

Abstract
Despite significant influence of secondary bile acids on human health and disease, limited structural and biochemical information is available for the key gut microbial enzymes catalyzing its synthesis. Herein, we report apo- and co-factor bound crystal structures of BaiA2, a short chain dehydrogenase/reductase from Clostridium scindens VPI 12708 that represent the first protein structure of this pathway. The structures elucidated the basis of co-factor specificity and mechanism of proton relay. A conformational restriction involving Glu42 located in the co-factor binding site seems crucial in determining co-factor specificity. Limited flexibility of Glu42 results in imminent steric and electrostatic hindrance with 2′-phosphate group of NADP(H). Consistent with crystal structures, steady-state kinetic characterization performed with both BaiA2 and BaiA1, a close homolog with 92% sequence identity, revealed specificity constant (kcat/KM) of NADP+ at least an order of magnitude lower than NAD+. Substitution of Glu42 with Ala improved specificity towards NADP+ by 10- fold compared to wild type. The co-factor bound structure uncovered a novel nicotinamide-hydroxyl ion (NAD+-OH−) adduct contraposing previously reported adducts. The OH− of the adduct in BaiA2 is distal to C4 atom of nicotinamide and proximal to 2′-hydroxyl group of the ribose moiety. Moreover, it is located at intermediary distances between terminal functional groups of active site residues Tyr157 (2.7 Å) and Lys161 (4.5 Å). Based on these observations we propose an involvement of NAD+-OH− adduct in proton relay instead of hydride transfer as noted for previous adducts.
Keywords: Short chain dehydrogenase reductase, steroid dehydrogenase, gut microbe mediated human metabolite, primary bile acid, secondary bile acid, nicotinamide-hydroxyl adduct, proton relay
INTRODUCTION
Deoxycholic acid (DCA) and lithocholic acid (LCA) are secondary bile acids that are generated in the gut by bacterial transformation of primary bile acids, cholic acid (CA) and chenodeoxycholic acid (CDCA), respectively 1. The proposed pathway for this transformation by Clostridium scindens (C. scindens) VPI 12708 involves a network of enzymes that catalyze the removal of C7-hydroxyl group from primary bile acids (Fig. 1). C. scindens and a few closely related organisms are the only microbes that are known to harbor biochemical pathway for synthesizing secondary bile acids 2. A recent metagenomic study identified C. scindens as a component of the “core microbiome”, a set of 57 bacteria which were present in >90% of the cohort in this study 3. Despite its ubiquitous presence in human microbiota, it is populated at a very low relative abundance as quantified by metagenomic surveys. Qin et al have found it to be one of the lowest abundant member of the “core microbiome”, present at approximately two orders of magnitude lower than the most copious microbes in human gut 3. Studies implicated secondary bile acids as effector molecules involved in human health influencing various signaling pathways associated with cancer and metabolism 4–6. Secondary bile acids are established as ligands for several cellular receptors such as TGR5, FXR and vitamin D receptor 7–9. In rodent models, LCA can even functionally supplement Vitamin D 10. It can hence be speculated that variation in population of a less abundant microbial species may influence the level of key metabolites in the human body and thereby human health. Hence, the microbial enzymes involved in this synthesis could represent potential drug targets for manipulating secondary bile acid level.
FIGURE 1.

Microbial synthesis of secondary bile acids from primary bile acids in the human distal gut. The oxidative and reductive arms of the pathway are enclosed within solid and dashed lines boxes, respectively. BaiB – bile acid CoA ligase; BaiA1/A2 - 3α-Hydroxysteroid Dehydrogenase; BaiCD - 3-oxo-Δ4-cholenoic acid oxidoreductase; BaiE - bile acid 7α dehydratase. Unidentified enzymes participating in the reductive arm are denoted by question mark.
The multistep 7α-dehydroxylation pathway of primary bile acids as proposed in C. scindens VPI 12708 involves a series of enzymes (Fig 1). The genes of these enzymes are localized in a bile acid inducible (bai) operon. In this study we report the structure-function characterization of the product of genes baiA1 and baiA2, BaiA1 and BaiA2 respectively. Both proteins are 3α-Hydroxysteroid Dehydrogenases with 92% sequence identity over their 249 amino acid length and catalyze the oxidation of C3-hydroxyl group of bile acids generating a C3-oxo bile acid intermediate (Fig. 2A) 11. The proteins belong to a large family of short chain dehydrogenase/reductase (SDR) that utilize either NAD or NADP as co-factor 12. Previous characterization using crude cell extracts of E. coli expressing BaiA1 revealed preferential utilization of Coenzyme A (CoA) – bile acid esters in presence of both co-factors 11. These studies did not detect any product formation from unconjugated primary bile acids, cholic acid (CA) and chenodeoxycholic acid (CDCA).
FIGURE 2.

Reaction and NMR characterization of substrates and products of BaiA2. (A) Reaction catalyzed by BaiA2. (B) Representative regions of 1H-NMR spectra showing the turnover of different bile acid substrates by BaiA2. Reactions were performed at 300°K in 20 mM d-TRIS pH 8.7 with 100 μM substrate and 100 μM co-factor. BaiA2 concentrations are noted at the bottom. Spectra were collected at approximately 0, 20 and 45 min after addition of enzyme. The label centered beneath the chemical shift axis denotes the substrate used in the reaction for that stacked plot and the dashed lines mark the location of selected peaks that appear during the reaction. Reference spectra of substrates and products acquired in the same buffer are shown on top and labeled as follows: A (CA), B (3-oxo-CA), C (GCA), D (C-CoA), and E (3-oxo-C-CoA). The assignments for selected peaks are noted by a number indicated in (A).
Herein we report the crystal structures of apo- (1.9 Å resolutions) and co-factor bound (2.0 Å resolutions) BaiA2 from C. scindens VPI 12708. Alongwith the structures we also report steady-state kinetic characterization of BaiA2 and BaiA1, a close homolog with 92% sequence identity. Our study represents first structural and steady-state kinetic characterization of enzymes involved in synthesis of secondary bile acids. The crystal structure of BaiA2 revealed the canonical Rossmann fold with distinct binding sites for substrate and NAD. The structures provided a basis of co-factor specificity that can be ascribed to the predicted restrictive conformational flexibility of residue Glu42. The restrictive flexibility of Glu42 makes the cofactor binding site incompatible with NADP due to steric and electrostatic hindrance of the Glu42-COO− side chain with the 2′-phospahte group. Consistent with the crystallographic observations steady-state kinetic experiments confirmed NAD as the preferred co-factor for both BaiA1 and BaiA2. In addition, analysis of the electron density map detected a residual electron density feature that can be accounted by an oxygen atom located at a distance of 1.8 Å from the C6 atom of the nicotinamide moiety and 2.9 Å from the 2′-hydroxy group of the nicotinamide ribose. Quantum chemical calculations predicted that the additional oxygen atom to be OH− and the nicotinamide ring possessing a positive charge, suggesting a nicotinamide-hydroxyl ion (NAD+-OH−) as a stable adduct. The OH− of the adduct is located intermediary between the terminal functional groups of active site residues Tyr157 and Lys161. Furthermore, the OH− is proximal to the 2′-hydroxyl group of the nicotinamide ribose, and distal to the C4 atom of nicotinamide moiety, which contrapose previously observed nicotinamide-hydroxyl adducts. Based on these observations we propose that the observed NAD+-OH− may be involved in proton relay instead of hydride transfer. Overall the reported structures and functional characterization provide new insights into the mechanism of bacterial transformation of primary bile acids to secondary bile acids in the human gut.
MATERIALS AND METHODS
Chemicals
All bile acid substrates are obtained from Steraloids (Newport, RI) except glycocholic acid (GCA) and taurocholic acid (TCA) that are obtained from Sigma Aldrich (St. Louis, MO).
Cloning, Expression and Purification
Clones were generated using the Polymerase Incomplete Primer Extension (PIPE) cloning method 13. Genomic DNA C. scindens VPI 12708 was a kind gift from Philip B. Hylemon, Virginia Commonwealth University. Briefly, baiA1 and baiA2 were amplified by polymerase chain reaction (PCR) from C. scindens VPI 12708 genomic DNA and cloned into the expression vector, pSpeedET, which encodes an amino-terminal tobacco etch virus (TEV) protease-cleavable His6- tag for purification of the following sequence: MGSDKIHHHHHHENLYFQ/G with cleavage site between Gln and Gly. Site-directed variants Arg16Ala and Glu42Ala were generated using the M-PIPE method 14. Recombinant clones encoding the entire 249 amino acid sequence were confirmed by DNA sequencing.
