

Abstract
Sorafenib has substantial clinical activity as third- or fourth-line treatment of imatinib- and sunitinib-resistant gastrointestinal stromal tumors (GIST). Because sorafenib targets both angiogenesis-related kinases (VEGFR) and the pathogenetic kinases found in GIST (KIT or PDGFRA), the molecular basis for sorafenib efficacy in this setting remains unknown. We sought to determine the spectrum of activity of sorafenib against different mutant kinases associated with drug-sensitive and drug-resistant GIST. We compared the activity of imatinib and sorafenib against transiently expressed mutant forms of KIT and PDGFRA, including various secondary mutations that have been identified in imatinib-resistant or sunitinib-resistant GISTs. We also examined these drugs against four GIST cell lines, three of which are imatinib resistant. In our in vitro studies, we determined that sorafenib inhibited imatinib-resistant mutations in exons encoding the ATP/drug-binding pocket and in exons encoding the activation loop, with the exception of substitutions at KIT codon D816 and PDGFRA codon 842. Notably our data indicate that sorafenib is more effective than imatinib or sunitinib for inhibiting the kinase activity of drug-resistant KIT mutants (as assessed by biochemical IC50). We hypothesize that a major determinant of the efficacy of sorafenib for treatment of advanced GIST is the activity of this agent against KIT or PDGFRA-mutant kinases. These results have implications for the further development of treatments for drug-resistant GIST.
Introduction
Activating mutations of receptor tyrosine kinases KIT or PDGFRA are key to the pathogenesis of most gastrointestinal stromal tumors (GIST). More than 80% of GISTs express mutated, constitutively active KIT receptors, another 5% to 7% express mutated PDGFRA, and the remaining 10% to 15% are wild-type GISTs (WT) lacking mutations in either of these kinases (1, 2).
Imatinib mesylate, a small molecule kinase inhibitor with potent activity against KIT and PDGFRA, has revolutionized GIST treatment and is now well established as front-line medical treatment for advanced disease. Despite very high rates of disease control with this agent, up to 50% of patients suffer disease progression within 2 years of initiating therapy, and the vast majority of patients eventually develop drug-resistant disease (3, 4). In the majority of cases, the tumor regrowth occurs after an initial response, becoming radiologically evident more than 6 months after beginning treatment (delayed or secondary resistance). However, 10% to 15% of patients have tumors that exhibit primary imatinib resistance, defined as progression with 3 to 6 months of starting therapy (5).
There are distinct molecular mechanisms underlying primary and secondary imatinib resistance. Primary resistance is typically found in GISTs with specific genotypes: KIT exon 9 mutation, PDGFRA D842V mutation, or WT GIST. Resistance in this setting is probably due to relative or absolute resistance of these kinases to imatinib at clinically achievable drug levels (6). In the case of exon 9–mutant GIST, evidence from phase III studies indicates that the probability of primary resistance can be reduced by using a higher dose of imatinib (4). Most cases of secondary resistance are associated with an acquired kinase mutation of the ATP/drug-binding pocket or the kinase activation loop on the same allele (cis-conformation) as the primary activating mutation (5, 7, 8). Such secondary mutations are rare in cases of primary imatinib resistance.
Sunitinib, another small-molecule tyrosine kinase inhibitor (TKI) with selectivity for KIT and PDGFRA [as well as PDGFRB, all 3 isoforms of VEGFR, FMS-like tyrosine kinase 3 (FLT3), and colony-stimulating factor 1 receptor (CSF-1R)] has shown clinical benefit in phase I–III trials with patients who were intolerant of imatinib or had disease that was imatinib resistant (6, 9, 10). Sunitinib has been approved for treatment of GIST patients for whom prior imatinib therapy failed because of disease progression or drug intolerance. Unfortunately, the median progression-free survival (PFS) with second-line sunitinib is only 6 to 7 months (9). Profiling of sunitinib against imatinib-resistant kinases (i.e., KIT with primary + secondary mutations) has shown that sunitinib potently inhibits imatinib-resistant KIT ATP/drug-binding pocket mutations but has little activity against imatinib-resistant KIT activation loop mutations (6, 11, 12). Thus, many imatinib-resistant mutations confer cross-resistance to sunitinib, thereby accounting for the relatively short PFS with second-line sunitinib. Currently, the presence of these multi-TKI–resistant clones limits the overall survival (OS) of patients with advanced GIST.
Various agents have been tested as salvage therapy for patients with imatinib- and sunitinib-resistant GIST. Sorafenib, a multitargeted kinase inhibitor with a spectrum of drug targets similar to sunitinib has been tested in the third- or fourth-line setting, yielding promising results in phase II studies. Notably, the PFS of patients with TKI-resistant GIST in these studies was similar to that seen with second-line sunitinib (13–15). Because sorafenib targets both angiogenesis-related kinases (VEGFR) and the pathogenetic kinases found in GIST (KIT or PDGFRA), the molecular basis for sorafenib efficacy in this setting remains unknown (16). In addition, only limited profiling of the activity of sorafenib against secondary mutations associated with drug-resistant GIST has been carried out to date (17, 18). Specifically, sorafenib has only been tested against the following KIT secondary mutations: V654A, T670I, and D820Y.
To explore the molecular mechanisms responsible for sorafenib clinical activity against TKI-resistant GIST, we profiled the drug against a panel of KIT- and PDGFRA-mutant kinases expressed by transient transfection of a reference cell line. For comparison, we tested the potency of imatinib in the same experiments. This approach allowed us to compare the results to our previous work in which we profiled sunitinib using the same methodology (6). To verify our results, we also carried out head-to-head comparisons of imatinib, sunitinib, and sorafenib in our isogenic cell models. In addition, we used imatinib-sensitive and imatinib-resistant GIST cell lines to further corroborate our results.
