of orthogonal ligand specificity in a single scaffold. Angewandte Chemie. 2009;48:7783–7786. doi: 10.1002/anie.200903413. [DOI] [PubMed] [Google Scholar]
- Jorgensen P, Nishikawa JL, Breitkreutz BJ, Tyers M. Systematic identification of pathways that couple cell growth and division in yeast. Science. 2002;297:395–400. doi: 10.1126/science.1070850. [DOI] [PubMed] [Google Scholar]
- Sopko R, et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol. cell. 2006;21:319–330. doi: 10.1016/j.molcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- Shimizu-Sato S, Huq E, Tepperman JM, Quail PH. A light-switchable gene promoter system. Nat. biotechnol. 2002;20:1041–1044. doi: 10.1038/nbt734. [DOI] [PubMed] [Google Scholar]
- Polstein LR, Gersbach CA. Light-inducible spatiotemporal control of gene activation by customizable zinc finger transcription factors. J. Am. Chem. Soc. 2012;134:16480–16483. doi: 10.1021/ja3065667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losi A, Gartner W. Old chromophores, new photoactivation paradigms, trendy applications: flavins in blue light-sensing photoreceptors. Photochem. Photobiol. 2011;87:491–510. doi: 10.1111/j.1751-1097.2011.00913.x. [DOI] [PubMed] [Google Scholar]
- Boyden ES, Zhang F, Bamberg E, Nagel G, Deisseroth K. Millisecond-timescale, genetically targeted optical control of neural activity. Nat. Neurosci. 2005;8:1263–1268. doi: 10.1038/nn1525. [DOI] [PubMed] [Google Scholar]
- Gradinaru V, Mogri M, Thompson KR, Henderson JM, Deisseroth K. Optical deconstruction of parkinsonian neural circuitry. Science. 2009;324:354–359. doi: 10.1126/science.1167093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milias-Argeitis A, et al. In silico feedback for in vivo regulation of a gene expression circuit. Nat. biotechnol. 2011;29:1114–1116. doi: 10.1038/nbt.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]