Protein expression was performed at 37°C under arabinose promoter in a selenomethionine-containing medium for structure determination and in Luria Bertani medium for kinetic studies. Cell pellets were obtained by centrifugation at the end of fermentation, and lysozyme was added to a final concentration of 250 μg/ml. The pellets were flash frozen for storage at −20°C. After thawing at room temperature the cell pellets were lyzed by using a microfluidizer. The N-terminal His6-tagged protein was purified from the E. coli extract by a single step Ni-NTA chromatography. Typically protein from a 500 ml expression culture was purified on a 1.5 ml bed volume of Ni-NTA. Proteins were eluted from the column by 20 mM HEPES pH 8.0, 300 mM imidazole, 10% (v/v) glycerol, 1 mM TCEP after a wash step with 1 column volume of the buffer containing 30 mM imidazole. The eluate was buffer exchanged into 20 mM HEPES pH 8.0, 1 mM TCEP using an Amicon Ultra (Millipore) centrifugal filter device. The molecular mass of the monomer was characterized by both LC-MS and denaturing polyacrylamide gel electrophoresis (SDS-PAGE). Oligomeric profile was determined by analytical size exclusion chromatography performed at room temperature on Agilent HP1100 HPLC system utilizing a Shodex 8 × 300mm Protein KW-802.5 column. The N-terminal purification tag was not cleaved.
Synthesis of Bile Acid CoA Thioester
Synthesis of bile acid - CoA thioesters were modified from an earlier procedure 15. A dichloromethane solution (6 mL, anhydrous) of γ-collidine (48 μL) and ethyl chloroformate (30 μL) was added to bile acid (~ 0.144 mmol) while stirring at 23 °C. Aliquots of the reaction mixture were taken out after 3 hours and reacted with methanol while monitoring the progress of mixed anhydride formation by RP-C18 HPLC. Solvent was evaporated by blowing dry N2 stream. The residue was dissolved in tetrahydrofuran (anhydrous, 6 mL × 2) and added to 500 mM sodium borate solution (6 mL) of Coenzyme A with vigorous stirring at 23 °C for 12 – 16 hours. The reaction was monitored by RP-C18 HPLC. The reaction mixture was lyophilized upon completion and the crude mixture was purified by reverse phase flash column chromatography using C18AQ column of 5.5 g bed weight. Gradient elution was performed with 10–100% methylcyanate/water at a flow rate of 18 mL/minute. Fractions of desired product were combined and lyophilized. The following bile acid-CoA esters were synthesized and were analyzed by MS: cholyl CoA (C-CoA), chenodeoxycholy CoA (CDC-CoA), lithocholyl CoA (LC-CoA), 3-oxo-cholyl CoA (3-oxo-C-CoA), 3-oxo-chenodeoxycholyl CoA (3-oxo-CDC-CoA) and 3-oxo-lithocholyl CoA (3-oxo-LC-CoA).
NMR Analysis of the BaiA2 reaction
1H-NMR spectra were acquired at 300 °K on a Bruker Avance instrument equipped with a 1H/13C/15N-TXI cryoprobe operating at 600 MHz with 16 scans and a recycle delay of 2 s employing excitation sculpting to suppress water signal 16. Isotope labeled compounds were purchased from Sigma-Aldrich/Isotec. All reaction samples contained 3-(trimethylsilyl)-2,2′,3,3′-tetradeuteropropionic acid as an internal standard that was referenced to 0ppm. The reaction was initiated by adding enzyme to a solution containing 100 μM substrate, 100 μM co-factor and 5 mM MgCl2 prepared in 20 mM deuterated Tris-HCl pH 8.7 with 10% D2O. Reaction mixtures containing bile acid – CoA esters did not include MgCl2. The enzyme concentration was 4.3 μM for cholic acid and glycocholic acid and 0.27 μM for all CoA-conjugated substrates. Samples were immediately transferred to the NMR instrument after addition of enzyme and allowed to equilibrate until the lock stabilized before initiating data collection. In order to accurately monitor the reactions, the following two-dimensional spectra were assigned: double quantum filtered COSY 17,18, non-decoupled heteronuclear multiple bond correlation (HMBC) 19,20, and rotating frame Overhauser enhancement spectroscopy (ROESY) with a 200 ms mix time 21. The compounds were dissolved in the appropriate deuterated solvent (either DMSO or methanol).
Steady-state kinetics
Kinetic measurements were performed on a Varian Cary 100 Bio UV-Visible spectrophotometer equipped with a thermojacketted cuvette holder. Steady state kinetic assays were performed at 37 °C by monitoring the formation or consumption of NADH measured at wavelength 340 nm with an extinction coefficient of 0.00622 μM−1 cm−1. Reactions were performed in 20 mM Tris-HCl pH 8.7 under fixed co-factor concentrations. The concentration of the substrate was varied below and beyond KM value of the respective substrates. Reactions were initiated by addition of enzyme to final concentration of 0.27 μM for CoA – bile acid substrates and 2.1 μM for other substrates. Reaction rates were obtained from the initial, linear region of the reaction progress curves. The data were fitted to Michelis-Menten equation by non-linear regression using the enzyme kinetics module in GraphPad Prism (GraphPad Software, La Jolla, CA).
Kinetic parameters for the co-factor were determined in a similar method as described above. The reaction rate was monitored in presence of 1.4 mM Glycocholic acid (GCA) at wavelength 340 nm at 37°C. The co-factor concentration varied below and beyond the KM value. Enzyme concentration was maintained at 0.27 μM in presence of NAD+ and 2.1 μM in presence of NADP+. The reaction rates and kinetic parameters were determined as described above.
Crystallization of BaiA2
Two crystal forms of BaiA2 were obtained in two different crystallization reagents from the JCSG Core Suite III (Qiagen Sciences, MD, USA) using the sitting nanodrop vapor diffusion method at 20°C utilizing standard Joint Center for Structural Genomics crystallization protocols 16,22. Drops of 0.2 μl volume, with 1:1 ratio of reservoir and protein (46 mg/ml in 20 mM HEPES pH 8.0) solution were equilibrated against 100 μl of reservoir solution. The orthorhombic crystal form was obtained in 0.1 M sodium acetate pH 4.5, 2.5 M sodium chloride, 0.2 M lithium sulfate. Monoclinic crystals were obtained in 30% polyethylene glycol 4000, 0.1M Tris-HCl pH 8.5, 0.2 M sodium acetate.
Structure Determination and Refinement
Diffraction data were collected on beamline 9-2 at the Stanford Synchrotron Radiation Lightsource (SSRL) at wavelengths corresponding to the high-energy remote (λ1), inflection (λ2) and peak (λ3) wavelengths of a selenium multi-wavelength anomalous diffraction (MAD) experiment. The datasets were collected at 100 K using a Rayonix MarMosaic MX-325 CCD detector using the Blu-Ice data-collection environment 23. The MAD data collected from P21 crystal form were integrated and reduced using XDS and then scaled with the program XSCALE 24,25. The datasets collected from I222 crystal form was integrated and reduced using MOSFLM and SCALA. An initial Se sub-structure solution was obtained with SHELXD 26 and the phases were refined using autoSHARP 27, which gave a mean figure of merit of 0.51 to 2 Å for the P21 crystal form and 0.43 to 1.9 Å for the I222 crystal form. Automated model building was performed with ARP/wARP 28. The crystal properties and statistics of data collection are enlisted (Table I). Model completion and refinement were performed with COOT 29 and REFMAC 30 in CCP4 program suite 31 and BUSTER32. Coordinates for NAD were generated using PRODRG 33. Stereochemical properties of the models and hydrogen bonding were analyzed by the JCSG QC server (http://smb.slac.stanford.edu/jcsg/QC/). Statistics of the refined models are indicated (Table I).
Table I.
