

Abstract
All cellular processes depend on the functionality of proteins. Although the functionality of a given protein is the direct consequence of its unique amino acid sequence, it is only realized by the folding of the polypeptide chain into a single defined three-dimensional arrangement or more commonly into an ensemble of interconverting conformations. Investigating the connection between protein conformation and its function is therefore essential for a complete understanding of how proteins are able to fulfill their great variety of tasks. One possibility to study conformational changes a protein undergoes while progressing through its functional cycle is hydrogen-1H/2H-exchange in combination with high-resolution mass spectrometry (HX-MS). HX-MS is a versatile and robust method that adds a new dimension to structural information obtained by e.g. crystallography. It is used to study protein folding and unfolding, binding of small molecule ligands, protein-protein interactions, conformational changes linked to enzyme catalysis, and allostery. In addition, HX-MS is often used when the amount of protein is very limited or crystallization of the protein is not feasible. Here we provide a general protocol for studying protein dynamics with HX-MS and describe as an example how to reveal the interaction interface of two proteins in a complex.
Keywords: Chemistry, Issue 81, Molecular Chaperones, mass spectrometers, Amino Acids, Peptides, Proteins, Enzymes, Coenzymes, Protein dynamics, conformational changes, allostery, protein folding, secondary structure, mass spectrometry
Introduction
The number of crystal structures of proteins and protein complexes increased rapidly in recent years. They present invaluable snapshots of the structural organization of these proteins and provide a basis for structure-function analysis. However, the dynamics of proteins and the conformational changes, which are essential for their functions, are rarely revealed by X-ray crystallography. Cryo-electronmicroscopy, on the other hand, is able to capture protein and protein complexes in different conformations but generally cannot resolve conformational changes down to secondary structure level1. Conformational dynamics of proteins in solution at atomic details can only be resolved by NMR, but this method is still restricted to proteins of relatively small sizes (generally ≤ 30 kDa) and needs high concentrations of proteins (≥ 100 μM), which hampers experiments with oligomerization or aggregation prone proteins2. One method that is able to bridge between high-resolution X-ray crystallography and cryo-electronmicroscopy and which is not limited by protein size or concentration is amide hydrogen-1H/2H-exchange (HX) in combination with mass spectrometry (MS). In recent years this method has developed to a valuable analytical tool for the analysis of protein dynamics, protein folding, protein stability and conformational changes3-5. The molecular basis of this method is the labile nature of backbone amide hydrogens in proteins, which will exchange with deuterium atoms when the protein is placed in a D2O solution. The subsequent increase in protein mass over time is measured with high-resolution MS.
In short unstructured peptides HX only depends on temperature, catalyst concentration (OH-, H3O+ i.e. pH, see Figure 3) and amino acid side chains of adjacent residues due to inductive, catalytic and steric effects. These effects on the intrinsic chemical exchange rate kch have been elegantly quantified by Bai et al.6 and a program is available (courtesy Z. Zhang), which calculates kch for each amino acid within a polypeptide dependent on pH and temperature. At neutral pH and ambient temperatures kch is in the order of 101-103 sec-1. In folded proteins HX can be 2-9 orders of magnitude slower mainly due to hydrogen bonding in the secondary structure and to a minor degree due to restricted access of hydrated OH- ions to the interior of a tightly folded protein. HX in native proteins therefore implicates partial or global unfolding, chemical exchange and refolding to the native state according to equation (1) and the observed exchange rates kobs depend on the opening rate kop, the closing rate kcl and the intrinsic chemical exchange rate kch according to equation (2).
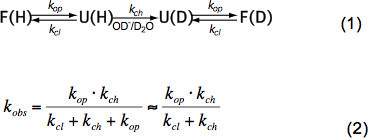 Under native state conditions kop is much smaller than kch and can be neglected in the denominator. There are two extreme exchange regimes called EX1 and EX2. If the kcl is much smaller than kch (EX1) the observed rate is practically equal to the opening rate and HX allows immediate observation of the unfolding of a structural element. Such an exchange regime, where all amide protons exchange at once upon opening of the structural element, is readily observable in MS by a bimodal distribution of the isotope peaks7. If kcl is much greater than kch (EX2) the observed rate is proportional to kch whereby the proportionality constant is equal to the folding-unfolding equilibriums constant Ku = kop/kcl. Under these conditions, many opening and closing events are necessary before all amide protons exchange for deuterons, leading to a gradual increase in average mass while the isotopic distribution remains roughly the same. The EX2 regime allows the determination of the free energy of unfolding ΔGu and therefore the stability of a structural element. Under native state condition the EX2 regime is most common. Increase of pH and addition of chaotropic agents can shift the exchange mechanism to EX1. Therefore, HX-MS can be used to explore thermodynamic as well as kinetic parameters of protein folding and conformational changes.
Under native state conditions kop is much smaller than kch and can be neglected in the denominator. There are two extreme exchange regimes called EX1 and EX2. If the kcl is much smaller than kch (EX1) the observed rate is practically equal to the opening rate and HX allows immediate observation of the unfolding of a structural element. Such an exchange regime, where all amide protons exchange at once upon opening of the structural element, is readily observable in MS by a bimodal distribution of the isotope peaks7. If kcl is much greater than kch (EX2) the observed rate is proportional to kch whereby the proportionality constant is equal to the folding-unfolding equilibriums constant Ku = kop/kcl. Under these conditions, many opening and closing events are necessary before all amide protons exchange for deuterons, leading to a gradual increase in average mass while the isotopic distribution remains roughly the same. The EX2 regime allows the determination of the free energy of unfolding ΔGu and therefore the stability of a structural element. Under native state condition the EX2 regime is most common. Increase of pH and addition of chaotropic agents can shift the exchange mechanism to EX1. Therefore, HX-MS can be used to explore thermodynamic as well as kinetic parameters of protein folding and conformational changes.
As mentioned above HX is intrinsically pH and temperature dependent and the exchange half-life of a completely solvent exposed proton of the backbone amide group is between 5-400 msec at physiological pH (pH 7.6) and 30 °C, but 10 min to >15 hr with an average of >2 hr at pH 2.9 and 0 °C (except for the proton of the first backbone amide bond of a polypeptide, which exchanges with a half-life of ca. 1-2 min). Under such slow exchanging conditions it is possible to digest the sample using proteases (e.g. pepsin) that are active under these conditions, with out losing all the information contained in the incorporated deuterons. Since the introduction of peptic digestion under slow exchanging conditions, not only the overall HX kinetics of full-length proteins can be analyzed but HX can be localized to specific regions8,9. Spatial resolution is currently limited to the size of the peptic fragments generated, which is in general between 10-30 residues. However, overlapping fragments created due to the nonspecific nature of cleavage by pepsin could lead to an increase in spatial resolution. In addition, several other proteases were found to be active under quench conditions, however, much less efficient than pepsin10. Further increase of spatial resolution can be reached by fragmentation of peptides in the gas phase by methods that preserved the deuteration pattern such as electron capture dissociation (ECD), electron transfer dissociation (ETD) and infrared multiphoton dissociation (IRMPD)11-13. These techniques prevent the loss of spatial resolution due to intramolecular proton migration ("scrambling"), which is observed by collision-induced dissociation (CID) the most commonly used fragmentation technique. However, these methods require optimization for every individual peptide and is thus still quite challenging.
