

Abstract
Parkinson’s disease (PD) is currently recognized as a multisystem disorder affecting several components of the central and peripheral nervous system. This new understanding of PD helps explain the complexity of the patients’ symptoms while challenges researchers to identify new diagnostic and therapeutic strategies. Cardiac neurodegeneration and dysautonomia affect PD patients and are associated with orthostatic hypotension, fatigue, and abnormal control of electrical heart activity. They can seriously impact daily life of PD patients, as these symptoms do not respond to classical anti-parkinsonian medications and can be worsened by them. New diagnostic tools and therapies aiming to prevent cardiac neurodegeneration and dysautonomia are needed. In this manuscript we critically review the relationship between the cardiovascular and nervous system in normal and PD conditions, current animal models of cardiac dysautonomia and the application of molecular imaging methods to visualize cardiac neurodegeneration. Our goal is to highlight current progress in the development of tools to understand cardiac neurodegeneration and dysautonomia and monitor the effects of novel therapies aiming for global neuroprotection.
Keywords: Positron emission tomography, SPECT, 18F-FDA, 11C-MHED, cardiac dysautonomia, sympathetic nervous system, Parkinson’s disease
Introduction
Parkinson’s disease (PD) is classically described as a movement disorder associated to the loss of dopaminergic neurons in the substantia nigra pars compacta [1]. Yet today, PD is recognized as a multisystem disorder affecting several components of the central and peripheral nervous system [2]. This new understanding of PD helps explain the complexity of the patients’ symptoms while challenges researchers to identify new diagnostic and therapeutic strategies.
Abnormal autonomic control of the heart or cardiac dysautonomia [3-6], and cardiac sympathetic neurodegeneration are frequently observed in PD patients, although until recently frequently under diagnosed and mistreated. Presently PD treatments focus on replacing nigral DA loss, which do not help nonmotor symptoms and, in the case of cardiac dysautonomia worsen them [7,8]. Therapies aiming to prevent or slow down central or peripheral neuronal loss are not currently available.
Central and peripheral neurodegeneration in PD has been linked to multiple pathogenic factors including alterations in mitochondrial bioenergetics, calcium homeostasis, toxic catecholaldehyde formation, accumulation of misfolded α-synuclein and other proteins (Lewy bodies) and neuroinflammation [9,10]. Animal models of cardiac dysautonomia present an opportunity to assess those neurodegenerative pathways, identify targets for disease-modifying strategies and develop unbiased methods of evaluation. In vivo imaging techniques are emerging as means to identify and study changes in cardiac innervation, and hold promise as biomarkers for diagnoses, disease progression and therapeutic efficacy.
In this manuscript we critically review the relationship between the cardiovascular and nervous system in normal and PD conditions, current animal models of cardiac dysautonomia and the application of molecular imaging methods to visualize cardiac neurodegeneration. Our goal is to highlight current progress in the development of tools to understand cardiac neurodegeneration and dysautonomia and monitor the effects of novel therapies aiming for global neuroprotection.
The heart-nervous system connection
Optimal cardiac function depends on the integrity of the heart-nervous system connection. The heart is innervated by the autonomic nervous system (ANS), which is part of the peripheral nervous system that involuntarily regulates visceral function. The ANS is classically divided into two components, the sympathetic and parasympathetic system. The sympathetic nervous system is responsible for fight-or-flight responses while the parasympathetic nervous system controls the rest-and-digest activities. These systems function in opposition, but work together to maintain homeostasis. Sympathetic and parasympathetic neural divisions play a major role in the regulation of cardiac function [11].
Parasympathetic innervation of the heart generally has an inhibitory effect, slowing down the heart rate and delaying the transmission of the atrial excitation to the ventricles [12]. The parasympathetic fibers originate in the nucleus ambiguous and the dorsal motor nucleus of the vagus. They reach the heart as the vagus nerve, which exits the medulla oblongata, passing through the jugular foramen and projects to the epicardial neural plexus to unevenly innervate all areas of the heart with dense innervation in the sinoatrial and atrioventricular nodes. Parasympathetic efferent neurons release the neurotransmitter acetylcholine that activates muscarinic receptors in cardiomyocytes and cardiac vasculature [13,14].
Cardiac sympathetic innervation accelerates heart rate and increase ventricular contractility. Fibers of the sympathetic preganglionic neurons travel down the intermediolateral column of the spinal cord from T1-T5 and synapse with neurons in the paravertebral sympathetic ganglia located in lower cervical and upper thoracic regions, referred to as superior, middle and inferior cervical ganglia, thoracic ganglia and stellate ganglion (comprised of the inferior cervical (C7) and first thoracic ganglia) [15]. Axons from the postganglionic neurons targeting the heart descend with the cardiopulmonary splanchnic nerves along the anterior surface of the trachea to pass through the cardiac plexus located in the superior mediastinum. From the cardiac plexus the sympathetic efferents travel to the heart, where they follow the greater coronary arteries and innervate the myocardium of the atria or ventricles either directly or through interneurons of surrounding cardiac ganglia. They release the neurotransmitter norepinephrine (NE) also known as noradrenaline.
Sympathetic cardiac imaging targets different molecules involved in NE metabolism for visualization (Figure 1). NE is synthesized from dopamine by dopamine-β-hydroxylase and packaged into type 2 vesicular monoamine transporters (VMAT2). Once NE is released into the presynaptic cleft it binds and activates α and β receptors in neighboring cardiac tissues, with β1 and β2 adrenergic receptors playing the major role in myocardial sympathetic control. NE can also be reuptaken into the presynaptic terminal by NE transporters (NET), referred to as uptake-1 or by surrounding non-neuronal cells or myocardium and termed uptake-2. The NE is reuptaken into the nerve terminal and repackaged into synaptic vesicles through vesicular monoamine transporters (VMAT2) or enzymatically degraded by monoamine oxidase (MAO) or catechol-O-methyl transferase (COMT) and released into the bloodstream [16].
Figure 1.

Graphic Representation of radioligands cellular target for visualization of cardiac sympathetic innervation. Norepinephrine (NE) is produced from dopamine by dopamine-β-hydroxylase (DβH) in neurosecretory vesicles of catecholaminergic neurons. After NE is released into the synaptic cleft it can be taken up into the sympathetic nerve terminal by norepinephrine transporters (NET), bind α2 or β1 adrenergic receptors (AR), enter cardiomyocytes or escape into the bloodstream. Similar to NE reuptake, radiotracers including 123/125I-MIBG, 18F-FDA, and 11C-MHED can enter the sympathetic terminal through NET (uptake-1). Once in the cardiac sympathetic nerve, NE and 18F-FDA are either metabolized by monoamine oxidase (MAO) into 3,4-dihydroxymandelic acid (DOMA)/metabolites or, like 123/125I-MIBG and MHED, packaged into synaptic vesicles entering through vesicular monoamine transporter 2 (VMAT2). Once the nerve is activated, NE and radiotracers that are not broken down by DβH (such as 18F-FDA) are released by exocytosis into the cleft. Alternatively, the radiotracers 123/125I-MIBG, 18F-FDA and 11C-MHED can be taken up (uptake-2) by non-neuronal cells, such as cardiomyocytes. Within these cells, NE and 18F-FDA can be metabolized by COMT. 99mTc-FBPBAT binds β1AR to activate the post-synaptic myocardial cell.
Cardiac sympathetic receptors are also activated by circulating catecholamines, which are mainly produced by chromaffin cells of the adrenal medulla [17]. These cells are derived from the embryonic neural crest that migrated to create the sympathetic chain [18-20]. They secrete catecholamines directly into the blood stream. The adrenal medulla is innervated by presynaptic sympathetic fibers that travel through the aorticorenal ganglion and directly synapse on the medulla. These fibers release acetylcholine that binds to local nicotinic receptors. Because the medulla is directly innervated by preganglionic fibers it has been referred to as a modified sympathetic ganglion. Generally, catecholamines (dopamine, NE and epinephrine) and their metabolites (dihydroxyphenylglycol (DHPG) and dihydroxyphenylacetic (DOPAC)) are measured in plasma to assess the health of the adrenals. It should be noted that concentration of plasma NE directly relate to cardiac performance.
The relationship between symptoms of cardiac neurodegeneration and dysautonomia in PD
Cardiac sympathetic neurodegeneration refers to loss of catecholaminergic innervation to heart tissues, and has been proposed as an underlying mechanism of abnormal autonomic control of the heart or cardiac dysautonomia [21]. Interestingly, PD patients present with varying levels of cardiac denervation and may not have measureable autonomic abnormalities [22]. This could be related to the sensitivity of the tools used to identify sympathetic loss vs. dysautonomia or to individual thresholds of neurodegeneration needed before symptoms can be detectable. Although cardiac neurodegeneration and dysautonomia in PD are intimately related, different symptoms have been linked to each component. Cardiac neurodegeneration in PD presents with orthostatic hypotension, fatigue, dysrhythmias, shortness of breath during exercise, and reduced time to peak heart rate [3-6]. Other more lethal consequences of cardiac denervation, such as arrhythmias, are not frequent causes of death in PD. Cardiac dysautonomia refers to the symptoms associated not only to cardiac nerve loss but also to the decline in circulating catecholamines. Specifically, cardiac dysautonomia symptoms reported in PD also include OH and fatigue, plus increased corrected QT intervals, reductions in heart rate variability and plasma NE [23,24]. Below we describe the main physiological manifestations of cardiac neurodegeneration and dysautonomia in PD (Table 1).
Table 1.
Differential diagnosis of cardiac dysautonomia symptoms in Parkinson’s disease (PD), multiple system atrophy (MSA), pure autonomic failure (PAF) and diabetes. Frequency and severity of cardiac dysautonomia symptoms vary by disorder
| Feature | PD | MSA | PAF | Diabetes |
|---|---|---|---|---|
| Orthostatic hypotension | x | x | x | x |
| Fatigue | x | x | x | x |
| Increased QT interval | x | x | x | x |
| Abnormal Valsalva Maneuver | x | x | x | x |
| Circulating catecholamines do not double following supine position | x | x | x | x |
| Basal low circulating catecholamines | x | - | x | - |
| Syncope | x | x | x | - |
| Denervation supersensitivity | x | - | x | x |
| Decreased HRV | x | x | x | x |
| Increased resting HR | - | - | - | x |
| Unresponsive HR to exercise, sleep (severe denervation) | - | - | - | x |
| Postganglionic sympathetic neurodegeneration | x | - | x | x |
| Preganglionic sympathetic neurodegeneration | - | x | - | - |
HR, heart rate; HRV, heart rate variability.
Orthostatic hypotension (OH) is defined as a drop in systolic and diastolic blood pressure of more than 20 and 10 mm Hg, respectively, after a 3-5 minute postural challenge from a supine position [25,26]. OH has been reported to occur in up to 58% of PD patients [27,28], though up to 38.5% were found asymptomatic [29]. In general, postural challenge of this nature can lead to symptoms as subtle as fatigue, weakness, impaired cognitive performance, neck and back pain or as disabling as dizziness, visual disturbances and syncope [30] which can increase risk of falls and greatly impair quality of life. Virtually all PD patients with OH present cardiac sympathetic denervation, yet of the remaining PD patients without OH, 50% also have cardiac denervation [31], suggesting that patients become symptomatic after surpassing a cardiac neurodegeneration threshold. Daily activities such as eating and exercise can exacerbate symptoms and some medications, including insulin, antihypertensives, tricyclic antidepressants and antiparkinsonian agents, can influence the prevalence or severity of OH [32]. OH can also be found due to non-neurogenic causes including low intravascular volume, reduced cardiac output, drug use, excessive vasodilatation, and cardiac impairment such as myocarditis [30,33,34].
Fatigue is present in over 40% of PD patients [35-37] and its presence correlates with cardiac dysautonomia and sympathetic denervation [38]. Central fatigue is found in many neurological disorders and encompasses both physical and mental features [39]. In a recent study [38], fatigued compared to non-fatigued PD patients (defined as a mean score higher than 3.3 on the Parkinson’s Fatigue Scale [40]), showed significantly more prevalence of cardiac denervation as detected by the sympathetic radiotracer 123I-metaiodobenzylguanidine and a trend of increased OH presence. Additionally, fatigued PD patients had significantly higher pressor responses to infusions of NE and dobutamine, a selective β1 stimulant, further confirmingthe loss of presynaptic sympathetic nerves and denervation supersensitivity [38]. Other studies have found a measureable relationship between fatigue and cardiac functioning as evaluated by stroke index and cardiac output index [41].
PD patients experience dysregulation in the electrical activity of the heart that can put them at risk to develop cardiac dysrhythmias. Prolongation in the corrected QT (QTc) interval, which describes ventricular depolarization and repolarization corrected for heart rate, predicts cardiovascular mortality and has been reported in PD [42,43]. It is not clear how this measurement is a predictor of arrhythmias in PD, and further longitudinal studies must be completed to understand the correlation with cardiovascular causes of death [44]. Similar to other physiological cardiac autonomic measurements, QTc correlates with Hoehn & Yahr (H&Y) PD staging and duration of disease [42]. Furthermore, anti-parkinsonian drugs did not have adverse effects on QT intervals [42,43], unlike other classes of drugs such as antiemetics, antidepressants and antibiotics [45].
Dysfunction in the electrical activity of the heart can also lead to changes in cardiac rhythmicity that is regulated by both sympathetic and parasympathetic mechanisms, referred to as cardiovagal function. This balance can be evaluated by the R-R variation or heart rate variability (HRV), and although there are many methods to analyze HRV it is most widely measured with power spectral analysis for time and frequency domains. PD has been associated with a decrease in HRV that worsens with disease severity and further supports the involvement of the peripheral sympathetic system in PD [46-49]. Importantly, diminished HRV may have life threatening consequences; in myocardial infarction it is considered a strong predictor of mortality [50,51]. Because of the association between HRV and cardiac denervation, HRV has been a suggestive measure of re-innervation [52-55].
Changes in cardiovagal function in PD can also be evaluated during the “Valsalva maneuver” in which the relationship between the interbeat interval and systolic blood pressure are analyzed after forceful exhalation against a closed airway. In PD, sympathetic cardiovascular responses are hampered following a decrease in stroke volume compared to controls, described by a drop in blood pressure during continued strain (phase II) and no overshoot after normal breathing (Phase IV) [4,23,56].
Decreased concentration of circulating plasma NE is another measure used to detect dysautonomia in PD, yet caution when analyzing the data is needed, as the patient’s condition during sample collection may affect the results. Multiple PD studies have confirmed that the level of plasma NE depends on the patient’s orthostatic response. At rest, reduced plasma NE has been reported in PD patients with OH compared to PD patients without OH [25,57,58]. However, other studies have shown no significant difference in NE concentrations at rest in PD with OH or even an increase in PD patients without OH compared to age-matched controls [25]. Normal levels of adrenaline and its metabolite metanephrine have been reported in PD patients at rest with OH and low NE, suggesting variability in adrenomedullary function [59]. Reduced plasma NE in PD patients has been linked to changes in the adrenal medulla, including decreased adrenal catecholamines and TH [60-62] and presence of Lewy body pathology [63].
Medications can affect the levels of plasma NE and should be controlled to accurately detect catecholamine production. For example, at rest PD patients with OH off-levodopa have been reported to have levels of plasma NE and DHPG lower than on-levodopa, while the latter had similar levels to healthy controls [64]. It should be noted that an earlier study from the same group found significantly decreased basal NE levels in PD with OH than without when excluding patients with high levels of levodopa [57].
Because of the variability found in plasma catecholamines at rest researchers have adopted to orthostatic increment measurements where patients stand for at least 5 minutes after being in a supine position for 15 minutes for blood sampling. Following the postural challenge, plasma NE increased more than 60% in 11 of the 18 PD patients lacking OH compared to 100% in controls, while PD with OH did not have similar increases to either group [57]. Controlling for antiparkinsonian treatments (e.g.: request off-levodopa overnight) is recommended for precise evaluations [64]).
Modeling cardiac sympathetic neurodegeneration and dysautonomia
Animal models of cardiac sympathetic neurodegeneration and dysautonomia are essential to study pathogenesis, validate therapeutic targets and assess novel imaging tools. Today there are several animal models available that present different advantages and disadvantages for preclinical evaluation (Table 2).
Table 2.
Animal models of cardiac sympathetic denervation
| Model | Species | Advantages | Disadvantages |
|---|---|---|---|
| Surgical Sympathectomy | rat | Immediate effect | Invasive procedure |
| dog | Functional effects of sympathetic neurodegeneration | Denervation is not specific | |
| sheep | Spontaneous recovery | ||
| Streptozotocine (STZ) | mouse | Cardiac nerve damage confirmed by IHC | Model of hyperglycemia that produces other peripheral effects |
| rat | Functional effects of sympathetic neurodegeneration | Time course of cardiac NE loss and progression is variable | |
| monkey | |||
| pig | |||
| dog | |||
| Reserpine | mouse | Reversible effects | NE cell loss is questionable |
| rat | Repetitive doses has irreversible effect | Produce other peripheral and central effects | |
| dog | |||
| rabbit | |||
| MPTP | mouse | Potential to cause both central and peripheral catecholamine effects | High doses required for denervation |
| rat | Spontaneous recovery | ||
| monkey | Safety precautions needed for handling toxin and animals | ||
| 6-OHDA | mouse | Sympathetic neurodegeneration confirmed by IHC | Intensive monitoring and care required during toxin delivery |
| rat | Decreases plasma catecholamines | ||
| rabbit | |||
| cat | |||
| dog | |||
| monkey | |||
| Genetic (Thy1α-Syn) | mouse | HRV and baroreflex abnormalities | Cardiac sympathetic innervation not yet evaluated |
| Central and cardiac α-synuclein accumulations |
NE, norepinephrine; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; 6-OHDA, 6-hydroxydopamine; IHC, immunohistochemistry; HRV, heart rate variability.
Surgical sympathectomy
Surgical sympathectomy models are created by total resection of unilateral or bilateral sympathetic stellate or upper thoracic ganglia, which produces partial cardiac denervation [65]. This procedure has been used clinically to manage congenital long QT syndrome [66,67], primary hyperhidrosis [68], and reduce ventricular tachyarrhythmias in patients with myocardial infarcts and ischemic or nonischemic cardiomyopathy [69,70]. Surgical sympathectomy has been tested in animals including rats [71,72], dogs [73,74] and sheep [75] with different degrees of success. While surgical sympathectomy has the advantage of producing an immediate effect, it can be technically challenging, as it requires extensive and invasive surgery. In addition, the regional innervation of the left ventricle by the right and left stellate ganglion is still unclear, and consequently the exact dispersion of denervation from a stellectomy is difficult to interpret. Another disadvantage is that spontaneous recovery is frequently observed after the procedure [76]. Functionally, electrocardiographic alterations were seen in dogs following unilateral stellate ganglionectomy including an increase in QT interval and prolonged refractory period [77]; and in humans, decreases in ejection fraction and resting and peak heart rates [78].
Streptozotocine (STZ)
STZ is a glucosamine-nitrosourea compound that is used to model type I diabetes mellitus and its consequences, including metabolic cardiac nerve damage [79,80]. STZ reaches into pancreatic β cells by glucose transport protein GLUT2, inducing cell death by DNA fragmentation, which can be perpetuated by STZ-induced reactive oxygen species and nitric oxide [81]. Its administration induces a diabetic-like state characterized by hyperglycemia, impaired glucose tolerance [82] and loss of β cell sensitivity. STZ concentrations, route of administration and exposure time vary depending on species (reviewed in [80]). Briefly, low dose intraperitoneal injections of STZ are more commonly used in rodents while intravenous administration is the preferred route for larger animals.