Crystal properties, data collection statistics and refinement of BaiA2.
| Apo | NAD | |||||||
|---|---|---|---|---|---|---|---|---|
| Crystal Properties | ||||||||
| Space Group | I222 | P21 | ||||||
| Unit Cell Parameters (Å) | a = 54.0, b = 93.2, c = 105.4 | a = 68.5, b = 92.9, c = 82.9 | ||||||
|
| ||||||||
| Data Collection | λ1 MAD-Se | λ2 MAD-Se | λ3 MAD-Se | λ1 MAD-Se | λ2 MAD-Se | λ3 MAD-Se | ||
| Wavelength (Å) | 0.91162 | 0.91162 | 0.97915 | 0.97936 | 0.91162 | 0.97919 | ||
| Resolution (Å) | 1.90 | 1.90 | 1.95 | 2.00 | 2.00 | 2.00 | ||
| No. of observations | 78306 | 78306 | 72365 | 260483 | 258916 | 260082 | ||
| No. of unique reflections | 21347 | 21347 | 19787 | 68715 | 68376 | 68754 | ||
| Completeness (%)(last shell)a | 99.9 (100) | 99.9 (100) | 99.9 (100) | 97.1 (89.0) | 97.2 (88.9) | 97.0 (88.7) | ||
| Mean I/Σ(I) (last shell) | 9.9 (1.7) | 9.9 (1.7) | 9.0 (1.6) | 12.1 (2.1) | 12.6 (2.3) | 12.5 (1.8) | ||
| Rmerge on I† (%)(last shell) | 6.3 (75.8) | 6.3 (75.8) | 6.9 (77.1) | 10.9 (73.7) | 9.1 (64.9) | 11.0 (85.5) | ||
| Rmeas on I‡ (%)(last shell) | 7.4 (88.7) | 7.4 (88.7) | 8.0 (90.1) | 12.7 (86.5) | 10.6 (76.1) | 12.8 (100.4) | ||
|
| ||||||||
| Model and refinement statistics | Data set used in refinement | Data set used in refinement | ||||||
| Resolution range (Å) | 29.81 – 1.90 | 29.01 – 2.00 | ||||||
| No. of reflections (total) § | 20248 | 68331 | ||||||
| No. of reflections (test) | 1097 | 3444 | ||||||
| Completeness (%) | 99.63 | 97.36 | ||||||
| Rcryst | 0.162 | 0.153 | ||||||
| Rfree | 0.190 | 0.178 | ||||||
|
| ||||||||
| Stereochemical parameters | ||||||||
| Restraints (RMSD observed) | ||||||||
| Bond angle (°) | 1.434 | 0.99 | ||||||
| Bond length (Å) | 0.011 | 0.010 | ||||||
| Average protein isotropic B-factor (Å2) †† (all atoms/protein residues only) | 45.18 | 30.52 | ||||||
| ESU based on Rfree (Å) ‡‡ | 0.109 | 0.127 | ||||||
| No. of protein atoms | 1662 | 7607 | ||||||
| No. of water molecules/NAD | 103/0 | 770/4 | ||||||
Rmerge = ΣhklΣi|Ii(hkl) − <I(hkl)>|/Σhkl ΣiIi(hkl).
Rmeas = Σhkl[N/(N−1)]1/2Σi|Ii(hkl) − <I(hkl)>|/ΣhklΣiIi(hkl).
Typically, the number of unique reflections used in refinement is slightly less than the total number that were integrated and scaled. Reflections are excluded owing to negative intensities and rounding errors in the resolution limits and unit-cell parameters.
Rcryst = Σhkl||Fobs| − |Fcalc||/Σhkl|Fobs|, where Fcalc and Fobs are the calculated and observed structure-factor amplitudes, respectively.
Rfree – same as Rcryst but for 4.9% of the total reflections chosen at random and omitted from refinement.
This value represents the total B that includes TLS and residual B components.
Estimated overall coordinate error (36).
The final model of the apo- structure (Rcryst/Rfree: 16.2/18.7) contained one protomer in the asymmetric unit with 215 residues built. The electron density of side chain and main chain atoms for residues in the N-terminal tag (-18-0), and 150–157, 193–214 and 246–249 is discontinuous in the structure. The co-factor bound structure (Rcryst/Rfree: 15.2/18.2) contained four protomers in the asymmetric unit without the N-terminal His6-tag modeled. The apo and cofactor bound structures superimpose within a rmsd of 0.61 Å over 206 Cα atoms. The co-factor observed in the structure originated from the expression system as it was not added in the purification or crystallization buffers. The unbiased (mFo-DFc) electron density map is incompatible with NADP as the 2′-phosphate group of adenine ribose cannot be modeled. The map is compatible with both oxidation states, NAD+ and NADH, where nicotinamide and adenine moieties adopt syn- and anti- conformation, respectively (Fig. 4A). Since the protein solution did not exhibit the characteristic absorption maxima of NADH at 340 nm the co-factor in the structure is refined as NAD+. Validated apo- and the co-factor bound coordinates of BaiA2 are deposited in the Protein Data Bank (PDB) with codes 4IS2 and 4IS3, respectively.
FIGURE 4.

Co-factor binding in BaiA2. (A) Unbiased (mFo-DFc) electron density maps: blue - before inclusion of NAD+ and OH− (contour level 0.17 e−/Å3 (3 rmsd)); green- before inclusion of OH− but after addition of NAD+ (contour level 0.14 e−/Å3 (3 rmsd)). C, N, O and P atoms are colored green, blue, red and orange, respectively. The O atom of OH− of the nicotinamide-OH− adduct is depicted as a purple sphere. (B) Interaction of OH− with the nicotinamide moiety. Numbers indicate distances in Å. (C) Comparison of the interaction involving a conserved Arg and the residue equivalent to Glu42 in other related SDRs. C atoms are colored gold, cyan and green, respectively for BaiA2, mannitol dehydrogenase (MtDh; PDB Code: 1H5Q) and 3α, 20β-hydroxysteroid dehydrogenase (PDB Code: 2HSD). Label of residues and distances of interaction are colored according to the carbon atoms of the respective structure. (D) Key interactions involving residue Glu42 in the adenine moiety binding site. Dashed black colored lines indicate hydrogen bonding interactions, whereas blue colored dashed lines indicate a salt bridge interaction. Numbers indicate distances in Å. N and O atoms are colored blue and red, respectively.
Quantum Chemical Calculations
Theoretical calculations were performed on a nicotinamide model system generated from the co-factor built in the crystal structure. The model was truncated containing only the nicotinamide and ribose moieties with the pyrophosphate group replaced by a hydrogen atom. Coordinates of the active site residues were also truncated to contain the terminal functional groups, OH-C2H5 (Tyr157) and NH3+-C2H5 (Lys161). The geometry of the nicotinamide moiety with added hydrogen atoms was optimized with constraints at Hartree Fock method with 6-31** basis set using GAMESS Quantum mechanics (QM) suite. We applied Cartesian constraints to the oxygen atom of OH−/water keeping it in a fixed position relative to the pyridine ring, ribose moiety and active site residues Tyr157 and Lys161, as observed in the crystal structure (Fig. 5A). Four different QM calculations were performed by combinatorial pairs of oxidized and reduced states of the nicotinamide moiety with water and hydroxyl ion (OH−).
FIGURE 5.