HX-MS has been used to analyze protein-ligand and protein-protein interactions including viral capsid assembly14-17. Protein unfolding and refolding as well as temperature induced conformational changes were investigated7,18,19. Phosphorylation and single amino acid mutation-related conformational changes16,20 and nucleotide-induced changes were analyzed21,22. Therefore, this method seems ideally suitable to analyze assembly and dynamics of molecular machines. One attractive candidate, whose mechanism is of great general interest, is the Hsp90 chaperone complex.
Protocol
1. Preparation of Buffers and Protein Samples
Prepare H2O buffer. Use Hsp90 standard buffer (40 mM HEPES/KOH, pH 7.5, 50 mM KCl, 5 mM MgCl2, 10% glycerol) as H2O buffer. Note: If the sample was dialyzed before analysis, use the dialysis buffer as H2O buffer. It is essential that the D2O buffer differs from the H2O buffer only in the hydrogen isotope. Volatile buffers like NH4CO3 or NH4-acetate or buffer components are not suitable!
Prepare D2O buffer by lyophilization of Hsp90 standard buffer using a vacuum concentrator. After complete evaporation of H2O add pure D2O to the tube to reach the initial total volume (e.g. 1 ml buffer with 15% glycerol require the addition of 850 μl D2O). Repeat the complete evaporation of the buffer and redissolve the buffer/salt components in D2O 2x.
Prepare quench buffer (0.4 M KH2PO4/H3PO4 pH 2.2). Note: To improve the efficiency of peptic digest of very stable proteins 4 M guanidine hydrochloride and 0.5 M Tris(2-carboxyethyl)phosphine (TCEP-HCl) can be added.
Prepare 100% control sample (6 M guanidine hydrochloride, D2O). Add guanidine hydrochloride to an Hsp90 aliquot to reach a final concentration of 6 M. Completely evaporate H2O from the sample and add D2O to the tube to reach the initial total volume (e.g. 100 μl sample with 10% glycerol require the addition of 90 μl D2O). Repeat the complete evaporation of the buffer and redissolve the buffer/salt components in D2O. Note: 20-100 pmol of sample are required for each injection. Prepare enough sample to have a 100% control for every day of HX-MS experiments.
Prepare 50 pmol Hsp90 in 5 μl Hsp90 standard buffer. Note: An amount of 20-100 pmol of sample is required for each point of raw data. The volume of sample in the reaction is 1-5 μl ideally. Adjust the concentration to fit these requirements. Any buffer can be used as long as it doesn't contain detergents or volatile components.
2. Preparation of Immobilized Pepsin on Aldehyde Activated Beads
Dissolve 80 mg fresh pepsin in 2 ml 50 mM sodium citrate (pH 5).
Dissolve 20 mg of sodium cyanoborohydride, handle with care (very toxic!) in 1 ml 2 M Na2SO4 and add to pepsin solution.
Incubate mixture for 10 min at room temperature while gently agitating (e.g. overhead shaker).
Add 600 mg beads with immobilized aldehyde groups to the mixture and incubate for 5-10 min at room temperature.
Add 2.2 ml of 2 M Na2SO4 (pH 5) in 100 μl aliquots every 3 min over one hour to slowly salt out the pepsin. Gently mix the sample between additions in an overhead shaker at room temperature.
Incubate the pepsin beads at 4 °C for 14-16 hr/overnight in an overhead shaker.
Quench the reaction by adding of 1 ml 1 M ethanolamine and incubation at room temperature for 2 hr.
Spin down the beads in a 50 ml falcon tube at 500 rpm, discard the supernatant and resuspend the beads in 0.1% formic acid. Repeat this step 2x. After the last centrifugation step, discard supernatant and estimate volume of beads. Add an equivalent volume of 0.1% formic acid and store at 4 °C.
3. Preparation of Columns for Amide Hydrogen-exchange
Use guard columns with an inner diameter of 1 mm for trapping columns and with 2 mm for pepsin columns.
Unscrew one side of the guard column and remove the filter. Tightly screw packing funnel onto the open end of the column. Use a 1/16 inch adapter and tubing to attach an empty syringe (5 ml) to the bottom outlet of the column. Make sure to fix it gas-tight to the guard column.
Apply a few drops of slurry bead material on top of the funnel. Pull the plunger of the syringe to suck the slurry through the funnel into the guard column. Apply more slurry bead material onto the funnel and continue the procedure until the guard column is completely filled with bead material. Remove the funnel and place filter and filter ring onto the open end. Tightly screw the column cap onto the guard column and remove the syringe from the other side. Close both ends of the guard columns with plugs to avoid drying out of column material.
4. Setting up the System for Hydrogen Exchange Mass Spectrometry (HX-MS)
Connect the trap column with the HPLC system (Figure 1). Equilibrate the column by setting the flow rate of pump A to 0.4 ml/min with 0.1% formic acid as solvent. Do not connect the pepsin column nor the analytical column yet.
Calibrate the mass spectrometer and connect the outlet of the HPLC to the source of the mass spectrometer.
5. Determination of the Dynamic Range of Exchange
Prepare ultrapure solvent A (0.1% formic acid in water) and ultrapure solvent B (0.1% formic acid in acetonitrile); ready mixed solvents are commercially available. Purge the HPLC pumps. Set up the chromatography and mass spectrometry methods in the control software by choosing a program with a step gradient after a short desalting step. For full-length Hsp90 use a 1-2 min desalting step followed by switching the 6-port valve from DESALT/LOAD to ELUTE and a step gradient from 90% solvent A/10% solvent B to 5% solvent A/95% solvent B. Before injection set the injection valve to LOAD and the 6-port valve to DESALT/LOADING position. Note: Do not use a pepsin or analytical column during this experiment.
Prepare 100-200 pmol of Hsp90 in 1-10 μl H2O buffer and incubate for 10 min at 30 °C. Add temperature adjusted D2O buffer to bring the sample volume up to 100 μl and incubate exactly for a defined period of time (e.g. 10 sec, 100 sec, 1,000 sec). Add 100 μl quench buffer and mix by pipetting up and down. Inject the 200 μl sample into the injection port of the injection valve with a Hamilton syringe. Start the chromatography program and switch the injection valve into INJECT position. After 2 min switch the 6-port valve from DESALT/LOADING to ELUTE. Repeat this for at least three time-points.
Repeat step 5.2 but add 2-3x excess of Sti1 to the Hsp90 sample prior to incubating for 10 min at 30 °C.
Determine the full-length protein masses by deconvolution of spectra in the mass spectrometry software. Calculate the number of incorporated deuterons by comparing the molecular weight of full-length Hsp90 with the observed mass after each run (e.g. 10 sec, 100 sec, 1,000 sec).