Cardiac effects of STZ (reviewed by [83]) have been observed in mice [84,85], rats [86-90], monkeys [91], pigs [92] and dogs [93]. A number of studies have investigated the NE concentration in the heart [90,94-96] and found significantly lower NE and β-adrenergic receptor levels with variation of loss across time between different STZ models. Yet, sympathetic ventricular denervation was confirmed by immunohistochemical staining where tyrosine hydroxylase was reduced in mice 6 months following STZ [97] and in rats 20 weeks after STZ [98]. STZ has produced functional effects such as decreased heart rate and HRV in rodents [96,99-103]. Few STZ studies have shown an increase in heart rate [97,104], which better matches the clinical presentation of tachycardia found in diabetic patients [105], but clashes with that found in denervated patients such as in PD. Together, this evidence suggests that the sympathetic activity of the heart can be affected in STZ models, and further characterization is needed to understand the extent of morphological and functional loss.
Other diabetic models with noradrenergic cardioneuropathy (reviewed by [83]) documented by molecular imaging are GK/Crj rats (a non-insulin dependent form; [106]), alloxan-treated rabbits [98] and BioBreeding rats [107], yet a discussion about these models is beyond the scope of this review.
Reserpine
Reserpine is an alkaloid used in the past as an antipsychotic and antihypertensive treatment [108]. It irreversibly blocks active and passive NE uptake into storage vesicles by blocking VMAT2, thereby depleting the vesicular NE storage [109,110]. Catecholamine depletion from a single dose of reserpine is temporary until VMAT2 can be replenished [111], but its repeated administration has a cumulative effect of profound and irreversible catecholamine loss [108].
The cardiac effects of reserpine have been tested in mice [112], rats [113], dogs [114] and rabbits [115]. In rats (4 mg/kg ip) a decline in endogenous catecholamines was found by 1 hour and a highest depletion at 4 hours, gradually increasing to baseline levels. In the heart, NE recovery was not seen until 15 days post reserpine dosing (4.5 mg/kg x 2). Directly after a single dose of reserpine, heart rate increases due to the release of NE from heart tissues, while after two daily doses (day 3 and 2 before analysis) increases in heart rate are not observed [114].
Reserpine also exerts its catecholamine depleting effects in the paraganglia [116], adrenal medulla [117-119], brain [120-122] and liver with the extent of loss and recovery subject to the dose, route of administration and blood flow to the organ [117]. The overall effect and time-course of recovery varies among tissues. For example, in order to achieve 25% reduction of catecholamines in the submaxillary gland, a dose had to be 2-10 times greater than the dose eliciting 25% loss in the heart. In a heart with 80-100% loss, there was 50% cardiac recovery after 7-15 days, while the superior cervical ganglia recovered almost completely after 7 days [123]. Reserpine’s side effects include sleepiness, diarrhea, impaired temperature regulation and hypersecretion of adrenocorticotropic hormone [108].
PD genetic models
PD patients with genetic mutations such as leucine-rich repeat kinase 2 (LRRK2) R1441G, T2356I and G2019S or triplication of the α-synuclein gene have cardiac neurodegeneration and dysautonomia [31,124-127]. Rodent models based on genetic mutations linked to PD have been developed [128], yet most of the research has focused on their central dopaminergic cell neurodegeneration and subsequent motor disturbances. Exceptions are mice that overexpress wild-type α-synuclein under the Thy-1 promoter (Thy1α-Syn or Thy1-ASO). These mice have proteinase-K resistant accumulations of α-synuclein in ventricular and atrial walls [129], and increased HRV [130] and impaired baroreflex at an early age [131]. Molecular imaging studies for cardiac innervation have not been performed in these models; therefore, it is not clear if these mice exhibit cardiac neurodegeneration.
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)
MPTP is a lipophilic neurotoxin, widely used to model PD nigral dopaminergic cell loss in mice [132-134] and monkeys [135,136]. MPTP central effects are due to its capacity to cross the blood brain barrier, but it also has peripheral effects including loss of cardiac sympathetic innervation [6,137-140].
MPTP active form, MPP+, is produced by monoamine oxidase (MAO) present in brain astrocytes and liver cells. MPP+ is preferentially taken up by nigral neurons through the dopamine transporter (DAT). Peripherally, it is thought that MPP+ selectively enters postganglionic sympathetic nerves through monoamine transporters [140,141]. In either case, MPP+ concentrates in mitochondria where it inhibits complex I, leading to catecholamine neuron degeneration by affecting energy production and increasing oxidative stress [142-144]. Although, MAO-B inhibitors prevent nigral dopaminergic cell loss, they do not protect against cardiac NE loss, suggesting that MPTP does not need to be converted to MPP+ for cardiac effects and that other factors may be involved in MPTP toxicity in the heart [145]. Peripherally, MPTP toxicity has selective effects in for catecholamine neurons in the heart and gut [146,147], but spares the loss of chromaffin cells of the adrenal medulla, although catecholamine production is altered [148].
MPTP-induced cardiac sympathetic nerve loss has been demonstrated in mice [6,140,149,150], rats [151], and monkeys [152]. Doses and regimens of intoxication vary between species to produce peripheral cardiac damage, yet the effects seem to be short term. Mice administered a single MPTP dose of 32 mg/kg ip had significantly decreased cardiac NE levels 24-48 hours after dosing, but returned to normal levels within 4-7 days [153]. In another study, rats were administered a daily dose of MPTP or MPP+ (20 mg/kg ip x 5 days) to produce 93.5-100% loss of cardiac NE compared to controls, and 10 days after the final MPTP/MPP+ dose, the cardiac NE levels had recovered to 40% loss [151]. Furthermore, immunohistochemical analysis 1 week after the administration of MPTP (30 mg/kg ip x 2) to mice revealed well preserved cardiac NE innervation, yet HPLC analysis resulted in decreased NE and dopamine heart content [154], suggesting functional cardiac sympathetic loss but not actual denervation. In monkeys systemic MPTP temporarily induced cardiac nerve loss as well as lower levels of circulating catecholamines [152].
6-Hydroxydopamine (6-OHDA)
6-OHDA is a neurotoxin widely used to model PD dopaminergic nigral cell loss in rodents by direct intracerebral delivery, as 6-OHDA does not cross the blood brain barrier [155]. 6-OHDA is also used to model cardiac sympathetic neurodegeneration by systemic dosing. Either in the central or peripheral nervous system, 6-OHDA enters the catecholaminergic neurons through monoamine transporters and induces sympathetic neuronal loss by increasing the production of reactive oxygen species (ROS), and disrupting energy metabolism and neuronal membranes [156]. It should be noted that a decrease in nocturnal heart rate has been described in rats given a unilateral injection of 6-OHDA into the medial forebrain bundle [157], but no other cardiac effects have been described after intracerebral 6-OHDA admini-stration.
The effects of systemic 6-OHDA in sympathetic cardiac denervation have been evaluated in mice [158,159], rats [160,161], rabbits [162,163], cats [164,165], dogs [76,166,167] and monkeys [152,168]. Dose and route of delivery of the neurotoxin vary between species. In general, 6-OHDA administration produces a strong sympathomimetic effect, as a result the method of preference is fractionated doses delivered slowly over a long period of time (hours) until an effective total dose is given. The 6-OHDA displacement of NE from its binding sites in sympathetic nerve terminals leads to an acute response including a marked rise in blood pressure, piloerection, mydriasis, and profuse salivation [76,160]. Tachyphylaxis of the sympathetic response occurs during 6-OHDA dosing. Blood pressure increases are much less pronounced after a partial dose of 6-OHDA (approximately 10 mg/kg in dogs and monkeys), since the sympathetic nerve terminals are destroyed and no longer able to release NE. Within days following 6-OHDA, non-cardiovascular signs such as miosis, and diarrhea have been reported [76]. A cumulative dose of 50 mg/kg intravenously to dogs reduced NE cardiac apical content by 97% and basal content by 90% 5 days later, yet NE content increased in the stellate ganglia. Other studies have shown limited effect on the ganglia suggesting that 6-OHDA destroys the terminals of sympathetic postganglionic neurons without affecting the cell bodies [169,170]. In rhesus monkeys 6-OHDA produces cardiac nerve loss that persists for up to 3 months and was associated with a decrease in circulating catecholamines compared to baseline [168].
In vivo visualization of cardiac sympathetic neurodegeneration in PD
The association between cardiac dysautonomia and sympathetic denervation has been established by correlating cardiac autonomic symptoms with cardiac sympathetic imaging. Both in animal models and patients, different molecular radiotracers have been designed to evaluate sympathetic functional integrity. Cardiac denervation, as determined by a reduction in radioligand uptake, has been confirmed postmortem in PD by markedly reduced immunohistochemical staining of tyrosine hydroxylase, neurofilament and S-100 protein in epicardial tissue [3,5,171]. The principal radioligands for the assessment of cardiac sympathetic innervation are 123/125I-Meta-iodobenzyl-guanidine (123/125I-MIBG), 99m-4-fluorobenzyl-4-(2-mercapto-2-methyl-4-aza-pentyl)-4-(2-mercapto-2-methylpropylamino)-piperidine[fluorobenzylpiperidinyl-bis-(aminoethanethiol) (99mTc-FBPBAT), 6-18F-fluorodopamine (18F-FDA), and 11C-meta-hydroxyephedrine (11C-MHED; Figure 2).
Figure 2.

Chemical structures of norepinephrine and related radiolabeled tracers for the imaging of cardiac catecholaminergic innervation. The asterisk (*) labels the position of the radioisotope.
Uptake of these tracers correlate with the density of sympathetic innervation assuming sufficient myocardial blood flow, which can be assessed using the radiotracers 201thallium or technetium-99m-MIBI (99mTc-MIBI) with SPECT (Single Photon Emission Computed Tomography) imaging or 13N-labelled ammonia (13N-NH3) with PET (Positron Emission Tomography) imaging. In this section we will briefly review the imaging technologies and radiotracers used to evaluate cardiac sympathetic innervation and the imaging results from both clinical and animal models of cardiac neurodegeneration with particular attention to identify a pattern of loss and how those results correspond to other dysautonomia symptoms.
SPECT vs. PET
While 123I-MIBG and 99mTc-FBPBAT are detected using SPECT technology, 18F-FDA and 11C-MHED radiotracers require detection by PET. Each imaging technique presents advantages and disadvantages for cardiac neuroimaging.
SPECT imaging uses a rotating gamma camera to directly detect gamma rays emitted from a radioisotope injected into the patient. This technique collects multiple 2D images that are reconstructed into a 3D structure. The compiled SPECT images have limited spatial resolution of 15 mm and increased occurrence of artifacts and noise. However spatial resolution has been improved with technological advances such as pinhole collimators [172]. The higher half-lives of SPECT agents allow patients to be observed for longer periods of time, lending a potential advantage to SPECT imaging. However, their half-lives also limit the number of follow-up studies that can be performed in the same day. SPECT analysis is traditionally regarded as non-quantitative, but recent developments are combining the technology with computed tomography to achieve quantitative image reconstruction [173]. Although SPECT cardiac imaging is more often used to evaluate myocardial perfusion and function for clinical management, it can also be used to detect cardiac neuronal architecture.
PET imaging differs from SPECT as it requires radionuclides that decay with positron emission creating two opposing photons that are coincidentally detected and processed to form an image corresponding to the concentration of radionuclide. PET radioisotopes generally have much lower half-lives than SPECT isotopes, and therefore must be synthesized near the PET scan facility. This can limit the use of PET scans since positron-emitting radionuclide production requires a cyclotron, which may not be easily available. To their advantage, PET isotopes result in lower radiation exposure to the patient and higher sensitivity, and do not limit the number of repeat tracer injections as SPECT. Spatial image resolution in clinical PET is as low as 3 mm full width at half maximum compared to 15 mm in SPECT for whole body scanning. Furthermore, attenuation correction is less challenging in PET analysis than in SPECT, minimizing artifacts [174]. Small animal PET scanners (microPET or Inveon) have very similar features as the clinical technologies, with the exception of improved detection efficiency to increase spatial resolution to 1-3 mm full width at half maximum [175].
123/125I-MIBG
MIBG is used to visualize cardiac sympathetic innervation in vivo and postmortem. MIBG acts as a false neurotransmitter (guanethidine) and is usually labeled with either 123I for in vivo or 125I for ex vivo evaluation. It is taken up into presynaptic terminals through NET by uptake-1 mechanisms with 50% selectivity relative to uptake-2 into non-neuronal cells [176] (Figure 1). It is then distributed within the neuronal cytoplasm and stored in neurosecretory vesicles [177]. As 123/125I-MIBG is also taken up by non-neuronal cells through uptake-2 mechanisms, it has been argued that it does not provide a true measurement of neuronal integrity, although cardiac transplant patients, which lack sympathetic innervation, have no cardiac 123/125I-MIBG accumulation [178]. Unlike NE and other catecholamines, 123/125I-MIBG cannot be catabolized by MAO or COMT, which facilitates its effective visual detection. 123I-MIBG is easily purchased from a manufacturer. It is one of the most widely used clinical sympathoneuronal radiotracer.
123/125I-MIBG is not positron-emitting therefore it is used as a SPECT radiotracer, although efforts are being made to develop a positron-emitting analogue. Heterogeneous 123I-MIBG uptake has been reported in the inferior wall of the left ventricle of normal subjects associated to physiological differences [179]. Further research must be completed to understand if the variation in the inferior wall is due to true physiological changes or due to attenuation or artifact correction methods that result in inaccurate assessments of regional distribution of sympathetic innervation [180]. 123I-MIBG is commonly analyzed by uptake and washout parameters, requiring early (15-20 minute) and delayed (2-4 hours) imaging. The routine semi-quantitative assessments for 123I-MIBG are the heart to mediastinum (H/M) ratio or washout rates. Both methods largely provide a global measure of sympathetic innervation and function; washout rates additionally provide information of catecholamine turnover.
Clinical application of 123I-MIBG
MIBG scintigraphy is used to aid in the differential diagnosis of PD, as H/M measurements are reduced in PD compared to multiple systems atrophy (MSA), progressive supranuclear palsy (PSP), vascular parkinsonism, essential tremor, and control patients (Table 3) [6,181-184]. A recent meta-analysis report on 123I-MIBG scintigraphy determined that the delayed H/M had 89% sensitivity to detect PD and 77% specificity to differentiate PD from MSA [185] and 91.4% sensitivity and 78% specificity when discriminating between PD and PSP [186]. Yet, the relationship between cardiac 123I-MIBG and PD severity and duration is still not clear. Some studies have found that H/M measurements negatively correlate with PD stage, yet up to 55% of early stage PD patients already exhibit low 123I-MIBG measurements [6,183]. Others have not found a clear association between H/M and PD stage or disease duration [184]. When evaluating central and cardiac cell loss, 123I-MIBG H/M from PD at all H&Y stages positively correlated with central dopamine loss in the striatum as detected by striatal uptake of N-(3-fluoropropyl)-2β-carbomethoxy-3β-(4-[(123)I]iodophenyl) nortropane (123I-FP-CIT), a SPECT radiotracer taken up by DAT and therefore visualizing the density of dopaminergic neurons [187]. 123I-MIBG only correlated with 123I-FP-CIT uptake in the putamen on the most effected side but no other striatal nuclei, with 123I-MIBG accumulation reduced 14% [188]. These studies suggest that the effects on the central and peripheral systems are not uniform.
Table 3.
Comparison of clinical reports evaluating cardiac innervation in PD using 123I-MIBG
| Reference | Number of Patients | Mean PD age (age range) | PD H&Y stage (disease duration yrs) | Methods to evaluate cardiac DYA | PD patient DYA description | Homogenous cardiac perfusion Y/N (radiotracer) | LV regions analyzed or uptake measurement | % of LV global uptake reduction | Region of maximal reduction (% of loss) | Significant loss of plasma catechol-amines Y/N | Comments | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||||||
| PD | MSA | PSP | Control | |||||||||||
| 123I-MIBG | ||||||||||||||
| Braune et al., 1998 | 10 | - | - | 8 | No mean defined (65-83) | No H&Y defined, 3-15 on Webster scale (<1-20) | Head tilt, OH evaluation after standing, plasma NE and Epi, valsalva | n=6 of 10 AF n=4 of 10 asymptom-atic | Y ([99mTc]MIBI) | H/M ratio | 47.5%b | - | N (NE reduced in 2 of 6 PD with AF at rest; majority have abnormal NE response after standing) | No correlation between DYA tests and plasma catecholamines or global MIBG uptake. |
| Orimo et al., 1999 | 46 | 7 | - | 10 | No mean defined (41-84) | H&Y 1, n=8; H&Y 2, n=3; H&Y 3, n=2; H&Y 4, n=9; H&Y 5, n=4 (disease duration not defined) | Head tilt, ECG (coefficient of variation RR interval) | n=13 of 46 OH with H&Y <3 | Y (201thallium) | H/M ratio | 80-84%a | - | - | H/M decreased with H&Y stage progression. Duration of PD correlated with early H/M, but not with delayed. PD with OH had lower mean H/M than other PD. |
| Takatsu et al., 2000 | 32 | 5 | - | 7 | 67 (No range defined) | H&Y 1, n=2; H&Y 2, n=9; H&Y 3, n=14; H&Y 4, n=6; H&Y 5, n=1 (disease duration not defined) | OH evaluation after standing 24 hr, ambulatory and nocturnal BP monitoring | n=21 of 26 PD evaluated for DYA have OH and abnormal nocturnal BP | - | H/M ratio | 34.7-48.8%b | - | - | Global MIBG normal in n=1 of 32 PD patients. Marked reduction found in H&Y I and 2, but significant reduction at H&Y 3-5. MIBG low in PD with or without OH/abnormal BP. |
| Taki et al., 2000 | 41 | 9 | 6 | 10 | 63 (No range defined) | H&Y 1, n=11; H&Y 2, n=15; H&Y 3, n=10; H&Y 4, n=2 (disease duration not defined) | No additional cardiac DYA evaluation | n=0 of 41 AF, n=3 of 41 DLB | Y ([99mTc]MIBI and 201thallium) | H/M ratio | 28.1-38%b | - | - | Global MIBG early and delayed H/M ratios significantly lower in PD than controls, MSA, and PSP. No significant differences in H/M between PD stages. |
| Courbon et al., 2003 | 18 | 10 | - | - | No mean defined (36-79) | H&Y 1, n=2; H&Y 2, n=11; H&Y 3, n=5 (3.6 non-DYA PD; 9.3 DYA PD) | OH evaluation after standing, ECG (BP and HR for RR interval), valsalva | n=10 of 18 PD with DYA and H&Y <2, n=8 of 18 PD non-DYA H&Y >3 | Y ([99mTc]MIBI) | H/M ratio. Anterior, inferior, lateral, septal walls and apex uptake assessed with a 3 pt color scale between PD non-DYA and MSA. | 27.4%b,E (non-DYA PD); 50.8%b,E (DYA PD) | Apex (85%c) in non-DYA; Inferior (70%c) | - | H/M of PD with DYA significantly lower than non-DYA. |
| Orimo et al., 2005 | 5 (Parkin n=2) | - | - | 3 | Parkin: 70 (66-73); non-Parkin: 81 (70-91) | No stage defined (Parkin: 38-49; non-Parkin: 10-15) | No additional cardiac DYA evaluation | - | - | H/M ratio | 3.1-13.3%b | - | - | Parkin have normal TH positive epicardial nerve fascicles. |
| Spiegel et al., 2007 | 102 | - | - | - | 57-63 (37-78) | H&Y 1, n=57; H&Y 2, n=22; H&Y 3 and 4, n=23 (disease duration not defined) | OH evaluation after standing (evaluated in n=19 H&Y 1 and n=22 H&Y 2-4) | n=6 of 19 H&Y 1 and n=10 of 22 H&Y 2-4 with OH | Y ([99mTc]MIBI) | H/M ratio | Overall PD, 28.6%b; H&Y 1, 25.5%b; H&Y 2, 30.5%b; H&Y 3, 35.5%b | - | - | PD with OH have reduced H/M compared to PD without OH (H&Y 1, 26.4%b and H&Y 2-4 18.9%b) |
| Ruiz-Martinez et al., 2011 | 90 (LRRK2 G2019S n=4 or R1441G n=23; LRRK2 non-carrier n=63) | - | - | - | LRRK2: 70, non-LRRK2: 70.3 (no range defined) | Mean LRRK2: 2.0, mean non-LRRK2: 2.1 (mean LRRK2: 9.2, mean non-LRRK2: 7.4) | - | - | - | H/M ratio | No global results provided | - | - | Overall PD, early H/M inversely related to the severity of PD (H&Y and UPDRS). 48-69% LRRK2 carriers have low H/M compared to 80-93% of PD non-carriers. |
| Chiaravalloti et al., 2012 | 37 | - | - | 21 | 62 (No range defined) | H&Y 1, n=15; H&Y 1.5, n=8; H&Y 2, n=7; H&Y 3, n=7 (mean 26) | No additional cardiac DYA evaluation | No further description included | - | H/M ratio | 14%b | - | - | H/M correlated with 123I-FP-CIT uptake in the putamen ipsilateral to the prevalently effected side, but did not correlate with any other striatal uptake. |
| Tijero et al., 2013 | 25 (LRRK2 G2019S n=6, and R1441G n=6; iPD n=13) | - | - | 10 | LRRK2: 56.4, iPD: 59.2 (No range defined) | Stage not defined (mean LRRK2: 8.77, mean iPD 6.47) | OH tilt test, Plasma NE, valsalva, deep breathing, autonomic question-naire | LRRK2, n=2 of 12 with OH; iPD, n=3 of 13 with OH | Y ([99mTc]MIBI) | H/M ratio | 12.58%b compared to LRRK2 | - | N | H/M correlated to bp response during valsalva phase IV in both iPD and LRRK2 groups. LRRK2 R1441G mutation had larger phase IV overshoot than G2019S (no other differences between mutations). |
Listed percentages describe loss of MIBG uptake compared to controls (unless otherwise noted) and reported originally in the reference (a) or calculated based on data (b numbers or c graph) from the publication. Ecompared to MSA group. 123/125I-MIBG, 123/125I-Meta-iodobenzylguanidine; AF, autonomic failure; BP, blood pressure; CBD, corticobasal degeneration; DYA, dysautonomia; Epi, epinephrine; H/M, heart to mediastium; H&Y, Hoehn & Yahr; HR, heart rate; LRRK2, leucine rich repeating kinase 2; LV, left ventricle; OH, orthostatic hypotension; MSA, multiple systems atrophy; NE, norepinephrine; PD, Parkinson’s disease; PSP, progressive supranuclear palsy; UPDRS, unified Parkinson’s disease rating scale.