Active site of BaiA2 and proposed catalytic mechanism. (A) Orientation of the active site residues. Dashed black colored lines indicate hydrogen bonding interactions. Non-hydrogen bonding interactions are indicated in dashed red lines. Numbers denote distances in Å. The O atom of OH−/water of the nicotinamide-OH− adduct is depicted as a purple sphere. (B) Comparison of the interaction between active site residues of BaiA2, MtDh (NADP-; PDB Code: 1H5Q), 3α, 20β-hydroxysteroid dehydrogenase (NAD-; PDB Code: 2HSD), 3β, 17β-hydroxysteroid dehydrogenase (apo-; PDB Code: 1HXH), and Rv2002 (ternary-; PDB Code: 1NFQ). Water molecules in BaiA2 and MtDh are depicted as red and orange spheres, respectively. Numbers indicate distances in Å from the terminal functional groups of Tyr169 and Lys173 of MtDh. (C) Proton relay network observed in BaiA2:NAD+ structure. O atoms of protein residues and bound water molecules are colored red and orange, respectively. Dashed black colored lines depict hydrogen bonding interactions. (D) Proposed mechanism of oxidation catalyzed by BaiA1/A2. Catalysis initiated by proton abstraction from Tyr157-OH group by nicotianmide-OH− adduct (denoted in purple). The terminal oxyanion of Tyr157 is re-protonated by the proton from the C3-hydroxyl group of the substrate and hydride transfer (dashed arrow) from C3 atom of the substrate to the C4 atom of the nicotinamide moiety. The proton abstracted by the nicotinamide-hydroxyl adduct is relayed through the 2′-hydroxyl group of nicotinamide ribose, Lys161 and a solvent channel denoted in panel C. Solid black arrows indicate direction of proton relay. Numbers indicate distances in Å observed in BaiA2:NAD+ crystal structure. The denoted distance of C3-OH group of the substrate from Tyr157-OH is based on the observed distance of water molecule, W680, in BaiA2:NAD+ structure that might represent the 3-oxygen atom of the substrate. In figures A, B, and C, C atoms of protein residues are colored gold (BaiA2), green (3β, 17β-hydroxysteroid dehydrogenase), cyan (3α, 20β-hydroxysteroid dehydrogenase), orange (MtDh) and gray (Rv2002). N atoms are colored blue. For clarity only the nicotinamide and the linked ribose moiety of NAD+ are shown. Oxygen atom of the nicotinamide-OH−/water adduct is denoted by purple sphere in figures A and C.
RESULTS
Substrate turnover detection by 1H-NMR
The spectra for several BaiA2 reactions utilizing different substrates are shown in Fig. 2B. The region in the figure contains peaks associated with the methyl protons of 18 and 19 (see Fig. 2A). These provided convenient sites for monitoring the reactions by measuring the decrease in substrate peak intensity and the appearance of product signals. During the cholic acid (CA) oxidation reaction (Fig. 2B) a clear decrease in the signal intensity of substrate peaks associated with methyl groups 18 and 19 (0.73 and 0.93 ppm respectively), accompanied by an increase in the peak area of the same groups in 3-oxo-cholic acid (3-oxo-CA) (0.77 and 1.03 ppm respectively) was observed. These changes were not isolated to the region displayed in the figure and were observed for all positions and confirm that the expected product was generated. The glycocholic acid (GCA) substrate showed similar results with a significant decrease in intensity of the peaks at 0.92 and 0.73 ppm with a corresponding increase at 1.03 and 0.77ppm. While there was no available product standard the magnitude of the chemical shift change is almost identical to that observed for 3-oxo-CA suggesting that the new peaks correspond to 3-oxo-glycocholic acid. The GCA reaction clearly proceeds at a much faster rate compared to CA. The forward and reverse reactions for cholyl-CoA (C-CoA) are also shown in Fig. 2B. The reaction rate for these substrates was significantly faster compared to the non-CoA bile acid substrates and it was necessary to decrease the enzyme concentration in order to collect spectra over a reasonable time series. Similar to the CA observations, the oxidation reaction showed an increase in signal intensity of methyl groups 19 and 18 from 3-oxo-cholyl CoA (3-oxo-C-CoA) (0.96 and 0.60 ppm respectively) accompanied by a decrease in the corresponding peaks of C-CoA (0.85 and 0.56ppm respectively). The opposite peak intensity changes were observed for the reverse reaction using the 3-oxo-C-CoA substrate. In all cases turnover of the bile acid substrate was accompanied by conversion of the co-factor. Comparable profiles were obtained for similar 1H-NMR studies conducted using BaiA1.
Steady-state kinetic analysis
Earlier characterization of this step of the pathway was performed with cell extract of E. coli expressing BaiA1 11. This study reports the first kinetic characterization of enzymes postulated to be involved in the first oxidation step of the pathway. Steady-state kinetic parameters for both BaiA1 and BaiA2 are listed (Table II). BaiA1 exhibited relatively higher substrate turnover rate constant (kcat) compared to BaiA2. Under the standard assay conditions the turnover rate constants (kcat) of CoA – bile acid esters were greater with substrate specificity constants (kcat/KM) 1 or 2 orders of magnitude higher than the corresponding bile acids. The substrate specificity constants of bile acids were comparable with the amino acid - bile acid esters, Glycocholic acid (GCA) and Taurocholic acid (TCA). Moreover, the steady-state kinetic parameters for substrates cholyl-CoA (C-CoA), chenodeoxycholyl-CoA (CDC-CoA), lithocholyl-CoA (LC-CoA) and deoxycholyl-CoA (DC-CoA) are comparable, with substrate specificity ranging between 0.7 and 3.3 μM−1 min−1 for BaiA1 and between 0.3 and 0.6 μM−1 min−1 for BaiA2. Steady-state kinetic parameters of CoA esters of primary and secondary bile acids deviated within same order of magnitude suggesting removal of C7-hydroxyl group did not have a profound effect. Similarly, the steady-state kinetic parameters were comparable for CoA esters of 3-oxo-primary bile acids (3-oxo-C-CoA, 3-oxo-CDC-CoA) and 3-oxo-secondary bile acids (3-oxo-LC-CoA and 3-oxo-DC-CoA). The steady-state kinetic parameters are also comparable for CA and CDCA for both enzymes. Steady-state kinetic parameters for lithocholic acid (LCA) are not determined because the substrate did not have adequate solubility. Moreover, steady-state kinetic parameters of 3-oxo bile acids, such as 3-oxo-CA and 3-oxo-DCA, are also not determined as no detectable turnover is observed.
Table II.
Steady-state kinetic parameters of BaiA1 and BaiA2 with different substrates †
| Substrates | BaiA1
|
BaiA2
|
||||
|---|---|---|---|---|---|---|
| kcat (min−1) | KM (μM) | kcat/KM (μM−1 min−1) | kcat (min−1) | KM (μM) | kcat/KM (μM−1 min−1) | |
| CA | 16 (0.85) | 1158 (106) | 0.014 | 5.4 (0.4) | 802.9 (114) | 0.007 |
| CDCA | 15 (0.71) | 555 (56) | 0.027 | 3.3 (0.34) | 333.8 (88) | 0.010 |
| GCA | 25 (1.1) | 383 (40) | 0.065 | 8.7 (0.56) | 204.6 (39) | 0.043 |
| TCA | 24 (1.1) | 329 (39) | 0.073 | 8.8 (0.33) | 201.4 (22) | 0.044 |
| C-CoA | 74 (2.2) | 106 (11) | 0.698 | 10 (0.38) | 32 (6.10) | 0.313 |
| CDC-CoA | 49 (0.95) | 53 (4.20) | 0.925 | 12 (0.34) | 23 (3.40) | 0.522 |
| LC-CoA | 44 (0.69) | 22 (1.90) | 2.000 | 11 (0.26) | 18 (2.50) | 0.611 |
| DC-CoA | 52 (1.10) | 16 (1.10) | 3.250 | 9.5 (0.11) | 17 (1.20) | 0.559 |
| 3-oxo-C-CoA | 33 (0.67) | 9.1 (0.57) | 3.626 | 8.3 (0.16) | 1.8 (0.19) | 4.611 |
| 3-oxo-CDC-CoA | 24 (0.24) | 3.3 (0.14) | 7.230 | 10 (0.18) | 1.1 (0.13) | 9.090 |
| 3-oxo-LC-CoA | 42 (1.70) | 8.3 (1.00) | 5.060 | 7.3 (0.10) | 1.3 (0.11) | 5.615 |
| 3-oxo-DC-CoA | 44 (1.20) | 5.6 (0.55) | 7.860 | 9.1 (0.47) | 1.5 (0.45) | 6.100 |
Experiments performed with final enzyme concentration of 2.1 μM for substrates CA, CDCA, GCA and TCA and 0.27 μM for all bile acid-CoA esters in 20 mM TRIS pH 8.7 at 37 °C. Values in parentheses indicate standard error.