Plot the incorporated deuterons for Hsp90 in absence and presence of Sti1 (y-axis) versus time (x-axis). Determine the time-point in the dynamic range where the difference between both curves is maximal. Use this value for the incubation time in D2O when identifying Hsp90-Sti1 interface and dynamics on peptide level.
6. Determination of Peptic Peptides Using MS/MS Spectra
Connect the pepsin column and analytical column to the system.
Set up the parameters for the chromatography and mass spectrometry in the control software by choosing gradient type and mass spectrometry method. Choose a long gradient (e.g. more than 90 min) to ensure good chromatographic resolution. Enable MS/MS spectra on the mass spectrometer. Note: Good resolution on the HPLC and high mass accuracy have highest priority in this step.
Prepare 100-200 pmol of Hsp90 in 100 μl H2O buffer. Add 100 μl quench buffer and mix by pipetting up and down. Inject the 200 μl sample into the injection port of the injection valve with a Hamilton syringe. Start the chromatography program and switch the injection valve into INJECT position. After 2 min switch the 6-port valve from DESALT/LOADING to ELUTE position. Note: If more than one protein is to be analyzed in HX-MS (i.e. protein-protein interaction interfaces) the peptides for each protein have to be determined individually.
Identify peptic peptides of Hsp90 by searching a database (i.e. Mascot) for the resulting peptides. Note: It is possible to use a custom database as the aim is to determine as many peptic peptides as possible and analysis is done with purified protein. Sample purity will have to be taken into account.
Repeat this step without MS/MS and with the gradient that will be used for the actual HX experiment. For Hsp90 and Sti1 use a 10 min gradient from 90% solvent A/10% solvent B to 45% solvent A/55% solvent B. Note: Gradients are normally between 5-15 min and highly depend on sample complexity and HX system specifications.
Determine the retention times of the peptic peptides identified in 6.4 in the gradient used in 6.5 and create a list comprising 1.) peptide sequence 2.) peptide charge state and 3.) retention time. This will be used to identify each peptide after HX experiments. Note: Be aware that peptides with small m/z differences, identical charge state and identical retention time can be a source of ambiguity.
7. Identification of Protein-protein Interaction Interfaces
Set up the gradient and mass spectrometry method in the control software. Use a 10 min linear gradient from 90% solvent A/10% solvent B to 45% solvent A/55% solvent B. Load a mass spectrometry method that is optimized for detection of masses between 300-1,500 m/z, although most of the peptides will be below 1,000 m/z. Set the injection valve into LOAD position, the 6-port valve in LOADING/DESALTING Position. Set the flow rate of the loading pump to 0.4 ml/min. Note: Length and type of gradient are sample dependent and may have to be optimized. Longer gradients improve the resolution of the chromatography but decrease the incorporation of deuterons into proteins due to back-exchange. The chosen methods need to be the same for all experiments to be compared.
For the unexchanged reference prepare 20-100 pmol of Hsp90 in 100 μl H2O buffer. Add 100 μl ice-cold quench buffer, pipette up and down twice and inject sample into the injection valve of the HPLC. Immediately start the chromatography program and switch the injection valve into INJECT position. After 2 min switch the 6-port valve from DESALT/LOADING to ELUTE position. Do this for Hsp90 and Sti1 individually and for the mixture of proteins.
Prepare 20-100 pmol of Hsp90 in a volume of 1-5 μl. Add temperature adjusted D2O buffer to bring the sample volume up to 100 μl and incubate exactly for a defined period of time (e.g. 30 sec; for conformational dynamics see Note). Add 100 μl of ice-cold quench buffer, pipette up and down twice and quickly inject the 200 μl into the injection valve of the HPLC. Immediately start the chromatography program and switch the injection valve into INJECT position. After 2 min switch the 6-port valve from DESALT/LOADING to ELUTE position. Do this for each protein individually, to determine the deuteron incorporation into each peptide in absence of the interacting protein. Note: When studying the dynamics of conformational changes, repeat the experiment with different incubation times of Hsp90 in D2O buffer. Try to cover a wide timescale by choosing incubation times logarithmically (e.g. 10 sec, 30 sec, 100 sec, 300 sec, 1,000 sec, etc.). D2O buffer and sample buffer must be exactly identical except for the hydrogen isotope.
Prepare an equal amount of 100% control sample (20-100 pmol) and add D2O buffer to bring the sample volume up to 100 μl. Add 100 μl of ice-cold quench buffer, pipette up and down twice and quickly inject the 200 μl into the injection valve of the HPLC. Immediately start the chromatography program and switch the injection valve into INJECT position. After 2 min switch the 6-port valve from DESALT/LOADING to ELUTE position.
To determine the interaction surface mix Hsp90 with an at least 2-fold excess of Sti1 to shift the equilibrium to the bound state (see Note) and incubate at the desired temperature until complex formation is at equilibrium. Add temperature adjusted D2O buffer to bring the sample volume up to 100 μl and incubate exactly for a defined period of time (e.g. 30 sec). Add 100 μl of ice-cold quench buffer, pipette up and down twice and quickly inject into the injection valve of the HPLC. After 2 min switch the 6-port valve from DESALT/LOADING to ELUTE position. Repeat this experiment with 20-100 pmol of Sti1 and excess of Hsp90. Note: The absolute concentrations depend on the dissociation equilibrium constant of the interaction. Ideally, after dilution into D2O the concentration of the protein added in excess should be at least 10x the KD (corresponding to >85% of the lower concentrated protein bound).
Analyze the acquired data with suitable software and use the retention times determined in step 6.5 to find each peptide in the analysis. Calculate the centroid of the isotope distribution for the unexchanged protein (step 7.2) and for the HX experiments (step 7.3). Note: Although the isotope patterns of exchanged peptides look different from their unexchanged counterparts, both the knowledge about charge state of the peptide and the high mass accuracy of the mass spectrometer allows for easy determination of the centroids.
Compare incorporation of deuterons of target protein alone and with excess of binding partner. This can be done automatically with commercial software or manually with a spreadsheet program. The 100% control sample values denote the maximal exchange for each peptide and can be used to determine the amount of D2O label lost during the experiment due to back exchange.
Representative Results
Hsp90 is a molecular chaperone in yeast and member of the Hsp90 chaperone family. By going through a complex ATPase cycle it assists late folding steps of many protein clients. Efficient folding requires transfer of clients from Hsp70 and interaction of the co-chaperone Sti1/Hop. Sti1 directly binds to Hsp90 and facilitates client binding by inhibition of Hsp90's ATPase activity. Interaction of Hsp90 with Sti1 was recently studied using HX-MS23. Here we present representative results of the underlying experiments following the above protocol.