Regional instead of global 123I-MIBG cardiac analysis may be the key to identify progressive changes and possibly PD severity. Only one 123I-MIBG study reported a regional analysis, in which 123I-MIBG uptake was visually assessed at both the early and delayed images with a 3-point color scale in the anterior, inferior, lateral, septal and apical cardiac areas between nondysautonomia PD and MSA [182]. No differences were found between the 2 planar images and the largest 123I-MIBG reductions were noted in the inferior (70%) and apical (85%) regions of the left ventricle. The relationship between the regional uptake and any marker for PD severity (stage or duration) was not evaluated. Clearly, mapping a pattern of loss with 123I-MIBG is limited and further investigation is warranted.
To assess the relationship between cardiac denervation and abnormal cardiac symptoms, several studies combined 123I-MIBG imaging and autonomic tests including head tilt test for OH, blood pressure monitoring, deep breathing and ECG for HRV and general heart health, as well as plasma catecholamine measurements. In general, PD patients with clinical dysautonomia had considerably reduced 123I-MIBG accumulation and increased washout rates, whereas those not exhibiting autonomic abnormalities had mixed 123I-MIBG results. For example, one study found 27.4% loss of 123I-MIBG uptake in nondysautonomia PD patients compared to 50.8% loss in dysautonomia PD [182]. Moreover, H/M was reduced by similar amounts in PD patients with OH at H&Y stage 1 and 2-4 compared to controls, 42.4% and 40.9%, respectively [189]. Yet those without OH have greater H/M loss at H&Y 2-4 (27.1%) than at H&Y 1 (21.7%) compared to controls. Interestingly, it has been reported that PD patients without OH and with normal circadian patterns in blood pressure (nocturnal drop of at least 10% from diurnal) also had significantly low H/M ratios [6], and patients deemed asymptomatic for autonomic failure (AF) as determined by patient reports, had abnormal autonomic function detected by study tests and reduced H/M ratios [181].
123I-MIBG has also been used to analyze cardiac innervation in PD patients with genetic mutations. LRRK2 G2019S or R1441G PD carriers have been reported to have decreased 123I-MIBG uptake compared to controls, although they have higher H/M ratios compared to idiopathic PD. Few of them have autonomic impairments [126,190]. In two PD Parkin carriers 123I-MIBG uptake was reported to be within normal range (Figure 3). This matched postmortem results of normal TH positive epicardial nerve fascicles and no evidence of Lewy body pathology [191].
Figure 3.
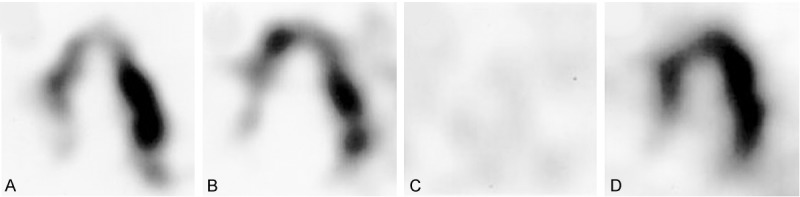
123/125I-MIBG SPECT long-axis views of the heart of Parkinson’s disease patients with a parkin mutation (A and B), idiopathic Parkinson’s disease (C), and a normal control (D). Reproduced with permission from [191] © John Wiley and Sons.
123/125I-MIBG use in preclinical studies
123/125I-MIBG uptake has been studied in rodents treated with STZ, reserpine, MPTP and 6-OHDA. Notably, many of the preclinical studies using smaller species conducted ex vivo autoradiographic studies after an injection of MIBG to live animals; few performed in vivo 123I-MIBG SPECT [85,107,192] (Table 4). Ex vivo measurements of 125I-MIBG were calculated as the percentage of dose per gram of tissue, whereas the in vivo evaluations were reported as the percentage of injected activity and not the common H/M measurement due to resolution limitations when working with rodents.
Table 4.
Mapping cardiac innervation in animal models of cardiac sympathetic neurodegeneration
| Reference | Specie | Animal Model (dose) | Number of subjects | Time of evaluation after model induction | Homogenous cardiac perfusion (Y/N) | Method of radiotracer analysis (In vivo/ex vivo) | % of LV global uptake reduction | LV regions/ ROI analyzed | Region of maximal reduction (% of loss) | Significant loss of plasma catechol-amines (Y/N) | |
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|||||||||||
| Exp | Control | ||||||||||
| 123/125 I-MIBG | |||||||||||
| Kiyono et al., 2001 | Rat | STZ (55 mg/kg iv) | 7 | 7 | 4 weeks | Y ([99mTc]MIBI) | ex vivo | No global results provided | Anterior and inferior walls | Inferior (13.1%b; 25.3%b compared to anterior wall in STZ) | - |
| Kiyono et al., 2005 | Rat | STZ (55 mg/kg iv) | 5 | 5 | 4 weeks | - | ex vivo | No global results provided | Anterior, inferior, lateral, and septal walls | Inferior (no values provided) | - |
| Kusmic et al., 2008 | Mouse | STZ (35 mg/kg x 5 ip) | 7 | 7 | 7 months | - | in vivo | No global loss (similar value to controls) | - | - | - |
| Goethals et al., 2009 | Rat | STZ (60 mg/kg iv) | 12 | 12 | 8 weeks | - | in vivo | 3.6-8.4%b | Anterior, anterolateral, inferolateral, inferior, inferoseptal and anteroseptal | Anteroseptal (9-10%c compared to inferoseptal and inferolateral in STZ) | - |
| Matsuki et al., 2010 | Rat | STZ (65 mg/kg ip) | 7 | 7 | 5 weeks | - | ex vivo | 26-33%c | Anterior, inferior, lateral, and septal walls | No differences found between regions | - |
| Takatsu et al., 2000 | Mouse | MPTP (5 mg/kg x 1 sc) | 7 | 7 | 1 week | - | ex vivo | 40%c | - | - | - |
| MPTP (5 mg/kg x 2 sc) | 6 | 66%c | |||||||||
| MPTP (50 mg/kg x 2 sc) | 10 | 73%c | |||||||||
| Takatsu et al., 2002 | Mouse | MPTP (50 mg/kg x 2 sc) | 10 | 10 | 1 week | - | ex vivo | 70%c | - | - | - |
| Fukumitsu et al., 2006 | Mouse | MPTP (10 mg/kg x 1 ip) | 10 | 10 | 1 week | - | ex vivo | 49.6%b | - | - | - |
| MPTP (10 mg/kg x 4 ip) | 10 | 61.1%b | |||||||||
| Fukumitsu et al., 2009 | Mouse | MPTP (40 mg/kg x 4 ip) | 14 | 14 | 1 day | - | ex vivo | 71.7%b | - | - | Y (NE) |
| MPTP (40 mg/kg x 4 ip) | 14 | 4 days | 57.6%b | N | |||||||
| MPTP (40 mg/kg x 4 ip) | 14 | 7 days | 49.5%b | N | |||||||
| MPTP (40 mg/kg x 4 ip) | 14 | 21 days | 47.3%b | N | |||||||
| MPTP (40 mg/kg at 7, 14, and 21 days ip) | 14 | 21 days | 60.3%b | N | |||||||
| Sisson et al., 1987 | Rat | 6-OHDA (100 mg/kg ip) | 11 | 9 | 5 days | - | ex vivo | 31%a | - | - | - |
| Nomura et al., 2006 | Rabbit | Reserpine (2 mg/kg ip) | 4 | 4 | 14 days | Y ([13N]NH3) | ex vivo | 18%b | - | - | - |
| 6-OHDA (100 mg/kg iv) | 4 | 90%a | |||||||||
| 18 F-FDA | |||||||||||
| Goldstein et al., 1990 | Dog | Reserpine (3 mg/kg po) | 3 | Same animals after 2 weeks | 24 hours | - | in vivo | No global results provided | Free wall and septal wall | No differences found between regions | Y(NE, DHPG) |
| Goldstein et al., 1991 | Dog | 6-OHDA (50 mg/kg iv) | 3 | 0 | 3 days | - | in vivo | >95%a | - | - | Y (DHPG, DOPAC) |
| Goldstein et al., 2003 | Monkey | MPTP (4 doses, dose not clear) | 1 | 2 | 2 weeks | - | in vivo | no loss, 70% increaseb | - | - | Y (NE, EPI, DHPG, DOPAC) |
| MPTP (4 doses x 2 separated by 2 weeks, dose not clear) | 1 | 6 weeks | 27.5%b | Y (NE, EPI, DHPG, DOPAC) | |||||||
| MPTP (4 doses x 2 separated by 2 weeks, dose not clear) | 1 | 2 years | no loss, 21.9% increaseb | N (only slightly decreased) | |||||||
| 6-OHDA (50 mg/kg iv) | 3 | 1 week | 60%b | Y (NE, EPI, DHPG, DOPAC) | |||||||
| 11 C-MHED | |||||||||||
| Schmid et al., 1999 | Rat | STZ (50 mg/kg ip) | 18 | 15 | 6 months | - | ex vivo | No global results provided | Proximal and distal segments | Distal (33%a) | - |
| STZ (50 mg/kg ip) | 11 | 11 | 9 months | Proximal (44%a) | |||||||
| Thackeray et al., 2011 | Rat | STZ (45 mg/kg ip) | 4 | 6 | 2 weeks | - | ex vivo | No change | Free wall and septal wall | No change | N |
| STZ (45 mg/kg ip) | 14 | 8 | 8 weeks | 13%a | Free wall and septal evenly decreased (13%a) | N (increased 2.5x) | |||||
| Nomura et al., 2006 | Rabbit | Reserpine (2 mg/kg ip) | 4 | 4 | 14 days | Y ([13N]NH3) | in vivo | 50.3%b | - | - | - |
| 6-OHDA (100 mg/kg iv) | 4 | 90%a | |||||||||
| Raffel et al., 2006 | Rat | 6-OHDA (7 mg/kg ip) | 5 | 5 | 24 hours | - | ex vivo | 28.2%b | - | - | - |
| 6-OHDA (11 mg/kg ip) | 5 | 60.1%b | |||||||||
| 6-OHDA (15 mg/kg ip) | 5 | 53.8%b | |||||||||
| 6-OHDA (22 mg/kg ip) | 5 | 72.5%b | |||||||||
| 6-OHDA (100 mg/kg ip) | 5 | 91.9%b | |||||||||
| Joers et al., 2013 | Monkey | 6-OHDA (50 mg/kg iv) | 5 | Same animals at baseline | 1, 4, 10, 14 weeks | - | in vivo | 51.1%a | Anterior, inferior, lateral, and septal walls | Inferior (49.5%b) | Y (NE, DOPAC) |
Listed percentages describe loss compared to controls (unless otherwise noted) and reported originally in the reference (a) or calculated based on data (b numbers or c graph) from the publication. 123/125I-MIBG, [123/125I]-Meta-iodobenzylguanidine; 6-OHDA, 6-hydroxydopamine; DHPG, Dihydroxyphenylglycine; DOPAC, dihydroxyphenylacetic acid; EPI, epinephrine; LDOPA, L-3,4-dihydroxyphenylalanine; LRRK2, leucine rich repeat kinase 2; LV, left ventricle; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NE, norepinephrine; NH3, ammonia; PD, Parkinson’s disease; STZ, streptozotocine.
In STZ-induced diabetic models global or heterogenous 123/125I-MIBG tracer reduction has been documented [85,89,192-194]. Rats treated with STZ (55 mg/kg iv) and evaluated with 125I-MIBG SPECT 4 weeks later showed a significant decrease of 25.3% in the inferior compared to the anterior wall of the left ventricle [193]. The heterogenous pattern of loss was not due to a change in the myocardial blood flow, as rats had normal 99mTc-MIBI uptake in both regions of the left ventricle. Similar preferential loss in the inferior wall was reported in a follow-up study where STZ rats were observed 4 weeks later. The animals had reduced 125I-MIBG in the inferior wall and comparable reduction in NET density measured by a desipramine binding assay [194]. In vivo measurements taken at later timepoints after STZ at either 8 weeks or 7 months, showed less global 123I-MIBG loss in the left ventricle [85,192]. Although less 123I-MIBG uptake was observed, high washout rates were found in both studies compared to controls, suggesting decreased 123I-MIBG reuptake and NET density. However these reports did not include postmortem histological studies to confirm pattern of 123/125I-MIBG uptake or NET density. One study had evaluated hearts of STZ-treated mice 6 months after diabetes onset with immunohistochemistry and found globally diminished TH. This study did not compare regions of the left ventricle in the postmortem analysis or conduct 123I-MIBG imaging [97]. Differences in the in vivo 123I-MIBG and ex vivo 125I-MIBG data may not be due to the selected timepoints, but simply to the method of evaluation.
When reserpine (2 mg/kg ip) was administered to rabbits 2 hours before 125I-MIBG injection, ligand uptake was not significantly affected (18% loss compared to controls) [163]. Yet, reserpine dosing 4 hours before 125I-MIBG injection significantly reduced tracer uptake up to 6 hours (63% loss; [195]). Similar results were found in reserpinized rats (4 mg/kg; [160]). These findings suggest that analysis of 125I-MIBG imaging data in reserpine-treated animals should consider the timing of dosing of both compounds, as well as their differential effects on uptake systems (e.g.: reserpine only affects intravesicular uptake, while 125I-MIBG can accumulate extra- and intra-vesicularly).
MPTP-treated animals, like STZ models, display impairments in cardiac 125I-MIBG uptake, but at earlier timepoints and with a greater magnitude. Mice administered subcutaneous MPTP at one dose of 5 mg/kg, or two doses of either 5 mg/kg or 50 mg/kg delivered 16 hours apart and evaluated 1 week after final MPTP dose had a dose-dependent reduction in 125I-MIBG uptake up to approximately 73% [6]. Similar dose-imaging responses were found in mice evaluated for ex vivo radioactivity of 125I-MIBG 1 week after intraperitoneal delivery of MPTP as a single dose of 10 mg/kg or a series of 4 doses totaling 40 mg/kg [140]. A comparison between reductions of cardiac 125I-MIBG uptake and a binding assay for NET showed a close relationship after MPTP. Interestingly, postmortem analysis of synaptophysin, TH and NET immunostaining in the hearts of MPTP-treated mice (30 mg/kg ip x 2, 12 hr apart) one week after neurotoxin challenge, showed no difference compared to controls [154] suggesting that MPTP may disrupt cardiac sympathetic function as observed with 123/125I-MIBG but not necessarily cause neurodegeneration. This is further supported by the finding of increased plasma NE and ex vivo 125I-MIBG cardiac radioactivity at 4, 7 and 21 days post MPTP compared to 1 day after toxin dosing with plasma NE comparable to baseline levels at 7 and 21 days [149].
6-OHDA dosing also decreases 125I-MIBG cardiac uptake. Ex vivo examination of cardiac rat tissue showed a 31% decrease in uptake of 125I-MIBG 5 days after 6-OHDA (100 mg/kg ip) compared to controls [196]. In rabbits dosing of 100 mg/kg iv of 6-OHDA produced a 90% 125I-MIBG global retention deficit 14 days after neurotoxin [163]; the animals’ heart rate and blood pressure showed no differences compared to controls. Regional analysis of 125I-MIBG cardiac uptake was not performed in reserpine-, MPTP- or 6-OHDA-based models.
99mTc-FBPBAT
The radiotracer 99mTc-FBPBAT binds to α(1)/β(1)-adrenoreceptors providing an assessment of postsynaptic myocardial autonomic function [197]. The properties of 99mTc-FBPBAT are advantageous for a clinical setting because of its long 6-hour half-life. Uptake of 99mTc-FBPBAT is temperature dependent and has shown greater uptake than the other SPECT adrenergic radiotracer, 123/125I-MIBG. To its disadvantage, this post-synaptic tracer has a rapid plasma clearance and high renal excretion with 40% of urine radioactivity unmetabolized and 50% converted to a metabolite of 99mTc-FBPBAT 6 hours after tracer injection. This tracer has been studied in rats [197] and pigs for feasibility and in humans for radiation dose estimation [198]. From these studies, authors suggest that 99mTc-FBPBAT binds to myocardium and is mostly taken up by post-synaptic adrenoreceptors. To the best of our knowledge, there are no reports of 99mTc-FBPBAT imaging in PD or animal models of cardiac neurodegeneration.
