

Abstract
In this article we describe a method for colorimetric detection of miRNA in the kidney through in situ hybridization with digoxigenin tagged microRNA probes. This protocol, originally developed by Kloosterman and colleagues for broad use with Exiqon miRNA probes1, has been modified to overcome challenges inherent in miRNA analysis in kidney tissues. These include issues such as structure identification and hard to remove residual probe and antibody. Use of relatively thin, 5 mm thick, tissue sections allowed for clear visualization of kidney structures, while a strong probe signal was retained in cells. Additionally, probe concentration and incubation conditions were optimized to facilitate visualization of microRNA expression with low background and nonspecific signal. Here, the optimized protocol is described, covering the initial tissue collection and preparation through the mounting of slides at the end of the procedure. The basic components of this protocol can be altered for application to other tissues and cell culture models.
Keywords: Basic Protocol, Issue 81, microRNA, in situ hybridization, kidney, renal tubules, microRNA probe
Introduction
MicroRNA are small (approximately 22 nucleotides long) noncoding RNAs that are endogenously produced. They generally function to suppress protein expression through translational repression or mRNA degradation. miRNAs bind to mRNA targets with incomplete complementarity, making it possible for a single miRNA to suppress multiple targets.
Understanding which cell types and structures express miRNAs is an important part of understanding the mechanisms through which alterations in miRNA expression influence cell and tissue phenotypes. While methods such as miRNA sequencing, qPCR and Northern blotting can be used for detection of miRNAs in whole tissues, this approach does not allow one to determine which specific cell type they came from within a given tissue. Dissection of cellular and structural components prior to analysis using these methods can be very difficult and the conditions needed to achieve adequate isolations can lead to alterations in gene expression or degradation of RNAs. microRNA in situ hybridization is a method used to visualize microRNA location and expression levels in tissues. This technique is especially valuable in tissues composed of heterogeneous structures, such as the kidney.
MicroRNAs have been shown to play an regulatory role in kidney development2,3 and physiology4. microRNA expression alterations have also been shown to be involved with renal pathology such as fibrosis5-10, diabetic nephropathy7, renal carcinoma11,12, and acute kidney injury13. In our research, we have found that optimizing microRNA in situ hybridization for kidney tissues was valuable for determining the exact structural locations of miRNA expression in both health and disease14. Determination of the tubular and cellular expression of different microRNAs is important because their regulation of targets may be dependent on cellular functions. In diseased states it is also important to determine how alterations in miRNA expression may be impacting function.
The goal of the method described here was to build upon existing ISH methodologies developed by Kloosterman et al.15, other investigators16,17, and those suggested by Exiqon1 and optimize the method for formalin fixed kidney tissues. We have successfully used this method to identify distinct regional differences in renal microRNA miR-382 expression with unilateral ureteral obstruction18. This approach can be used with other tissues as well, with additional optimization.
Protocol
1. Kidney Tissue Sections
Formalin-fixed (10%) paraffin-embedded kidney sections are cut onto microscope slides at 5 mm thickness using a Microm HM 355 S. The slides can be stored for several months before analysis.
2. Solution Preparation
Prepare 1 L of 1x PBS in ddH2O in an autoclavable screw top bottle. Fill another bottle with 1 L of ddH2O. Add 1 ml of DEPC to each bottle, cover and shake vigorously. Let the solutions sit overnight at room temperature to destroy RNAses and then autoclave. Add 400 ml Tween-20 to 1 L of 1x PBS to make PBS-T.
Prepare proteinase K buffer (50 mMTris-HCl, pH 8.0 and 1 mM CaCl2 in DEPC treated water).
Prepare a solution of 0.2% glycine in PBS-T.
3. Tissue Preparation
Prepare a large volume of hybridization buffer (50-100 ml) composed of 50% Formamide, 5x SSC, 0.1% Tween, 9.2 mM citric acid for adjustment to pH 6, 50 µg/ml heparin, and 500 µg/ml yeast RNA in DEPC treated ddH2O. Divide into 1 ml aliquots and store at -80 ºC.
Remove the paraffin from the tissue by washing 3x in fresh xylene for 5 min each.
Rehydrate the tissue sections by 5 min incubations in decreasing concentrations of ethanol (100%, 75%, 50%, and 25%) in ddH2O.
Treat the slides with enough DEPC to completely cover the tissue sections (approximately 0.4-0.5 ml) for 1 min. Make sure tissues do not dry out.