The direct protein-protein interaction was tested by labeling the protein complex in equilibrium with D2O and comparing the Sti1 peptide spectra in absence and presence of Hsp90.The resulting difference plot can be seen in Figure 5A (data from23). The peptides are oriented from top to bottom starting at the N-terminus. Mostly peptides in TPR2a and TPR2b show a strong protection in the presence of Hsp90 (negative values for D+Hsp90 - D-Hsp90), indicating that these regions interact with Hsp90. The protection in TPR2a can easily be rationalized from the crystal structure of Sti1-TPR2a-TPR2b in complex with a peptide representing the C-terminal MEEVD-motif of Hsp90 (Figure 5B). Such a clear protection in protein-protein interactions is not always observed. The protection in Hsp90 in the presence of Sti1 is not as pronounced and not as localized as in Sti1 (Figures 5C and 5D). All peptides show slightly increased protection from deuteron incorporation in presence of Sti1. Sti1 obviously stabilizes Hsp90 globally as no region of the protein shows more flexibility in presence of Sti1. We can however not distinguish, if this reduced deuteron incorporation originates from direct interaction with Sti1 or through allosteric effects on the conformation of Hsp90. The information obtained reduces the putative interaction site to a few peptides (e.g. peptide 43-62 of NBD). Additional experiments such as crosslinking are commonly used to confirm the binding sites after HX-MS23. Of note, the sequence coverage of Hsp90 is decreased when measuring the Hsp90-Sti1 complex. Since Sti1 was added in 3.5-fold excess the theoretical overall amount of peptides in the analysis was some fourfold higher than with Hsp90 alone. This increases the risk of Sti1 peptides overshadowing or overlapping Hsp90 peptides during analysis. To optimize for higher sequence coverage better chromatographic separation seems to be the most promising approach.
One possibility to study the dynamics of different protein regions is the use of continuous labeling HX-MS. It involves the incubation of equilibrated protein sample in D2O for different periods of time and therefore allows predictions about the stability of individual protein segments. Figure 6 illustrates the spectra of peptide 43-62 from the NBD of Hsp90 in presence and absence of a 3.5-fold excess of Sti1. The incubation times were chosen on a logarithmic scale (10 sec, 100 sec, and 1,000 sec) to get good coverage over a wide time window. The peptide spectra show a time dependent incorporation of deuterium into the protein that increases for longer incubation times. For peptide 43-62 exchange follows the EX2 mechanism where the kint << kcl. The degree of protection changes throughout the incubation time, showing the major difference at shorter incubation time and almost similar exchange rate at the longest time point. This suggests a lower degree stabilization or a region of dynamic interactions with frequent dissociation and reassociation. Either the observed protection is due to an overall reduced exchange rate of amide protons in the peptide or the effect can be assigned to one or two specific amide protons that exchange more slowly in presence of Sti1. In this case the latter is much more probable as the Hsp90 crystal structure revealed this region to be an exposed loop rather than a regular secondary structure like α-helices or β-sheets.
It is important to mention that the back exchange has not been subtracted from the data shown implying that the observed effects will be more pronounced once normalized to the 100% control.
 Figure 1. HPLC setup for HX-MS. a) Typical configuration of a HX-MS set up. The different colored areas represent sections maintained at different temperatures. The sample is injected into the sample loop of the injection valve (in LOAD mode, cyan) and after switching the valve into "inject mode" (black) pumped through the pepsin column into the 6-port valve. The pepsin column is maintained roughly at 10 °C to improve enzymatic activity of pepsin. The 6-port valve has two modes, "Loading/Desalting Mode" and "Elution Mode" (see b). After desalting of the sample the peptides are chromatographically separated with an acetonitrile gradient on an analytical reversed-phase column. The peptides are then sprayed into the mass spectrometer with an ESI-source. b) Modes of the 6-port valve. Directly after HX the 6-port valve is in "Loading/Desalting Position" to trap the peptic peptides on the trap column. This is continued for up to three minutes to remove any salts or compounds of the reaction buffers that could interfere with mass spectrometry. After this the 6-port valve switches into "Elution Mode" putting the loaded trap column into the flow path between gradient pump and analytical column. The trapped peptic peptides now elute from the trap column and become separated in a gradient of 0.1% formic acid/0.1% formic acid in acetonitrile.
Figure 1. HPLC setup for HX-MS. a) Typical configuration of a HX-MS set up. The different colored areas represent sections maintained at different temperatures. The sample is injected into the sample loop of the injection valve (in LOAD mode, cyan) and after switching the valve into "inject mode" (black) pumped through the pepsin column into the 6-port valve. The pepsin column is maintained roughly at 10 °C to improve enzymatic activity of pepsin. The 6-port valve has two modes, "Loading/Desalting Mode" and "Elution Mode" (see b). After desalting of the sample the peptides are chromatographically separated with an acetonitrile gradient on an analytical reversed-phase column. The peptides are then sprayed into the mass spectrometer with an ESI-source. b) Modes of the 6-port valve. Directly after HX the 6-port valve is in "Loading/Desalting Position" to trap the peptic peptides on the trap column. This is continued for up to three minutes to remove any salts or compounds of the reaction buffers that could interfere with mass spectrometry. After this the 6-port valve switches into "Elution Mode" putting the loaded trap column into the flow path between gradient pump and analytical column. The trapped peptic peptides now elute from the trap column and become separated in a gradient of 0.1% formic acid/0.1% formic acid in acetonitrile.
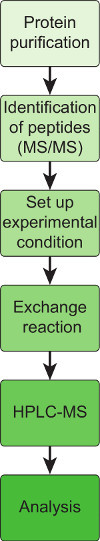 Figure 2. Flow scheme of an HX experiment. 1) Target protein is purified and supplied in standard buffers without detergent. 2) The protein is enzymatically digested with pepsin and the peptic peptides are identified and characterized by tandem mass spectrometry (MS/MS). Only after obtaining sequence, charge state and retention time of as many peptides as possible HX-MS can be successfully performed. 3) The experimental conditions are set up such as incubation of the protein under specific conditions (addition of ligand/chemical compound or shift in temperature or pH. 4) HX is performed by diluting the sample 1:10 or higher in D2O buffer. The D2O buffer must be identical to the sample buffer to avoid buffer effects. Then the sample is incubated exactly for a defined period of time. 5) After incubation the reaction is quenched and injected into the HPLC-MS system. The sample is enzymatically cleaved and the resulting peptic peptides are separated in an organic solvent gradient and injected into the mass spectrometer. 6) Analysis of the obtained peptide spectra. The centroids of the peptide spectra are compared under different reaction conditions. This can be done manually with suitable software or automatically by an HX analysis program.
Figure 2. Flow scheme of an HX experiment. 1) Target protein is purified and supplied in standard buffers without detergent. 2) The protein is enzymatically digested with pepsin and the peptic peptides are identified and characterized by tandem mass spectrometry (MS/MS). Only after obtaining sequence, charge state and retention time of as many peptides as possible HX-MS can be successfully performed. 3) The experimental conditions are set up such as incubation of the protein under specific conditions (addition of ligand/chemical compound or shift in temperature or pH. 4) HX is performed by diluting the sample 1:10 or higher in D2O buffer. The D2O buffer must be identical to the sample buffer to avoid buffer effects. Then the sample is incubated exactly for a defined period of time. 5) After incubation the reaction is quenched and injected into the HPLC-MS system. The sample is enzymatically cleaved and the resulting peptic peptides are separated in an organic solvent gradient and injected into the mass spectrometer. 6) Analysis of the obtained peptide spectra. The centroids of the peptide spectra are compared under different reaction conditions. This can be done manually with suitable software or automatically by an HX analysis program.
