

Abstract
Background
White matter (WM) injury is common after cardiopulmonary bypass or deep hypothermic circulatory arrest (DHCA) in neonates who have cerebral immaturity secondary to in utero hypoxia. The mechanism remains unknown. We investigated effects of preoperative-hypoxia on DHCA-induced WM-injury using a combined experimental paradigm in rodents.
Methods
Mice were exposed to hypoxia (Pre-Hypoxia). Oxygen-glucose deprivation (OGD) was performed under three temperatures to simulate brain conditions of DHCA including ischemia-reperfusion/reoxygenation under hypothermia.
Results
WM-injury in Pre-Normoxia was identified after 35°C-OGD. In Pre-Hypoxia, injury was displayed in all groups. Among oligodendrocyte stages, the pre-oligodendrocyte was the most susceptible while the oligodendrocyte progenitor was resistant to insult. When effects of Pre-Hypoxia were assessed, injury of mature oligodendrocytes and oligodendrocyte progenitors in Pre-Hypoxia significantly increased compared with Pre-Normoxia, indicating that mature oligodendrocytes and progenitors which had developed under hypoxia had greater vulnerability. Conversely, damage of oligodendrocyte progenitors in Pre-Hypoxia were not identified after 15°C-OGD, suggesting that susceptible oligodendrocytes exposed to hypoxia are protected by deep hypothermia.
Discussion
Developmental alterations due to hypoxia result in an increased WM susceptibility to injury. Promoting WM regeneration by oligodendrocyte progenitors after earlier surgery using deep hypothermia is the most promising approach for successful WM development in CHD patients.
The hospital mortality of severe/complex congenital heart disease (CHD) has been dramatically reduced in the last 2 decades (1). Over this period, however, it has been recognized that neurological deficits frequently occur in CHD patients (2,3). In particular, many clinical studies have now demonstrated a high incidence (25% to 55%) of newly-developed white matter (WM)-injury after cardiac surgery in neonates/infants with severe/complex CHD (4–8). The major brain insults during surgery include cardiopulmonary bypass (CPB) and deep hypothermic circulatory arrest (DHCA) (3,9). DHCA is a unique and specific pathological condition for patients undergoing cardiac surgery, which exposes them to ischemia-reperfusion/reoxygenation under hypothermia. Importantly it has been recently identified that postoperative WM-injury is significantly associated with preoperative WM-injury (7,8). An additional association of postoperative WM-injury is brain immaturity secondary to CHD-induced abnormal cerebral blood flow and reduced oxygen saturation in utero (7,8,10,11). Thus it is possible that management strategies of CPB/DHCA that are optimal for cerebral protection in the normally developing brain are damaging to the brain that has developed in the presence of restricted oxygen supply. Therefore, more complete refinement of cardiac surgery management will be necessary to investigate the cellular effects of CPB/DHCA-induced insults on the developing brain with preoperative WM-injury and WM-immaturity due to preoperative hypoxia.
Large animal models such as piglets have been widely used for the laboratory investigation of CPB/DHCA (12–16). In addition, we recently developed a unique brain slice model in which living brain slices are transferred to a closed circulation system perfused by artificial cerebrospinal fluid (aCSF) to replicate specific brain conditions of CPB/DHCA including ischemia-reperfusion/reoxygenation under hypothermia (17). This experimental paradigm has demonstrated temperature-dependent WM-injury similar to that observed in a large animal DHCA model (17). Reproducing abnormal fetal cerebral blood flow and oxygen saturation observed in CHD patients is still challenging in large laboratory animals. However in rodents there is a well-established model of chronic hypoxia during development that is the equivalent time period to the 3rd trimester in humans (18). Importantly the cellular/molecular mechanisms producing diffuse WM-injury and WM-immaturity caused by hypoxia have been identified in this model (19).
In the present study we performed brain slice experiments after exposing mice to chronic hypoxia to replicate brain conditions in neonates with severe/complex CHD. The combined experimental paradigm in rodents allowed us to investigate the cellular effects of preoperative hypoxia on WM-injury associated with CPB/DHCA. We focused on oligodendrocytes which we previously identified as a likely cellular target for WM protection (17).
Results
Preoperative hypoxia increases developing WM vulnerability to ischemia-reperfusion/reoxygenation injury
To replicate the preoperative-hypoxic state of the fetus with severe/complex CHD, we used a model of chronic sublethal hypoxia, where hypoxic rearing began on post-natal day 3 (P3) and continued until P11 in a hypoxic chamber system (Pre-Hypoxia, Figure 1a). The maturation stage from P3 to P11 in the mouse WM is the equivalent time period to the 3rd trimester in humans (Figure 1a) (20). A recent MRI study has identified that during the third trimester there is a progressive and significant decline in gestational age–adjusted total brain volume in CHD fetuses relative to normal controls (Pre-Normoxia) (11). To investigate whether preoperative hypoxia results in further caspase activated cell death of oligodendrocytes, we compared the number of caspase3+ cells in Pre-Normoxia and Pre-Hypoxia WM at 20hours after OGD-induced ischemia-reperfusion/reoxygenation injury (Figure 1b).