Reaction rate profiles measured over pHs 7.3, 8.7 and 9.9 using constant ionic strength buffers indicated that the preference of catalyzing the direction of reaction is pH dependent, (Table III). At pH 7.3, clearly the reduction reaction is preferred with an order of magnitude higher velocity than the oxidation reaction. At pH 8.7, the velocities of the oxidation and reduction reaction are comparable. However, at pH 9.9 there is no marked difference between the velocities of the oxidation and reduction reaction. The reaction velocities at pH 9.9 are comparable with the velocity of the reduction reaction being 60% of oxidation reaction. This suggests that both BaiA1 and BaiA2 may prefer the reduction reaction compared to the oxidation reaction. Consistent with this observation, under standard assay conditions both BaiA1 and BaiA2 exhibited at least 1 or 2 orders of magnitude higher substrate affinity (KM) and 1 order of magnitude higher kcat/KM of C3-oxo bile acid – CoA compared to C3-hydroxyl bile acid – CoA esters, (Table II).
Table III.
Reaction rate profile of turnover of bile acid – CoA esters by BaiA1 and BaiA2 at three different pHs‡
| pH | BaiA1
|
BaiA2
|
||||||
|---|---|---|---|---|---|---|---|---|
| Oxidation CDC-CoA |
Reduction 3-oxo-CDC-CoA |
Oxidation DC-CoA |
Reduction 3-oxo-DC-CoA |
Oxidation CDC-CoA |
Reduction 3-oxo-CDC-CoA |
Oxidation DC-CoA |
Reduction 3-oxo-DC-CoA |
|
| 7.3 | 3.5 | 10.1 | 4.8 | 20.8 | 0.8 | 2.2 | 0.7 | 3.2 |
| 8.7 | 5.2 | 5.2 | 6.9 | 9.1 | 1.0 | 0.4 | 1.2 | 0.6 |
| 9.9 | 5.6 | 3.4 | 5.4 | 3.2 | 3.3 | 1.7 | 3.3 | 1.8 |
Unit of reaction rate, μM min−1
Earlier studies suggested that BaiA1 could utilize either NAD+ and NADP+ as the co-factor 11. Standard assays performed in presence of bile acid substrates suggest that both BaiA1 and BaiA2 possess exclusive co-factor specificity towards NAD. No change in either NADP(H) or bile acid substrate peaks were observed in NMR experiments (data not shown). The specificity constants, kcat/KM, for NADP+ were at least an order of magnitude lower compared to NAD+ for both BaiA1 and BaiA2 (Table IV). The specificity constants of NADP+ were 0.008 μM−1 min−1 and 0.018 μM−1 min−1 with KM 883 μM and 84 μM, respectively, for BaiA1 and BaiA2. Similarly, the turnover rate of the reduction reaction in presence of NADPH was significantly low (data not shown). Steady-state kinetic studies performed with Glu42Ala variant of BaiA2 showed improved utilization of NADP. The specificity constant is ~10-fold higher for BaiA2-Glu42Ala than the wild type with ~6-fold higher affinity (KM) towards NADP (Table IV). Kinetic studies performed with the Arg16Ala variant of BaiA2 revealed impaired substrate turnover (data not shown).
Table IV.
Steady state kinetic parameters of BaiA1 and BaiA2 for co-factors
| Co-Factors | BaiA1
|
BaiA2
|
BaiA2_E42A
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| kcat (min−1) | KM (μM) | kcat/KM (μM−1 min−1) | kcat (min−1) | KM (μM) | kcat/KM (μM−1 min−1) | kcat (min−1) | KM (μM) | kcat/KM (μM−1 min−1) | |||
|
|
|
|
|||||||||
| NAD+ | 18 | 17 | 1.06 | 5.3 | 6.7 | 0.79 | 19 | 17 | 1.11 | ||
| NADP+ | 6.8 | 883 | 0.008 | 1.5 | 84 | 0.018 | 3 | 15 | 0.2 | ||
Monomeric Structure and Quaternary Architecture
The overall fold of BaiA2 is similar to structures of SDR family of proteins with top matches in both FATCAT (score: 593.34) and DALI (Z score: 36.7) 34. BaiA2 is a single domain protein bearing the characteristic Rossmann fold with a doubly wound βαβαβ motif (Fig. 3A). The central β sheet composed of 7 parallel strands sandwiched between 3 helices on either side; helices α1, α2 and α9 on one side and helices α3, α5 and α7 on the opposite side. The strand topology is β3-β2-β1-β4-β5-β6-β7 with a long crossover between strands β3 and β4 involving helix α3. The substrate binding pocket and co-factor binding site are separate (Fig. 3B). The postulated catalytic triad residues in the active site, Ser144, Tyr157 and Lys161 are located at the interface of substrate and co-factor binding sites. The co-factor binding site is located at the interface of the C-terminal ends of the first six strands of the central β sheet and the amino- terminal end of helix α1. The substrate binding site comprised of residues 95–101, helix α6 (145–150), residues 151–154, four N-terminal residues of helix α7 and residues 199–210 of helix α8. In the apo- structure, the side chain of residues 145–158 and 191–215 of the substrate binding site exhibited poor electron density. Commensurate with this observation, residues of helix α8 exhibit high B-factor values ranging between 40 and 61 Å2 compared to mean B-factor value of 30 Å2 (Fig. 3A and B). This suggests conformational flexibility of helix α8 and may be essential for accommodating the substrate.
FIGURE 3.
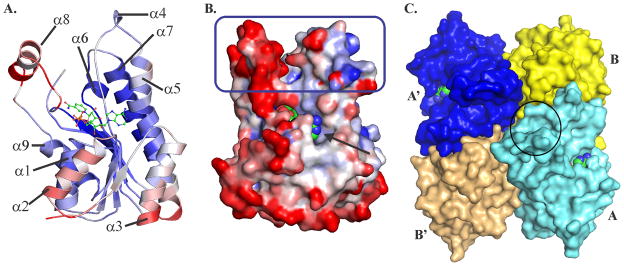
Monomeric and quaternary structure of BaiA2. (A) Characteristic Rossmann fold observed in the BaiA2:NAD+ monomer. (B) Surface representation of BaiA2 monomer depicting discrete substrate binding site (blue rectangle) and co-factor binding site (black arrow). Conformational flexibility depicted in (A) and (B) according to B-value of Cα atoms from blue (lowest 18.0 Å2) to red (highest 59.7 Å2). (C) Tetrameric assembly observed in the P21 space group crystal with the bound NAD+. Entry to one of the substrate binding pocket is indicated by the black circle on the monomer colored cyan. The bound NAD+ depicted in ball and sticks in (A), and spheres in (B) and (C). The atoms of NAD+ are colored as follows: carbon – green, nitrogen – blue, oxygen – red and phosphorus – orange.
Analytical size exclusion chromatography indicates that both BaiA1 and BaiA2 are tetramers in solution (data not shown). Both the apo- and NAD+ - bound structures of BaiA2 revealed a tetrameric assembly with 222 (D2) point group symmetry (Fig. 3C). The tetramer is composed of two dimers AB and A′B′ related by two-fold symmetry. The two interfaces, AB and AB′, observed in the crystal cover an area of 1691 Å2. and 1282 Å2, respectively. All four protomers in the asymmetric unit of NAD+ - bound structure have highly similar structures with rmsd ranging between 0.15 and 0.44 Å for the superposed Cα atoms.
Nicotinamide-Hydroxyl Adduct
After refinement a residual positive electron density peak (7.2 – 8.2 rmsd) is observed proximal to C6 atom of the nicotinamide moiety in all four monomers (Fig. 4A). The residual electron density can be modeled as an oxygen atom (labeled UNL in the PDB file) located at distances 1.8 Å, 2.2 Å and 2.9 Å, respectively from C6 and N1 atoms of the nicotinamide ring and 2′-hydroxyl group of ribose moiety as observed in Chain A (Fig. 4B). The additional oxygen atom is located at distances ranging between 2.3 and 2.6 Å from the C5 atom of the nicotinamide moiety. Quantum chemical calculations based on the deviation of the nicotinamide geometry from the refined coordinates in the crystal structure suggest that the oxygen atom is likely to be OH−. RMSD of the optimized NAD(H)-OH− is comparatively lower than NAD(H)-water (Table V). The rmsd value of NAD+-OH− reached 0.24 Å in comparison to 0.46 Å for NAD+-water. Similarly, the rmsd value of the optimized NADH-OH− (0.19 Å) is also lower than NADH-water (0.23 Å). Consistently, lower side chain rmsd values for Tyr157 and Lys161 are noted in NAD(H)-OH− calculations reaching as low as 0.14 Å, in comparison to NAD(H)-water where the highest rmsd value is 0.93 Å. Based on the refinement trials the hydroxyl moiety in the adduct is unlikely to interact through a covalent bond with the C6 atom of the nicotinamide moiety. Refining the nicotinamide moiety with single bond constraints on C5–C6 (mimicking covalently bonded adduct) raises the B-factor value of OH− to as high as 60 Å2 and displacing the oxygen atom from its electron density map. Restraining the above bond as a double bond (mimicking non-covalent addition) limits the B-factor value of OH− within 40 Å2. Based on these observations it is likely that the residual electron density is due to a non-covalently adducted OH−.