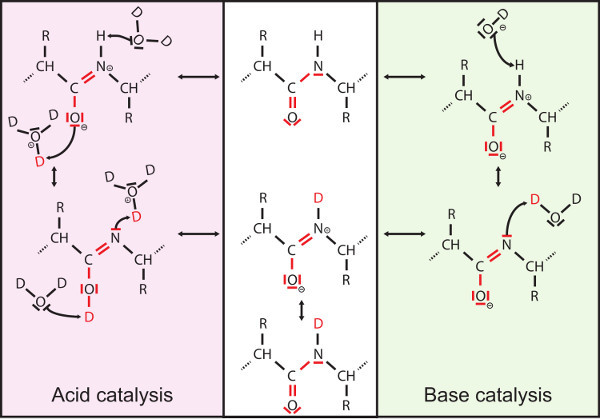 Figure 3. Hydrogen exchange mechanism. The reaction of hydrogen exchange can be catalyzed by base- or acid and therefore is pH dependent with a minimal exchange rate between pH 2.5 and 3 depending on the side chains of the residues flanking the amide bond6. During desalting and chromatographic separation of peptides, which is performed in H2O-containing solvents, incorporated deuterons are exchanged back for protons according to the same mechanism ("back exchange"). Therefore, optimization of the system and used solvents is crucial to reduce the loss of incorporated deuterons.
Figure 3. Hydrogen exchange mechanism. The reaction of hydrogen exchange can be catalyzed by base- or acid and therefore is pH dependent with a minimal exchange rate between pH 2.5 and 3 depending on the side chains of the residues flanking the amide bond6. During desalting and chromatographic separation of peptides, which is performed in H2O-containing solvents, incorporated deuterons are exchanged back for protons according to the same mechanism ("back exchange"). Therefore, optimization of the system and used solvents is crucial to reduce the loss of incorporated deuterons.
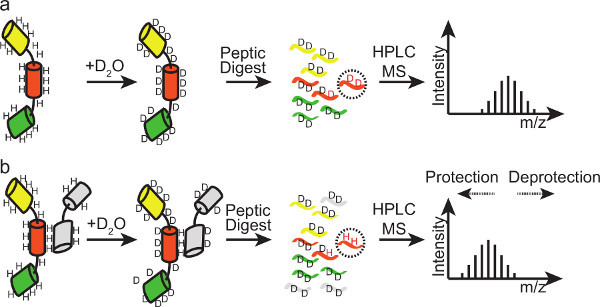 Figure 4. Principle of HX-MS. Binding of a ligand to the protein results in protection of the amide backbone hydrogens at the binding site from the surrounding solvent. This decreases the exchange rates of affected protons and decreases the incorporation of deuterons. Because the weight difference between proton and deuteron is 1 Da, peptides derived from this region of the protein will show a lower m/z in mass spectrometry. Comparing the peptide spectra of the experiments in absence and presence of binding partner will reveal a shift of the centroid of the spectra to lower m/z values (protection event). Regions that exhibit prominent protection after addition of binding partner are potential binding sites. Depending on the sequence coverage and the availability of overlapping peptides binding sites can be narrowed down to a few amino acids.
Figure 4. Principle of HX-MS. Binding of a ligand to the protein results in protection of the amide backbone hydrogens at the binding site from the surrounding solvent. This decreases the exchange rates of affected protons and decreases the incorporation of deuterons. Because the weight difference between proton and deuteron is 1 Da, peptides derived from this region of the protein will show a lower m/z in mass spectrometry. Comparing the peptide spectra of the experiments in absence and presence of binding partner will reveal a shift of the centroid of the spectra to lower m/z values (protection event). Regions that exhibit prominent protection after addition of binding partner are potential binding sites. Depending on the sequence coverage and the availability of overlapping peptides binding sites can be narrowed down to a few amino acids.
 Figure 5. Interaction of Hsp90 and Sti1. a, Difference plot of deuteron incorporation into Sti1 in the presence of binding partner Hsp90 minus deuteron incorporation into Sti1 in the absence of Hsp90 (Data from23). b, Cartoon representation of a fragment of Sti1 comprising TPR2a and TPR2b (PDB code 3UQ3) in complex with a peptide, which represents the C-terminal MEEVD-motif of Hsp90. The cartoon is colored according to the number of amide protons protected from deuteron incorporation as indicated. c, Difference plot of Hsp90 in presence of binding partner Sti1. Observed deuteron incorporation into Hsp90 after 10 min preincubation with 3.5x excess Sti1 at 30 °C and 30 sec; HX at the same temperature. The experiment was carried out in triplicate and standard error of the mean is shown for each peptide. Global negative values denote reduced incorporation of deuterons and therefore higher overall stability of Hsp90 in presence of Sti1. Back exchange was not considered in this experiment. d, Cartoon representation of yeast Hsp90 (PDB code 2CG9) colored according to the number of amide protons protected from deuteron incorporation as indicated in b. Click here to view larger figure.
Figure 5. Interaction of Hsp90 and Sti1. a, Difference plot of deuteron incorporation into Sti1 in the presence of binding partner Hsp90 minus deuteron incorporation into Sti1 in the absence of Hsp90 (Data from23). b, Cartoon representation of a fragment of Sti1 comprising TPR2a and TPR2b (PDB code 3UQ3) in complex with a peptide, which represents the C-terminal MEEVD-motif of Hsp90. The cartoon is colored according to the number of amide protons protected from deuteron incorporation as indicated. c, Difference plot of Hsp90 in presence of binding partner Sti1. Observed deuteron incorporation into Hsp90 after 10 min preincubation with 3.5x excess Sti1 at 30 °C and 30 sec; HX at the same temperature. The experiment was carried out in triplicate and standard error of the mean is shown for each peptide. Global negative values denote reduced incorporation of deuterons and therefore higher overall stability of Hsp90 in presence of Sti1. Back exchange was not considered in this experiment. d, Cartoon representation of yeast Hsp90 (PDB code 2CG9) colored according to the number of amide protons protected from deuteron incorporation as indicated in b. Click here to view larger figure.