Figure 1.

The combined experimental paradigm using a rodent hypoxic and brain slice model to investigate the cellular effects of preoperative hypoxia on WM oligodendrocyte injury. (a) Study design and equivalent time period for WM development between the mouse and human. (b) Perfusion protocol for oxygen-glucose deprivation under hypothermia. (c) Antibody markers used to immunostain distinct developmental stages of oligodendrocyte lineage cells.
In Pre-Normoxia group, a significant increase in caspase3+ cells was identified after OGD at 35°C (P = 0.02), but not 15°C and 25°C (Figure 2a–d). However, the number in Pre-Hypoxia group was significantly increased in all OGD groups including 15°C-OGD (Figure 2e–h,i). The results are consistent with recent clinical findings where postoperative WM-injury was associated with preoperative WM-injury due to hypoxia in utero (7,8). Before the OGD experiment, there was a significant increase in CNP+caspase3+ cell number in Pre-Hypoxia group compared with those in Pre-Normoxia group (P < .05) (19). Therefore, for direct comparison between pre-Normoxia and Pre-Hypoxia, we used changes of caspase3+ cell number from Control in each group (brain slice experiment without OGD). The changes were significantly different among OGD groups (P < .001) and between Pre-Normoxia and Pre-Hypoxia (p < .001), independently (P < .001). The percentage in Pre-Hypoxia was significantly higher than in Pre-Normoxia in all OGD groups (Figure 2j). These findings indicate that Preoperative hypoxic conditions increase developing WM vulnerability to ischemia-reperfusion/reoxygenation injury.
Figure 2.

Preoperative hypoxia increases developing WM vulnerability to ischemia-reperfusion/reoxygenation injury. (a–h) WM caspse3+ cells in different OGD groups after Pre-Normoxia and Pre-Hypoxia. (i) Caspase3+ cell number after OGD experiments following Pre-Hypoxia (n=6–12 each). (j) Changes of caspase3+ cells from Control after OGD experiments between Pre-Normoxia (white columns) and Pre-Hypoxia (gray columns; n=6–9 each). *P < .01, **P < .001, vs. Control by one-way ANOVA with Bonferroni comparisons. † P ≤ .01, ‡P < .001 by student t-test. Scale bar = 50μm.
The pre-oligodendrocyte is the most susceptible stage within the oligodendrocytes lineage
Our previous study has identified the developing oligodendrocyte lineage as an important cellular target for potential protection of immature WM (17). Based on these earlier findings, we investigated the response of each developmental stage of the oligodendrocyte lineage against OGD-induced insults in both Pre-Normoxia and Pre-Hypoxia groups (Figure 1c). In order to identify the most vulnerable oligodendrocyte stage, double immunostaining was performed using different stage-specific antibodies (Figure 3a,b). CNP+CC1− oligodendrocytes displayed significantly greater susceptibility in Pre-Normoxia and Pre-Hypoxia groups compared with other oligodendrocyte developmental stages (Figure 3c). Since the damage of CNP+PDGFRa+ oligodendrocyte progenitor cells (OPCs) only contributed to approximately 35% of the total cellular damage in CNP+CC1− oligodendrocytes (Figure 3c), the results suggest that the pre-oligodendrocyte is the most susceptible to ischemia-reperfusion/reoxygenation injury among the oligodendrocyte developmental stages. When we compared percent change of CNP+CC1− caspase3+ after OGD-insults, however, there were no significant differences in the change between Pre-Normoxia and Pre-Hypoxia in all OGD groups (Figure 3d–l). This indicates that Pre-Hypoxia did not change cellular responses of the pre-oligodendrocyte, which is the most susceptible stage within oligodendrocyte lineage to ischemia-reperfusion/reoxygenation injury.
Figure 3.

The pre-oligodendrocyte is the most susceptible stage within the oligodendrocytes lineage. (a) CNP+PDGFRa+Caspase3+ OPCs (rectangular box). (b) CNP+CC1−Caspase3+ immature oligodendrocytes (upper rectangular box) and CNP+CC1+Caspase3+ mature oligodendrocytes (lower rectangular box). (c) Caspase3+ cell number after OGD with different oligodendrocyte lineage antibody (n=22–23, each). (d–k) CNP+CC1−Caspase3+ oligodendrocytes after different OGD experiments following Pre-Normoxia and Pre-Hypoxia. (l) OGD-induced change of CNP+CC1−Caspase3+ cell number from Control between Pre-Normoxia (white columns) and Pre-Hypoxia (gray columns, n=5–9 each). *P < .001 by one-way ANOVA with Bonferroni comparisons. Scale bar = 50μm.