Our results indicate that sorafenib has good IC50 potency against all imatinib-resistant mutations, with the exception of mutations involving KIT codon 816 (exon 17, activation loop) and PDGFRA codon 842 (exon 18, activation loop). These results provide a potential mechanistic basis for the activity of sorafenib against drug-resistant GIST. In addition, our data suggest that the use of sorafenib in the first- or second-line setting might circumvent or prevent drug resistance mutations and yield improved clinical results.
Materials and Methods
Reagents and antibodies
Imatinib and sorafenib were purchased from LC Laboratories. Polyclonal rabbit antibody to KIT was from Dako. Polyclonal rabbit antibodies to phospho-KIT Y721 (catalog #3391), phospho-KIT Y703 (catalog #3073), phospho-AKT S473 (catalog #9271), total AKT (catalog #9272), phospho-S6 S235/236 (catalog #2211), and mouse monoclonal antibody (mAb) to total S6 (catalog #2317) were from Cell Signaling. Mouse mAb to actin (catalog #A4700) was from Sigma. For PDGFRA immunoprecipitation experiments, we used rabbit polyclonal anti-PDGFRA antibody (Santa Cruz Biotechnology, SC-20) and Protein A/G beads (Santa Cruz). For detection of phospho-PDGFRA and total PDGFRA, we used anti-phosphotyrosine mAb (PY-20, 1:500; BD Transduction Labs) and anti-PDGFRA rabbit polyclonal antibody (SC-20, 1:500; Santa Cruz), respectively. KIT immunoprecipitation experiments were carried out as previously described (6). For detection of phospho- and total-KIT, we used rabbit polyclonal antibody to P-KIT Tyr 719 (catalog #3391, 1:500; Cell Signaling Technology) or total KIT (C-19, 1:500; Santa Cruz), respectively.
Sensitivity of kinase mutants to TKIs in vitro
Chinese hamster ovary (CHO) cells were purchased from the American Tissue Type Collection at the start of the study. CHO cells were transiently transfected with mutated KIT or PDGFRA cDNA constructs and treated with various concentrations of sorafenib, sunitinib, or imatinib as previously described (6). The common exon 11 mutation V560D was selected as a prototypic primary KIT exon 11 mutation, and representative exon 13, 14, and 17 mutations were chosen as secondary mutations. Experiments involving recombinant DNA were conducted using biosafety level 2 conditions in accordance with published guidelines.
Several GIST cell lines were established by the study authors (GIST882, GIST48, GIST430, and GIST-T1/829 by J.A.F, and GIST-T1 parental line by T.T). At the start of this study, all lines were credentialed by Sanger sequencing, confirming presence of unique KIT mutations that differ between the lines and were also validated by 250K Nsp SNP profiling, showing identity of origin with the primary tumor cultures (GIST882, GIST430, and GIST48) or early-passage cell line cultures (GIST-T1) from which these immortal cell lines were established. The GIST-T1 cell line was established from an untreated GIST, and the GIST48 and GIST430 cell lines were established from imatinib-resistant clinically progressing GISTs as previously described (5, 19). The cell lines were recredentialed using the above procedures every 3 months during the study. GIST-T1/829 is a novel subline established by culturing GIST-T1 in incrementally increasing concentrations of imatinib: this subline contains the same KIT exon 11 mutation as parental GIST-T1, coupled in cis with a secondary A829P kinase domain mutation. Protein lysates from transfected CHO cells or GIST cell lines were prepared and subjected to immunoprecipitation using anti-KIT or anti-PDGFRA antibodies followed by sequential immunoblotting for phospho-KIT or total KIT, or phosphotyrosine or total PDGFRA, respectively, as previously reported (6, 20, 21). Densitometry was carried out to measure drug effect, using Photoshop CS4 software to quantify the level of phospho-KIT or phospho-PDGFRA normalized to total protein. Densitometry data were analyzed using Calcusyn 2.1 software (Biosoft) to mathematically determine the IC50 values. The Wilcoxon rank sum test was used to compare the IC50 values of imatinib and sorafenib for a given mutation.
Results
In vitro measure of IC50 activity of sorafenib or imatinib against specific mutants
We first assessed the potency of sorafenib against the 3 most common KIT mutant kinases found in GIST: KIT exon 11 mutant, KIT exon 9 mutant, and WT KIT. For purposes of comparison with previous publications and standardization of our subsequent experiments with double mutant isoforms, we chose the common exon 11 mutation V560D as a prototypic primary KIT mutant kinase and used imatinib as the comparator TKI.
Sorafenib potently inhibited the KIT exon 11 V560D kinase with an IC50 of less than 100 nmol/L. Sorafenib was significantly less potent against ligand-activated wild-type KIT with an IC50 value of approximately 2,700 nmol/L, which was similar to the IC50 of 3,400 nmol/L obtained for imatinib (Fig. 1, panel A, Table 1). Sorafenib inhibited the KIT exon 9–mutant kinase with an IC50 of approximately 1,800 nmol/L, whereas the IC50 for imatinib was approximately 3,970 nmol/L (Fig. 1, panel B, Table 1).
Figure 1.

Comparative biochemical activity of imatinib versus sorafenib for inhibiting KIT activity in transiently transfected cells. CHO cells were transiently transfected and protein lysates from transfected cells were prepared and subjected to immunoprecipitation using anti-KIT antibody followed by sequential immunoblotting for phospho-KIT (P-KIT) or total KIT (KIT), respectively, as previously reported. Representative results from a minimum of 3 replicate experiments for the comparative activity of imatinib or sorafenib for inhibiting WT (unmutated, ligand-stimulated KIT), isolated KIT exon 11 mutation (V560D), isolated KIT exon 17 mutation (D816V), or compound mutations of V560D with a secondary imatinib-resistance mutation are shown.