Table V.
RMSD values of the optimized nicotinamide moiety with respect to the starting coordinates taken from either BaiA2:NAD+ or BaiA2:NADH crystal structures
| System | RMSD Nicotinamide moiet Ref: BaiA2:NAD+ |
RMSD Tyr/Lys moiety Ref: BaiA2:NAD+ |
RMSD Nicotinamide moiety Ref: BaiA2:NADH |
RMSD Tyr/Lys moiety Ref: BaiA2:NADH |
|---|---|---|---|---|
|
|
|
|||
| NADH/OH− | 0.504 | 0.220 | 0.187 | 0.138 |
| NADH/H2O | 0.663 | 0.300 | 0.232 | 0.463 |
| NAD+/OH− | 0.239 | 0.272 | 0.274 | 0.515 |
| NAD+/H2O | 0.460 | 0.930 | 0.481 | 0.829 |
The observed NAD+-OH− in BaiA2:NAD+ is significantly different from the previously reported nicotinamide-hydroxyl adduct in horse liver alcohol dehydrogenase (LADH). In LADH, the hydroxyl group is distal to the 2′-hydroxyl group of the nicotinamide-ribose located at a distance of 4.1 Å (PDB Code: 1HET 35). In contrast, the OH− ion of the adduct in BaiA2:NAD+ as noted above is proximal to 2′-hydroxyl group of the nicotinamide ribose. A similar nicotinamide-hydroxyl adduct is observed in an alcohol dehydrogenase from Shewanella denitrificans OS-217 (PDB code: 3RF7) where the additional hydroxyl group is observed within a distance of 2.8 Å from the 2′-hydroxyl group of the nicotinamide moiety. Furthermore, another noted difference is the location of the additional hydroxyl group relative to the C4 atom of the nicotinamide ring, the site of hydride transfer. In LADH the additional hydroxyl group of the adduct is located within a distance of 2.8 Å from the C4 atom contrasting a distance of 3.1 Å and 3.7 Å observed in BaiA2:NAD+ and PDB code: 3RF7, respectively.
Structural Basis of Co-Factor Specificity
The enzyme:co-factor interaction observed at the adenine binding subsite in the structure explains the observed exclusive co-factor specificity towards NAD(H) by both BaiA1 and BaiA2. Modeling a 2′-phospahte group on the adenine ribose results both steric and electrostatic hindrances with the side chain of residue Glu42, which is located on helix α2. Least square superposition of NADP bound structure of mannitol dehydrogenase (MtDh) from Agaricus bisporus onto BaiA2:NAD+ also revealed a similar electrostatic and steric clash (Fig. 4C). Least squares superposition of protein Cα atoms of residues interacting with the adenine moiety places the 2′-phosphate group of NADP within 1.8 Å from Glu42 carboxyl group (Fig. 4C). The above mentioned hindrance with the 2′-phosphate group can be avoided by orienting the carboxyl group of Glu42 away from the co-factor binding site. This is possible by either Glu42 assuming the conformation of rotamer mm-40° (incompatible with the electron density) or attaining an orientation similar to Asp47 observed in the MtDh:NADP structure through main chain conformational flexibility. However, several key interactions of Glu42 with proximal residues in the binding site of adenine moiety observed in NAD bound structure seem to restrict both main chain and side chain conformational flexibility (Fig. 4D). The main chain conformational flexibility around Glu42 seems restricted due to hydrogen bonding interactions involving the backbone amide of Glu42 and side chain Thr39 hydroxyl group (3.2 Å) and the backbone Val43 amide and backbone Thr39 amide groups (3.2 Å). The side chain conformational flexibility of Glu42 is restricted due to hydrogen bonding interaction with backbone Glu38 amide (3.2 Å) and a salt bridge interaction with the guanidinium group of Arg16 (3.2 Å). Residue Arg16 is located on helix α1 and is one residue prior to the second Gly of the characteristic co-factor binding motif, Gly-(X)2-Gly-X-Gly in SDR. In NADP utilizing SDRs the residue prior to second Gly in the motif is usually conserved as Arg. The salt bridge interaction is also observed in apo- structures of BaiA2 although some apo- structures obtained from different crystallization condition revealed relatively weaker electron density of the terminal guanidium group of Arg16. A similar salt bridge interaction is also observed in 3α,20β-hydroxysteroid dehydrogenase where the equivalent Arg and Glu residues are interacting at a distance of 2.8 Å (Fig. 4C). These interactions may restrict the conformational flexibility of the Glu42 side chain and “lock” the carboxylate side chain into an orientation that projects into the co-factor binding site and conforming Glu42 to rotamer mt-10° (compatible with electron density). Modeling other rotamers of Glu generates steric clashes with proximal residues in the adenine binding site. Consistent with the crystallographic observations kinetic studies performed with BaiA2-Glu42Ala mutant revealed improved utilization of NADP compared to wild type, see above. Residues Arg16 and Glu42 are conserved in BaiA1 and kinetic studies also revealed similar cofactor utilization. Hence, the restricted conformational flexibility of Glu42 guides preferential utilization of co-factor in both BaiA1 and BaiA2.
Active Site and Mechanism of Catalysis
The active site residues, Ser144, Tyr157 and Lys161 are located at the interface of the substrate and co-factor binding sites. It is postulated that catalysis by SDRs invokes a Tyr-Lys coupled ionization pair mechanism with a proton relay network involving the 2′-hydroxyl group of nicotinamide ribose 36,37. Analysis of the active site of BaiA2 in co-factor bound structure suggests a deviation from such a mechanism. Comparison with other representative SDR structures by least square superposition over four Cα atoms between active site Tyr and Lys residues suggest Tyr157 has a different orientation (Fig. 5B). Tyr157-Oη atom is oriented away from Lys161-Nζ atom and 2′-hydroxyl group of the nicotinamide ribose by a distance of 6.6 Å and 5.6 Å, respectively (Fig. 5A). Moreover, in the apo- structure both Tyr157 and Lys161 exhibited conformational flexibility. As noted above the electron density for stretch of residues 150–157 is poor and no residues could be built in that part of the structure. Furthermore, comparison with co-factor bound BaiA2, Lys161 in the apo- structure attained significantly deviant conformation – being oriented away from the active site. The distance between Tyr157-Oη (BaiA2:NAD+) and Lys161-Nζ atoms (apo- ) after least square superposition also revealed a distance of 6.6 Å. In previously reported apo-, co-factor bound and ternary complex structures of SDRs, active site Tyr-Oη atom interact with both Lys-Nζ atom and 2′-hydroxyl group of nicotinamide ribose within distances 4.6 Å and 2.8 Å, respectively (Fig. 5B 12,36,38–42). The interacting distances of Tyr157 with Lys161 and nicotinamide ribose is hence, too large to promote an effective proton relay during catalysis. However, the proton relay can be mediated through the nicotinamide-OH− adduct, which is located at intermediary distances of 2.8 Å and 4.5 Å, respectively from Tyr157-Oη and Lys161-Nζ atoms (Fig. 5A). Because of its location the nicotinamide-OH− adduct may be able to influence the pKa value of Tyr157-OH group during catalysis. It can deprotonate and protonate the Tyr157-Oη atom respectively, during oxidation and reductive reaction. In absence of the nicotinamide-OH− adduct the proton relay network with 2′-hydroxyl group of nicotinamide ribose and Lys161 can be generated only through conformational flexibility of Tyr157 and/or Lys161. The accompanying proton relay during catalysis is mediated through 2′-hydroxyl group of nicotinamide ribose and Lys161 (Fig. 5D). Lys161 interacts within hydrogen bonding distances with the hydroxyl groups of nicotinamide ribose (3.0 Å), a water molecule (3.1 Å), and backbone carbonyl group of Thr142 (2.9 Å) through the terminal amino group. The interaction leads to a solvent channel comprising six water molecules (Fig. 5C).