Pre-operative hypoxia increases the vulnerability of oligodendrocyte progenitors and mature oligodendrocytes to ischemia-reperfusion/reoxygenation injury
We next assessed the response of CNP+CC1+ mature oligodendrocytes to OGD-induced insults. There were no significant differences in CNP+CC1+caspase3+ apoptotic mature oligodendrocytes number after three OGD conditions in Pre-Normoxia (F = 0.9, P = .47). However, CNP+CC1+caspase3+ cell number after 25°C- and 35°C-OGD following Pre-Hypoxia significantly increased compared with Control (Figure 4a–d,i). In contrast to the response in pre-oligodendrocytes, percentage changes in caspase3+ mature oligodendrocytes number after 25°C- and 35°C-OGD following Pre-Hypoxia significantly increased compared with Pre-Normoxia (Figure 4j). These results indicate that oligodendrocytes that developed under hypoxic conditions had greater vulnerability to ischemia/reperfusion-reoxygenation injury.
Figure 4.

Pre-operative hypoxia increases the vulnerability of OPCs and mature oligodendrocytes to ischemia-reperfusion/reoxygenation; however, susceptible oligodendrocytes exposed to preoperative hypoxia are protected by deep hypothermia. (a–d) CNP+CC1+Caspase3+ oligodendrocytes in different OGD groups after Pre-Hypoxia. (e–h) CNP+PDGFRa+Caspase3+ OPCs in different OGD groups after Pre-Hypoxia. (i) CNP+CC1+Caspase3+ cell number after OGD experiments following Pre-Hypoxia (n=6–12, each). (j) OGD-induced change of CNP+CC1+Caspase3+ cell number from Control between Pre-Normoxia (white columns) and Pre-Hypoxia (gray columns, n=5–9 each). (k) CNP+PDGFRa+Caspase3+ cell number after OGD experiments in Pre-Hypoxia group (n=5–9 each). (l) OGD-induced change of CNP+PDGFRa+Caspase3+ cell number from Control between Pre-Normoxia (white columns) and Pre-Hypoxia (gray columns, n=5–9 each). *P < .05, **P < .001 vs. Control and 15°C OGD by one-way ANOVA with Bonferroni comparisons. §P < .001 by one-way ANOVA with Bonferroni comparisons. †P < .05, ‡P <. 001 by student t-test. Scale bar = 50μm.
Our previous study identified OPCs as the most resistant stage within the oligodendrocyte lineage to ischemia-reperfusion/reoxygenation injury (16). Consistent with the result in mature oligodendrocytes, in Pre-Normoxia group there was no significant difference in caspase3+ OPC number after OGD insults compared with Control (F = 2.6, P = .07). However, the number of caspase3+ OPCs after 25°C- and 35°C-OGD insults following Pre-Hypoxia was significantly increased compared with the number in Control (Figure 4e–h,k). The percent change of caspase3+ OPCs after 25°C- and 35°C-OGD in Pre-Hypoxia significantly increased compared with Pre-Normoxia (Figure 4l), demonstrating that Pre-Hypoxia also results in an increase in the vulnerability of OPC to ischemia-reperfusion/reoxygenation injury.
Susceptible oligodendrocyte progenitors and mature oligodendrocytes exposed to preoperative hypoxia are protected by deep hypothermia
Our results demonstrate that pre-Hypoxia results in increased susceptibility of both OPCs and mature oligodendrocytes. Caspase3+ OPC and mature oligodendrocyte cell numbers increased after 25°C- and 35°C-OGD insults (Figure 4j,l). However, the numbers after 15°C-OGD following Pre-Hypoxia did not increase compared with Control (Figure 4i,k). Between Pre-Normoxia and Pre-Hypoxia, there were no significant differences in percent changes of CNP+CC1+caspase3+ and CNP+PDGFRa+caspase3+ cell numbers after 15°C-OGD (Figure 4j,l). This indicates that although the susceptibility of mature oligodendrocytes and OPCs to damage increased due to Pre-Hypoxia, deep hypothermia is able to protect these oligodendrocyte stages against ischemia-reperfusion/reoxygenation injury.
There was a significant increase in WM caspase3+ cells after 15°C-OGD following Pre-Hypoxia (Figure 2i); however, caspase3+ OPC and immature/mature oligodendrocyte cell numbers did not increase after 15°C-OGD (Figure 3l and 4i,k). To identify the contributions of oligodendrocyte lineages to WM-injury after deep hypothermic OGD, total CNP+caspase3+ cell numbers after OGD following Pre-Hypoxia were assessed. Interestingly, in contrast with significant increases of CNP+caspase3+ cell number after 25°C- and 35°C-OGD, there was no significant difference in the number between Control and 15°C-OGD (Figure 5a–e). The results suggest that damaged cell populations are different between 15°C-OGD and 25/35°C-OGD. Since 15°C-OGD likely replicates cerebral conditions under DHCA, it is possible that other cell populations contribute to WM-injury after DHCA in the brain that has developed under hypoxic conditions.
Figure 5.