18F-FDA
The radioligand 18F-FDA targets sympathetic nerves by acting as the catecholamine precursor dopamine. It is taken up into sympathetic neurons via NET, where 18F-FDA is packaged into endogenous neurotransmitter vesicles. NET uptake-1 specificity for 18F-FDA is 79% relative to uptake-2, and VMAT2 selectivity is 90%, demonstrating 18F-FDA ability to reflect the uptake and storage of dopamine [199]. 18F-FDA acts as a catecholamine precursor, thus it is considered to have greater similarity to NE and greater biological relevance than other catecholamine analogue tracers. Yet, as 18F-FDA is degraded by MAO or COMT, its visualization is time-limited. 18F-FDA is not commercially available and as other PET radioligands requires cyclotron production. 18F-FDA has a biological half-life of 90 minutes and additional radiotracers can be used later in the day if multiple tracers need to be evaluated [200]. Cardiac 18F-FDA PET images are quantified from time averaged 18F-FDA concentrations from multiple regions of interest within the left ventricle. Time points for data acquisition and the quantity and location of regions of interest vary across studies.
Clinical application of 18F-FDA
Similar to 123I-MIBG, 18F-FDA uptake is globally reduced in PD patients compared to controls [4,31,127,201-204] and MSA patients [4,201] (Table 5). Patients with diffuse ventricular loss were significantly older (average 66 years) than those PD with localized loss (57 years) [4]. The localized loss was found in the free wall and apex of the left ventricle compared to the septal wall. 13N-NH3 perfusion studies confirmed that the regional differences were due to changes in sympathetic function [204]. PD patients exhibiting symptoms of dysautonomia compared to those without sympathetic abnormalities consistently had reduced cardiac 18F-FDA [4,31,201-204]. It should be noted that normal 18F-FDA uptake was reported in 4 out of 20 PD patients with chronic OH or abnormal responses to the Valsalva maneuver [4]. Additionally, when cardiac 18F-FDA uptake was tracked for 1-4 years in PD patients with no signs of dysautonomia, a progressive loss of uptake was observed overtime (Figure 4). The global loss averaged 23%, while the regional analysis showed a maximal 31% loss in the free wall and 16% in the septal wall [203]. These data suggest that sympathetic denervation can occur without clinically measurable symptoms. In that regard, changes in cardiac 18F-FDA uptake seem to predate the onset of motor dysfunction associated with nigrostriatal dopaminergic loss. In a case report reduced 18F-FDA plus exercise and orthostatic intolerance was found 4 years before the onset of PD motor symptoms [202].
Table 5.
Comparison of clinical reports evaluating cardiac innervation in PD using 18F-FDA
| Reference | Number of Patients | Mean PD age (age range) | PD H&Y stage (disease duration yrs) | Methods to evaluate cardiac DYA | PD patient DYA descripttion | Homogenous cardiac perfusion Y/N (radiotracer) | LV regions analyzed | % of LV global uptake reduction | Region of maximal reduction (% of loss) | Significant loss of plasma catechol-amines Y/N | Comments | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||||||
| PD | MSA | PSP | Control | |||||||||||
| 18F-FDA | ||||||||||||||
| Goldstein et al., 1997 | 2 | 9 with SNF 4 w/o SNF | - | 22 | No age defined | Stage and disease duration not defined | NE spillover, plasma DHPG, LDOPA, DOPAC | n=2 of 2 PD with SNF (No detectable NE spillover) | - | Septal and free wall | Undetect-able levels | - | No detectable increments | PD FDA uptake reduced compared to MSA. |
| Goldstein et al., 2000 | 29 | 24 | - | 33 | 70 (No range defined) | Average H&Y 2.4 (disease duration not defined) | OH evaluation after standing, ECG (BP and HR for RR interval), plasma NE spillover, plasma DHPG and LDOPA, valsalva | n=9 of 29 PD with SNF (decreased NE spillover), n=20 of 29 without SNF (12 of 20 had abnormal valsalva) | - | Septal wall (free wall not easily identified). Apex and free wall analyzed by visual inspection. | 70%b | Apex or free wall (no % provided; n=5 of 20 PD without SNF) | Y (DHPG/ LDOPA in 9 of 29) | All PD have reduced septal FDA compared to MSA. Septal FDA lower in PD with SNF compared to PD without SNF. Of 20 PD without SNF, 4 had normal FDA. |
| Li et al., 2002 | 9 | - | - | - | 60 (No range defined) | H&Y 1-3; average 2.2, (disease duration not defined) | Plasma NE and Epi and valsalva only conducted at T=1. | 2 timepoints (T1, T2) 1-4 yrs apart. Abnormal valsalva and no OH at T1 or T2. | - | Septal and lateral walls and apex | 23%a,D | Lateral (31%a,D loss) | N | T1, 2 of 9 had normal FDA and 7 of 9 had decreased FDA confined to lateral wall or apex. T2, all 9 PD have reduced FDA. |
| Singleton et al., 2004 | 2 (triplication of α-syn) | - | - | 1 | 5th decade of life | Stage not defined (1-10) | OH evaluation after standing, plasma NE and Epi, valsalva | n=2 of 2 have abnormal valsalva, n=1 of 2 OH | Y (13N-NH3) | Septal wall | 81.6%b | - | N | Patients with triplication of α-synuclein have similar FDA uptake as iPD. |
| Tipre et al., 2005 | 26 | - | - | 12 | No mean defined (30-70) | Stage and disease duration not defined | No additional cardiac DYA evaluations | n=26 of 26 PD with OH | Y (13N-NH3) | Septal wall | No global results provided | - | - | FDA uptake significantly reduced in septal wall of PD and PAF compared to controls. |
| Goldstein et al., 2007a | 1 | - | - | - | 56 | T1=not yet diagnosed with PD; T2=Stage not defined | Plasma NE and Epi, valsalva | 2 timepoints (T1, T2) 4 yrs apart. Abnormal valsalva and exercise and orthostatic intolerance. | - | Septal wall | 85%c | - | N | Severely decreased global FDA uptake at both T1 and T2. |
| Goldstein et al., 2007b | 1 (LRRK2 T2356I mutation) | - | - | - | 63 | No stage defined (10) | OH evaluation after standing, plasma NE and DHPG, valsalva | Abnormal valsalva but no OH | Y (13N-NH3) | Septal wall | Undetectable levels | - | Y (DHPG was low during OH evaluation; normal NE) | Brain FDA PET showed decreased uptake in the striatum and nigra. |
Listed percentages describe loss of 18F-FDA uptake compared to controls (unless otherwise noted) and reported originally in the reference (a) or calculated based on data (b numbers or c graph) from the publication. Dcompared across timepoints. 13N-NH3, [13N]-ammonium; 18F-FDA, [18F]-dopamine; AF, autonomic failure; BF, baroreflex failure; BP, blood pressure; DHPG, Dihydroxyphenylglycine; DOPAC, dihydroxyphenylacetic acid; DYA, dysautonomia; Epi, epinephrine; H&Y, Hoehn & Yahr; iPD, idiopathic PD; LDOPA, L-3,4-dihydroxyphenylalanine; LRRK2, leucine rich repeating kinase 2; LV, left ventricle; MSA, multiple systems atrophy; NE, norepinephrine; OH, orthostatic hypotension; PAF, pure autonomic failure; PD, Parkinson’s disease; PSP, progressive supranuclear palsy; SNF, sympathetic neurocirculatory failure.
Figure 4.
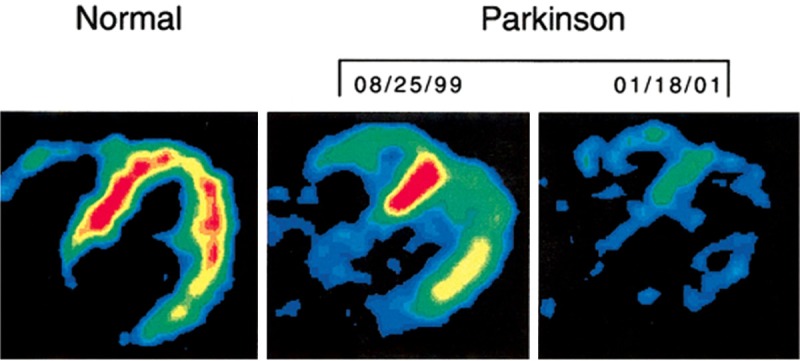
18F-FDA uptake in a normal control patient (left) and the progressive loss of uptake in Parkinson’s disease patient within 2 years. Reproduced with permission from [203] © John Wiley and Sons.
18F-FDA has also been used to assess cardiac sympathetic loss in PD patients with known genetic mutations. A single case of LRRK2 T2356l mutation [31] and 2 patients with triplication of the α-synuclein gene [127] have shown virtually absent cardiac 18F-FDA uptake. This suggests severe cardiac sympathetic loss. The 3 patients had abnormal Valsalva response, although only 1 triplication case exhibited OH; the 3 of them had normal levels of circulating NE.
18F-FDA use in preclinical studies
18F-FDA PET has been used for evaluation of cardiac sympathetic loss induced by reserpine, 6-OHDA and MPTP intoxication in dogs and nonhuman primates (Table 4). Dogs treated with reserpine (3 mg/kg po) 24 hours before 18F-FDA PET imaging, showed decreased 18F-FDA retention at 30 minutes and accelerated 18F-FDA clearance compared to untreated animals [205]. Authors proposed that reserpine did not inhibit 18F-FDA initial uptake, and in the axoplasm it was quickly metabolized to 18F-DOPAC and then released into the blood stream. In a follow up study, dogs injected with 6-OHDA (total dose of 50 mg/kg iv) and imaged with 18F-FDA at least 3 days later had a decrease of more than 95% in cardiac uptake [166], suggesting 18F-FDA uptake was limited due to the destruction of the sympathetic terminals. The cardiac radioactivity clearance was similar to that in reserpine-treated dogs.
In nonhuman primates, 18F-FDA imaging [152] was used to compare a single 6-OHDA-treated rhesus monkey (total dose 50 mg/kg) to 1 monkey that received a series of 4 MPTP doses (actual doses were not specified) and did not show motor PD signs (acute phase; n=1) to another animal that had a series of 8 doses to induce severe parkinsonism (subacute phase; n=1) within 2 and 6 weeks, respectively. The 6-OHDA treated monkey imaged 1 week post-toxin showed decreased global cardiac 18F-FDA concentration. Plasma catecholamines (NE and epinephrine) and metabolites (DHPG and DOPAC) were markedly lower in this monkey than untreated animals. Approximately 6 weeks after the last MPTP dose the monkey in acute phase had increased cardiac 18F-FDA concentrations by 70% compared to controls, and decreased rate of 18F-FDA loss as compared with the accelerated 18F-FDA loss and overall 60% reduction in 18F-FDA uptake in the 6-OHDA-treated monkey. However, the acute MPTP treatment decreased circulating catecholamines compared to untreated monkeys, and the values were comparable to subacute phase and 6-OHDA monkeys. The subacute MPTP monkey had decreases of heart 18F-FDA radioactivity levels of 27.5%, but not as low as the 6-OHDA-treated monkey. When 3 other MPTP-treated monkeys (undefined toxin doses) were evaluated 2 years after intoxication, both 18F-FDA radioactivity and plasma catecholamines returned to values similar to untreated animals suggesting spontaneous recovery. Cardiac regional uptake of 18F-FDA was not evaluated in the monkey or dog models.
11C-MHED
The radioligand 11C-MHED acts as a false neurotransmitter that binds to NET and is packaged into neurotransmitter vesicles [206]. An advantage of 11C-MHED compared to 123I-MIBG and 18F-FDA, is its higher specificity to neuronal uptake-1 mechanisms (92%). 11C-MHED also has greater metabolic stability than 18F-FDA due to it is resistance to MAO and COMT metabolism [207]. For these reasons, 11C-MHED more accurately measures the integrity of sympathetic noradrenergic nerves [176,208]. 11C-MHED has a myocardial half-life of 240 minutes in humans [209]. Clinical studies with 11C-MHED PET usually use the uptake index calculated as the 11C-MHED uptake during the final frame (30-40 minutes) divided by the integral of blood time-activity at 40 minutes [210,211] or an irreversible model to derive the influx of 11C-MHED with an ROI in the center of the left ventricle chamber as an input function. These methods of analysis have limitations as 11C-MHED retention index depends on the re-uptake by NET. 11C-MHED is quickly taken up by NET and released through diffusion for re-uptake into the neurons; therefore, these methods are limited by the rate of diffusion out of the neuron and do not reflect the true amount of NET. Others have suggested that 11C-MHED PET data may be best analyzed using a reversible model [168,212,213]. Clinical MHED regional analysis of the left ventricle has commonly evaluated the perfusion areas the left circumflex artery (LCX), left anterior descending artery (LAD) and right coronary artery (RCA). LAD, LCX and RCA distributions overlap segments of the anterior, inferior, lateral or septal walls of the left ventricle (Figure 5).
Figure 5.

Standard American Heart Association polar map model used for imaging of the heart. Map displays the myocardial segments corresponding to the perfusion regions of the left anterior descending artery (LAD; blue), left circumflex coronary artery (LCX; orange) and right coronary artery (RCA; red). AA, apical anterior; AI, apical inferior; AL, apical lateral; AS, apical septal; BA, basal anterior; BAS, basal anteroseptal; BI, basal inferior; BIS, basal inferoseptal; BAL, basal anterolateral; BIL, basal inferolateral; MA, mid-anterior; MAS, mid-anteroseptal; MIS, mid-inferoseptal; MI, mid-inferior; MIL, mid-inferolateral; MAL, mid-anterolateral. Modified from [222].
Clinical application of 11C-MHED
There have been only few reports evaluating cardiac innervation in PD with 11C-MHED [214-216]. In these reports the PD population varied within the first stages of the disease (reported as H&Y 1-2.5 stages or duration years) (Table 6). In these studies, global cardiac denervation was present in 44 to 85% of PD patients (Figure 6). The global loss varied between studies from 23-36% [214], 26% [161] to 50% [216]. A clear association between loss of cardiac 11C-MHED uptake and disease duration, mean age or H&Y stage was not found.
Table 6.
Comparison of clinical reports evaluating cardiac innervation in PD using 11C-MHED
| Reference | Number of Patients | Mean PD age (age range) | PD H&Y stage (disease duration yrs) | Methods to evaluate cardiac DYA | PD patient DYA description | Homogenous cardiac perfusion Y/N (radio-tracer) | LV regions analyzed | % of LV global uptake reduction | Region of maximal reduction (% of abnormal sectors - AS) | Significant loss of plasma catechol-amines Y/N | Comments | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||||||
| PD | MSA | PSP | Control | |||||||||||
| 11C-MHED | ||||||||||||||
| Berding et al., 2003 | 5 | 2 | - | - | No mean defined (51-69) | No stage defined (2-16) | OH evaluation with Schellong test | n=3 of 5 PD with OH, n=2 of 5 probable/ possible PD without OH | - | Polar maps created for global MHED influx values. | 23.8-36.4%b,E | - | - | MHED uptake lower in PD and OH (41.4-59.3%b,E of loss) than PD without OH (+2.5-2.1%b,E) compared to MSA. |
| Raffel et al., 2006 | 9 | 10 | 8 | 10 | No mean defined (35-77) | H&Y 1.5-2.5 (disease duration not defined) | No additional cardiac DYA evaluations | n=1 of 9 PD with OH | - | LCX, LAD, and RCA. Polar maps with 480 sectors created. Regional effects are calculated as % of 480 sectors that are abnormal. | 26%b (96%a of 480 sectors have abnormal values) | LCX and RCA (100% AS) | - | MHED retention was abnormal in 4 of 9 PD patients and were the only subjects analyzed and reported. No correlation between MHED uptake and disease duration. |
| Wong et al., 2012 | 27 | - | - | 33 | 62 (50-74) | H&Y 1-2.5 (1-14) | No additional cardiac DYA evaluations | No further description included | - | LCX, LAD, and RCA. Anterior, inferior, lateral and septal walls in respect to proximal and distal areas. Polar maps with 480 sectors created. Regional effects calculated as % of 480 sectors that are abnormal. | 50%b (61.7%a of 480 sectors have abnormal values) | Lateral (distal 68%a AS and proximal 74%a AS); Inferior (distal 63% AS and proximal 69% AS); LCX (70.9%b AS) | - | 23 of 27 PD patients have some global deficit. Of those with abnormal uptake, regional analysis showed abnormal uptake in LCX (7-100%a AS), LAD (5-100%a AS) and RCA (14-100%a AS) perfusion areas. |
Listed percentages describe loss of 18F-FDA uptake compared to controls (unless otherwise noted) and reported originally in the reference (a) or calculated based on data (b numbers or c graph) from the publication. Ecompared to MSA group. 11C-MHED, [11C]-meta-hydroxyephedrine; AS, abornmal sectors; DYA, dysautonmia; H&Y, Hoehn & Yahr; LAD, left anterior descending artery; LCX, left circumflex artery; LV, left ventricle; MSA, multiple systems atrophy; NE, norepinephrine; OH, orthostatic hypotension; PD, Parkinson’s disease; PSP, progressive supranuclear palsy; RCA, right coronary artery.
Figure 6.

Short axis (SA), ventral long axis (VLA), and horizontal long axis (HLA) views of MHED radioactivity in the heart of a control (NC 3) and idiopathic Parkinson’s disease patient (IPD 6). Polar map displays 11C-MHED retention index of left ventricle (A, anterior; L, lateral; S, septal). This research was originally published in JNM. [161] © by the Society of Nuclear Medicine and Molecular Imaging, Inc.
In 2 of the 3 studies 11C-MHED uptake was reported as the number of abnormal sectors in polar maps representing the LAD, LCX and RCA perfusion areas [161,216]. These studies categorize the patients by the extent of their global denervation into normal, mild to moderate and severe denervation. The LCX perfusion area was the most affected, with the number of abnormal sectors reaching up to 100% [161] and 70.9% [216]. One of the studies further compared regional 11C-MHED uptake in the apex and the anterior, septal, inferior and lateral walls of the left ventricle in proximal and distal segments. The most severe denervation was found in the proximal lateral and inferior walls (loss of 74% and 69%, respectively [216]). Presence of OH symptoms was only assessed in one of the 3 studies, without finding a correlation [214]. The other 2 studies [161,216] also assessed striatal dihydrotetrabenazine ([11C]DTBZ), a radioligand that binds to vesicular monoamine transporter (VMAT2) and reflects nigral dopaminergic neuron activity. The investigators found no correlation between sympathetic nigrostriatal and cardiac loss.
11C-MHED use in preclinical studies
11C-MHED PET has been used to image cardiac sympathetic innervation in rats, rabbits and monkeys after STZ, reserpine or 6-OHDA-intoxication (Table 4).
Two studies in STZ-induced diabetic rats evaluated 11C-MHED uptake [90,217] at different timepoints. In the first study, diabetic rats (50 mg/kg ip STZ) were assessed with 11C-MHED PET at 6 and 9 months [90]. At 6 months, proximal left ventricular segments were not significantly different (9% loss) compared to controls, yet distal segments were significantly decreased (33%). At 9 months, proximal and distal regions were significantly decreased (44% and 40%, respectively). However, NE quantification by HPLC showed no significant difference in either proximal or distal segments at 6 or 9 months in STZ rats compared to controls. In the second study, diabetic rats (45 mg/kg ip STZ) also showed 11C-MHED uptake reduction throughout the entire heart (13-26%) and equal loss (13%) in the septum and free wall of the left ventricle compared to controls at 8 but not at 2 weeks after hyperglycemic induction [217]. However, at 8 weeks postmortem cardiac TH staining was similar to controls. Plasma and left ventricle NE were increased by 2.5 times and 1.2 times, respectively, compared to controls, suggesting normalization of cardiac sympathetic innervation. The 11C-MHED uptake data suggests that STZ takes over 2 weeks to produce cardiac neurodegeneration, lasting up to 9 months. Since different regions were analyzed between studies a pattern of STZ-induced loss is difficult to identify. Additionally, the postmortem data did not confirm sympathetic nerve loss, and instead suggest a dysfunctional cardiac sympathetic system.