Rinse the slides in DEPC treated PBS-T twice for 5 min, collecting the DEPC containing waste for proper disposal. All reagents should be DEPC treated to eliminate RNAses from this point on.
Prewarm Proteinase K buffer and add proteinase K to a final concentration of 10 μg/ml. Cover tissue sections with proteinase K solution and incubate at 37 ºC for 5 min.
Quickly remove the proteinase K solution and add 0.2% glycine in PBS-T for 30 sec.
Rinse slides in DEPC treated PBS-T twice for 30 sec each.
Fix sections in freshly made 4% paraformaldehyde for 10 min.
4. Hybridization
Add 50 µl of hybridization buffer (50% Formamide, 5x SSC, 0.1% Tween, 9.2 mM citric acid for adjustment to pH 6, 50 µg/ml heparin, 500 µg/ml yeast RNA) per tissue section and cover with RNAse free HybriSlips cut just larger than the tissue sections.
Prehybridize sections in humidity controlled hybridization oven for 2 hr at hybridization temperature determined for the 3’ digoxigenin labeled probe (usually DNA Tm of the probe -21 °C, ( i.e. 54 °C for miR-382, probe DNA Tm= 75 °C), using tissue lab wipes soaked in 50% formamide/50% 5x SSC to keep the chamber humidified.
Dilute the miRNA probe, positive control (U6, small nuclear RNA) and negative control (scrambled sequence) to 40 nM in hybridization buffer (75 µl of buffer is needed per section).
Heat the probe in hybridization buffer at 65 ºC for 5 min.
Remove slides from hybridization oven and remove coverslips and excess hybridization buffer from prehybridization.
Add 75 µl of probe containing buffer per tissue section, directly to tissue. Cover tissue with HybriSlips just large enough to cover the tissue sections.
Keeping slide holder level, return to hybridization oven for overnight incubation at temperature from step 4.2 (approximately 16 hr).
5. Stringency Wash
Add 200 µl 2x SSC, heated to the hybridization temperature, just under one edge of the coverslips to help release the coverslip from the tissue. Remove the coverslip by tilting the slide.
Wash the slides in 200 µl of 50% formamide, 50% 2x SSC at hybridization temperature 3x for 30 min each.
Rinse the slides 5x in PBS-T at room temperature for 5 min each on orbital shaker on low speed.
6. Detection
Incubate slide in blocking buffer (2% goat or horse serum, 2 mg/ml BSA in PBS-T) for 1 hr at room temperature on orbital shaker on low speed.
Dilute anti-DIG-AP Fab fragments in blocking buffer at 1:100. Add 100 µl per tissue section and cover with HybriSlips cut just large enough to cover the section.
Incubate antibody in a humidified chamber at 4 °C overnight (approximately 16 hr).
Wash the slides in PBS-T 7x, for 5 min each on orbital shaker at low speed.
Wash the slides in AP buffer (100 mM TrisHCl pH 9.5, 50 mM MgCl2,100mM NaCl, 0.1% Tween 20) 3x, for 5 min each.
Add NBT/BCIP solution to AP buffer (200 µl/10 ml buffer)
Add 400-500 ml of diluted NBT/BCIP solution to each slide to completely cover all tissue sections. Develop in a dark, level, humidified chamber for 4-5 hr.
7. Mounting Slides
Dehydrate sections in increasing concentrations of ethanol (25%, 50%, 75%, 100%) for 5 min each.
Incubate in xylene 5x in order to clear sections.
Mount slides in Permount mounting medium and dry overnight.
Representative Results
The areas of a tissue section that become dried out during the hybridizations, incubations or wash steps generally end up staining more darkly during the NBT/BCIP development. Figure 1 shows a portion of a kidney section in which the HybriSlip slipped off of the edge of the tissue, allowing it to become partially dehydrated. Despite rehydration and coverage in the remaining steps, the signal in the dehydrated portion is artificially high.
The importance of controlling the time of NBT-BCIP development is demonstrated in Figures 2A and 2B. Development periods of longer than 4-5 hr result in pronounced nonspecific color development in scrambled-miR control probed tissues throughout the kidney (Figure 2A). Kidneys probed for high abundance targets, such as the U6 small nuclear RNA control, do not exhibit improved signals with incubations longer than 4-5 hours (Figure 2B).