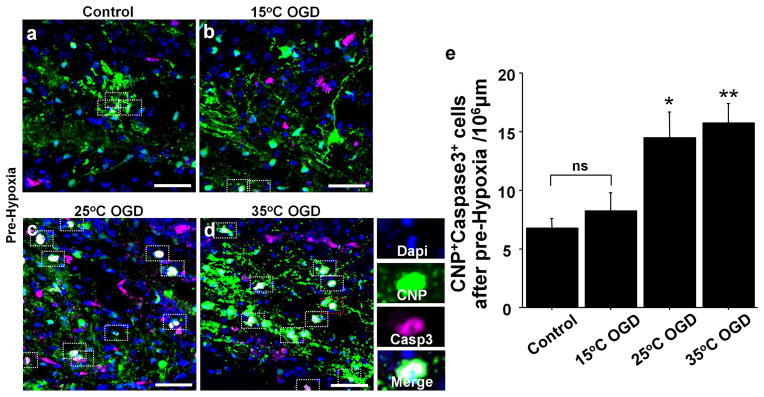
Other cell populations contribute to WM injury after DHCA in the brain that has developed under hypoxic conditions. (a–d) CNP+Caspase3+ oligodendrocytes in different OGD groups after Pre-Hypoxia. (e) CNP+Caspase3+ cell number after OGD experiments following Pre-Hypoxia (n=6–12, each). *P < .05, **P < .01 vs. Control and 15°C OGD by one-way ANOVA with Bonferroni comparisons. Scale bar = 50μm.
Discussion
This study is the first to develop a combined experimental paradigm using rodent hypoxia and brain slice model, identifying WM-injury after hypothermic ischemia-reperfusion/reoxygenation in the brain following preoperative hypoxia. This finding is very similar to that observed in neonates with severe/complex CHD, which suggests that the model enables us to replicate the conditions in CHD patients. Our analysis has identified a uniquely susceptible cellular target of OGD-induced WM-injury within the oligodendrocyte lineage. Our studies have also defined the cellular effects of preoperative hypoxia on each oligodendrocyte developmental stage. Finally we have demonstrated that deep hypothermia is able to protect susceptible oligodendrocyte stages against ischemia-reperfusion/reoxygenation injury.
Due to improved mortality after cardiac surgery for neonates/infants with severe/complex CHD, a current challenge is to reduce long-term sequelae, particularly neurodevelopmental delays (1,3). Recent clinical studies suggest that abnormal cerebral blood flow and oxygen saturation in the CHD fetus cause immature brain development and preoperative WM-injury (5,10,11). Preoperative WM-injury also increases the susceptibility and is associated with newly developed WM-injury after cardiac surgery (7,8). A significant correlation between neurodevelopmental deficits in CHD patients and WM-injury determined by conventional imaging techniques has not been identified (21,22). A recent study demonstrated altered WM microstructure using diffusion tensor imaging in regions without obvious WM abnormality on routine anatomic brain MRI in adolescents with CHD (23). Although further research is required to examine how these alterations in WM microstructure correlate with measured cognitive and behavioral function in children with CHD, deficits of these brain functions have been well documented in this cohort (2). It has been known in premature infants that alterations of the WM microstructure persist into later life (24). Thus, in order to reduce long-term neurodevelopmental delays, it will require an improved understanding of the effects of the pathological state caused by CHD on WM development. The development of innovative image-processing technology is now enabling us to capture and quantify the growth as well as organizational and remodeling processes that occur during brain development in the CHD fetus and neonate (25). These advances in imaging techniques will improve our knowledge of the correlation between alterations of WM development and neurodevelopmental deficits observed in CHD patients.
The cellular/molecular mechanisms producing brain immaturity and preoperative WM-injury in CHD neonates remain largely unexplored. A rodent model of diffuse WM-injury exposing neonatal mice to chronic hypoxia recently demonstrated cellular/molecular alterations in oligodendrocyte development resulting in WM hypomyelination, and including oligodendrocyte death, delayed oligodendrocyte differentiation and abnormal myelination (19). This experimental paradigm is well established to mimic brain injury in premature infants (18,19,26). Importantly, clinical findings of imaging in premature infants such as lower ratios of N-acetylaspartate to choline, higher average diffusivity, and lower WM fractional anisotropy are similar to findings in CHD newborns (5,27) suggesting that cellular/molecular alterations observed in infants with severe/complex CHD are similar to those of premature infants. Using the rodent hypoxia model, promising target molecules have been identified for promoting oligodendrogenesis in WM-injury and for restoring normal uptake of glutamate after injury (19,26). Therefore, further cellular/molecular investigations using a rodent hypoxia model may assist in developing novel therapeutic approaches to improving brain immaturity and reducing preoperative WM-injury in CHD neonates.