Table 1.
Biochemical IC50 values for inhibition of KIT kinase activity in transfected cells
| 1° Mutation | 2° Mutation | Imatinib | Sorafenib | Sunitinib |
|---|---|---|---|---|
| V560D | None | <100 | <100 | <100 |
| V560D | V654A | 1,600 | 560a | <100 |
| V560D | T670I | >5,000 | 200a | <100 |
| V560D | D816H | 1,990 | 1,930 | >1,000 |
| V560D | D820A | 4,000 | <100 | ND |
| V560D | D820G | 3,255 | 480a | >1,000 |
| V560D | N822K | 2,820 | 480a | >1,000 |
| V560D | Y823D | 2,100 | 280a | >1,000 |
| WT | None | 3,400 | 2,700 | <100 |
| D816V | None | >5,000 | >5,000 | >1,000 |
| KIT insAY502-503 | None | 3,970 | 1,829a | <100 |
| KIT insAY502-503 | V654A | 3,100 | 590a | 100 |
| KIT insAY502-503 | D816H | 4,930 | 1,820a | >1,000 |
| KIT insAY502-503 | D820G | 2,640 | 1,380a | ND |
NOTE: The values for imatinib, sorafenib, and sunitinib represent the biochemical IC50 expressed in nmol/L units for the listed single mutant or compound mutant kinases. WT indicates the results for wild-type (unmutated), KIT ligand-stimulated KIT kinase. The IC50 values for imatinib and sorafenib are from direct head-to-head comparison from a minimum of 3 replicate experiments. The IC50 values for sunitinib are from a previous publication from our group (6).
Abbreviation: ND, not determined.
P < 0.05 sorafenib IC50 compared with imatinib IC50.
Sorafenib potently inhibited the phosphorylation of KIT exon 11 double mutants in which the second mutation occurred in the drug/ATP-binding site of the receptor, including V560D + V654A (exons 11 + 13) and V560D + T670I (exons 11 + 14). These double mutants were strongly or completely resistant to imatinib (Fig. 1, panel A, Table 1).
KIT exon 11–mutant kinases with a secondary mutation in the activation loop had strong in vitro resistance to imatinib, with IC50 values ranging from 1,990 to 4,000 nmol/L. In contrast, with the exception of V560D + D816H, all of these compound mutant kinases were sensitive to sorafenib, with IC50 values ranging from 100 to 480 nmol/L. The IC50 for V560D + D816H was 1,930 nmol/L. Resistance to sorafenib was also seen with KIT D816V, which is a primary activating mutation in AML and mast cell neoplasms, and an uncommon secondary mutation in GIST. The isolated D816V mutation was extremely resistant to imatinib and sorafenib, with IC50 values of more than 5,000 nmol/L for both drugs (Fig. 1, panel A, Table 1).
Although some KIT exon 9–mutant GISTs exhibit primary imatinib resistance, in many cases secondary drug resistance develops through the acquisition of mutations that are identical to those seen in imatinib-resistant GISTs with KIT exon 11 mutations. We tested the potency of sorafenib against KIT exon 9–mutant kinases with secondary V654A, D820G, or D816H mutations and found IC50 values of 590, 1,380, and 1,820 nmol/L, respectively. Consistent with previous results, the IC50 for imatinib against exon 9–mutant kinases with secondary V654A, D820G, or D816H mutations was 3,100, 2,640, and 4,930 nmol/L, respectively (Fig. 2A, Table 1).
Figure 2.

Comparative biochemical activity of imatinib versus sorafenib for extracellular KIT mutants or PDGFRA-mutant kinases in transiently transfected cells. CHO cells were transiently transfected and protein lysates from transfected cells were prepared and subjected to immunoprecipitation using anti-KIT or anti-PDGFRA antibodies followed by sequential immunoblotting for phospho-KIT (P-KIT) or total KIT (KIT), or phosphotyrosine (P-PDGFRA) or total PDGFRA (PDGFRA), respectively, as previously reported. A, representative results from a minimum of 3 replicated experiments of the comparative activity of imatinib or sorafenib for inhibiting the isolated KIT exon 9 mutation insertion 502–503 AY or compound mutations of KIT exon 9 with a secondary imatinib-resistant mutation are shown. B, representative results from a minimum of 3 replicate experiments of the comparative activity of imatinib or sorafenib for inhibiting WT (unmutated, PDGF-AA ligand-stimulated PDGFRA), isolated PDGFRA exon 12 (V561D) or exon 18–mutant kinases (D842V, deletion DIMH842-845), or the compound mutant kinase V561D + D842V are shown.
We also tested the potency of sorafenib in inhibiting the phosphorylation of wild-type PDGFRA, as well as the mutations PDGFRA V561D and deletion DIMH842-845 (Fig. 2B, Table 2). V561D is a relatively common primary PDGFRA mutation located in the receptor juxtamembrane domain encoded by exon 12 and is homologous to KIT V560D. The DIMH842-845 deletion is located in the activation loop of PDGFRA. Imatinib and sorafenib have similar potencies against these 3 PDGFRA isoforms (IC50 < 100 nmol/L).
Table 2.