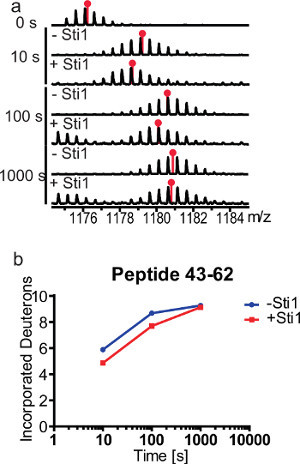 Figure 6. Time course of deuteron incorporation into yeast Hsp90 in the absence and presence of Sti1. a, Unprocessed peptide spectra of peptide 43-62 from the nucleotide binding domain of Hsp90 after continuous labeling hydrogen exchange. 20 μM Hsp90 were incubated with 70 μM Sti1 for 10 min at 30 °C to reach equilibrium and D2O labeling was performed at 30 °C for 10 sec, 100 sec, and 1,000 sec. Red pinpoint markers indicate the calculated centroids for each peptide. b, Exchange kinetics of peptide 43-62 in presence and absence of 3.5x excess Sti1. Back exchange was not considered in this experiment.
Figure 6. Time course of deuteron incorporation into yeast Hsp90 in the absence and presence of Sti1. a, Unprocessed peptide spectra of peptide 43-62 from the nucleotide binding domain of Hsp90 after continuous labeling hydrogen exchange. 20 μM Hsp90 were incubated with 70 μM Sti1 for 10 min at 30 °C to reach equilibrium and D2O labeling was performed at 30 °C for 10 sec, 100 sec, and 1,000 sec. Red pinpoint markers indicate the calculated centroids for each peptide. b, Exchange kinetics of peptide 43-62 in presence and absence of 3.5x excess Sti1. Back exchange was not considered in this experiment.
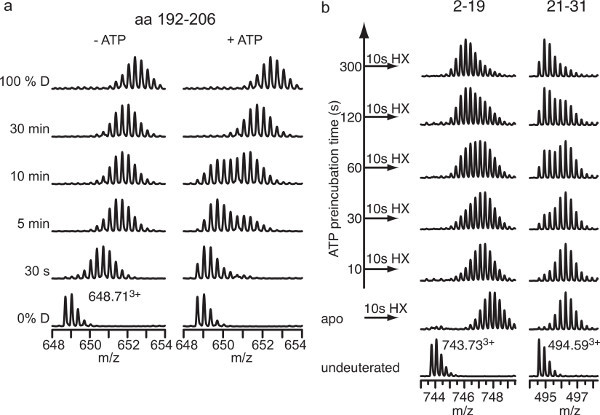 Figure 7. Examples for peptide spectra with bimodal distribution. a, Deuteron incorporation into E. coli Hsp90 in the absence and presence of ATP (data taken from 22 suppl. Figure 3). Spectra of peptide residues 192-206 are shown before incubation in D2O (0% D), after 30 sec, 5 min, 10 min, and 30 min incubation in D2O, and the 100% control (100% D). In the absence of ATP (-ATP) a typical EX2 exchange behavior is observed. In the presence of ATP (+ATP) a strong protection is observed at 30 sec and at 5 min and 10 min bimodal distributions of the isotopic peaks, indicating two populations of proteins, one population similar to the ATP bound state and a second similar to the nucleotide-free state. At 30 min no difference between absence and presence of ATP is observed. The bimodal distribution is most likely caused by ATP hydrolysis and transition through the nucleotide-free state with higher flexibility in this protein segment. These spectra resemble a classical EX1 exchange regime. b, Bimodal distribution of isotopic peaks in pulse-labeling HX experiments. E. coli Hsp90 was incubated in the absence of ATP or in the presence of ATP for 10-300 sec and subsequently pulse-labeled for 10 sec in D2O as indicated (data taken from22
Figure 4A). The bimodal distributions indicate again two coexisting populations of molecules, one exchanging more rapidly amide protons for deuterons than the other. ATP induces a slow transition from the faster exchanging into the slower exchanging conformation.
Figure 7. Examples for peptide spectra with bimodal distribution. a, Deuteron incorporation into E. coli Hsp90 in the absence and presence of ATP (data taken from 22 suppl. Figure 3). Spectra of peptide residues 192-206 are shown before incubation in D2O (0% D), after 30 sec, 5 min, 10 min, and 30 min incubation in D2O, and the 100% control (100% D). In the absence of ATP (-ATP) a typical EX2 exchange behavior is observed. In the presence of ATP (+ATP) a strong protection is observed at 30 sec and at 5 min and 10 min bimodal distributions of the isotopic peaks, indicating two populations of proteins, one population similar to the ATP bound state and a second similar to the nucleotide-free state. At 30 min no difference between absence and presence of ATP is observed. The bimodal distribution is most likely caused by ATP hydrolysis and transition through the nucleotide-free state with higher flexibility in this protein segment. These spectra resemble a classical EX1 exchange regime. b, Bimodal distribution of isotopic peaks in pulse-labeling HX experiments. E. coli Hsp90 was incubated in the absence of ATP or in the presence of ATP for 10-300 sec and subsequently pulse-labeled for 10 sec in D2O as indicated (data taken from22
Figure 4A). The bimodal distributions indicate again two coexisting populations of molecules, one exchanging more rapidly amide protons for deuterons than the other. ATP induces a slow transition from the faster exchanging into the slower exchanging conformation.
Discussion
Binding of an interaction partner to a protein inevitably causes changes in solvent accessibility on the binding site. Additionally, many proteins undergo dynamic conformational changes upon binding, which affect other regions than the actual binding interface. HX-MS is a robust method to monitor these changes and is even capable of revealing conformational changes in proteins on timescales that other methods cannot cover.
To successfully perform HX-MS three points are critical: 1) an optimal system setup; 2) precise execution of sample processing and 3) careful data analysis when assigning peptide spectra and interpreting results.
The system setup
Since mass spectrometry is used for detecting the deuteron incorporation into the peptides, the same precautions regarding sample preparation apply for HX-MS. Detergents, salts and other compounds that interfere with mass spectrometry should be avoided or removed before analysis. While HX-MS allows analysis of large protein complexes, the higher complexity demands increased quality of peptide separation and mass spectrometry. Optimization of chromatographic conditions is key to obtaining good quality data. This involves the choice of the high quality organic solvents, reversed-phase material as well as optimization of gradients and solvents. Washing of columns with organic solvents can be done between experiments and overnight to reduce background signal. Mass spectrometry methods should be optimized for detection of peptic peptides (between 300-1,200 m/z). High-resolution mass spectrometers greatly facilitate data analysis because partially overlapping isotopic peak clusters originating from different peptides can be distinguished.
The processing of samples is crucial for HX-MS. Usually our buffers and samples are spun down for 10 min at 13,000 rpm (microfuge) to remove aggregates and particles. In addition, several inline filters can be installed in the HPLC system to prevent clogging of columns. Minor deviations in D2O incubation times can have large effects. For very flexible protein regions a difference of only a few seconds can make the difference between no deuteron incorporation at all and full deuteration. Since protein dynamics as well as the intrinsic exchange rate are a function of temperature, incubation of sample and buffers are tightly temperature controlled. It is advantageous to always repeat the exact sequence of steps for every experiment. If executed correctly the error between two experiments will be relatively small. Today, automated systems for HX-MS are commercially available and reduce the challenges of sample processing.