11C-MHED PET has also been used to evaluate cardiac innervation after 6-OHDA intoxication in rats [215], rabbits [163] and monkeys [152,168]. In rats, 11C-MHED PET performed 24 hours after varying doses of 6-OHDA (7, 11, 15, 22 and 100 mg/kg ip) [215] showed a dose-dependent decrease in radioligand uptake, with the highest dose creating an average whole-heart 11C-MHED reduction of 92%. Middle-range doses induced variable loss of 11C-MHED uptake, yet it was highly correlated with NET density as determined postmortem (30 minutes after 11C-MHED injection) with a 3H-mazindol saturation binding assay. In 6-OHDA-treated rabbits (100 mg/kg iv), 11C-MHED PET 2 weeks post intoxication showed a 90% reduced cardiac uptake compared to controls. This loss was greater than the one induced by 2 mg/kg ip reserpine (50.3%; [163]). Rhesus monkeys given 6-OHDA (50 mg/kg iv) had significantly lower 11C-MHED distribution values at 1, 4, 10 and 14 weeks than at baseline, with some recovery that stabilized between 10 and 14 weeks (Figure 7). At 14 weeks, there was a 51% global reduction of 11C-MHED uptake [168] and regional analysis showed a maximal loss in the inferior wall of the left ventricle (49.5%) compared to the anterior wall. Circulating catecholamines and their metabolites were also reduced and positively correlated with 11C-MHED uptake. Postmortem quantification of cardiac TH and PGP9.5 immunoreactivity confirmed sympathetic cardiac loss [218].
Figure 7.

Horizontal long axis view of the heart shows 11C-MHED uptake at baseline (A) and 10 weeks after systemic 6-OHDA (B) in a rhesus monkey. Polar maps display 11C-MHED distribution volume in left ventricle with the greatest deficit in the apex and inferior walls 10 weeks after toxin (D) compared to baseline (C). A, anterior; I, inferior; L, lateral; S, septal.
Conclusions and future steps
The impact of cardiac neurodegeneration and dysautonomia in PD disability cannot be underestimated. The symptoms of fatigue, OH, shortness of breath and changes in cardiac electrical activity complicate typical PD motor symptoms, are part of the nonmotor symptom spectrum and should be considered when planning a patient treatment strategy. Advances in molecular imaging have allowed progress in the identification and characterization of cardiac sympathetic innervation, its role in dysautonomia in PD, and the evaluation of animal models, yet much work is still needed.
Molecular imaging using 123/125I-MIBG, 18F-FDA or 11C-MHED has demonstrated that PD patients have loss of cardiac sympathetic innervation. This has proven useful as a tool for differential diagnosis between PD and MSA or PSP, as the two latter diseases may present with similar symptoms, but unlike PD, they usually have normal cardiac postganglionic sympathetic uptake. Interestingly, the amount and distribution of sympathetic loss in PD seems to vary among individuals and it has been reported as either diffuse, at times extensively, or following a nonuniform pattern with maximal loss in the apex and the inferior and lateral walls of the left ventricle. Progression of the neurodegeneration overtime seems to influence the depth and extension of the sympathetic loss. For example, 9 PD patients with H&Y stages ranging from 1-3 showed after 2 years a 31% reduction in 11C-MHED uptake in the left ventricular free wall and 16% in the septum while a more uniform loss is seen in advanced stages of the disease [203]. This suggests that regional quantification may be more sensitive than global data to evaluate progression and, potentially, recovery. An obstacle against drawing a final conclusion on patterns of loss in PD is that only few studies, with small number of cases each, reported regional analysis of cardiac sympathetic innervation. Increased efforts towards regional mapping are needed to help characterize a universal PD pattern of sympathetic cardiac loss.
The use of sympathetic radiotracers has established that PD patients exhibiting autonomic symptoms such as OH have some degree of cardiac sympathetic neurodegeneration. Yet at this time, based on the accumulated evidence predicting extent of nerve loss based on symptoms alone is unreliable. First, depending on the radioligand, reports may be based on a few patients, and although they provide valuable data to strengthen the importance of cardiac neurodegeneration in PD, the scope of the conclusions that can be drawn from them is limited [201,202]. Second, the definition of autonomic dysfunction and failure used as the criteria for including patients in different groups varies between articles. For example, some patients were considered to have autonomic failure by assessment of OH abnormalities [203,204] while others defined it by sympathetic neurocirculatory failure or the presence of both OH and blood pressure defects [201]. Third, evaluation of dysautonomia symptoms, like head tilt, OH after standing, ECG for variation in RR intervals, plasma catecholamines and Valsalva, vary between reports or is missing. Further research is needed to assess the measurements predictive value, sensitivity and specificity toward specific disorders. Although overall autonomic scoring systems exist, including Composite Autonomic Symptom Score (COMPASS) and Autonomic Symptom Profile (ASP), these scales have limited evaluation of cardiac dysautonomia, and are questionnaire-based, restricting their physiological relevance [219-221]. The development of a composite rating scale evaluating multiple cardiac autonomic measurements with levels of severity and PD H&Y stage that can be correlated with imaging measures may be sensitive enough to estimate cardiac neurodegeneration, yet this scale does not currently exist.
Identification of clusters of patients with gene mutations associated to PD, yet asymptomatic, present an opportunity to study prodomal signs including cardiac dysautonomia and may allow to answer the question if dysautonomia precedes motor symptoms or not. Imaging of cardiac sympathetic denervation with different radioligands has shown varying levels of loss in cases of PD with LRRK2 G2019S, R1441G and T2356l mutations and triplication of α-synuclein. Further research needs to be conducted to better understand cardiac dysautonomia and denervation in PD linked to genetics.
Preclinical evaluation with molecular imaging presents advantages for clinical translation as the outcome measure and analysis can be easily transferred between species. In that regard, studies in animal models allow, for example, the isolation of specific conditions in order to identify the magnitude of their impact in the overall health, as well as time-dependent correlations with actual biochemical and morphological data. During the evaluation of current available models of cardiac neurodegeneration in PD some emerged as better suited for preclinical testing. STZ seems to have several limitations for modeling cardiac sympathetic neurodegeneration. 125I-MIBG studies suggest that STZ induces inconsistent lesions that also vary depending on the timeline of STZ administration, while 11C-MHED studies demonstrate a reduction in tracer uptake, but it does not match the biochemical analyses. Overall, STZ-induced neuronal death does not result as quickly as some of the other toxin based models and produces hyperglycemia, a confounding factor for PD research, and should be considered when selecting appropriate models. MPTP-based models do not seem to induce long-term cardiac sympathetic denervation, but instead develop cardiac dysfunction that recovers overtime, making it a poor system to study the full cardiac effects found in PD. Additionally, subacute MPTP dosing to monkeys, which produces cardiac abnormalities, is associated with severe parkinsonism that requires extensive animal care and increased measures of personnel safety, making it a difficult model to use. In comparison systemic 6-OHDA-based models seem to be more reliable with less human risk. In that regard the 6-OHDA monkey model has a lesion that is stabilized between 2-3 months, with a reproducible pattern of loss where the left ventricular inferior wall retains a significant deficit compared to the inferior wall, similar to PD. In addition, 6-OHDA also induces a reduction in plasma NE as often found in PD patients with other dysautonomia symptoms and when medications are restricted. Genetic models have begun to be evaluated for cardiac pathologies and hold potential to unravel the etiologies of cardiac dysautonomia. Thy1-αSyn mice have already shown α-synuclein accumulation and cardiac autonomic dysregulation. Studies to evaluate the relationship between protein accumulation and cardiac innervation in Thy1-αsyn mice as well as evaluation of transgenic animals with other PD related mutations such as with LRRK2 are warranted as they can help identify and test targets for neuroprotective strategies.
The concept of cardiac imaging as a biomarker for disease progression has already been tested to monitor reduction of cardiac 18F-FDA uptake after 4-years. A natural follow up to this study would be to assess the effects of disease-modifying strategies, once one is identified. A challenge for neuroprotective studies is defining the time to begin treatment for cardiac neurodegeneration that would increase the chances for success. Ideally, it would be when there are enough cardiac ganglia neurons to protect and/or to stimulate reinnervation. Identification of subtle neuronal loss, as well as evaluation of neuroprotective therapies, requires methods that enable researchers to interpret changes overtime. 11C-MHED PET imaging has demonstrated sympathetic nerve restoration in heart transplant patients [55] by defining a reinnervation threshold based on the 11C-MHED uptake/min. Since then, spatial resolution has increased in PET technologies. Although advances in SPECT imaging are expected to improve quantification of the images, the spatial resolution is still too poor to detect what we expect would be slow and small changes in reinnervation following treatment in PD. At this time it is not possible to distinguish epicardial versus endocardial layers of the heart with molecular imaging. Progress to improve the use of these techniques may assist the differentiation of PD from other parkinsonian disorders or identify disease progression. Further development in sympathoneuronal radiotracers may also strengthen the management of cardiac neurodegeneration and guidance for future therapies. In addition a more comprehensive assessment where presynaptic and postsynaptic imaging is combined may give clinicians a better understanding to the cardiac deficits and better predict outcomes. Equally important will be the development and application of radioligands aiming to visualize candidate neurodegenerative pathways (e.g.: α-synuclein, inflammation).
To conclude, molecular imaging is an effective tool to detect cardiac sympathetic neurodegeneration in PD and animal models and holds promise as a tool to detect disease progression and treatment efficacy. With the advancement of these technologies, molecular imaging may better identify patterns of cardiac sympathetic loss and correlate with clinical cardiac dysautonomia features.
Acknowledgements
This project was supported in part by the Clinical and Translational Science Award (CTSA) program, previously through the National Center for Research Resources (NCRR) grant 1UL1RR025011, and now by the National Center for Advancing Translational Sciences (NCATS), grant 9U54TR000021; WNPRC NIH base grant P51RR000167 and Awards from the Graduate School, University of Wisconsin, Madison. The authors are grateful to Dr. Jim Holden for his comments and Mr. Shahrose Rahman for computer graphics. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Disclosure of conflict of interest
The authors declare no conflict of interest.
References
- 1.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 2.Langston JW. The Parkinson’s complex: parkinsonism is just the tip of the iceberg. Ann Neurol. 2006;59:591–596. doi: 10.1002/ana.20834. [DOI] [PubMed] [Google Scholar]
- 3.Amino T, Orimo S, Itoh Y, Takahashi A, Uchihara T, Mizusawa H. Profound cardiac sympathetic denervation occurs in Parkinson disease. Brain Pathol. 2005;15:29–34. doi: 10.1111/j.1750-3639.2005.tb00097.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldstein DS, Holmes C, Li ST, Bruce S, Metman LV, Cannon RO 3rd. Cardiac sympathetic denervation in Parkinson disease. Ann Intern Med. 2000;133:338–347. doi: 10.7326/0003-4819-133-5-200009050-00009. [DOI] [PubMed] [Google Scholar]
- 5.Orimo S, Amino T, Itoh Y, Takahashi A, Kojo T, Uchihara T, Tsuchiya K, Mori F, Wakabayashi K, Takahashi H. Cardiac sympathetic denervation precedes neuronal loss in the sympathetic ganglia in Lewy body disease. Acta Neuropathol. 2005;109:583–588. doi: 10.1007/s00401-005-0995-7. [DOI] [PubMed] [Google Scholar]
- 6.Takatsu H, Nishida H, Matsuo H, Watanabe S, Nagashima K, Wada H, Noda T, Nishigaki K, Fujiwara H. Cardiac sympathetic denervation from the early stage of Parkinson’s disease: clinical and experimental studies with radiolabeled MIBG. J Nucl Med. 2000;41:71–77. [PubMed] [Google Scholar]
- 7.Linden D, Diehl RR, Berlit P. Sympathetic cardiovascular dysfunction in long-standing idiopathic Parkinson’s disease. Clin Auton Res. 1997;7:311–314. doi: 10.1007/BF02267723. [DOI] [PubMed] [Google Scholar]
- 8.Reid JL, Calne DB, George CF, Vakil SD. The action of L(-)-dopa on baroreflexes in Parkinsonism. Clin Sci. 1972;43:851–859. doi: 10.1042/cs0430851. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein DS, Sewell L, Sharabi Y. Autonomic dysfunction in PD: a window to early detection? J Neurol Sci. 2011;310:118–122. doi: 10.1016/j.jns.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Hirsch EC, Jenner P, Przedborski S. Pathogenesis of Parkinson’s disease. Mov Disord. 2013;28:24–30. doi: 10.1002/mds.25032. [DOI] [PubMed] [Google Scholar]
- 11.Loewy AD, Spyer LK. Central Regulation of Autonomic Functions. New York: Oxford University Press; 1990. [Google Scholar]
- 12.Higgins CB, Vatner SF, Braunwald E. Parasympathetic control of the heart. Pharmacol Rev. 1973;25:119–155. [PubMed] [Google Scholar]
- 13.Brodal A. [Nobel prize in physiology and medicine 1981. Brain research] . Tidsskr Nor Laegeforen. 1981;101:1901–1904. [PubMed] [Google Scholar]
- 14.Brodde OE, Bruck H, Leineweber K, Seyfarth T. Presence, distribution and physiological function of adrenergic and muscarinic receptor subtypes in the human heart. Basic Res Cardiol. 2001;96:528–538. doi: 10.1007/s003950170003. [DOI] [PubMed] [Google Scholar]
- 15.Ellison JP, Williams TH. Sympathetic nerve pathways to the human heart, and their variations. Am J Anat. 1969;124:149–162. doi: 10.1002/aja.1001240203. [DOI] [PubMed] [Google Scholar]
- 16.Cooper J, Bloom F, Roth R. The Biochemical Basis of Neuropharmacology. New York: Oxford University Press; 1991. [Google Scholar]
- 17.Perlman RL, Chalfie M. Catecholamine release from the adrenal medulla. Clin Endocrinol Metab. 1977;6:551–576. doi: 10.1016/s0300-595x(77)80071-6. [DOI] [PubMed] [Google Scholar]
- 18.Anderson DJ, Axel R. A bipotential neuroendocrine precursor whose choice of cell fate is determined by NGF and glucocorticoids. Cell. 1986;47:1079–1090. doi: 10.1016/0092-8674(86)90823-8. [DOI] [PubMed] [Google Scholar]
- 19.Anderson DJ, Carnahan JF, Michelsohn A, Patterson PH. Antibody markers identify a common progenitor to sympathetic neurons and chromaffin cells in vivo and reveal the timing of commitment to neuronal differentiation in the sympathoadrenal lineage. J Neurosci. 1991;11:3507–3519. doi: 10.1523/JNEUROSCI.11-11-03507.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Le Douarin NM, Teillet MA. Experimental analysis of the migration and differentiation of neuroblasts of the autonomic nervous system and of neurectodermal mesenchymal derivatives, using a biological cell marking technique. Dev Biol. 1974;41:162–184. doi: 10.1016/0012-1606(74)90291-7. [DOI] [PubMed] [Google Scholar]
- 21.Jain S, Goldstein DS. Cardiovascular dysautonomia in Parkinson disease: from pathophysiology to pathogenesis. Neurobiol Dis. 2012;46:572–580. doi: 10.1016/j.nbd.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haensch CA, Lerch H, Jorg J, Isenmann S. Cardiac denervation occurs independent of orthostatic hypotension and impaired heart rate variability in Parkinson’s disease. Parkinsonism Relat Disord. 2009;15:134–137. doi: 10.1016/j.parkreldis.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 23.Goldstein DS. Dysautonomia in Parkinson’s disease: neurocardiological abnormalities. Lancet Neurol. 2003;2:669–676. doi: 10.1016/s1474-4422(03)00555-6. [DOI] [PubMed] [Google Scholar]
- 24.Post KK, Singer C, Papapetropoulos S. Cardiac denervation and dysautonomia in Parkinson’s disease: a review of screening techniques. Parkinsonism Relat Disord. 2008;14:524–531. doi: 10.1016/j.parkreldis.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 25.Goldstein DS, Holmes C, Sharabi Y, Brentzel S, Eisenhofer G. Plasma levels of catechols and metanephrines in neurogenic orthostatic hypotension. Neurology. 2003;60:1327–1332. doi: 10.1212/01.wnl.0000058766.46428.f3. [DOI] [PubMed] [Google Scholar]