Biochemical IC50 values for inhibition of PDGFRA kinase activity in transfected cells
| 1° Mutation | 2° Mutation | Imatinib | Sorafenib | Sunitinib |
|---|---|---|---|---|
| WT | None | <100 | <100 | <100 |
| V561D | None | <100 | <100 | <100 |
| V561D | D842V | 3,100 | 3,100 | >1,000 |
| Del DIMH842-845 | None | <100 | <100 | ND |
| D842V | None | 1,700 | 1,280 | >1,000 |
NOTE: The values for imatinib, sorafenib, and sunitinib represent the biochemical IC50 expressed in nmol/L units for the listed single mutant or compound mutant kinases. WT indicates the results for wild-type (unmutated), PDGF-AA ligand-stimulated PDGFRA kinase. The IC50 values for imatinib and sorafenib are from direct head-to-head comparisons from a minimum of 3 replicate experiments. The IC50 values for sunitinib are from a previous publication from our group (6). The IC50 for imatinib was not significantly different than the IC50 for sorafenib for any of these mutant kinases.
Abbreviation: ND, not determined.
D842V, which is the most common primary PDGFRA mutation in GISTs, resides in the activation loop encoded by exon 18 and confers imatinib resistance when present as either a primary or a secondary mutation. Both the primary D842V and the V561D + D842V–mutant kinases were insensitive to sorafenib in our in vitro experiments (IC50 of 1,280–3,100 nmol/L, Fig 1, panel C, Table 2). This level of activity was similar to that seen for imatinib.
We have previously evaluated the activity of imatinib or sunitinib against our panel of mutations using the same experimental methods (6). Notably, the imatinib IC50 values in the present studies are identical to those we previously reported. To further confirm our current results, we directly compared the potency of imatinib, sunitinib, or sorafenib in the same experiment. For these confirmatory experiments, we expressed the following mutant kinases: V560D + V654A, V560D + T670I, V560D + D816H, and V560D + N822K. Both sorafenib and sunitinib were potent inhibitors of V560D + V654A with IC50 values of less than 100 and 560 nmol/L, respectively, whereas imatinib was significantly less effective (IC50 1,570 nmol/L; data not shown). Both sorafenib and sunitinib also had potent activity against V560D + T670I (IC50 values of 100–200 nmol/L), whereas imatinib had no activity against this particular mutant kinase (IC50 > 5,000 nmol/L). V560D + D816H was resistant to all drugs, with IC50 values of more than 1,000, 2,000, and 2,000 nmol/L, respectively, for sunitinib, sorafenib, and imatinib. Another activation loop mutant V560D + N822K was very resistant to sunitinib (IC50 of >1,000 nmol/L) and imatinib (IC50 2,820 nmol/L), but sensitive to sora- fenib (IC50 480 nmol/L).
These results for imatinib and sunitinib are very similar to our previous published results (6). Therefore, for comparative purpose, we have tabulated our current imatinib and sorafenib data with our previously reported sunitinib data to allow the relative comparison of the potency of these 3 TKIs against GIST-associated mutations, including isolated primary as well as primary + secondary mutations (Tables 1 and 2).
Comparative biochemical activity of imatinib and sorafenib in GIST cell lines
To confirm these findings in a GIST cell context, we tested the relative potency with which imatinib and sorafenib inhibited KIT kinase activity and downstream signaling in GIST cell lines. These included GIST-T1 cells with a primary exon 11 mutation alone (GIST-T1) and GISTs with primary exon 11 mutations coupled with secondary kinase domain mutations V654A (GIST430), D820A (GIST48), and A829P (GIST-T1/829; Fig. 3; refs. 6, 19)
Figure 3.
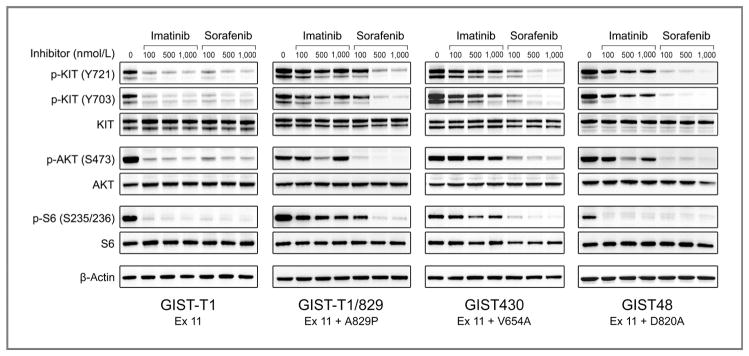
Comparative activity of imatinib and sorafenib for inhibition of KIT kinase and activation of downstream signaling pathways in GIST cell lines. GIST cell lines were treated with imatinib or sorafenib for 6 hours and then harvested for protein lysates. Whole-cell lysates were immunoblotted and the membrane was probed with antibodies to activated (p-KIT, p-AKT, and p-S6) and total forms of KIT, AKT, and S6. The bottom frame contains the results for β-actin, which was used as a loading control.
Consistent with our results against the isolated exon 11 V560D mutation expressed in CHO cells, both imatinib and sorafenib had similar potency against the KIT exon 11–mutant GIST-T1 cells (IC50 < 100 nmol/L). In addition, imatinib and sorafenib were equally potent at inhibiting activation of KIT-dependent signaling pathways, as assessed by inhibition of phosphorylation of AKT (serine 473) and S6 (S253/236). By contrast, KIT and AKT were potently inhibited by sorafenib (IC50 < 100) but not imatinib (IC50 > 1,000) in a GIST-T1 subline with the known imatinib-resistance secondary mutation, A829P.
The GIST430 cell line, derived from an imatinib-resistant clinical specimen, is heterozygous for a KIT exon 11 deletion mutation and the exon 13 V654A substitution (both on the same allele; ref. 5). The IC50 for imatinib was approximately 750 nmol/L. Notably, this concentration is almost 10-fold higher than that necessary to inhibit exon 11–mutant KIT kinase in the GIST-T1 cell line. In contrast, sorafenib was a substantially more potent inhibitor of the KIT oncoproteins in GIST430 cells, with an IC50 of less than 100 nmol/L. Consistent with these results, sorafenib was also more potent than imatinib for inhibition of AKT and S6.