Analysis of data
The most critical step in HX-MS is analysis of the data, in particular the assignment of peptide spectra and determination of deuteration levels. The output of HX-MS is an elution profile of the chromatographic gradient containing a great number of peptide spectra. The operators task is to assign these spectra to the peptides identified by MS/MS in step 6) of the protocol. This can be somewhat difficult as the spectra can significantly shift towards higher m/z after HX. Also the partial overlapping of spectra may complicate the assignment of peptides. In this case the use of high-resolution mass spectrometers with high mass accuracy pays off because they often can resolve overlapping peptides. Differences in deuteron incorporation are calculated based on the centroids of peptide spectra. The centroids can be determined: 1) manually by marking each isotopic peak of a peptide spectrum and transferring its values for intensity and m/z into a spreadsheet program (e.g. Excel, Microsoft) and calculating the centroid or 2) with software designed to assign the isotopic peaks belonging to an identified peptide and to calculate the centroids automatically (e.g. HDExaminer, Sierra Analytics or HeXicon24,25).
Optimization of HX-MS
If the number of evaluable peptides is too low, there are several other possibilities to optimize the experiment: 1) optimizing the chromatography (solvents, length/shape of gradient, use of different reversed-phase material) or 2) changing the enzymatic cleavage of the protein (alternative protease, longer/shorter incubation time with pepsin, addition of denaturing agents to the quench buffer). Both parameters change the elution behavior of peptides as they either alter the peptide pool itself or HPLC separation. Both optimization approaches come at the price of the need to recharacterize peptides. Other proteases will produce different peptides that have to be identified by MS/MS and characterized as described in step 6.6). Changing the chromatography conditions will not alter the peptide pool but the retention times of the individual peptides. The retention times then have to be redetermined as in step 6.5). It should be mentioned that higher retention times always will decrease the amount of detectable label as back exchange increases at longer retention times. Normalization of the data to the 100% control is the best way to deal with back exchange. High back exchange leads to a loss of incorporated deuterons and therefore the signal. It is strongly recommended to reduce the amount of back exchange to a minimum. It should be noted that different HX-MS setups will have diverging rates of back exchange, depending on the hardware, the composition of used buffers and solvents and the type of gradients that are used26.
Interpreting the Data
When analyzing data, the intensity distribution of the isotopic peaks needs to be evaluated carefully since it can give indications to the exchange mechanism. For peptides with a mass of less than 2,000 Da the intensities of the isotopic peaks of the unexchanged sample is generally asymmetric with higher intensities for the monoisotope or the first higher isotope peak. For the EX2 exchange regime this asymmetric intensity distribution becomes Gaussian-like with increasing time in D2O and increasing shift to higher m/z and the width of the isotopic cluster increase by about 50% (Figure 7). As mentioned in the introduction EX2 is most commonly observed in HX-MS under native conditions and the observed rate of exchange is proportional to the equilibrium unfolding constant Ku and therefore to the thermodynamic stability of the protein. In contrast, for the EX1 exchange regime the width distribution of the isotopic cluster increases significantly and bimodal or multimodal intensity distributions are observed (see Figure 7)25,27. Bimodal isotope distributions always indicate two or more different molecule species in the analysis, which are potentially interesting. However, there are two types of pitfalls. First, the isotope cluster may be composed of peaks originating from two different peptides. It is therefore mandatory to check the spectra of the unexchanged sample for peptides with similar m/z and identical charge. Such peptides usually differ slightly in retention time and the intensity distribution of the isotopic peaks varies with elution time. In high-resolution mass spectrometers, isotopic peaks that do not belong to the same peptide may also be distinguished by the decimals of the m/z values. Second, carryover from a previous HPLC-MS run is observed28. Such carryover appears as nonexchanged species and can be eliminated by blank HPLC gradient runs between analysis runs. There are many reasons for apparent EX1 exchange regime. 1) In HX experiments in the presence of denaturants EX1 is frequently observed29. 2) Spontaneous or induced, reversible or irreversible local or global unfolding of proteins causes EX1 behavior7,30,31. 3) Conformational transitions that are slow as compared to HX caused by e.g. substrate turnover, ATP hydrolysis cause bimodal isotope distributions resembling EX1 exchange regime (Figure 7a)22. 4) Ligand-induced slow conformational transitions in pulse-labeling experiments also exhibit an EX1-like signature (Figure 7b)22.
The interpretation of obtained data is eventually what makes HX-MS such a challenging but also powerful technique. A broad knowledge of protein dynamics is required to get all the way from the observed protection/deprotection patterns to a mechanistic model of protein dynamics. Nonetheless, producing high quality HX-MS data can be done within relatively short time and is highly reproducible. The interpretation of data clearly benefits from any additional information on the structure of the protein investigated. On the other side, HX-MS can indicate which parts of a protein are flexible, which may have to be removed to facilitate crystallization for structure determination.
HX-MS is a method that greatly profits by innovations in mass spectrometry and chromatography. A recent advance in HX-MS is the use of lipid nanodiscs for analysis of membrane proteins that further broadens the use of this technique32. Also the use of electron transfer dissociation (ETD) and electron capture dissociation (ECD) show strong potential for HX-MS as it increases the resolution of incorporated deuteron to the level of single amino acids11,12 33.
Disclosures
We have nothing to disclose.
Acknowledgments
We thank M. Boysen for comments on the manuscript. This project was funded by the Deutsche Forschungsgemeinschaft (SFB638 and MA 1278/4-1 to M.P.M., and Cluster of Excellence: CellNetworks EXC 81/1). M.P.M. is investigator of the Cluster of Excellence: CellNetworks.