For further refinement of the management in pediatric cardiac surgery it will be necessary to investigate cellular effects of CPB/DHCA-induced insults on the brain with preoperative WM-injury and WM-immaturity. In the present study, we used a combined experimental approach with rodent hypoxia and a brain slice model. Our results demonstrate that CNP+CC1− oligodendrocytes exhibit significant vulnerability to OGD-induced ischemia-reperfusion/reoxygenation after both Pre-Normoxia and Pre-Hypoxia, suggesting that the pre-oligodendrocyte should be a target cell type for WM protection. The susceptibility of pre-oligodendrocytes to oxidative stress, hypoxia, and CPB-induced insults has been well recognized in other animal models such as rodents, fetal sheep, or piglets (16,28). The expression level of glutamate receptors on pre-oligodendrocytes is a possible cause of this susceptibility (29); however, the mechanism is not fully understood. An understanding of the cellular/molecular mechanisms of pre-oligodendrocyte susceptibility to damage will lead to improved brain protections during CPB/DHCA. Rodent brain slice models have been used widely for the development of pharmacological therapies for neurological disorders (30). This model will be useful for the study of adjunctive pharmacological approaches to protect pre-oligodendrocytes during cardiac surgery.
Although pre-oligodendrocytes are the most vulnerable stage to damage, there was no change in the susceptibility to OGD-induced insults after prolonged hypoxia. In contrast, we observed that OPCs and mature oligodendrocytes developed under hypoxic condition increased vulnerability to ischemia-reperfusion/reoxygenation injury. It has been demonstrated that arrested oligodendrocyte maturation due to hypoxia conferred enhanced susceptibility to recurrent hypoxia (31); however, there is little information regarding the increased vulnerability of OPCs and mature oligodendrocytes after hypoxia. Our results also suggested that damage of other cell population, but not oligodendrocytes, contributes to WM-injury after deep hypothermic OGD following hypoxia. Compared with preclinical studies using other experimental models, this is an unexpected observation and may represent a unique feature of the brain injury seen in CHD individuals. Further research using this combined experimental paradigm will allow us to identify the specific features of CHD-induced alterations of WM development.
The brain slice model certainly differs from the in vivo practice of CPB/DHCA and is not suitable for investigating the effects of hemodynamic changes in the cerebral circulation or blood-brain barrier disruption (14,15). Compared with brain conditions in neonates/infants during CPB/DHCA, this ex vivo system differs in extracellular volume or diffusion characteristics and constituents of extracellular brain volume. For investigation of these effects, an in vivo model using a large animal such as the piglet is beneficial for laboratory study (12–16). Neurodevelopmental deficits observed in CHD children are complex, multi-etiological, and cumulative (3,6). In contrast to an in vivo model, the brain slice model allows us to study each factor individually and helps in our understanding of complex and multifactorial events on the developing brain, such as our investigation of effects of pre-operative conditions on ischemia-reperfusion/reoxygenation under hypothermia in this study. Future study using both rodent brain slice model and an in vivo large animal model will help our further understanding of cellular/molecular mechanisms underlying neurological deficits related to CHD and subsequent cardiac surgery.
OPCs retain mitotic and differentiation potential throughout their lifespan (32). Recently endogenous WM repair mechanisms, including promotion of remyelination by enhanced signaling systems and functional recovery of WM axons by OPCs migrating from the subventricular zone, have been described in rodent models (32–34). Our previous study has suggested that earlier cardiac repair potentially provides successful WM development because OPC number decreases with age (16). Although OPC vulnerability to damage increased after pre-hypoxia, hypothermia at 15°C protected OPCs from OGD-induced insults in the present study. Abnormal cerebral circulation due to CHD results in significant WM abnormalities by the time of birth (5,7,8,10,11). Therefore, our results suggest that promoting WM regeneration after normalization of cerebral circulation by earlier cardiac surgery using deep hypothermia is the most promising cellular based approach for successful WM development in CHD patients. Our previous results from in vivo animal models demonstrated that temperature of the perfusion interacts with pH and duration to determine the safe management of hypothermic circulatory arrest and selective cerebral perfusion (12,13). Further research will be required as to how these interactions correlate with cellular damage of oligodendrocyte lineages, in particular OPCs and pre-oligodedrocytes, for further refinement of the management of pediatric cardiac surgery.
In conclusion, developmental alterations due to hypoxia result in an increased WM susceptibility to injury. Further understanding of the effects of hypoxia will help develop adjunctive protection during cardiac surgery in the brain with WM immaturity and preoperative injury. Endogenous WM repair mediated by OPCs is a potential approach for successful WM development in CHD patients.
Methods
Animal
We used a 2′,3′-cyclic nucleotide 3′ phosphodiesterase (CNP) - human enhanced green fluorescent protein (EGFP) mouse to identify oligodendrocytes (Figure 1c). In CNP-EGFP mice, various stages of the oligodendrocyte lineage have been visualized based on EGFP expression (35). The effects of WM oligodendrocyte injury were investigated in P11 which have WM maturation equivalent to the human full-term newborn (Figure 1a) (20). We performed all experiments in compliance with the NIH Guide for the Care and Use of Laboratory animals. The study was approved by the Animal Care and Use Committee of the Children’s National Medical Center.