The GIST48 cell line, also derived from an imatinib-resistant clinical specimen, is homozygous for a primary KIT exon 11 V560D mutation and heterozygous for a secondary exon 17 D820A mutation (5). The imatinib IC50 for this cell line was approximately 150 nmol/L but even at a dose of 1,000 nmol/L, KIT was only 60% inhibited. Sorafenib was significantly more potent than imatinib, with 85% inhibition of KIT phosphorylation using doses as low as 100 nmol/L. Likewise, sorafenib treatment was substantially more effective than imatinib at inhibiting phosphorylation of AKT.
Discussion
Molecular mechanisms resulting in imatinib resistance have been delineated in GIST patients, and these mechanisms differ between cases of primary (or early) versus secondary (or delayed) resistance (5, 8, 22, 23). Primary imatinib resistance, defined as progression during the first 6 months of treatment, is typically found in GIST with following genotypes: KIT exon 9–mutant, PDGFRA D842V–mutant GIST, or WT GIST. In the case of KIT exon 9–mutant or PDGFRA D842V–mutant GIST, this is likely due to relative or absolute resistance of these kinases to imatinib at clinically achievable drug levels. WT GIST is a heterogeneous set of tumors, and sensitivity to imatinib is likely related, in part, to the degree that the tumor is actually dependent upon KIT activation for growth and survival (23, 24).
Secondary imatinib resistance, defined as resistance that develops after 6 months or more of therapy, is much more common and is typically associated with acquisition of additional KIT kinase mutations. Notably, these mutations tend to cluster in 2 distinct domains of the protein: the ATP/drug-binding pocket encoded by exons 13 and 14, and the extended activation loop encoded by exons 17 and 18. These mutations are invariably found on the same allele as the primary KIT activation mutation. Mutation of KIT exons 13 or 14 confers extreme imatinib resistance. Various mutations of KIT exon 17 and 18 cause a range of resistance, from mild/moderate to extreme/absolute resistance (5, 7, 8, 22, 23, 25, 26).
Sunitinib has been shown to be clinically effective for the treatment of imatinib-resistant GIST and is now established as second-line therapy (9). Sunitinib, like imatinib, potently inhibits KIT and PDGFRs. One major difference between these 2 drugs is that sunitinib also targets VEGFR, whereas imatinib does not (27). Theoretically, the activity of sunitinib against imatinib-resistant tumors might be because of targeting KIT and/or VEGFR. Sunitinib potently targets KIT-mutant kinases with secondary mutations of the ATP/drug-binding pocket. However, sunitinib has little significant clinical activity against KIT-mutant kinases with secondary mutations of the activation loop (6). To date, the only generally recognized mechanism of sunitinib resistance in GIST is the acquisition of KIT activation loop mutations (6, 12). The contribution of VEGFR inhibition to the overall clinical activity of sunitinib against imatinib-resistant GIST remains unknown.
Sorafenib, a biaryl urea small molecular kinase inhibitor (Fig. 4A), has potent activity against KIT, VEGFRs, FLT3, and PDGFRA/B tyrosine kinases. In addition, sorafenib has weak activity against certain intracellular serine/threonine kinases, including RAF1 and BRAF (16). The similar target profiles of sunitinib and sorafenib, as well as the commercial availability of sorafenib, has helped drive several phase II clinical studies of sorafenib for treatment of imatinib-resistant GIST.
Figure 4.

A, chemical structures of sorafenib and regorafenib. B, graphical comparison of the potency of imatinib, sunitinib, and sorafenib for imatinib-resistant KIT mutations (cellular IC50). The left-most stick figure depicts the protein structure of the KIT protein, with location and frequency of primary GIST mutations to the left of the figure and the location of critical functional domains indicated on the right side of the figure (JM, juxtamembrane domain). The right-most stick figure is an exploded view of a portion of the cytoplasmic domain, with labeling to indicate the location of critical exons (colored rectangles). The location and composition of amino acid substitutions associated with secondary KIT kinase mutations are indicated on the right of the exploded stick figure (e.g., V654A). The traffic light boxes to the right of the figure indicate sensitivity of the indicated mutations to imatinib (IM), sunitinib (SU), or sorafenib (SOR). Drug sensitive mutations were defined as those with a cellular IC50 in our model system of less than 200 nmol/L for sunitinib and less than 1,000 nmol/L for imatinib and sorafenib.
Kindler and colleagues reported the final results of a phase II consortium study of sorafenib for drug-resistant GIST. The original study was for patients with documented progression on imatinib. However, following U.S. Food and Drug Administration approval of sunitinib as a second-line treatment for GIST, the study protocol was amended to require tumor progression after both imatinib and sunitinib therapy. All patients were treated with sorafenib 400 mg twice daily as the target starting dose. Overall, 38 patients were enrolled at 6 centers (6 imatinib resistant, 32 imatinib/sunitinib resistant). The disease control rate was 68% (13% PR: 1 imatinib resistant, 4 imatinib + sunitinib resistant; 55% SD: 3 imatinib resistant, 18 imatinib+ sunitinib resistant). Median progression-free and OS was 5.2 and 11.6 months, respectively. This treatment regimen was associated with a greater than 50% rate of grade 3–4 toxicity (e.g., hand–foot syndrome 45%) requiring at least one dose reduction in 63% of patients (13). Park and colleagues reported the results of a phase II study of sorafenib as third-line treatment. The overall disease control rate was 65% (10% PR, 55% SD). The median PFS was 4.6 months and the median OS had not been reached with a median follow-up of 7.5 months (15).