The applicability of the ISH method described here to miRNA detection in kidney tissue is demonstrated in Figure 3, a figure reproduced from Kriegel et al.18 In that study we found that unilateral ureteral obstruction (UUO) induced a nearly 3-fold increase in qPCR detected miR-382 expression in homogenized kidney tissue when compared to sham operated animals. This increase was blocked by intravenous deliver of anti-miR-382 (data not shown). The use of ISH on these kidney tissues indicated that the miR-382 detection provided by this probe was sensitive enough to provide quantifiable results that mirrored qPCR based expression analysis. Additionally, ISH allowed us to measure distinct regional differences in miRNA expression within the kidney.
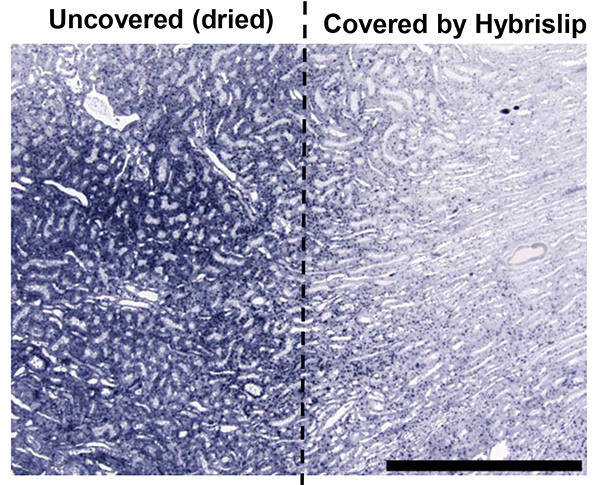 Figure 1.The effect of the partial dehydration of a rat kidney section during hybridization step. Calibration bar =100 mm. Click here to view larger image.
Figure 1.The effect of the partial dehydration of a rat kidney section during hybridization step. Calibration bar =100 mm. Click here to view larger image.
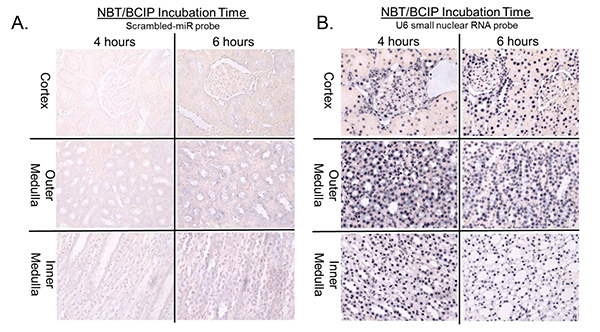 Figure 2.Optimization of the duration of NBT/BCIP development. Allowing the NBT/BCIP development to continue for more than 4-5 hr results in significant background staining in scrambled-miR probed tissues (Figure 2A), while providing no further improvement in the signal in the positive control, U6 small nuclear RNA probed samples (Figure 2B). Click here to view larger image.
Figure 2.Optimization of the duration of NBT/BCIP development. Allowing the NBT/BCIP development to continue for more than 4-5 hr results in significant background staining in scrambled-miR probed tissues (Figure 2A), while providing no further improvement in the signal in the positive control, U6 small nuclear RNA probed samples (Figure 2B). Click here to view larger image.
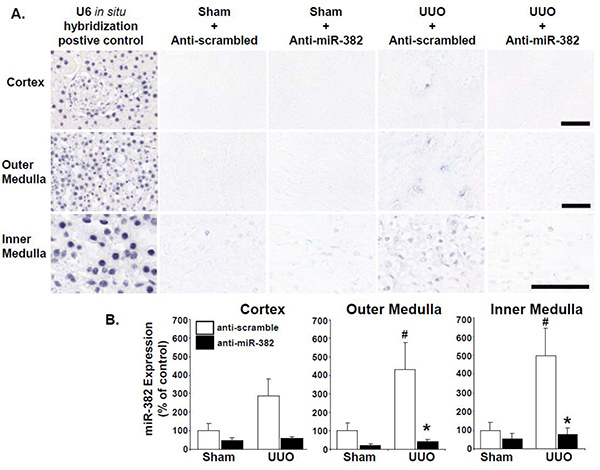 Figure 3.Representative images of miR-382 expression in mouse kidney sections obtained by in situ hybridization. Calibration bar = 50 µm. (B) miR-382 expression as percent average IOD of sham + anti-scrambled treated mice. This figure has been modified from Kriegel et al., (Figure 5), which describes the details of treatment groups, methods, and results18. Anti-scrambled treated n= 10/group; anti-miR-382 n= 8/group; *significantly different from anti-scrambled treated mice with same surgery (P<0.05), # significantly different from sham operated animals receiving the same anti-miR treatment. Click here to view larger image.