Hypoxic experiment to replicate in utero hypoxia
The hypoxic rearing began on P3 and continued until P11 in a hypoxic chamber system to reproduce the preoperative hypoxic state of the CHD fetus (Pre-Hypoxia, Figure 1a). During the experiment the oxygen concentration was maintained, monitored and recorded continuously with sensors placed inside the chamber to achieve a level of 10.5 ± 0.5 % (19,26). Strain-matched and age-matched mice reared in normal oxygen levels were used to simulate preoperative normoxic condition (Pre-Normoxia).
Brain slice preparation
The brains were collected at P11 and coronal sections, 400um, were cut using a vibratome. Perfusate temperature was then gradually increased up to 36°C as is the practice for electrophysiological studies using murine brain slices (36). The slices were transferred to a customized tissue chamber (CellMicro, Norfork, VA), and were continuously perfused (2ml/min) with aCSF saturated with 95% O2/5% CO2 (PO2, 648 ± 14mmHg; PCO2, 46.2 ± 0.9mmHg; O2 content, 1.94 ± 0.04ml/dl). A heating and cooling device (CellMicro, Norfork, VA) controlled the temperature of the perfusing aCSF. The details of the preparation and methods have been described previously (17).
Oxygen-glucose deprivation
In order to replicate DHCA, slices were exposed to oxygen-glucose deprivation (OGD) as is widely applied for the study of brain ischemia (though in standard brain slice and cell culture models at normothermia) (37). OGD was achieved by changing the perfusion solution from aCSF saturated with 95% O2/5% CO2 to glucose-free aCSF saturated with 95% N2/5% CO2 (PO2, 54.8 ± 2.8 mmHg; PCO2, 48.3 ± 1.2 mmHg; O2 content, 0.16 ± 0.01 ml/dl). The slice was reperfused with glucose-containing aCSF saturated with 95% O2/5% CO2 after the period of OGD, providing ischemia-reperfusion/reoxygenation to the brain slices. The OGD was performed under three hypothermic conditions (Figure 1b) (17).
Perfusion protocol
Each experiment was assigned to one of three different temperature groups for OGD (15°C-, 25°C-, and 35°C-OGD). After the initial recovery period following the slice procedure, brain slices were cooled to a temperature for 30min according to the protocol (Figure 1b). The OGD was then performed for 60min. After the OGD, slices were reperfused with glucose containing aCSF saturated with 95% O2/5% CO2 for 5min, followed by a 30min rewarming period (Figure 1b). This replicates DHCA-induced hypothermic ischemia-reperfusion/reoxygenation. Damage to WM oligodendrocytes in brain slices was assessed at 20hours after rewarming (Figure 1c). Control slices which were perfused at 36°C with aCSF saturated with 95% O2/5% CO2 for the same duration (no OGD) were used for comparison with the OGD groups. A similar perfusion protocol has been utilized in an in vivo CPB model including 60min of circulatory arrest (12–16). The results of the previous study demonstrating temperature-dependent injury after 60min OGD were similar to observations in a porcine DHCA model (12), suggesting that this model is suitable for investigation of brain injury resulting from hypothermic circulatory arrest.
Immunohistochemistry
Twenty-μm sections were obtained from each 400um brain slice. OPCs and mature oligodendrocytes were identified by each marker of the lineage (Figure 1c): antibody to PDGFRa and CC1, respectively. An antibody to cleaved-Caspase3 was utilized to identify apoptosis in the WM (16,17). WM-injury in oligodendrocyte lineage cells was assessed by counting CNP+caspase-3+, CNP+PDGFRa+caspase-3+, and CNP+CC1+caspase-3+ cell numbers (Figure 3a,b). To determine cell density, the antibody positive cells were quantified in 3 microscopic fields from the corpus callosum of each brain slice as previously described (16,17,33).
Statistical analysis
One-way ANOVA with Bonferroni post-hoc comparisons was used to detect differences in the caspase3+ cell number in the experimental groups. Two-way ANOVA with Bonferroni comparisons was used to evaluate changes in the caspase3+ cell number among OGD groups and between Pre-Normoxia and Pre-Hypoxia. Two-group comparisons were performed by unpaired Student t-tests. All data are expressed as mean ± SEM. Reported p-values are two-tailed. Analysis of the data was performed using SPSS version 19.0 (SPSS Inc./IBM, Chicago, IL).
Acknowledgments
We thank Dr. David Zurakowski for assistance with statistical analysis.
Financial Support: This work was supported in part by National Institutes of Health (Bethesda, MD) grants R01HL104173 (R.A.J.), R01NS045702 (V.G.), P30HD040677 (V.G.), P01 NS0626860 (V.G.), and K08NS073793 (J.S.).