There is also experience with using sorafenib as a fourth-line therapy after progression or intolerance during prior imatinib, sunitinib, and nilotinib therapy. In a retrospective report, 32 patients were treated with standard dose sorafenib (400 mg twice daily). The overall tumor control rate was 63% (19% PR, 44% SD). Median PFS and OS were 4.6 and 10.4 months, respectively (14).
Notably, the disease control rate and survival data for sorafenib in the third- or fourth-line settings are comparable with those reported for sunitinib in the second-line setting (sunitinib disease control rate 65%, median PFS 5.5 months, median OS 17.2 months; ref. 9). Regorafenib, a close analog of sorafenib (Fig. 4A), is also effective for treating imatinib- and sunitinib-resistant GISTs, with a PFS of 10 months in a phase II study (28).
The most common grade 3–4 adverse events in GIST patients treated with sorafenib included hand–foot skin reaction (16%), anemia (7%), elevated liver enzymes or bilirubin (7%), hypertension (5%), and fatigue (5%). This spectrum and frequency of adverse events is similar to that reported with use of sorafenib in other malignancies.
We hypothesized that sorafenib would have a broader spectrum of activity against drug-resistant KIT mutations than imatinib or sunitinib, thus potentially explaining the activity of sorafenib in the treatment of advanced, drug-resistant GIST. Using an isogenic model to express single or compound KIT or PDGFRA mutations, we profiled the IC50 activity of sorafenib across a broad panel of clinically relevant mutations. Sorafenib was found to be a potent inhibitor of KIT exon 11–mutant kinase with a potency that was equal to or greater than imatinib or sunitinib. Sorafenib was significantly more potent than imatinib against KIT kinase with secondary mutation of the ATP/drug-binding pocket (V654A or T670I) and would be predicted to be clinically active against these mutations. However, sunitinib was the most potent of the 3 inhibitors against these particular mutations. Sorafenib was potent against secondary KIT mutations involving the activation loop with the notable exception of mutations involving codon 816 (D816H or D816V). Among the 3 inhibitors that we have profiled, sorafenib had the broadest spectrum of activity against drug-resistant mutations (see Fig. 4B for comparative IC50 activity).
Our results in the isogenic transient transfection model are in substantial agreement with those of Guo and colleagues and Guida and colleagues who profiled sorafenib against 3 secondary resistance mutation isoforms (V654A, T670I, and D820Y) in non-GIST models (17, 18). In addition, we obtained confirmatory results for the relative potency of imatinib and sorafenib using 4 different GIST cell models. On the basis of our results, we believe that inhibition of KIT kinase activity is a major determinant of clinical activity of sorafenib against multi-TKI–resistant GIST.
We also profiled the activity of sorafenib against primary and secondary PDGFRA mutations associated with GIST. Imatinib and sorafenib had equivalent potency against PDGFRA WT, V561D, or deletion DIMH842-845 isoforms. However, neither drug has significant activity against PDGFRA D842V, either as an isolated primary mutation or when combined with a primary V561D mutation. These results are consistent with reports by Lierman and colleagues and von Bubnoff and colleagues using clinical samples or in vitro cell models to test the activity of sorafenib- against imatinib-resistant FIP1L1-PDGFRA mutants (29–31).
In most reported series of imatinib-resistant GIST, there is an approximately equal frequency of ATP-binding pocket and activation loop mutations (23). Importantly, individual patients often have multiple drug-resistant tumors, and these lesions can differ in their secondary mutations (25). Thus, sunitinib may only be effective against half of the lesions in any given patient (i.e., the cells with secondary ATP-binding pocket mutations). On the basis of our studies, sorafenib would be predicted to be active against the majority of imatinib-resistant tumors, with the exception of those lesions with secondary mutation of KIT codon 816. Presumably, the lack of activity of sorafenib against KIT D816V and PDGFRA D842V is due to a lack of drug binding to these kinases when they are strongly stabilized in the active confirmation by mutation of these aspartic acid residues.
Therefore, our data suggest that sorafenib might be a better agent for second-line treatment of imatinib-resistant GIST than the current standard agent, sunitinib. This prediction is also supported by the reported clinical activity of sorafenib in the third- or fourth-line setting that is comparable with that seen with sunitinib in the second-line setting.
Despite the promising activity of sorafenib for drug-resistant GIST, the manufacturers of sorafenib have chosen to suspend testing of this agent in GIST and instead proceed with clinical development of the highly related compound, regorafenib (28). This switch in drug development strategy occurred during the course of this study. Regorafenib has now been tested in a phase II study in which a 55% clinical benefit rate was seen (9% PR, 46% SD). The median PFS was 10 months (28). On the basis of these results, regorafenib has been studied in a randomized, double-blind, placebo-controlled phase III study. This study is fully accrued and analysis of the results is expected during the latter half of 2012. Because sorafenib and regorafenib only differ by the presence of one side chain fluorine molecule, our studies with sorafenib may also be relevant to the interpretation of ongoing phase II and III studies of regorafenib (Fig. 4A).
Our data suggest that sorafenib (or related compounds such as regorafenib) might be superior to imatinib in the front-line setting, as these agents would be predicted to suppress many of the drug-resistant clones that lead to clinical imatinib resistance. On the basis of experimental models and clinical studies of TKI-treatment of CML, front-line treatment with second-generation TKIs that suppress the development of drug resistance clones may contribute to more durable disease control than front-line imatinib (32–34). However, sorafenib and regorafenib are associated with a higher incidence of grade 3–4 adverse events requiring dose interruption and/or dose reduction than imatinib. Comparative clinical trials of sorafenib or structurally related compounds in the first- or second-line treatment of advanced GIST could be considered to help further optimize medical therapy for this disease.