References
- Saibil HR. Conformational changes studied by cryo-electron microscopy. Nat. Struct. Biol. 2000;7(9):711–714. doi: 10.1038/78923. [DOI] [PubMed] [Google Scholar]
- Mittermaier A, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312(5771):224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- Hoofnagle AN, Resing KA, Ahn NG. Protein analysis by hydrogen exchange mass spectrometry. Annu. Rev. Biophys. Biomol. Struct. 2003;32:1–25. doi: 10.1146/annurev.biophys.32.110601.142417. [DOI] [PubMed] [Google Scholar]
- Wales TE, Engen JR. Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass. Spectrom. Rev. 2006;25(1):158–170. doi: 10.1002/mas.20064. [DOI] [PubMed] [Google Scholar]
- Konermann L, Pan J, Liu Y-H. Hydrogen exchange mass spectrometry for studying protein structure and dynamics. Chem Soc. Rev. 2011;40(3):1224–1210. doi: 10.1039/c0cs00113a. [DOI] [PubMed] [Google Scholar]
- Bai Y, Milne JS, Mayne L, Englander SW. Primary structure effects on peptide group hydrogen exchange. Proteins. 1993;17(1):75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rist W, Jørgensen TJD, Roepstorff P, Bukau B, Mayer MP. Mapping temperature-induced conformational changes in the Escherichia coli heat shock transcription factor sigma 32 by amide hydrogen exchange. The Journal of biological chemistry. 1074;278(51):51415–51421. doi: 10.1074/jbc.M307160200. [DOI] [PubMed] [Google Scholar]
- Englander JJ, Rogero JR, Englander SW. Protein hydrogen exchange studied by the fragment separation method. Anal Biochem. 1985;147(1):234–244. doi: 10.1016/0003-2697(85)90033-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2(4):522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cravello L, Lascoux D, Forest E. Use of different proteases working in acidic conditions to improve sequence coverage and resolution in hydrogen/deuterium exchange of large proteins. Rapid Commun. Mass Spectrom. : RCM. 2003;17(21):2387–2393. doi: 10.1002/rcm.1207. [DOI] [PubMed] [Google Scholar]
- Rand KD, Zehl M, Jensen ON, Jørgensen TJD. Protein hydrogen exchange measured at single-residue resolution by electron transfer dissociation mass spectrometry. Anal chem. 2009;81(14):5577–5584. doi: 10.1021/ac9008447. [DOI] [PubMed] [Google Scholar]
- Pan J, Han J, Borchers CH, Konermann L. Electron capture dissociation of electrosprayed protein ions for spatially resolved hydrogen exchange measurements. J. Am. Chem. Soc. 2008;130(35):11574–11575. doi: 10.1021/ja802871c. [DOI] [PubMed] [Google Scholar]
- Yamada N, Suzuki E-I, Hirayama K. Identification of the interface of a large protein-protein complex using H/D exchange and Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 2002;16(4):293–299. doi: 10.1002/rcm.579. [DOI] [PubMed] [Google Scholar]
- Lee T, Hoofnagle AN, et al. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol. Cell. 2004;14(1):43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- Hasan A, Smith DL, Smith JB. Alpha-crystallin regions affected by adenosine 5'-triphosphate identified by hydrogen-deuterium exchange. Biochem. 2002;41(52):15876–15882. doi: 10.1021/bi026568x. [DOI] [PubMed] [Google Scholar]
- Lanman J, Lam TT, Emmett MR, Marshall AG, Sakalian M, Prevelige PE. Key interactions in HIV-1 maturation identified by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 2004;11(7):676–677. doi: 10.1038/nsmb790. [DOI] [PubMed] [Google Scholar]
- Wang L, Lane LC, Smith DL. Detecting structural changes in viral capsids by hydrogen exchange and mass spectrometry. Protein Sci. 2001;10(6):1234–1243. doi: 10.1110/ps.100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Raza AS, Smith DL. Equilibrium and kinetic folding of rabbit muscle triosephosphate isomerase by hydrogen exchange mass spectrometry. J. Mol. Biol. 2004;336(5):1251–1263. doi: 10.1016/j.jmb.2003.12.076. [DOI] [PubMed] [Google Scholar]
- Mazon H, Marcillat O, Forest E, Smith DL, Vial C. Conformational dynamics of the GdmHCl-induced molten globule state of creatine kinase monitored by hydrogen exchange and mass spectrometry. Biochem. 2004;43(17):5045–5054. doi: 10.1021/bi049965b. [DOI] [PubMed] [Google Scholar]
- Lanman J, Lam TT, et al. Identification of novel interactions in HIV-1 capsid protein assembly by high-resolution mass spectrometry. J. Mol. Biol. 2003;325(4):759–772. doi: 10.1016/s0022-2836(02)01245-7. [DOI] [PubMed] [Google Scholar]
- Rist W, Graf C, Bukau B, Mayer MP. Amide hydrogen exchange reveals conformational changes in hsp70 chaperones important for allosteric regulation. J. Biol. chem. 2006;281(24):16493–16501. doi: 10.1074/jbc.M600847200. [DOI] [PubMed] [Google Scholar]
- Graf C, Stankiewicz M, Kramer G, Mayer MP. Spatially and kinetically resolved changes in the conformational dynamics of the Hsp90 chaperone machine. EMBO J. 2009;28(5):602–613. doi: 10.1038/emboj.2008.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-T, Graf C, Mayer FJ, Richter SM, Mayer MP. Dynamics of the regulation of Hsp90 by the co-chaperone Sti1. EMBO J. 2012;31(6):1518–1528. doi: 10.1038/emboj.2012.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Kirchner M, et al. Deuteration distribution estimation with improved sequence coverage for HX/MS experiments. Bioinformatics. 2010;26(12):1535–1541. doi: 10.1093/bioinformatics/btq165. [DOI] [PubMed] [Google Scholar]
- Kreshuk A, Stankiewicz M, Lou X, Kirchner M, Hamprecht FA, Mayer MP. Automated detection and analysis of bimodal isotope peak distributions in H/D exchange mass spectrometry using HeXicon. Intl. J. mass spectrom. 2011;302(1-3):125–131. [Google Scholar]
- Walters BT, Ricciuti A, Mayne L, Englander SW. Minimizing back exchange in the hydrogen exchange-mass spectrometry experiment. J. Am. Soc. Mass Spectrom. 2012;23(12):2132–2139. doi: 10.1007/s13361-012-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis DD, Wales TE, Engen JR, Hotchko M, Ten Eyck LF. Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J. Am. Soc. Mass Spectrom. 2006;17(11):1498–1509. doi: 10.1016/j.jasms.2006.05.014. [DOI] [PubMed] [Google Scholar]
- Fang J, Rand KD, Beuning PJ, Engen JR. False EX1 signatures caused by sample carryover during HX MS analyses. Intl. J. Mass Spectrom. 2011;302(1-3):19–25. doi: 10.1016/j.ijms.2010.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Smith DL. Identification of unfolding domains in large proteins by their unfolding rates. Biochem. 1998;37(18):6256–6262. doi: 10.1021/bi972711o. [DOI] [PubMed] [Google Scholar]
- Wales TE, Engen JR. Partial unfolding of diverse SH3 domains on a wide timescale. J. Mol. Biol. 2006;357(5):1592–1604. doi: 10.1016/j.jmb.2006.01.075. [DOI] [PubMed] [Google Scholar]
- Pan Y, Piyadasa H, O'Neil JD, Konermann L. Conformational dynamics of a membrane transport protein probed by h/d exchange and covalent labeling: the glycerol facilitator. J. Mol. Biol. 2012;416(3):400–413. doi: 10.1016/j.jmb.2011.12.052. [DOI] [PubMed] [Google Scholar]
- Hebling CM, Morgan CR, Stafford DW, Jorgenson JW, Rand KD, Engen JR. Conformational analysis of membrane proteins in phospholipid bilayer nanodiscs by hydrogen exchange mass spectrometry. Anal chem. 2010;82(13):5415–5419. doi: 10.1021/ac100962c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abzalimov RR, Kaplan DA, Easterling ML, Kaltashov IA. Protein conformations can be probed in top-down HDX MS experiments utilizing electron transfer dissociation of protein ions without hydrogen scrambling. J. Am. Soc. Mass Spectrom. 2009;20(8):1514–1517. doi: 10.1016/j.jasms.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]