Footnotes
Disclosures: None
References
- 1.Kaltman JR, Andropoulos DB, Checchia PA, et al. Report of the pediatric heart network and national heart, lung, and blood institute working group on the perioperative management of congenital heart disease. Circulation. 2010;121:2766–72. doi: 10.1161/CIRCULATIONAHA.109.913129. [DOI] [PubMed] [Google Scholar]
- 2.Bellinger DC, Wypij D, Rivkin MJ, et al. Adolescents with d-transposition of the great arteries corrected with the arterial switch procedure: neuropsychological assessment and structural brain imaging. Circulation. 2011;124:1361–9. doi: 10.1161/CIRCULATIONAHA.111.026963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wernovsky G. Current insights regarding neurological and developmental abnormalities in children and young adults with complex congenital cardiac disease. Cardiol Young. 2006;16 (Suppl 1):92–104. doi: 10.1017/S1047951105002398. [DOI] [PubMed] [Google Scholar]
- 4.Gaynor JW. Periventricular leukomalacia following neonatal and infant cardiac surgery. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2004;7:133–40. doi: 10.1053/j.pcsu.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 5.Miller SP, McQuillen PS, Hamrick S, et al. Abnormal brain development in newborns with congenital heart disease. N Engl J Med. 2007;357:1928–38. doi: 10.1056/NEJMoa067393. [DOI] [PubMed] [Google Scholar]
- 6.McQuillen PS, Miller SP. Congenital heart disease and brain development. Ann N Y Acad Sci. 2010;1184:68–86. doi: 10.1111/j.1749-6632.2009.05116.x. [DOI] [PubMed] [Google Scholar]
- 7.Andropoulos DB, Hunter JV, Nelson DP, et al. Brain immaturity is associated with brain injury before and after neonatal cardiac surgery with high-flow bypass and cerebral oxygenation monitoring. J Thorac Cardiovasc Surg. 2010;139:543–56. doi: 10.1016/j.jtcvs.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beca J, Gunn JK, Coleman L, et al. New white matter brain injury after infant heart surgery is associated with diagnostic group and the use of circulatory arrest. Circulation. 2013;127:971–9. doi: 10.1161/CIRCULATIONAHA.112.001089. [DOI] [PubMed] [Google Scholar]
- 9.Jonas RA. Hypothermia, reduced flow and circulatory arrest. In: Jonas RA, editor. Comprehensive Surgical Management of Congenital Heart Disease. 1. London, England: A Hodder Arnold Publication; 2004. pp. 161–184. [Google Scholar]
- 10.Licht DJ, Shera DM, Clancy RR, et al. Brain maturation is delayed in infants with complex congenital heart defects. J Thorac Cardiovasc Surg. 2009;137:529–36. doi: 10.1016/j.jtcvs.2008.10.025. discussion 36–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Limperopoulos C, Tworetzky W, McElhinney DB, et al. Brain volume and metabolism in fetuses with congenital heart disease: evaluation with quantitative magnetic resonance imaging and spectroscopy. Circulation. 2010;121:26–33. doi: 10.1161/CIRCULATIONAHA.109.865568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakamoto T, Zurakowski D, Duebener LF, et al. Interaction of temperature with hematocrit level and pH determines safe duration of hypothermic circulatory arrest. J Thorac Cardiovasc Surg. 2004;128:220–32. doi: 10.1016/j.jtcvs.2003.11.070. [DOI] [PubMed] [Google Scholar]
- 13.Anttila V, Hagino I, Zurakowski D, et al. Specific bypass conditions determine safe minimum flow rate. Ann Thorac Surg. 2005;80:1460–7. doi: 10.1016/j.athoracsur.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 14.Okamura T, Ishibashi N, Kumar TS, et al. Hypothermic circulatory arrest increases permeability of the blood brain barrier in watershed areas. Ann Thorac Surg. 2010;90:2001–8. doi: 10.1016/j.athoracsur.2010.06.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishibashi N, Iwata Y, Okamura T, Zurakowski D, Lidov HG, Jonas RA. Differential neuronal vulnerability varies according to specific cardiopulmonary bypass insult in a porcine survival model. J Thorac Cardiovasc Surg. 2010;140:1408–15. e3. doi: 10.1016/j.jtcvs.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishibashi N, Scafidi J, Murata A, et al. White matter protection in congenital heart surgery. Circulation. 2012;125:859–71. doi: 10.1161/CIRCULATIONAHA.111.048215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murata A, Agematsu K, Korotcova L, Gallo V, Jonas RA, Ishibashi N. Rodent brain slice model for the study of white matter injury. J Thorac Cardiovasc Surg. 2013 doi: 10.1016/j.jtcvs.2013.02.071. S0022-5223(13)00269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scafidi J, Fagel DM, Ment LR, Vaccarino FM. Modeling premature brain injury and recovery. Int J Dev Neurosci. 2009;27:863–71. doi: 10.1016/j.ijdevneu.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jablonska B, Scafidi J, Aguirre A, et al. Oligodendrocyte regeneration after neonatal hypoxia requires FoxO1-mediated p27(Kip1) expression. J Neurosci. 2012;32:14775–93. doi: 10.1523/JNEUROSCI.2060-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Craig A, Ling Luo N, Beardsley DJ, et al. Quantitative analysis of perinatal rodent oligodendrocyte lineage progression and its correlation with human. Exp Neurol. 2003;181:231–40. doi: 10.1016/s0014-4886(03)00032-3. [DOI] [PubMed] [Google Scholar]