Acknowledgments
M.C. Heinrich’s COI are managed by OHSU and Portland VA COI committees.
Grant Support
This work was supported in part from funding from a Veterans Affairs Merit Review Grant (M.C. Heinrich), the Life Raft Group (M.C. Heinrich, J. A. Fletcher), the GIST Cancer Research Fund (M.C. Heinrich, J.A. Fletcher), GI SPORE 1P50CA127003 (J.A. Fletcher), BP Lester Foundation (M.C. Heinrich), the Virginia and Daniel K. Ludwig Trust for Cancer Research (J.A. Fletcher), Fundacion Alfonso Martín Escudero (A. Marino-Enriquez), and Cathay General Hospital Research Fund (C.-W. Liang).
Footnotes
Disclosure of Potential Conflicts of Interest
M.C. Heinrich is a recipient of commercial research grant from Novartis, AROG, Imclone, and Ariad; has received honoraria from Speakers Bureau of Novartis and has ownership interest (including patents) from MolecularMD. He is also a consultant and advisory board member of Novartis.
Authors’ Contributions
Conception and design: M.C. Heinrich, J.A. Fletcher
Development of methodology: M.C. Heinrich, J.A. Fletcher
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): M.C. Heinrich, A. Marino-Enriquez, D.J. Griffith, A. McKinley, J. Patterson, C.-W. Liang, J.A. Fletcher
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): M.C. Heinrich, A. Marino-Enriquez, D.J. Griffith, A. McKinley, J. Patterson, J.A. Fletcher
Writing, review, and/or revision of the manuscript: M.C. Heinrich, J.A. Fletcher
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): M.C. Heinrich, A. Presnell, C.-W. Liang, J.A. Fletcher
Study supervision: M.C. Heinrich, J.A. Fletcher
References
- 1.Corless CL, Heinrich MC. Molecular pathobiology of gastrointestinal stromal sarcomas. Annu Rev Pathol. 2008;3:557–86. doi: 10.1146/annurev.pathmechdis.3.121806.151538. [DOI] [PubMed] [Google Scholar]
- 2.Rubin BP, Heinrich MC, Corless CL. Gastrointestinal stromal tumour. Lancet. 2007;369:1731–41. doi: 10.1016/S0140-6736(07)60780-6. [DOI] [PubMed] [Google Scholar]
- 3.Blanke CD, Demetri GD, von MM, Heinrich MC, Eisenberg B, Fletcher JA, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26:620–5. doi: 10.1200/JCO.2007.13.4403. [DOI] [PubMed] [Google Scholar]
- 4.Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247–53. doi: 10.1200/JCO.2009.24.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol. 2006;24:4764–74. doi: 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- 6.Heinrich MC, Maki RG, Corless CL, Antonescu CR, Harlow A, Griffith D, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26:5352–9. doi: 10.1200/JCO.2007.15.7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wardelmann E, Merkelbach-Bruse S, Pauls K, Thomas N, Schildhaus HU, Heinicke T, et al. Polyclonal evolution of multiple secondary KIT mutations in gastrointestinal stromal tumors under treatment with imatinib mesylate. Clin Cancer Res. 2006;12:1743–9. doi: 10.1158/1078-0432.CCR-05-1211. [DOI] [PubMed] [Google Scholar]
- 8.Antonescu CR, Besmer P, Guo T, Arkun K, Hom G, Koryotowski B, et al. Acquired resistance to imatinib in gastrointestinal stromal tumor occurs through secondary gene mutation. Clin Cancer Res. 2005;11:4182–90. doi: 10.1158/1078-0432.CCR-04-2245. [DOI] [PubMed] [Google Scholar]
- 9.Demetri GD, Van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–38. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 10.Demetri GD, Heinrich MC, Fletcher JA, Fletcher CD, Van den Abbeele AD, Corless CL, et al. Molecular target modulation, imaging, and clinical evaluation of gastrointestinal stromal tumor patients treated with sunitinib malate after imatinib failure. Clin Cancer Res. 2009;15:5902–9. doi: 10.1158/1078-0432.CCR-09-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gajiwala KS, Wu JC, Christensen J, Deshmukh GD, Diehl W, DiNitto JP, et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc Natl Acad Sci U S A. 2009;106:1542–7. doi: 10.1073/pnas.0812413106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishida T, Takahashi T, Nishitani A, Doi T, Shirao K, Komatsu Y, et al. Sunitinib-resistant gastrointestinal stromal tumors harbor cis-mutations in the activation loop of the KIT gene. Int J Clin Oncol. 2009;14:143–9. doi: 10.1007/s10147-008-0822-y. [DOI] [PubMed] [Google Scholar]
- 13.Kindler HL, Campbell NP, Wroblewski K, Maki RG, D’Adamo D, Chow WA, et al. Sorafenib (SOR) in patients (pts) with imatinib (IM) and sunitinib (SU)-resistant (RES) gastrointestinal stromal tumors (GIST): Final results of a University of Chicago Phase II Consortium trial. J Clin Oncol. 2011;29:abstr 10009. [Google Scholar]
- 14.Reichardt P, Montemurro M, Gederblom H, Blay J, Rutkowski P, Bui B, et al. Sorafenib fourth-line treatment in imatinib-, sunitinib-, and nilotinib-resistant metastatic GIST: A retrospective analysis. J Clin Oncol. 2011;27:abstr 10564. [Google Scholar]