- 26.Schatz IJ. Farewell to the “Shy-Drager syndrome”. Ann Intern Med. 1996;125:74–75. doi: 10.7326/0003-4819-125-1-199607010-00012. [DOI] [PubMed] [Google Scholar]
- 27.Allcock LM, Ullyart K, Kenny RA, Burn DJ. Frequency of orthostatic hypotension in a community based cohort of patients with Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2004;75:1470–1471. doi: 10.1136/jnnp.2003.029413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldstein DS. Orthostatic hypotension as an early finding in Parkinson’s disease. Clin Auton Res. 2006;16:46–54. doi: 10.1007/s10286-006-0317-8. [DOI] [PubMed] [Google Scholar]
- 29.Senard JM, Rai S, Lapeyre-Mestre M, Brefel C, Rascol O, Rascol A, Montastruc JL. Prevalence of orthostatic hypotension in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1997;63:584–589. doi: 10.1136/jnnp.63.5.584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathias CJ, Kimber JR. Clonidine-growth-hormone testing in Parkinson’s disease. Lancet. 1999;354:339–340. doi: 10.1016/s0140-6736(05)75248-x. [DOI] [PubMed] [Google Scholar]
- 31.Goldstein DS, Imrich R, Peckham E, Holmes C, Lopez G, Crews C, Hardy J, Singleton A, Hallett M. Neurocirculatory and nigrostriatal abnormalities in Parkinson disease from LRRK2 mutation. Neurology. 2007;69:1580–1584. doi: 10.1212/01.wnl.0000268696.57912.64. [DOI] [PubMed] [Google Scholar]
- 32.Luther JM. In: Drug-induced autonomic dysfunction. Robertson D, Biaggioni I, Burnstock G, Low PA, Paton JFR, editors. San Diego: Elsevier; 2012. pp. 511–514. [Google Scholar]
- 33.Mathias CJ, Kimber J, Watson L, Muthane U. Is clonidine-growth hormone stimulation a good test to differentiate multiple system atrophy from idiopathic Parkinson’s disease? J Neurol. 2002;249:488–489. doi: 10.1007/s004150200047. [DOI] [PubMed] [Google Scholar]
- 34.Freeman R. Clinical practice. Neurogenic orthostatic hypotension. N Engl J Med. 2008;358:615–624. doi: 10.1056/NEJMcp074189. [DOI] [PubMed] [Google Scholar]
- 35.Friedman JH, Friedman H. Fatigue in Parkinson’s disease: a nine-year follow-up. Mov Disord. 2001;16:1120–1122. doi: 10.1002/mds.1201. [DOI] [PubMed] [Google Scholar]
- 36.Herlofson K, Ongre SO, Enger LK, Tysnes OB, Larsen JP. Fatigue in early Parkinson’s disease. Minor inconvenience or major distress? Eur J Neurol. 2012;19:963–968. doi: 10.1111/j.1468-1331.2012.03663.x. [DOI] [PubMed] [Google Scholar]
- 37.Karlsen K, Larsen JP, Tandberg E, Jorgensen K. Fatigue in patients with Parkinson’s disease. Mov Disord. 1999;14:237–241. doi: 10.1002/1531-8257(199903)14:2<237::aid-mds1006>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 38.Nakamura T, Hirayama M, Hara T, Hama T, Watanabe H, Sobue G. Does cardiovascular autonomic dysfunction contribute to fatigue in Parkinson’s disease? Mov Disord. 2011;26:1869–1874. doi: 10.1002/mds.23744. [DOI] [PubMed] [Google Scholar]
- 39.Chaudhuri A, Behan PO. Fatigue in neurological disorders. Lancet. 2004;363:978–988. doi: 10.1016/S0140-6736(04)15794-2. [DOI] [PubMed] [Google Scholar]
- 40.Brown RG, Dittner A, Findley L, Wessely SC. The Parkinson fatigue scale. Parkinsonism Relat Disord. 2005;11:49–55. doi: 10.1016/j.parkreldis.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 41.Nelesen R, Dar Y, Thomas K, Dimsdale JE. The relationship between fatigue and cardiac functioning. Arch Intern Med. 2008;168:943–949. doi: 10.1001/archinte.168.9.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deguchi K, Sasaki I, Tsukaguchi M, Kamoda M, Touge T, Takeuchi H, Kuriyama S. Abnormalities of rate-corrected QT intervals in Parkinson’s disease-a comparison with multiple system atrophy and progressive supranuclear palsy. J Neurol Sci. 2002;199:31–37. doi: 10.1016/s0022-510x(02)00079-5. [DOI] [PubMed] [Google Scholar]
- 43.Oka H, Mochio S, Sato H, Katayama K. Prolongation of QTc interval in patients with Parkinson’s disease. Eur Neurol. 1997;37:186–189. doi: 10.1159/000117432. [DOI] [PubMed] [Google Scholar]
- 44.Ishizaki F, Harada T, Yoshinaga H, Nakayama T, Yamamura Y, Nakamura S. [Prolonged QTc intervals in Parkinson’s disease--relation to sudden death and autonomic dysfunction] . No To Shinkei. 1996;48:443–448. [PubMed] [Google Scholar]
- 45.Malek NM, Grosset KA, Stewart D, Macphee GJ, Grosset DG. Prescription of drugs with potential adverse effects on cardiac conduction in Parkinson’s disease. Parkinsonism Relat Disord. 2013;19:586–589. doi: 10.1016/j.parkreldis.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 46.Bordet R, Broly F, Destee A, Libersa C. Debrisoquine hydroxylation genotype in familial forms of idiopathic Parkinson’s disease. Adv Neurol. 1996;69:97–100. [PubMed] [Google Scholar]
- 47.Devos D, Kroumova M, Bordet R, Vodougnon H, Guieu JD, Libersa C, Destee A. Heart rate variability and Parkinson’s disease severity. J Neural Transm. 2003;110:997–1011. doi: 10.1007/s00702-003-0016-8. [DOI] [PubMed] [Google Scholar]
- 48.Haapaniemi TH, Pursiainen V, Korpelainen JT, Huikuri HV, Sotaniemi KA, Myllyla VV. Ambulatory ECG and analysis of heart rate variability in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2001;70:305–310. doi: 10.1136/jnnp.70.3.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mastrocola C, Vanacore N, Giovani A, Locuratolo N, Vella C, Alessandri A, Baratta L, Tubani L, Meco G. Twenty-four-hour heart rate variability to assess autonomic function in Parkinson’s disease. Acta Neurol Scand. 1999;99:245–247. doi: 10.1111/j.1600-0404.1999.tb07355.x. [DOI] [PubMed] [Google Scholar]
- 50.Bigger JT Jr, Fleiss JL, Steinman RC, Rolnitzky LM, Kleiger RE, Rottman JN. Frequency domain measures of heart period variability and mortality after myocardial infarction. Circulation. 1992;85:164–171. doi: 10.1161/01.cir.85.1.164. [DOI] [PubMed] [Google Scholar]
- 51.Kleiger RE, Miller JP, Bigger JT Jr, Moss AJ. Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am J Cardiol. 1987;59:256–262. doi: 10.1016/0002-9149(87)90795-8. [DOI] [PubMed] [Google Scholar]
- 52.Cornelissen VA, Vanhaecke J, Aubert AE, Fagard RH. Heart rate variability after heart transplantation: a 10-year longitudinal follow-up study. J Cardiol. 2012;59:220–224. doi: 10.1016/j.jjcc.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 53.Fagard R, Macor F, Vanhaecke J. Signs of functional efferent reinnervation of the heart in patients after cardiac transplantation. Acta Cardiol. 1995;50:369–380. [PubMed] [Google Scholar]
- 54.Uberfuhr P, Frey AW, Reichart B. Vagal reinnervation in the long term after orthotopic heart transplantation. J Heart Lung Transplant. 2000;19:946–950. doi: 10.1016/s1053-2498(00)00181-9. [DOI] [PubMed] [Google Scholar]
- 55.Uberfuhr P, Frey AW, Ziegler S, Reichart B, Schwaiger M. Sympathetic reinnervation of sinus node and left ventricle after heart transplantation in humans: regional differences assessed by heart rate variability and positron emission tomography. J Heart Lung Transplant. 2000;19:317–323. doi: 10.1016/s1053-2498(00)00060-7. [DOI] [PubMed] [Google Scholar]
- 56.Oka H, Toyoda C, Yogo M, Mochio S. Cardiovascular dysautonomia in de novo Parkinson’s disease without orthostatic hypotension. Eur J Neurol. 2011;18:286–292. doi: 10.1111/j.1468-1331.2010.03135.x. [DOI] [PubMed] [Google Scholar]
- 57.Goldstein DS, Holmes CS, Dendi R, Bruce SR, Li ST. Orthostatic hypotension from sympathetic denervation in Parkinson’s disease. Neurology. 2002;58:1247–1255. doi: 10.1212/wnl.58.8.1247. [DOI] [PubMed] [Google Scholar]
- 58.Senard JM, Rascol O, Durrieu G, Tran MA, Berlan M, Rascol A, Montastruc JL. Effects of yohimbine on plasma catecholamine levels in orthostatic hypotension related to Parkinson disease or multiple system atrophy. Clin Neuropharmacol. 1993;16:70–76. doi: 10.1097/00002826-199302000-00008. [DOI] [PubMed] [Google Scholar]
- 59.Senard JM, Valet P, Durrieu G, Berlan M, Tran MA, Montastruc JL, Rascol A, Montastruc P. Adrenergic supersensitivity in parkinsonians with orthostatic hypotension. Eur J Clin Invest. 1990;20:613–619. doi: 10.1111/j.1365-2362.1990.tb01909.x. [DOI] [PubMed] [Google Scholar]
- 60.Riederer P, Rausch WD, Birkmayer W, Jellinger K, Seemann D. CNS Modulation of adrenal tyrosine hydroxylase in Parkinson’s disease and metabolic encephalopathies. J Neural Transm Suppl. 1978:121–131. [PubMed] [Google Scholar]
- 61.Stoddard SL. The adrenal medulla and Parkinson’s disease. Rev Neurosci. 1994;5:293–307. doi: 10.1515/revneuro.1994.5.4.293. [DOI] [PubMed] [Google Scholar]
- 62.Stoddard SL, Tyce GM, Ahlskog JE, Zinsmeister AR, Carmichael SW. Decreased catecholamine content in parkinsonian adrenal medullae. Exp Neurol. 1989;104:22–27. doi: 10.1016/0014-4886(89)90004-6. [DOI] [PubMed] [Google Scholar]
- 63.Fumimura Y, Ikemura M, Saito Y, Sengoku R, Kanemaru K, Sawabe M, Arai T, Ito G, Iwatsubo T, Fukayama M, Mizusawa H, Murayama S. Analysis of the adrenal gland is useful for evaluating pathology of the peripheral autonomic nervous system in lewy body disease. J Neuropathol Exp Neurol. 2007;66:354–362. doi: 10.1097/nen.0b013e3180517454. [DOI] [PubMed] [Google Scholar]
- 64.Goldstein DS, Eldadah BA, Holmes C, Pechnik S, Moak J, Saleem A, Sharabi Y. Neurocirculatory abnormalities in Parkinson disease with orthostatic hypotension: independence from levodopa treatment. Hypertension. 2005;46:1333–1339. doi: 10.1161/01.HYP.0000188052.69549.e4. [DOI] [PubMed] [Google Scholar]
- 65.Moak JP, Eldadah B, Holmes C, Pechnik S, Goldstein DS. Partial cardiac sympathetic denervation after bilateral thoracic sympathectomy in humans. Heart Rhythm. 2005;2:602–609. doi: 10.1016/j.hrthm.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 66.Schwartz PJ, Locati EH, Moss AJ, Crampton RS, Trazzi R, Ruberti U. Left cardiac sympathetic denervation in the therapy of congenital long QT syndrome. A worldwide report. Circulation. 1991;84:503–511. doi: 10.1161/01.cir.84.2.503. [DOI] [PubMed] [Google Scholar]
- 67.Turley AJ, Hunter S, Stewart MJ. A cardiac paraganglioma presenting with atypical chest pain. Eur J Cardiothorac Surg. 2005;28:352–354. doi: 10.1016/j.ejcts.2005.04.038. [DOI] [PubMed] [Google Scholar]
- 68.Askari A, Kordzadeh A, Lee GH, Harvey M. Endoscopic thoracic sympathectomy for primary hyperhidrosis: a 16-year follow up in a single UK centre. Surgeon. 2013;11:130–133. doi: 10.1016/j.surge.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 69.Ajijola OA, Wisco JJ, Lambert HW, Mahajan A, Stark E, Fishbein MC, Shivkumar K. Extracardiac neural remodeling in humans with cardiomyopathy. Circ Arrhythm Electrophysiol. 2012;5:1010–1116. doi: 10.1161/CIRCEP.112.972836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schwartz PJ, La Rovere MT, Vanoli E. Autonomic nervous system and sudden cardiac death. Experimental basis and clinical observations for post-myocardial infarction risk stratification. Circulation. 1992;85:I77–91. [PubMed] [Google Scholar]
- 71.Goto K, Longhurst PA, Cassis LA, Head RJ, Taylor DA, Rice PJ, Fleming WW. Surgical sympathectomy of the heart in rodents and its effect on sensitivity to agonists. J Pharmacol Exp Ther. 1985;234:280–287. [PubMed] [Google Scholar]
- 72.Yoshimoto M, Wehrwein EA, Novotny M, Swain GM, Kreulen DL, Osborn JW. Effect of stellate ganglionectomy on basal cardiovascular function and responses to beta1-adrenoceptor blockade in the rat. Am J Physiol Heart Circ Physiol. 2008;295:H2447–2454. doi: 10.1152/ajpheart.00958.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brouha L, Cannon WB, Dill DB. The heart rate of the sympathectomized dog in rest and exercise. J Physiol. 1936;87:345–359. doi: 10.1113/jphysiol.1936.sp003411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stramba-Badiale M, Lazzarotti M, Schwartz PJ. Development of cardiac innervation, ventricular fibrillation, and sudden infant death syndrome. Am J Physiol. 1992;263:H1514–1522. doi: 10.1152/ajpheart.1992.263.5.H1514. [DOI] [PubMed] [Google Scholar]
- 75.Goodall M. Studies of adrenaline and noradrenaline in mammalian heart and suprarenals. Acta Physiol Scand Suppl. 1951;24:7–51. [PubMed] [Google Scholar]
- 76.Gauthier P, Nadeau R, De Champlain J. Acute and chronic cardiovascular effects of 6-hydroxydopamine in dogs. Circ Res. 1972;31:207–217. doi: 10.1161/01.res.31.2.207. [DOI] [PubMed] [Google Scholar]
- 77.Yanowitz F, Preston JB, Abildskov JA. Functional distribution of right and left stellate innervation to the ventricles. Production of neurogenic electrocardiographic changes by unilateral alteration of sympathetic tone. Circ Res. 1966;18:416–428. doi: 10.1161/01.res.18.4.416. [DOI] [PubMed] [Google Scholar]
- 78.Cruz JM, Fonseca M, Pinto FJ, Oliveira AG, Carvalho LS. Cardiopulmonary effects following endoscopic thoracic sympathectomy for primary hyperhidrosis. Eur J Cardiothorac Surg. 2009;36:491–496. doi: 10.1016/j.ejcts.2009.03.059. [DOI] [PubMed] [Google Scholar]
- 79.Felten SY, Peterson RG, Shea PA, Besch HR Jr, Felten DL. Effects of streptozotocin diabetes on the noradrenergic innervation of the rat heart: a longitudinal histofluorescence and neurochemical study. Brain Res Bull. 1982;8:593–607. doi: 10.1016/0361-9230(82)90086-7. [DOI] [PubMed] [Google Scholar]
- 80.Sakata N, Yoshimatsu G, Tsuchiya H, Egawa S, Unno M. Animal models of diabetes mellitus for islet transplantation. Exp Diabetes Res. 2012;2012:256707. doi: 10.1155/2012/256707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Szkudelski T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol Res. 2001;50:537–546. [PubMed] [Google Scholar]
- 82.Portha B, Picon L, Rosselin G. Chemical diabetes in the adult rat as the spontaneous evolution of neonatal diabetes. Diabetologia. 1979;17:371–377. doi: 10.1007/BF01236272. [DOI] [PubMed] [Google Scholar]
- 83.Stables CL, Glasser RL, Feldman EL. Diabetic cardiac autonomic neuropathy: Insights from animal models. Auton Neurosci. 2013;177:74–80. doi: 10.1016/j.autneu.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 84.Kramp RC, Congdon CC, Smith LH. Isogeneic or allogeneic transplantation of duct-ligated pancreas in streptozotocin diabetic mice. Eur J Clin Invest. 1975;5:249–258. doi: 10.1111/j.1365-2362.1975.tb00452.x. [DOI] [PubMed] [Google Scholar]
- 85.Kusmic C, Morbelli S, Marini C, Matteucci M, Cappellini C, Pomposelli E, Marzullo P, L’Abbate A, Sambuceti G. Whole-body evaluation of MIBG tissue extraction in a mouse model of long-lasting type II diabetes and its relationship with norepinephrine transport protein concentration. J Nucl Med. 2008;49:1701–1706. doi: 10.2967/jnumed.108.054361. [DOI] [PubMed] [Google Scholar]
- 86.Ballinger WF, Lacy PE. Transplantation of intact pancreatic islets in rats. Surgery. 1972;72:175–186. [PubMed] [Google Scholar]
- 87.Ballinger WF, Lacy PE, Scharp DW, Kemp CB, Knight M. Isografts and allografts of pancreatic islets in rats. Br J Surg. 1973;60:313. [PubMed] [Google Scholar]
- 88.Joffe II, Travers KE, Perreault-Micale CL, Hampton T, Katz SE, Morgan JP, Douglas PS. Abnormal cardiac function in the streptozotocin-induced non-insulin-dependent diabetic rat: noninvasive assessment with doppler echocardiography and contribution of the nitric oxide pathway. J Am Coll Cardiol. 1999;34:2111–2119. doi: 10.1016/s0735-1097(99)00436-2. [DOI] [PubMed] [Google Scholar]
- 89.Matsuki A, Nozawa T, Igarashi N, Sobajima M, Ohori T, Suzuki T, Fujii N, Igawa A, Inoue H. Fluvastatin attenuates diabetes-induced cardiac sympathetic neuropathy in association with a decrease in oxidative stress. Circ J. 2010;74:468–475. doi: 10.1253/circj.cj-09-0402. [DOI] [PubMed] [Google Scholar]
- 90.Schmid H, Forman LA, Cao X, Sherman PS, Stevens MJ. Heterogeneous cardiac sympathetic denervation and decreased myocardial nerve growth factor in streptozotocin-induced diabetic rats: implications for cardiac sympathetic dysinnervation complicating diabetes. Diabetes. 1999;48:603–608. doi: 10.2337/diabetes.48.3.603. [DOI] [PubMed] [Google Scholar]
- 91.Scharp DW, Murphy JJ, Newton WT, Ballinger WF, Lacy PE. Transplantation of islets of Langerhans in diabetic rhesus monkeys. Surgery. 1975;77:100–105. [PubMed] [Google Scholar]
- 92.Sheehy A, Hsu S, Bouchard A, Lema P, Savard C, Guy LG, Tai J, Polyakov I. Comparative vascular responses three months after paclitaxel and everolimus-eluting stent implantation in streptozotocin-induced diabetic porcine coronary arteries. Cardiovasc Diabetol. 2012;11:75. doi: 10.1186/1475-2840-11-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Matas AJ, Sutherland DE, Najarian JS. Current status of islet and pancreas transplantation in diabetes. Diabetes. 1976;25:785–795. doi: 10.2337/diab.25.9.785. [DOI] [PubMed] [Google Scholar]
- 94.Akiyama N, Okumura K, Watanabe Y, Hashimoto H, Ito T, Ogawa K, Satake T. Altered acetylcholine and norepinephrine concentrations in diabetic rat hearts. Role of parasympathetic nervous system in diabetic cardiomyopathy. Diabetes. 1989;38:231–236. doi: 10.2337/diab.38.2.231. [DOI] [PubMed] [Google Scholar]