- 21.Andropoulos DB, Easley RB, Brady K, et al. Neurodevelopmental outcomes after regional cerebral perfusion with neuromonitoring for neonatal aortic arch reconstruction. Ann Thorac Surg. 2013;95:648–54. doi: 10.1016/j.athoracsur.2012.04.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soul JS, Robertson RL, Wypij D, et al. Subtle hemorrhagic brain injury is associated with neurodevelopmental impairment in infants with repaired congenital heart disease. J Thorac Cardiovasc Surg. 2009;138:374–81. doi: 10.1016/j.jtcvs.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rivkin MJ, Watson CG, Scoppettuolo LA, et al. Adolescents with d-transposition of the great arteries repaired in early infancy demonstrate reduced white matter microstructure associated with clinical risk factors. J Thorac Cardiovasc Surg. 2013;146:543–9. e1. doi: 10.1016/j.jtcvs.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ment LR, Hirtz D, Huppi PS. Imaging biomarkers of outcome in the developing preterm brain. Lancet Neurol. 2009;8:1042–55. doi: 10.1016/S1474-4422(09)70257-1. [DOI] [PubMed] [Google Scholar]
- 25.Clouchoux C, Limperopoulos C. Novel applications of quantitative MRI for the fetal brain. Pediatr Radiol. 2012;42 (Suppl 1):S24–32. doi: 10.1007/s00247-011-2178-0. [DOI] [PubMed] [Google Scholar]
- 26.Raymond M, Li P, Mangin JM, Huntsman M, Gallo V. Chronic Perinatal Hypoxia Reduces Glutamate-Aspartate Transporter Function in Astrocytes through the Janus Kinase/Signal Transducer and Activator of Transcription Pathway. J Neurosci. 2011;31:17864–71. doi: 10.1523/JNEUROSCI.3179-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller SP, Vigneron DB, Henry RG, et al. Serial quantitative diffusion tensor MRI of the premature brain: development in newborns with and without injury. Journal of magnetic resonance imaging: J Magn Reson Imaging. 2002;16:621–32. doi: 10.1002/jmri.10205. [DOI] [PubMed] [Google Scholar]
- 28.Riddle A, Luo NL, Manese M, et al. Spatial heterogeneity in oligodendrocyte lineage maturation and not cerebral blood flow predicts fetal ovine periventricular white matter injury. J Neurosci. 2006;26:3045–55. doi: 10.1523/JNEUROSCI.5200-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manning SM, Talos DM, Zhou C, et al. NMDA receptor blockade with memantine attenuates white matter injury in a rat model of periventricular leukomalacia. J Neurosci. 2008;28:6670–8. doi: 10.1523/JNEUROSCI.1702-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pena F. Organotypic cultures as tool to test long-term effects of chemicals on the nervous system. Curr Med Chem. 2010;17:987–1001. doi: 10.2174/092986710790820679. [DOI] [PubMed] [Google Scholar]
- 31.Segovia KN, McClure M, Moravec M, et al. Arrested oligodendrocyte lineage maturation in chronic perinatal white matter injury. Ann Neurol. 2008;63:520–30. doi: 10.1002/ana.21359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- 33.Etxeberria A, Mangin JM, Aguirre A, Gallo V. Adult-born SVZ progenitors receive transient synapses during remyelination in corpus callosum. Nat Neurosci. 2010;13:287–9. doi: 10.1038/nn.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gallo V, Armstrong RC. Myelin repair strategies: a cellular view. Curr Opin Neurol. 2008;21:278–83. doi: 10.1097/WCO.0b013e3282fd1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan X, Chittajallu R, Belachew S, Anderson S, McBain CJ, Gallo V. Expression of the green fluorescent protein in the oligodendrocyte lineage: a transgenic mouse for developmental and physiological studies. J Neurosci Res. 2002;70:529–45. doi: 10.1002/jnr.10368. [DOI] [PubMed] [Google Scholar]
- 36.Mangin JM, Li P, Scafidi J, Gallo V. Experience-dependent regulation of NG2 progenitors in the developing barrel cortex. Nat Neurosci. 2012;15:1192–4. doi: 10.1038/nn.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tekkok SB, Goldberg MP. Ampa/kainate receptor activation mediates hypoxic oligodendrocyte death and axonal injury in cerebral white matter. J Neurosci. 2001;21:4237–48. doi: 10.1523/JNEUROSCI.21-12-04237.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]