- 15.Park SH, Ryu MH, Ryoo BY, Im SA, Kwon HC, Lee SS, et al. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of Korean gastrointestinal stromal tumors study group. Invest New Drugs. 2012 Jan 25; doi: 10.1007/s10637-012-9795-9. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 16.Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109. doi: 10.1158/0008-5472.CAN-04-1443. [DOI] [PubMed] [Google Scholar]
- 17.Guida T, Anaganti S, Provitera L, Gedrich R, Sullivan E, Wilhelm SM, et al. Sorafenib inhibits imatinib-resistant KIT and platelet-derived growth factor receptor beta gatekeeper mutants. Clin Cancer Res. 2007;13:3363–9. doi: 10.1158/1078-0432.CCR-06-2667. [DOI] [PubMed] [Google Scholar]
- 18.Guo T, Agaram NP, Wong GC, Hom G, D’Adamo D, Maki RG, et al. Sorafenib inhibits the imatinib-resistant KITT670I gatekeeper mutation in gastrointestinal stromal tumor. Clin Cancer Res. 2007;13:4874–81. doi: 10.1158/1078-0432.CCR-07-0484. [DOI] [PubMed] [Google Scholar]
- 19.Taguchi T, Sonobe H, Toyonaga S, Yamasaki I, Shuin T, Takano A, et al. Conventional and molecular cytogenetic characterization of a new human cell line, GIST-T1, established from gastrointestinal stromal tumor. Lab Invest. 2002;82:663–5. doi: 10.1038/labinvest.3780461. [DOI] [PubMed] [Google Scholar]
- 20.Corless CL, Schroeder A, Griffith D, Town A, McGreevey L, Harrell P, et al. PDGFRA mutations in gastrointestinal stromal tumors: frequency, spectrum and in vitro sensitivity to imatinib. J Clin Oncol. 2005;23:5357–64. doi: 10.1200/JCO.2005.14.068. [DOI] [PubMed] [Google Scholar]
- 21.Heinrich MC. Molecular basis for treatment of gastrointestinal stromal tumours. Eur J Cancer Suppl. 2006;4:10–8. [Google Scholar]
- 22.Debiec-Rychter M, Cools J, Dumez H, Sciot R, Stul M, Mentens N, et al. Mechanisms of resistance to imatinib mesylate in gastrointestinal stromal tumors and activity of the PKC412 inhibitor against imatinib-resistant mutants. Gastroenterology. 2005;128:270–9. doi: 10.1053/j.gastro.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 23.Gramza AW, Corless CL, Heinrich MC. Resistance to tyrosine kinase inhibitors in gastrointestinal stromal tumors. Clin Cancer Res. 2009;15:7510–8. doi: 10.1158/1078-0432.CCR-09-0190. [DOI] [PubMed] [Google Scholar]
- 24.Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–78. doi: 10.1038/nrc3143. [DOI] [PubMed] [Google Scholar]
- 25.Liegl B, Kepten I, Le C, Zhu M, Demetri G, Heinrich M, et al. Heterogeneity of kinase inhibitor resistance mechanisms in GIST. J Pathol. 2008;216:64–74. doi: 10.1002/path. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen LL, Trent JC, Wu EF, Fuller GN, Ramdas L, Zhang W, et al. A missense mutation in KIT kinase domain 1 correlates with imatinib resistance in gastrointestinal stromal tumors. Cancer Res. 2004;64:5913–9. doi: 10.1158/0008-5472.CAN-04-0085. [DOI] [PubMed] [Google Scholar]
- 27.Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM. SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther. 2003;2:471–8. [PubMed] [Google Scholar]
- 28.George S, von Mehren M, Heinrich MC, Wang Q, Corless CL, Butrynski JE, et al. A multicenter phase II study of regorafenib in patients (pts) with advanced gastrointestinal stromal tumor (GIST), after therapy with imatinib (IM) and sunitinib (SU) J Clin Oncol. 2011;29:abstr 10007. [Google Scholar]
- 29.Lierman E, Folens C, Stover EH, Mentens N, Van MH, Scheers W, et al. Sorafenib is a potent inhibitor of FIP1L1-PDGFRalpha and the imatinib-resistant FIP1L1-PDGFRalpha T674I mutant. Blood. 2006;108:1374–6. doi: 10.1182/blood-2006-02-004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lierman E, Michaux L, Beullens E, Pierre P, Marynen P, Cools J, et al. FIP1L1-PDGFRalpha D842V, a novel panresistant mutant, emerging after treatment of FIP1L1-PDGFRalpha T674I eosinophilic leukemia with single agent sorafenib. Leukemia. 2009;23:845–51. doi: 10.1038/leu.2009.2. [DOI] [PubMed] [Google Scholar]
- 31.von Bubnoff N, Gorantla SP, Engh RA, Oliveira TM, Thone S, Aberg E, et al. The low frequency of clinical resistance to PDGFR inhibitors in myeloid neoplasms with abnormalities of PDGFRA might be related to the limited repertoire of possible PDGFRA kinase domain mutations in vitro. Oncogene. 2011;30:933–43. doi: 10.1038/onc.2010.476. [DOI] [PubMed] [Google Scholar]
- 32.Kantarjian HM, Hochhaus A, Saglio G, De SC, Flinn IW, Stenke L, et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011;12:841–51. doi: 10.1016/S1470-2045(11)70201-7. [DOI] [PubMed] [Google Scholar]
- 33.O’Hare T, Walters DK, Stoffregen EP, Jia T, Manley PW, Mestan J, et al. In vitro activity of Bcr-Abl inhibitors AMN107 and BMS-354825 against clinically relevant imatinib-resistant Abl kinase domain mutants. Cancer Res. 2005;65:4500–5. doi: 10.1158/0008-5472.CAN-05-0259. [DOI] [PubMed] [Google Scholar]
- 34.Kantarjian H, Shah NP, Hochhaus A, Cortes J, Shah S, Ayala M, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–70. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]