Figure 3.Representative images of miR-382 expression in mouse kidney sections obtained by in situ hybridization. Calibration bar = 50 µm. (B) miR-382 expression as percent average IOD of sham + anti-scrambled treated mice. This figure has been modified from Kriegel et al., (Figure 5), which describes the details of treatment groups, methods, and results18. Anti-scrambled treated n= 10/group; anti-miR-382 n= 8/group; *significantly different from anti-scrambled treated mice with same surgery (P<0.05), # significantly different from sham operated animals receiving the same anti-miR treatment. Click here to view larger image.
Discussion
The goal of this article was to describe a protocol for miRNA in situ hybridization that works well in formalin fixed kidney tissues. While working out this protocol several important sources of staining artifact have been identified. Careful attention to these points can help avoid staining artifact and increase the likelihood of a successful ISH run.
One of the most avoidable causes of staining artifact can occur when tissue sections become dried out during long incubations. When this occurs the portion of the tissue that becomes dried will stain very darkly during the NBT/BCIP development (Figure 1). Throughout this protocol it is critical that the tissue sections remain completely covered by liquid throughout all incubations and that the slides are kept completely level. Additionally, the tissue sections should remain completely covered by HybriSlips during long incubations such as hybridization steps and the antibody incubation, to help prevent liquid loss. If partial or complete drying of a tissue section is observed it is unlikely that the samples will yield optimal staining, which will not allow miRNA expression to be interpreted in those samples.
Other steps in the protocol where liquid coverage is essential are during the proteinase K digestion and paraformaldehyde treatment. This digestion is needed to permit access of the miRNA probe into the cells. If the coverage of the tissue is incomplete during this short incubation, the areas that are not exposed to the enzyme digestion will not permit as much probe to enter and bind, and will ultimately not stain as darkly as other as other areas of the tissue. When applying this protocol to other tissue types, the duration of proteinase K digestion may need to be optimized. The subsequent paraformaldehyde fixation is also essential to prevent loss of miRNAs following permeabilization.
The duration of NBT/BCIP development is also an important variable to control. We found that NBT/BCIP incubations of longer than 4 or 5 hr significant background artifact begins to develop in the tissues probed with the scrambled-miR control probes (Figure 2A). Four hours of NBT/BCIP development is sufficient for detection of probe targets expressed in relatively high abundance, such as the U6 small RNA positive control (Figure 2B). Longer incubations do not appear to allow for additional low level expression to be detected, rather this may simply results in increased background artifact.
This leads to the important issue of the detectability of lower expressed miRNAs. Though we have found the 5’ digoxigenin conjugated probes to provide us with sufficient detection sensitivity for our applications in kidney. The alkaline phosphatase based color development allows for multiple signal particles to be measured for each molecule of probe, however some low level miRNA still may not be detectable. Exiqon had developed miRNA probes that are labeled on both the 5’ and 3’ ends allowing for the potential for twice the colorimetric development per microRNA molecule. While our lab has not tested these probes, their use in kidney tissues with a similar protocol has been reported8.
This basic protocol can be adapted to other tissue types and miRNA probes by optimizing proteinase K digestion time, miRNA probe concentrations, hybridization temperatures, and NBT/BCIP development times. It is also possible to use the first portion of the protocol in combination with fluorescent antibodies in tissues where autofluorescence is appropriately low.
Disclosures
There is nothing to disclose.
Acknowledgments
This work was supported by US National Institutes of Health grants HL082798 and HL111580.