DISCUSSION
In this study we provided a structure-function correlation of co-factor utilization and catalysis. The apo- and putative NAD+-OH− adduct bound structure of BaiA2 provided a structural basis of co-factor specificity and involvement of the adduct in proton relay.
The exclusive preferential NAD(H) utilization by both BaiA1 and BaiA2 can be explained by restrictive conformational flexibility of Glu42 in the co-factor binding site. The co-factor binding site in both BaiA1 and BaiA2 is incompatible with NADP due to a predicted steric and electrostatic hindrance involving Glu42 and the 2′-phosphate group. Consistent with this observation replacement of Glu42 with Ala in BaiA2 improved NADP utilization by 10-fold compared to the wild type.
Our work has uncovered a novel nicotinamide-OH− adduct, which we propose to be involved in proton relay instead of hydride transfer as noted earlier. In LADH the nictoniamide-hydroxyl adduct is proposed to participate in the “activation process” for hydride transfer because of its proximity to the C4 atom of the nicotinamide ring. Analysis of nicotinamide-hydroxyl adducts in other structures also led to such a hypothesis 43. However, in BaiA2 the additional OH− in the observed adduct may be poised to facilitate proton transfer because of its proximity to 2′-hydroxyl group of ribose moiety. As noted earlier the large distance between the terminal functional groups of Tyr157 and Lys161 observed in the crystal structures of BaiA2 seem to hinder both the proton relay network and the Tyr-Lys coupled ionization pair. The OH− ion in the adduct is distal to the C4 atom and is located at intermediary distances between Tyr157 and Lys161. The observed NAD+-OH− is hence suitable to facilitate proton relay during catalysis.
Steady state kinetic characterization of BaiA1 and BaiA2 established that bile acid-CoA esters are the preferred substrates corroborating previous observation 11. In comparison to bile acids and bile acid-amino acid esters, the substrate specificity constants (kcat/KM) of bile acid-CoA esters were 2 orders of magnitude higher. Although, the monomeric and tetrameric structures of BaiA2 did not reveal a separate binding pocket of CoA, however significantly higher kinetic parameters observed for bile acid-CoA esters suggests a possible interaction of the CoA moiety with the protein. Binding of the bile acid-CoA ester may also generate an induced pocket to accommodate the large CoA moiety. In 3-oxo-Δ4,6- lithocholyl-CoA bound crystal structure of bile acid 7α dehydratase (BaiE) from Clostridium hiranonis DSM 13275, which catalyzes the removal of C7-hydroxyl group, we observed a binding pocket generated between monomers of different oligomeric assembly to accommodate the large CoA moiety (to be published). Similar pocket may also be formed during catalysis of BaiA1/A2 to interact with the CoA moiety.
Comparison of steady-state kinetic characterization performed at pHs 7.3 and 8.7 established preferential catalysis of both BaiA1 and BaiA2 towards the reduction reaction. Substrate specificity (kcat/KM) and affinity (KM) constants of both BaiA1 and BaiA2 towards C3-oxo bile acid-CoA esters are an order of magnitude higher than the corresponding bile acid-CoA esters. This suggests that BaiA1 and BaiA2 may have dual activity where the enzymes can potentially participate in both the reductive and oxidative arm of the pathway. The dual activity of these enzymes enables the bacteria to efficiently generate secondary bile acids without the need to produce separate enzymes.
In summary this study provides the first structural and steady-state kinetic characterization of any enzymes that are proposed to be involved in secondary bile acid synthesis by Clostridium sp. 44. There is currently a significant interest in the field about the role of gut microbes impacting human health and disease. However, the biochemical details underlying these events are largely unknown. In that regard, the structural and biochemical studies of the enzymes involved in secondary bile acid synthesis will provide a platform to elucidate how gut microbes influence human health and homeostasis and to indirectly manipulate the overall human metabolism.
Acknowledgments
We thank Dr. Phillip B. Hylemon, Virginia Commonwealth University for providing us the genomic DNA of Clostridium scindens VPI 12708. We thank the members of the JCSG high-throughput structural biology pipeline for their contribution to this work. We thank Dr. Marc Elsliger, The Scripps Research Institute for critical review of the paper. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (NIH), Award Number U54 GM094586. Stanford Synchrotron Radiation Lightsource (SSRL) Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393) and the National Center for Research Resources (P41RR001209). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Center for Research Resources, or the National Institutes of Health.
Abbreviations used
- SDR
Short Chain Dehydrogenase/Reductase
- CoA
Coenzyme A
- CA
cholic acid, 3α,7α,12α-trihydroxy-5β-cholanoic acid
- CDCA
chenodeoxycholic acid, 3α,7α-dihydroxy-5β-cholanoic acid
- LCA
lithocholic acid, 3α-hydroxy-5β-cholanoic acid
- DCA
deoxycholic acid, 3α,12α-dihydroxy-5β-cholanoic acid
- GCA
glycocholic acid, 3α,7α,12α-trihydroxy-5β-cholanoyl-glycine
- TCA
taurocholic acid, 3α,7α,12α-trihydroxy-5β-cholanyl-taurine
- 3-oxo-CA
3-oxo-cholic acid, 3-oxo-7α,12α-dihydroxy-5β-cholanoic acid
- C-CoA
cholyl-CoA, 3α,7α,12α-trihydroxy-5β-cholanoyl-CoA
- CDC-CoA
chenodeoxycholyl-CoA, 3α,7α-dihydroxy-5β-cholanoyl-CoA
- DC-CoA
deoxycholyl-CoA, 3α,12α-dihydroxy-5β-cholanoyl-CoA
- LC-CoA
lithocholyl-CoA, 3α-hydroxy-5β-cholanoyl-CoA
- 3-oxo-C-CoA
3-oxo-cholyl-CoA, 3-oxo-7α,12α-dihydroxy-5β-cholanoyl-CoA
- 3-oxo-CDC-CoA
3-oxo-chenodeoxycholyl-CoA, 3-oxo-7α-hydroxy-5β-cholanoyl-CoA
- 3-oxo-LC-CoA
3-oxo-lithocholyl-CoA, 3-oxo-5β-cholanoyl-CoA
- 3-oxo-DC-CoA
3-oxo-deoxycholyl-CoA, 3-oxo-12α-hydroxy-5β-cholanoyl-CoA
References
- 1.Wells JE, Hylemon PB. Identification and Characterization of a Bile Acid 7α-Dehydroxylation Operon in Clostridium sp. Strain TO-931, a Highly Active 7α-Dehydroxylating Strain Isolated from Human Feces. Applied and Environmental Microbiology. 2000;66(3):1107–1113. doi: 10.1128/aem.66.3.1107-1113.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doerner KC, Takamine F, LaVoie CP, Mallonee DH, Hylemon PB. Assessment of fecal bacteria with bile acid 7 alpha-dehydroxylating activity for the presence of bai-like genes. Applied and Environmental Microbiology. 1997;63(3):1185–1188. doi: 10.1128/aem.63.3.1185-1188.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto J-M, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Bork P, Ehrlich SD, Wang J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464(7285):59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tu H, Okamoto AY, Shan B. FXR, a Bile Acid Receptor and Biological Sensor. Trends in Cardiovascular Medicine. 2000;10(1):30–35. doi: 10.1016/s1050-1738(00)00043-8. [DOI] [PubMed] [Google Scholar]
- 5.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7(8):678–693. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 6.Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res. 2009;50(8):1509–1520. doi: 10.1194/jlr.R900007-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a Nuclear Receptor for Bile Acids. Science. 1999;284(5418):1362–1365. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 8.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, Hinuma S, Fujisawa Y, Fujino M. A G Protein-coupled Receptor Responsive to Bile Acids. Journal of Biological Chemistry. 2003;278(11):9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 9.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, Haussler MR, Mangelsdorf DJ. Vitamin D Receptor As an Intestinal Bile Acid Sensor. Science. 2002;296(5571):1313–1316. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 10.Nehring JA, Zierold C, DeLuca HF. Lithocholic acid can carry out in vivo functions of vitamin D. Proceedings of the National Academy of Sciences. 2007;104(24):10006–10009. doi: 10.1073/pnas.0703512104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mallonee DH, Lijewski MA, Hylemon PB. Expression in Escherichia coli and characterization of a bile acid-inducible 3α-hydroxysteroid dehydrogenase from Eubacterium sp. strain VPI 12708. Current Microbiology. 1995;30(5):259–263. doi: 10.1007/BF00295498. [DOI] [PubMed] [Google Scholar]