- 95.Paulson DJ, Light KE. Elevation of serum and ventricular norepinephrine content in the diabetic rat. Res Commun Chem Pathol Pharmacol. 1981;33:559–562. [PubMed] [Google Scholar]
- 96.Savarese JJ, Berkowitz BA. beta-Adrenergic receptor decrease in diabetic rat hearts. Life Sci. 1979;25:2075–2078. doi: 10.1016/0024-3205(79)90200-5. [DOI] [PubMed] [Google Scholar]
- 97.Kellogg AP, Converso K, Wiggin T, Stevens M, Pop-Busui R. Effects of cyclooxygenase-2 gene inactivation on cardiac autonomic and left ventricular function in experimental diabetes. Am J Physiol Heart Circ Physiol. 2009;296:H453–461. doi: 10.1152/ajpheart.00678.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang Y, Xuan YL, Hu HS, Li XL, Xue M, Cheng WJ, Suo F, Yan SH. Risk of ventricular arrhythmias after myocardial infarction with diabetes associated with sympathetic neural remodeling in rabbits. Cardiology. 2012;121:1–9. doi: 10.1159/000336148. [DOI] [PubMed] [Google Scholar]
- 99.Howarth FC, Chandler NJ, Kharche S, Tellez JO, Greener ID, Yamanushi TT, Billeter R, Boyett MR, Zhang H, Dobrzynski H. Effects of streptozotocin-induced diabetes on connexin43 mRNA and protein expression in ventricular muscle. Mol Cell Biochem. 2008;319:105–114. doi: 10.1007/s11010-008-9883-5. [DOI] [PubMed] [Google Scholar]
- 100.Howarth FC, Jacobson M, Naseer O, Adeghate E. Short-term effects of streptozotocin-induced diabetes on the electrocardiogram, physical activity and body temperature in rats. Exp Physiol. 2005;90:237–245. doi: 10.1113/expphysiol.2004.029439. [DOI] [PubMed] [Google Scholar]
- 101.Howarth FC, Jacobson M, Shafiullah M, Adeghate E. Long-term effects of streptozotocin-induced diabetes on the electrocardiogram, physical activity and body temperature in rats. Exp Physiol. 2005;90:827–835. doi: 10.1113/expphysiol.2005.031252. [DOI] [PubMed] [Google Scholar]
- 102.Yang B, Chon KH. Assessment of diabetic cardiac autonomic neuropathy in type I diabetic mice. Conf Proc IEEE Eng Med Biol Soc. 2011;2011:6560–6563. doi: 10.1109/IEMBS.2011.6091618. [DOI] [PubMed] [Google Scholar]
- 103.Lo Giudice P, Careddu A, Magni G, Quagliata T, Pacifici L, Carminati P. Autonomic neuropathy in streptozotocin diabetic rats: effect of acetyl-L-carnitine. Diabetes Res Clin Pract. 2002;56:173–180. doi: 10.1016/s0168-8227(01)00375-8. [DOI] [PubMed] [Google Scholar]
- 104.Mesangeau D, Laude D, Elghozi JL. Early detection of cardiovascular autonomic neuropathy in diabetic pigs using blood pressure and heart rate variability. Cardiovasc Res. 2000;45:889–899. doi: 10.1016/s0008-6363(99)00406-x. [DOI] [PubMed] [Google Scholar]
- 105.Pop-Busui R. What do we know and we do not know about cardiovascular autonomic neuropathy in diabetes. J Cardiovasc Transl Res. 2012;5:463–478. doi: 10.1007/s12265-012-9367-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kiyono Y, Iida Y, Kawashima H, Ogawa M, Tamaki N, Nishimura H, Saji H. Norepinephrine transporter density as a causative factor in alterations in MIBG myocardial uptake in NIDDM model rats. Eur J Nucl Med Mol Imaging. 2002;29:999–1005. doi: 10.1007/s00259-002-0798-1. [DOI] [PubMed] [Google Scholar]
- 107.Schneider R, Welt K, Aust W, Kluge R, Loster H, Fitzl G. Cardiovascular autonomic neuropathy in spontaneously diabetic rats with and without application of EGb 761. Histol Histopathol. 2010;25:1581–1590. doi: 10.14670/HH-25.1581. [DOI] [PubMed] [Google Scholar]
- 108.Alper MH. Where do we stand with reserpine and anesthesia? J Am Dent Soc Anesthesiol. 1965;12:241–243. [PMC free article] [PubMed] [Google Scholar]
- 109.Carlsson A, Jonasson J, Rosengren E. Time Correlation between the Effects of Reserpine on Behaviour and Storage Mechanism for Arylalkylamines. Acta Physiol Scand. 1963;59:474–477. doi: 10.1111/j.1748-1716.1963.tb02763.x. [DOI] [PubMed] [Google Scholar]
- 110.Roth RH, Stone EA. The action of reserpine on noradrenaline biosynthesis in sympathetic nerve tissue. Biochem Pharmacol. 1968;17:1581–1590. doi: 10.1016/0006-2952(68)90218-9. [DOI] [PubMed] [Google Scholar]
- 111.Haggendal J, Dahlstrom A. The recovery of noradrenaline in adrenergic nerve terminals of the rat after reserpine treatment. J Pharm Pharmacol. 1971;23:81–89. doi: 10.1111/j.2042-7158.1971.tb08617.x. [DOI] [PubMed] [Google Scholar]
- 112.Sharman DF, Vanov S, Vogt M. Noradrenaline content in the heart and spleen of the mouse under normal conditions and after administration of some drugs. Br J Pharmacol Chemother. 1962;19:527–533. doi: 10.1111/j.1476-5381.1962.tb01458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Iversen LL, Glowinski J, Axelrod J. The uptake and storage of H3-norepinephrine in the reserpine-pretreated rat heart. J Pharmacol Exp Ther. 1965;150:173–183. [PubMed] [Google Scholar]
- 114.Paasonen MK, Krayer O. The release of norepinephrine from the mammalian heart by reserpine. J Pharmacol Exp Ther. 1958;123:153–160. [PubMed] [Google Scholar]
- 115.Rutledge CO, Weiner N. The effect of reserpine upon the synthesis of norepinephrine in the isolated rabbit heart. J Pharmacol Exp Ther. 1967;157:290–302. [PubMed] [Google Scholar]
- 116.Pinon M, Racotta IS, Ortiz-Butron R, Racotta R. Catecholamines in paraganglia associated with the hepatic branch of the vagus nerve: effects of 6-hydroxydopamine and reserpine. J Auton Nerv Syst. 1999;75:131–135. doi: 10.1016/s0165-1838(98)00184-2. [DOI] [PubMed] [Google Scholar]
- 117.Martinez-Olivares R, Villanueva I, Racotta R, Pinon M. Depletion and recovery of catecholamines in several organs of rats treated with reserpine. Auton Neurosci. 2006;128:64–69. doi: 10.1016/j.autneu.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 118.Weiner N, Cloutier G, Bjur R, Pfeffer RI. Modification of norepinephrine synthesis in intact tissue dy drugs and during short-term adrenergic nerve stimulation. Pharmacol Rev. 1972;24:203–221. [PubMed] [Google Scholar]
- 119.Zallakian M, Knoth J, Metropoulos GE, Njus D. Multiple effects of reserpine on chromaffin-granule in membranes. Biochemistry. 1982;21:1051–1055. doi: 10.1021/bi00534a035. [DOI] [PubMed] [Google Scholar]
- 120.Kannari K, Tanaka H, Maeda T, Tomiyama M, Suda T, Matsunaga M. Reserpine pretreatment prevents increases in extracellular striatal dopamine following L-DOPA administration in rats with nigrostriatal denervation. J Neurochem. 2000;74:263–269. doi: 10.1046/j.1471-4159.2000.0740263.x. [DOI] [PubMed] [Google Scholar]
- 121.Ponzio F, Achilli G, Calderini G, Ferretti P, Perego C, Toffano G, Algeri S. Depletion and recovery of neuronal monoamine storage in rats of different ages treated with reserpine. Neurobiol Aging. 1984;5:101–104. doi: 10.1016/0197-4580(84)90038-1. [DOI] [PubMed] [Google Scholar]
- 122.Yamaguchi M, Yoshitake T, Fujino K, Kawano K, Kehr J, Ishida J. Determination of norepinephrine in microdialysis samples by microbore column liquid chromatography with fluorescence detection following derivatization with benzylamine. Anal Biochem. 1999;270:296–302. doi: 10.1006/abio.1999.4079. [DOI] [PubMed] [Google Scholar]
- 123.Wakade AR. A comparison of rates of depletion and recovery of noradrenaline stores of peripheral and central noradrenergic neurones after reserpine administration: importance of neuronal activity. Br J Pharmacol. 1980;68:93–98. doi: 10.1111/j.1476-5381.1980.tb10703.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Goldstein DS, Li ST, Kopin IJ. Sympathetic neurocirculatory failure in Parkinson disease: Evidence for an etiologic role of alpha-synuclein. Ann Intern Med. 2001;135:1010–1011. doi: 10.7326/0003-4819-135-11-200112040-00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 126.Ruiz-Martinez J, Gorostidi A, Goyenechea E, Alzualde A, Poza JJ, Rodriguez F, Bergareche A, Moreno F, Lopez de Munain A, Marti Masso JF. Olfactory deficits and cardiac 123I-MIBG in Parkinson’s disease related to the LRRK2 R1441G and G2019S mutations. Mov Disord. 2011;26:2026–2031. doi: 10.1002/mds.23773. [DOI] [PubMed] [Google Scholar]
- 127.Singleton A, Gwinn-Hardy K, Sharabi Y, Li ST, Holmes C, Dendi R, Hardy J, Singleton A, Crawley A, Goldstein DS. Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain. 2004;127:768–772. doi: 10.1093/brain/awh081. [DOI] [PubMed] [Google Scholar]
- 128.Chesselet MF, Richter F, Zhu C, Magen I, Watson MB, Subramaniam SR. A progressive mouse model of Parkinson’s disease: the Thy1-αSyn (“Line 61”) mice. Neurotherapeutics. 2012;9:297–314. doi: 10.1007/s13311-012-0104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hallett PJ, McLean JR, Kartunen A, Langston JW, Isacson O. alpha-Synuclein overexpressing transgenic mice show internal organ pathology and autonomic deficits. Neurobiol Dis. 2012;47:258–267. doi: 10.1016/j.nbd.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fleming SM, Holden JG, Sioshansi PC, Jordan MC, Masliah E, Roos KP. Society of Neuroscience. Chicago, IL: 2009. Alterations in heart rate variability in transgenic mice over-expressing human wildtype alpha synuclein. No. 531.9, K8. [Google Scholar]
- 131.Fleming SM, Jordan MC, Mulligan CK, Masliah E, Holden JG, Millard RW, Chesselet MF, Roos KP. Impaired baroreflex function in mice overexpressing alpha-synuclein. Front Neurol. 2013;4:103. doi: 10.3389/fneur.2013.00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Heikkila RE, Cabbat FS, Manzino L, Duvoisin RC. Effects of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine on neostriatal dopamine in mice. Neuropharmacology. 1984;23:711–713. doi: 10.1016/0028-3908(84)90170-9. [DOI] [PubMed] [Google Scholar]
- 133.Kopin IJ, Markey SP. MPTP toxicity: implications for research in Parkinson’s disease. Annu Rev Neurosci. 1988;11:81–96. doi: 10.1146/annurev.ne.11.030188.000501. [DOI] [PubMed] [Google Scholar]
- 134.Sundstrom E, Stromberg I, Tsutsumi T, Olson L, Jonsson G. Studies on the effect of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) on central catecholamine neurons in C57BL/6 mice. Comparison with three other strains of mice. Brain Res. 1987;405:26–38. doi: 10.1016/0006-8993(87)90986-3. [DOI] [PubMed] [Google Scholar]
- 135.Bankiewicz KS, Oldfield EH, Chiueh CC, Doppman JL, Jacobowitz DM, Kopin IJ. Hemiparkinsonism in monkeys after unilateral internal carotid artery infusion of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Life Sci. 1986;39:7–16. doi: 10.1016/0024-3205(86)90431-5. [DOI] [PubMed] [Google Scholar]
- 136.Przedborski S, Jackson-Lewis V, Popilskis S, Kostic V, Levivier M, Fahn S, Cadet JL. Unilateral MPTP-induced parkinsonism in monkeys. A quantitative autoradiographic study of dopamine D1 and D2 receptors and re-uptake sites. Neurochirurgie. 1991;37:377–382. [PubMed] [Google Scholar]
- 137.Ambrosio S, Blesa R, Mintenig GM, Palacios-Araus L, Mahy N, Gual A. Acute effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) on catecholamines in heart, adrenal gland, retina and caudate nucleus of the cat. Toxicol Lett. 1988;44:1–6. doi: 10.1016/0378-4274(88)90122-1. [DOI] [PubMed] [Google Scholar]
- 138.Fuller RW, Hahn RA, Snoddy HD, Wikel JH. Depletion of cardiac norepinephrine in rats and mice by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Biochem Pharmacol. 1984;33:2957–2960. doi: 10.1016/0006-2952(84)90593-8. [DOI] [PubMed] [Google Scholar]
- 139.Wallace RA, Boldry R, Schmittgen T, Miller D, Uretsky N. Effect of 1-methyl-4-phenyl- 1,2,3,6 tetrahydropyridine (MPTP) on monoamine neurotransmitters in mouse brain & heart. Life Sci. 1984;35:285–291. doi: 10.1016/0024-3205(84)90112-7. [DOI] [PubMed] [Google Scholar]
- 140.Fukumitsu N, Suzuki M, Fukuda T, Kiyono Y, Kajiyama S, Saji H. Reduced 125I-meta-iodobenzylguanidine uptake and norepinephrine transporter density in the hearts of mice with MPTP-induced parkinsonism. Nucl Med Biol. 2006;33:37–42. doi: 10.1016/j.nucmedbio.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 141.Fritz RR, Abell CW, Patel NT, Gessner W, Brossi A. Metabolism of the neurotoxin in MPTP by human liver monoamine oxidase B. FEBS Lett. 1985;186:224–228. doi: 10.1016/0014-5793(85)80713-4. [DOI] [PubMed] [Google Scholar]
- 142.Przedborski S, Kostic V, Jackson-Lewis V, Naini AB, Simonetti S, Fahn S, Carlson E, Epstein CJ, Cadet JL. Transgenic mice with increased Cu/Zn-superoxide dismutase activity are resistant to N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity. J Neurosci. 1992;12:1658–1667. doi: 10.1523/JNEUROSCI.12-05-01658.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36:2503–2508. doi: 10.1016/0024-3205(85)90146-8. [DOI] [PubMed] [Google Scholar]
- 144.Mizuno Y, Sone N, Saitoh T. Effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 1-methyl-4-phenylpyridinium ion on activities of the enzymes in the electron transport system in mouse brain. J Neurochem. 1987;48:1787–1793. doi: 10.1111/j.1471-4159.1987.tb05737.x. [DOI] [PubMed] [Google Scholar]
- 145.Fuller RW, Hemrick-Luecke SK. Depletion of norepinephrine in mouse heart by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mimicked by 1-methyl-4-phenylpyridinium (MPP+) and not blocked by deprenyl. Life Sci. 1986;39:1645–1650. doi: 10.1016/0024-3205(86)90161-x. [DOI] [PubMed] [Google Scholar]
- 146.Chaumette T, Lebouvier T, Aubert P, Lardeux B, Qin C, Li Q, Accary D, Bezard E, Bruley des Varannes S, Derkinderen P, Neunlist M. Neurochemical plasticity in the enteric nervous system of a primate animal model of experimental Parkinsonism. Neurogastroenterol Motil. 2009;21:215–222. doi: 10.1111/j.1365-2982.2008.01226.x. [DOI] [PubMed] [Google Scholar]
- 147.Natale G, Kastsiushenka O, Fulceri F, Ruggieri S, Paparelli A, Fornai F. MPTP-induced parkinsonism extends to a subclass of TH-positive neurons in the gut. Brain Res. 2010;1355:195–206. doi: 10.1016/j.brainres.2010.07.076. [DOI] [PubMed] [Google Scholar]
- 148.Edwards AV, Jones CT. Adrenal cortical and medullary responses to acetylcholine and vasoactive intestinal peptide in conscious calves. J Physiol. 1993;468:515–527. doi: 10.1113/jphysiol.1993.sp019785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fukumitsu N, Suzuki M, Fukuda T, Kiyono Y. Multipoint analysis of reduced (125)I-meta-iodobenzylguanidine uptake and norepinephrine turnover in the hearts of mice with 1-methyl-4-phenyl-1,2,3,6-tetrahydroxypyridine-induced parkinsonism. Nucl Med Biol. 2009;36:623–629. doi: 10.1016/j.nucmedbio.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 150.Takatsu H, Wada H, Maekawa N, Takemura M, Saito K, Fujiwara H. Significant reduction of 125 I-meta-iodobenzylguanidine accumulation directly caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydroxypyridine, a toxic agent for inducing experimental Parkinson’s disease. Nucl Med Commun. 2002;23:161–166. doi: 10.1097/00006231-200202000-00008. [DOI] [PubMed] [Google Scholar]
- 151.Algeri S, Ambrosio S, Garofalo P, Gerli P. Peripheral effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and its main metabolite 1-methyl-4-phenylpyridinium ion (MPP+) in the rat. Eur J Pharmacol. 1987;141:309–312. doi: 10.1016/0014-2999(87)90277-9. [DOI] [PubMed] [Google Scholar]
- 152.Goldstein DS, Li ST, Holmes C, Bankiewicz K. Sympathetic innervation in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine primate model of Parkinson’s disease. J Pharmacol Exp Ther. 2003;306:855–860. doi: 10.1124/jpet.103.051714. [DOI] [PubMed] [Google Scholar]
- 153.Fuller RW, Steranka LR. Central and peripheral catecholamine depletion by 1-methyl-4-phenyl-tetrahydropyridine (MPTP) in rodents. Life Sci. 1985;36:243–247. doi: 10.1016/0024-3205(85)90066-9. [DOI] [PubMed] [Google Scholar]
- 154.Amino T, Uchihara T, Tsunekawa H, Takahata K, Shimazu S, Mizusawa H, Orimo S. Myocardial nerve fibers are preserved in MPTP-treated mice, despite cardiac sympathetic dysfunction. Neurosci Res. 2008;60:314–318. doi: 10.1016/j.neures.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 155.Perese DA, Ulman J, Viola J, Ewing SE, Bankiewicz KS. A 6-hydroxydopamine-induced selective parkinsonian rat model. Brain Res. 1989;494:285–293. doi: 10.1016/0006-8993(89)90597-0. [DOI] [PubMed] [Google Scholar]
- 156.Kostrzewa RM, Jacobowitz DM. Pharmacological actions of 6-hydroxydopamine. Pharmacol Rev. 1974;26:199–288. [PubMed] [Google Scholar]
- 157.Slack K, Billing R, Matthews S, Allbutt HN, Einstein R, Henderson JM. Subtle cardiovascular dysfunction in the unilateral 6-hydroxydopamine-lesioned rat. Parkinsons Dis. 2010;2010:427810. doi: 10.4061/2010/427810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Porter CC, Totaro JA, Burcin A. The relationship between radioactivity and norepinephrine concentrations in the brains and hearts of mice following administration of labeled methyldopa or 6-hydroxydopamine. J Pharmacol Exp Ther. 1965;150:17–22. [PubMed] [Google Scholar]
- 159.Porter CC, Totaro JA, Stone CA. Effect of 6-hydroxydopamine and some other compounds on the concentration of norepinephrine in the hearts of mice. J Pharmacol Exp Ther. 1963;140:308–316. [PubMed] [Google Scholar]
- 160.Arbab AS, Koizumi K, Araki T. Uptake and washout of I-123-MIBG in neuronal and non-neuronal sites in rat hearts: relationship to renal clearance. Ann Nucl Med. 1996;10:211–217. doi: 10.1007/BF03165394. [DOI] [PubMed] [Google Scholar]