References
- Nagalakshmi VK, Ren Q, Pugh MM, Valerius MT, McMahon AP, Yu J. Dicer regulates thedevelopment of nephrogenic and ureteric compartments in the mammalian kidney. Kidney Int. 2011;79(3):317–330. doi: 10.1038/ki.2010.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho J, Pandey P, Schatton T, Sims-Lucas S, Khalid M, Frank MH, Hartwig S, Kreidberg JA. The pro-apoptotic protein Bim is a MicroRNA target in kidney progenitors. J. Am. Soc. Neph. 2011;22(6):1053–1063. doi: 10.1681/ASN.2010080841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Z, Greene AS, Pietrusz JL, Matus IR, Liang M. MicroRNA-target pairs in the rat kidney identified by microRNA microarray, proteomic, and bioinformaticanalysis. Genome Res. 2008;18(3):404–411. doi: 10.1101/gr.6587008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mladinov D, Liu Y, Mattson DL, Liang M. MicroRNAs contribute to the maintenance of cell-type-specific physiological characteristics: miR-192 targets Na+/K+-ATPase β1. Nucleic Acids Res. 2013;41(2):1273–1283. doi: 10.1093/nar/gks1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Taylor NE, Lu L, Usa K, Cowley AW, Ferreri NR, Yeo NC, Liang M. Renal medullary microRNAs in Dahl salt-sensitive rats: miR-29b regulates several collagens and related genes. Hypertension. 2010;55(4):974–982. doi: 10.1161/HYPERTENSIONAHA.109.144428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Zhang J, Wang M, Lanting L, Yuan H, Rossi JJ, Natarajan R. MicroRNA-192 in diabetic kidney glomeruliand its function in TGF-b-induced collagen expression via inhibition of E-box repressors. Proc. Natl. Acad. Sci. U.S.A. 2007;104(4):3432–3437. doi: 10.1073/pnas.0611192104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupa A, Jenkins R, Luo DD, Lewis A, Phillips A, Fraser D. Loss of microRNA-192 promotes fibrogenesis in diabetic nephropathy. J. Am. Soc. Neph. 2010;21(3):438–447. doi: 10.1681/ASN.2009050530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Herman-Edelstein M, Koh P, Burns W, Jandeleit-Dahm K, Watson A, Saleem M, Goodall GJ, Twigg SM, Cooper ME, Kantharidis P. E-cadherin expression is regulated by miR-192/215 by a mechanism that is independent of the profibrotic effects of transforming growth factor-β. Diabetes. 2010;59(7):1794–1802. doi: 10.2337/db09-1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Arce L, Wang M, Putta S, Lanting L, Natarajan R. A microRNA circuit mediates transforming growth factor-β1 autoregulation in renal glomerular mesangial cells. Kidney Int. 2011;80(4):358–368. doi: 10.1038/ki.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottardo F, Liu CG, Ferracin M, Calin GA, Fassan M, Bassi P, Sevignani C, Byrne D, Negrini M, Pagano F, Gomella LG, Croce CM, Baffa R. Micro-RNA profiling in kidney and bladder cancers. Urol. Oncol. 2007;25(5):387–392. doi: 10.1016/j.urolonc.2007.01.019. [DOI] [PubMed] [Google Scholar]
- Juan D, Alexe G, Antes T, Liu H, Madabhushi A, Delisi C, Ganesan S, Bhanot G, Liou LS. Identification of a microRNA panel for clear-cell kidney cancer. Urol. 2010;75(4):835–841. doi: 10.1016/j.urology.2009.10.033. [DOI] [PubMed] [Google Scholar]
- Xu X, Kriegel AJ, Liu Y, Usa K, Mladinov D, Liu H, Fang Y, Ding X, Liang M. Delayed ischemic preconditioning contributes to renal protection by upregulation of miR-21. Kidney Int. 2012;82(11):1167–1175. doi: 10.1038/ki.2012.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegel AJ, Mladinov D, Liang M. Translational study of microRNAs and its application in kidney disease and hypertension. 2012;122(10):439–447. doi: 10.1042/CS20110159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloosterman WP, Wienholds E, de Bruijn E, Kauppinen S, Plasterk RH. In situ detection of miRNAs in animal embryos using LNA-moidified oligonucleotide probes. Nat. Methods. 2006;3(1):27–29. doi: 10.1038/nmeth843. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Baldwin DA, Kloosterman WP, Kauppinen S, Plasterk RH, Mourelatos Z. RAKE and LNA-ISH reveal microRNA expression and localization in archival human brain tissue. RNA. 2006;12(2):187–191. doi: 10.1261/rna.2258506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obernosterer G, Martinez J, Alenius M. Locked nucleic acid-based in situ detection of microRNAs in mouse tissue sections. Nat. Protocols. 2007;2(6):1508–1514. doi: 10.1038/nprot.2007.153. [DOI] [PubMed] [Google Scholar]
- Kriegel AJ, Liu Y, Cohen B, Usa K, Liu Y, Liang M. MiR-382 targeting of kallikrein 5 contributes to renal inner medullary interstitial fibrosis. Physiol. Genomics. 2012;44(4):259–267. doi: 10.1152/physiolgenomics.00173.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]