- 12.Parkkinen T, Boer H, Jänis J, Andberg M, Penttilä M, Koivula A, Rouvinen J. Crystal Structure of Uronate Dehydrogenase from Agrobacterium tumefaciens. Journal of Biological Chemistry. 2011;286(31):27294–27300. doi: 10.1074/jbc.M111.254854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klock HE, Koesema EJ, Knuth MW, Lesley SA. Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts. Proteins: Structure, Function, and Bioinformatics. 2008;71(2):982–994. doi: 10.1002/prot.21786. [DOI] [PubMed] [Google Scholar]
- 14.Klock HE, Lesley SA. The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods in Molecular Biology: High Throughput Protein Expression and Purification. 2009:498. doi: 10.1007/978-1-59745-196-3_6. [DOI] [PubMed] [Google Scholar]
- 15.Ikegawa S, Ishikawa H, Oiwa H, Nagata M, Goto J, Kozaki T, Gotowda M, Asakawa N. Characterization of Cholyl-Adenylate in Rat Liver Microsomes by Liquid Chromatography/Electrospray Ionization–Mass Spectrometry. Analytical Biochemistry. 1999;266(1):125–132. doi: 10.1006/abio.1998.2944. [DOI] [PubMed] [Google Scholar]
- 16.Elsliger M-A, Deacon AM, Godzik A, Lesley SA, Wooley J, Wuthrich K, Wilson IA. The JCSG high-throughput structural biology pipeline. Acta Crystallographica Section F. 2010;66(10):1137–1142. doi: 10.1107/S1744309110038212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shaw AA, Salaun C, Dauphin J-F, Ancian B. Artifact-Free PFG-Enhanced Double-Quantum-Filtered COSY Experiments. Journal of Magnetic Resonance, Series A. 1996;120(1):110–115. [Google Scholar]
- 18.Ancian B, Bourgeois I, Dauphin J-F, Shaw AA. Artifact-Free Pure Absorption PFG-Enhanced DQF-COSY Spectra Including a Gradient Pulse in the Evolution Period. Journal of Magnetic Resonance. 1997;125(2):348–354. [Google Scholar]
- 19.Willker W, Leibfritz D, Kerssebaum R, Bermel W. Gradient selection in inverse heteronuclear correlation spectroscopy. Magnetic Resonance in Chemistry. 1993;31(3):287–292. [Google Scholar]
- 20.Bax A, Summers MF. Proton and carbon-13 assignments from sensitivity-enhanced detection of heteronuclear multiple-bond connectivity by 2D multiple quantum NMR. Journal of the American Chemical Society. 1986;108(8):2093–2094. [Google Scholar]
- 21.Bax A, Davis DG. Practical aspects of two-dimensional transverse NOE spectroscopy. Journal of Magnetic Resonance (1969) 1985;63(1):207–213. [Google Scholar]
- 22.Santarsiero BD, Yegian DT, Lee CC, Spraggon G, Gu J, Scheibe D, Uber DC, Cornell EW, Nordmeyer RA, Kolbe WF, Jin J, Jones AL, Jaklevic JM, Schultz PG, Stevens RC. An approach to rapid protein crystallization using nanodroplets. Journal of Applied Crystallography. 2002;35(2):278–281. [Google Scholar]
- 23.McPhillips TM, McPhillips SE, Chiu H-J, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. Journal of Synchrotron Radiation. 2002;9(6):401–406. doi: 10.1107/s0909049502015170. [DOI] [PubMed] [Google Scholar]
- 24.Kabsch W. XDS. Acta Crystallographica Section D. 2010;66(2):125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallographica Section D. 2010;66(2):133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sheldrick G. A short history of SHELX. Acta Crystallographica Section A. 2008;64(1):112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- 27.Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated Structure Solution With autoSHARP. 2007;364:215–230. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- 28.Cohen SX, Morris RJ, Fernandez FJ, Ben Jelloul M, Kakaris M, Parthasarathy V, Lamzin VS, Kleywegt GJ, Perrakis A. Towards complete validated models in the next generation of ARP/wARP. Acta Crystallographica Section D. 2004;60(12 Part 1):2222–2229. doi: 10.1107/S0907444904027556. [DOI] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallographica Section D. 2004;60(12 Part 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Murshudov GN, Vagin AA, Dodson EJ. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallographica Section D. 1997;53(3):240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 31.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallographica Section D. 2011;67(4):235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. BUSTER Version 2.10.0. Cambridge, United Kingdom: Global Phasing Limited; [Google Scholar]
- 33.Schuttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallographica Section D. 2004;60(8):1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 34.Holm L, Rosenström P. Dali server: conservation mapping in 3D. Nucleic Acids Research. 2010;38(suppl 2):W545–W549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meijers R, Morris RJ, Adolph HW, Merli A, Lamzin VS, Cedergren-Zeppezauer ES. On the Enzymatic Activation of NADH. Journal of Biological Chemistry. 2001;276(12):9316–9321. doi: 10.1074/jbc.M010870200. [DOI] [PubMed] [Google Scholar]
- 36.Filling C, Berndt KD, Benach J, Knapp S, Prozorovski T, Nordling E, Ladenstein R, Jörnvall H, Oppermann U. Critical Residues for Structure and Catalysis in Short-chain Dehydrogenases/Reductases. Journal of Biological Chemistry. 2002;277(28):25677–25684. doi: 10.1074/jbc.M202160200. [DOI] [PubMed] [Google Scholar]
- 37.Ladenstein R, Winberg JO, Benach J. Medium- and short-chain dehydrogenase/reductase gene and protein families. Cellular and Molecular Life Sciences. 2008;65(24):3918–3935. doi: 10.1007/s00018-008-8590-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ghosh D, Wawrzak Z, Weeks CM, Duax WL, Erman M. The refined three-dimensional structure of 3α,20β-hydroxysteroid dehydrogenase and possible roles of the residues conserved in short-chain dehydrogenases. Structure. 1994;2(7):629–640. doi: 10.1016/s0969-2126(00)00064-2. [DOI] [PubMed] [Google Scholar]
- 39.Hörer S, Stoop J, Mooibroek H, Baumann U, Sassoon J. The Crystallographic Structure of the Mannitol 2-Dehydrogenase NADP+ Binary Complex from Agaricus bisporus. Journal of Biological Chemistry. 2001;276(29):27555–27561. doi: 10.1074/jbc.M102850200. [DOI] [PubMed] [Google Scholar]
- 40.Yang JK, Park MS, Waldo GS, Suh SW. Directed evolution approach to a structural genomics project: Rv2002 from Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences. 2003;100(2):455–460. doi: 10.1073/pnas.0137017100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sogabe S, Yoshizumi A, Fukami TA, Shiratori Y, Shimizu S, Takagi H, Nakamori S, Wada M. The Crystal Structure and Stereospecificity of Levodione Reductase from Corynebacterium aquaticum M-13. Journal of Biological Chemistry. 2003;278(21):19387–19395. doi: 10.1074/jbc.M208146200. [DOI] [PubMed] [Google Scholar]
- 42.Tang Y, Lee HY, Tang Y, Kim C-Y, Mathews I, Khosla C. Structural and Functional Studies on SCO1815: A β-Ketoacyl-Acyl Carrier Protein Reductase from Streptomyces coelicolor A3(2)∞. Biochemistry. 2006;45(47):14085–14093. doi: 10.1021/bi061187v. [DOI] [PubMed] [Google Scholar]
- 43.Meijers R, Cedergren-Zeppezauer E. A variety of electrostatic interactions and adducts can activate NAD(P) cofactors for hydride transfer. Chemico-Biological Interactions. 2009;178(1–3):24–28. doi: 10.1016/j.cbi.2008.10.043. [DOI] [PubMed] [Google Scholar]
- 44.Ridlon JM, Kang D-J, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. Journal of Lipid Research. 2006;47(2):241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]