- 161.Raffel DM, Koeppe RA, Little R, Wang CN, Liu S, Junck L, Heumann M, Gilman S. PET measurement of cardiac and nigrostriatal denervation in Parkinsonian syndromes. J Nucl Med. 2006;47:1769–1777. [PubMed] [Google Scholar]
- 162.Anzai T, Yoshikawa T, Baba A, Nishimura H, Shiraki H, Nagami K, Suzuki M, Wainai Y, Ogawa S. Myocardial sympathetic denervation prevents chamber-specific alteration of beta-adrenergic transmembrane signaling in rabbits with heart failure. J Am Coll Cardiol. 1996;28:1314–1322. doi: 10.1016/S0735-1097(96)00305-1. [DOI] [PubMed] [Google Scholar]
- 163.Nomura Y, Matsunari I, Takamatsu H, Murakami Y, Matsuya T, Taki J, Nakajima K, Nekolla SG, Chen WP, Kajinami K. Quantitation of cardiac sympathetic innervation in rabbits using 11C-hydroxyephedrine PET: relation to 123I-MIBG uptake. Eur J Nucl Med Mol Imaging. 2006;33:871–878. doi: 10.1007/s00259-006-0105-7. [DOI] [PubMed] [Google Scholar]
- 164.Haeusler G, Haefely W, Thoenen H. Chemical sympathectomy of the cat with 6-hydroxydopamine. J Pharmacol Exp Ther. 1969;170:50–61. [PubMed] [Google Scholar]
- 165.Laverty R, Sharman DF. Modification by Drugs of the Metabolism of 3,4-Dihydroxyphenylethylamine, Noradrenaline and 5-Hydroxytryptamine in the Brain. Br J Pharmacol Chemother. 1965;24:759–772. doi: 10.1111/j.1476-5381.1965.tb01632.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Goldstein DS, Grossman E, Tamrat M, Chang PC, Eisenhofer G, Bacher J, Kirk KL, Bacharach S, Kopin IJ. Positron emission imaging of cardiac sympathetic innervation and function using 18F-6-fluorodopamine: effects of chemical sympathectomy by 6-hydroxydopamine. J Hypertens. 1991;9:417–423. doi: 10.1097/00004872-199105000-00005. [DOI] [PubMed] [Google Scholar]
- 167.Stone CA, Stavorski JM, Ludden CT, Wenger HC, Ross CA, Totaro JA, Porter CC. Comparison of Some Pharmacologic Effects of Certain 6-Substituted Dopamine Derivatives with Reserpine, Guanethidine and Metaraminol. J Pharmacol Exp Ther. 1963;142:147–156. [PubMed] [Google Scholar]
- 168.Joers V, Seneczko K, Goecks NC, Kamp TJ, Hacker TA, Brunner KG, Engle JW, Barnhart TE, Nickles RJ, Holden JE, Emborg ME. Nonuniform cardiac denervation observed by 11C-meta-hydroxyephedrine PET in 6-OHDA-treated monkeys. PLoS One. 2012;7:e35371. doi: 10.1371/journal.pone.0035371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tranzer JP, Thoenen H. An electron microscopic study of selective, acute degeneration of sympathetic nerve terminals after administration of 6-hydroxydopamine. Experientia. 1968;24:155–156. doi: 10.1007/BF02146956. [DOI] [PubMed] [Google Scholar]
- 170.de Champlain J. Degeneration and regrowth of adrenergic nerve fibers in the rat peripheral tissues after 6-hydroxydopamine. Can J Physiol Pharmacol. 1971;49:345–355. doi: 10.1139/y71-038. [DOI] [PubMed] [Google Scholar]
- 171.Orimo S, Oka T, Miura H, Tsuchiya K, Mori F, Wakabayashi K, Nagao T, Yokochi M. Sympathetic cardiac denervation in Parkinson’s disease and pure autonomic failure but not in multiple system atrophy. J Neurol Neurosurg Psychiatry. 2002;73:776–777. doi: 10.1136/jnnp.73.6.776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Weber DA, Ivanovic M. Ultra-high-resolution imaging of small animals: implications for preclinical and research studies. J Nucl Cardiol. 1999;6:332–344. doi: 10.1016/s1071-3581(99)90046-6. [DOI] [PubMed] [Google Scholar]
- 173.Bailey DL, Willowson KP. Quantitative SPECT/CT: SPECT joins PET as a quantitative imaging modality. Eur J Nucl Med Mol Imaging. 2013 doi: 10.1007/s00259-013-2542-4. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 174.Rahmim A, Zaidi H. PET versus SPECT: strengths, limitations and challenges. Nucl Med Commun. 2008;29:193–207. doi: 10.1097/MNM.0b013e3282f3a515. [DOI] [PubMed] [Google Scholar]
- 175.Visser EP, Disselhorst JA, Brom M, Laverman P, Gotthardt M, Oyen WJ, Boerman OC. Spatial resolution and sensitivity of the Inveon small-animal PET scanner. J Nucl Med. 2009;50:139–147. doi: 10.2967/jnumed.108.055152. [DOI] [PubMed] [Google Scholar]
- 176.Langer O, Halldin C. PET and SPET tracers for mapping the cardiac nervous system. Eur J Nucl Med Mol Imaging. 2002;29:416–434. doi: 10.1007/s002590100640. [DOI] [PubMed] [Google Scholar]
- 177.Bengel FM. Imaging targets of the sympathetic nervous system of the heart: translational considerations. J Nucl Med. 2011;52:1167–1170. doi: 10.2967/jnumed.110.084228. [DOI] [PubMed] [Google Scholar]
- 178.Glowniak JV, Turner FE, Gray LL, Palac RT, Lagunas-Solar MC, Woodward WR. Iodine-123 metaiodobenzylguanidine imaging of the heart in idiopathic congestive cardiomyopathy and cardiac transplants. J Nucl Med. 1989;30:1182–1191. [PubMed] [Google Scholar]
- 179.Morozumi T, Kusuoka H, Fukuchi K, Tani A, Uehara T, Matsuda S, Tsujimura E, Ito Y, Hori M, Kamada T, Nishimura T. Myocardial iodine-123-metaiodobenzylguanidine images and autonomic nerve activity in normal subjects. J Nucl Med. 1997;38:49–52. [PubMed] [Google Scholar]
- 180.Travin MI. Cardiac neuronal imaging at the edge of clinical application. Cardiol Clin. 2009;27:311–327. doi: 10.1016/j.ccl.2008.12.007. Table of Contents. [DOI] [PubMed] [Google Scholar]
- 181.Braune S, Reinhardt M, Bathmann J, Krause T, Lehmann M, Lucking CH. Impaired cardiac uptake of meta-[123I] iodobenzylguanidine in Parkinson’s disease with autonomic failure. Acta Neurol Scand. 1998;97:307–314. doi: 10.1111/j.1600-0404.1998.tb05958.x. [DOI] [PubMed] [Google Scholar]
- 182.Courbon F, Brefel-Courbon C, Thalamas C, Alibelli MJ, Berry I, Montastruc JL, Rascol O, Senard JM. Cardiac MIBG scintigraphy is a sensitive tool for detecting cardiac sympathetic denervation in Parkinson’s disease. Mov Disord. 2003;18:890–897. doi: 10.1002/mds.10461. [DOI] [PubMed] [Google Scholar]
- 183.Orimo S, Ozawa E, Nakade S, Sugimoto T, Mizusawa H. (123)I-metaiodobenzylguanidine myocardial scintigraphy in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 1999;67:189–194. doi: 10.1136/jnnp.67.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Taki J, Nakajima K, Hwang EH, Matsunari I, Komai K, Yoshita M, Sakajiri K, Tonami N. Peripheral sympathetic dysfunction in patients with Parkinson’s disease without autonomic failure is heart selective and disease specific. taki@med.kanazawa-u.ac.jp. Eur J Nucl Med. 2000;27:566–573. doi: 10.1007/s002590050544. [DOI] [PubMed] [Google Scholar]
- 185.Treglia G, Cason E, Stefanelli A, Cocciolillo F, Di Giuda D, Fagioli G, Giordano A. MIBG scintigraphy in differential diagnosis of Parkinsonism: a meta-analysis. Clin Auton Res. 2012;22:43–55. doi: 10.1007/s10286-011-0135-5. [DOI] [PubMed] [Google Scholar]
- 186.Orimo S, Suzuki M, Inaba A, Mizusawa H. 123I-MIBG myocardial scintigraphy for differentiating Parkinson’s disease from other neurodegenerative parkinsonism: a systematic review and meta-analysis. Parkinsonism Relat Disord. 2012;18:494–500. doi: 10.1016/j.parkreldis.2012.01.009. [DOI] [PubMed] [Google Scholar]
- 187.Spiegel J, Mollers MO, Jost WH, Fuss G, Samnick S, Dillmann U, Becker G, Kirsch CM. FP-CIT and MIBG scintigraphy in early Parkinson’s disease. Mov Disord. 2005;20:552–561. doi: 10.1002/mds.20369. [DOI] [PubMed] [Google Scholar]
- 188.Chiaravalloti A, Stefani A, Pierantozzi M, Stanzione P, Schillaci O. Does 123I-MIBG scintigraphy really assist the diagnosis of Parkinson’s disease? Parkinsonism Relat Disord. 2013;19:772–773. doi: 10.1016/j.parkreldis.2013.04.015. [DOI] [PubMed] [Google Scholar]
- 189.Spiegel J, Hellwig D, Farmakis G, Jost WH, Samnick S, Fassbender K, Kirsch CM, Dillmann U. Myocardial sympathetic degeneration correlates with clinical phenotype of Parkinson’s disease. Mov Disord. 2007;22:1004–1008. doi: 10.1002/mds.21499. [DOI] [PubMed] [Google Scholar]
- 190.Tijero B, Gomez Esteban JC, Somme J, Llorens V, Lezcano E, Martinez A, Rodriguez T, Berganzo K, Zarranz JJ. Autonomic dysfunction in parkinsonian LRRK2 mutation carriers. Parkinsonism Relat Disord. 2013;19:906–909. doi: 10.1016/j.parkreldis.2013.05.008. [DOI] [PubMed] [Google Scholar]
- 191.Orimo S, Amino T, Yokochi M, Kojo T, Uchihara T, Takahashi A, Wakabayashi K, Takahashi H, Hattori N, Mizuno Y. Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord. 2005;20:1350–1353. doi: 10.1002/mds.20594. [DOI] [PubMed] [Google Scholar]
- 192.Goethals LR, Weytjens CD, De Geeter F, Droogmans S, Caveliers V, Keyaerts M, Vanhove C, Van Camp G, Bossuyt A, Lahoutte T. Regional quantitative analysis of small animal myocardial sympathetic innervation and initial application in streptozotocin induced diabetes. Contrast Media Mol Imaging. 2009;4:174–182. doi: 10.1002/cmmi.278. [DOI] [PubMed] [Google Scholar]
- 193.Kiyono Y, Iida Y, Kawashima H, Tamaki N, Nishimura H, Saji H. Regional alterations of myocardial norepinephrine transporter density in streptozotocin-induced diabetic rats: implications for heterogeneous cardiac accumulation of MIBG in diabetes. Eur J Nucl Med. 2001;28:894–899. doi: 10.1007/s002590100550. [DOI] [PubMed] [Google Scholar]
- 194.Kiyono Y, Kajiyama S, Fujiwara H, Kanegawa N, Saji H. Influence of the polyol pathway on norepinephrine transporter reduction in diabetic cardiac sympathetic nerves: implications for heterogeneous accumulation of MIBG. Eur J Nucl Med Mol Imaging. 2005;32:993–997. doi: 10.1007/s00259-005-1865-1. [DOI] [PubMed] [Google Scholar]
- 195.Matsunari I, Bunko H, Taki J, Nakajima K, Muramori A, Kuji I, Miyauchi T, Tonami N, Hisada K. Regional uptake of iodine-125-metaiodobenzylguanidine in the rat heart. Eur J Nucl Med. 1993;20:1104–1107. doi: 10.1007/BF00173491. [DOI] [PubMed] [Google Scholar]
- 196.Sisson JC, Wieland DM, Sherman P, Mangner TJ, Tobes MC, Jacques S Jr. Metaiodobenzylguanidine as an index of the adrenergic nervous system integrity and function. J Nucl Med. 1987;28:1620–1624. [PubMed] [Google Scholar]
- 197.Samnick S, Scheuer C, Munks S, El-Gibaly AM, Menger MD, Kirsch CM. Technetium-99m labeled 1-(4-fluorobenzyl)-4-(2-mercapto-2- methyl-4-azapentyl)-4-(2-mercapto-2-methylprop ylamino)-piperidine and iodine-123 metaiodobenzylguanidine for studying cardiac adrenergic function: a comparison of the uptake characteristics in vascular smooth muscle cells and neonatal cardiac myocytes, and an investigation in rats. Nucl Med Biol. 2004;31:511–522. doi: 10.1016/j.nucmedbio.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 198.Richter S, Schaefer A, Menger MD, Kirsch CM, Samnick S. Mapping of the cardiac sympathetic nervous system by single photon emission tomography with technetium-99m-labelled fluorobenzylpiperidine derivative (99mTc-FBPBAT): result of a feasibility study in a porcine model and an initial dosimetric estimation in humans. Nucl Med Commun. 2005;26:361–368. doi: 10.1097/00006231-200504000-00009. [DOI] [PubMed] [Google Scholar]
- 199.Chang PC, Szemeredi K, Grossman E, Kopin IJ, Goldstein DS. Fate of tritiated 6-fluorodopamine in rats: a false neurotransmitter for positron emission tomographic imaging of sympathetic innervation and function. J Pharmacol Exp Ther. 1990;255:809–817. [PubMed] [Google Scholar]
- 200.Goldstein DS, Eisenhofer G, Dunn BB, Armando I, Lenders J, Grossman E, Holmes C, Kirk KL, Bacharach S, Adams R, et al. Positron emission tomographic imaging of cardiac sympathetic innervation using 6-[18F] fluorodopamine: initial findings in humans. J Am Coll Cardiol. 1993;22:1961–1971. doi: 10.1016/0735-1097(93)90786-z. [DOI] [PubMed] [Google Scholar]
- 201.Goldstein DS, Holmes C, Cannon RO 3rd, Eisenhofer G, Kopin IJ. Sympathetic cardioneuropathy in dysautonomias. N Engl J Med. 1997;336:696–702. doi: 10.1056/NEJM199703063361004. [DOI] [PubMed] [Google Scholar]
- 202.Goldstein DS, Sharabi Y, Karp BI, Bentho O, Saleem A, Pacak K, Eisenhofer G. Cardiac sympathetic denervation preceding motor signs in Parkinson disease. Clin Auton Res. 2007;17:118–121. doi: 10.1007/s10286-007-0396-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Li ST, Dendi R, Holmes C, Goldstein DS. Progressive loss of cardiac sympathetic innervation in Parkinson’s disease. Ann Neurol. 2002;52:220–223. doi: 10.1002/ana.10236. [DOI] [PubMed] [Google Scholar]
- 204.Tipre DN, Goldstein DS. Cardiac and extracardiac sympathetic denervation in Parkinson’s disease with orthostatic hypotension and in pure autonomic failure. J Nucl Med. 2005;46:1775–1781. [PubMed] [Google Scholar]
- 205.Goldstein M, Lieberman AN, Takasugi N, Shimizu Y, Kuga S. The antiparkinsonian activity of dopamine agonists and their interaction with central dopamine receptor subtypes. Adv Neurol. 1990;53:101–106. [PubMed] [Google Scholar]
- 206.Langer O, Dolle F, Valette H, Halldin C, Vaufrey F, Fuseau C, Coulon C, Ottaviani M, Nagren K, Bottlaender M, Maziere B, Crouzel C. Synthesis of high-specific-radioactivity 4- and 6-[18F] fluorometaraminol-PET tracers for the adrenergic nervous system of the heart. Bioorg Med Chem. 2001;9:677–694. doi: 10.1016/s0968-0896(00)00286-8. [DOI] [PubMed] [Google Scholar]
- 207.Rosenspire KC, Haka MS, Van Dort ME, Jewett DM, Gildersleeve DL, Schwaiger M, Wieland DM. Synthesis and preliminary evaluation of carbon-11-meta-hydroxyephedrine: a false transmitter agent for heart neuronal imaging. J Nucl Med. 1990;31:1328–1334. [PubMed] [Google Scholar]
- 208.Berding G, Schrader CH, Peschel T, van den Hoff J, Kolbe H, Meyer GJ, Dengler R, Knapp WH. [N-methyl 11C] meta-Hydroxyephe-drine positron emission tomography in Parkinson’s disease and multiple system atrophy. Eur J Nucl Med Mol Imaging. 2003;30:127–131. doi: 10.1007/s00259-002-1019-7. [DOI] [PubMed] [Google Scholar]
- 209.Munch G, Nguyen NT, Nekolla S, Ziegler S, Muzik O, Chakraborty P, Wieland DM, Schwaiger M. Evaluation of sympathetic nerve terminals with [(11)C] epinephrine and [(11)C] hydroxyephedrine and positron emission tomography. Circulation. 2000;101:516–523. doi: 10.1161/01.cir.101.5.516. [DOI] [PubMed] [Google Scholar]
- 210.DeGrado TR, Bergmann SR, Ng CK, Raffel DM. Tracer kinetic modeling in nuclear cardiology. J Nucl Cardiol. 2000;7:686–700. doi: 10.1067/mnc.2000.111127. [DOI] [PubMed] [Google Scholar]
- 211.Schwaiger M, Muzik O. Assessment of myocardial perfusion by positron emission tomography. Am J Cardiol. 1991;67:35D–43D. doi: 10.1016/s0002-9149(05)80006-2. [DOI] [PubMed] [Google Scholar]
- 212.DeGrado TR, Hutchins GD, Toorongian SA, Wieland DM, Schwaiger M. Myocardial kinetics of carbon-11-meta-hydroxyephedrine: retention mechanisms and effects of norepinephrine. J Nucl Med. 1993;34:1287–1293. [PubMed] [Google Scholar]
- 213.Logan J, Fowler JS, Volkow ND, Wolf AP, Dewey SL, Schlyer DJ, MacGregor RR, Hitzemann R, Bendriem B, Gatley SJ, et al. Graphical analysis of reversible radioligand binding from time-activity measurements applied to [N-11C-methyl] -(-)-cocaine PET studies in human subjects. J Cereb Blood Flow Metab. 1990;10:740–747. doi: 10.1038/jcbfm.1990.127. [DOI] [PubMed] [Google Scholar]
- 214.Berding G, Brucke T, Odin P, Brooks DJ, Kolbe H, Gielow P, Harke H, Knoop BO, Dengler R, Knapp WH. [123I] beta-CIT SPECT imaging of dopamine and serotonin transporters in Parkinson’s disease and multiple system atrophy. Nuklearmedizin. 2003;42:31–38. [PubMed] [Google Scholar]
- 215.Raffel DM, Chen W, Sherman PS, Gildersleeve DL, Jung YW. Dependence of cardiac 11C-meta-hydroxyephedrine retention on norepinephrine transporter density. J Nucl Med. 2006;47:1490–1496. [PMC free article] [PubMed] [Google Scholar]
- 216.Wong KK, Raffel DM, Koeppe RA, Frey KA, Bohnen NI, Gilman S. Pattern of cardiac sympathetic denervation in idiopathic Parkinson disease studied with 11C hydroxyephedrine PET. Radiology. 2012;265:240–247. doi: 10.1148/radiol.12112723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Thackeray JT, Parsa-Nezhad M, Kenk M, Thorn SL, Kolajova M, Beanlands RS, DaSilva JN. Reduced CGP12177 binding to cardiac beta-adrenoceptors in hyperglycemic high-fat-diet-fed, streptozotocin-induced diabetic rats. Nucl Med Biol. 2011;38:1059–1066. doi: 10.1016/j.nucmedbio.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 218.Joers V, Dilley K, Rahman S, Simmons H, Emborg ME. Society of Neuroscience. San Diego, CA: 2013. Morphological evaluation of a monkey model of cardiac dysautonomia. No. 137.02, G11. [Google Scholar]
- 219.Sletten DM, Suarez GA, Low PA, Mandrekar J, Singer W. COMPASS 31: a refined and abbreviated Composite Autonomic Symptom Score. Mayo Clin Proc. 2012;87:1196–1201. doi: 10.1016/j.mayocp.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Suarez GA, Opfer-Gehrking TL, Offord KP, Atkinson EJ, O’Brien PC, Low PA. The Autonomic Symptom Profile: a new instrument to assess autonomic symptoms. Neurology. 1999;52:523–528. doi: 10.1212/wnl.52.3.523. [DOI] [PubMed] [Google Scholar]
- 221.Low PA. Composite autonomic scoring scale for laboratory quantification of generalized autonomic failure. Mayo Clin Proc. 1993;68:748–752. doi: 10.1016/s0025-6196(12)60631-4. [DOI] [PubMed] [Google Scholar]
- 222.Imaging guidelines for nuclear cardiology procedures, part 2. American Society of Nuclear Cardiology. J Nucl Cardiol. 1999;6:G47–84. [PubMed] [Google Scholar]