

Abstract
The global prevalence of dementia is as high as 24 million, and has been predicted to quadruple by the year 2050. In the US alone, Alzheimer disease (AD) – the most frequent cause of dementia characterized by a progressive decline in cognitive function in particular the memory domain -causes estimated health-care costs of $172 billion per year. Key neuropathological hallmarks of the AD brain are diffuse and neuritic extracellular amyloid plaques—often surrounded by dystrophic neurites—and intracellular neurofibrillary tangles. These pathological changes are frequently accompanied by reactive microgliosis and loss of neurons, white matter and synapses. The etiological mechanisms underlying these neuropathological changes remain unclear, but are probably caused by both environmental and genetic factors. In this review article, we provide an overview of the epidemiology of AD, review the biomarkers that may be used for risk assessment and in diagnosis, and give suggestions for future research
Introduction
The global prevalence of dementia, which is characterized by progressive deterioration in cognition, function and behavior, places a considerable burden on society. Currently, the prevalence is estimated to amount to 24 million and predicted to quadruple by the year 2050. In the US alone, Alzheimer disease (AD) – the most frequent cause of dementia- is associated with estimated health-care costs of $172 billion per year.(1)
The key pathological changes observed in AD brain tissue are amyloid-β (Aβ) peptide deposited extracellularly in diffuse and neuritic plaques, and hyperphosphorylated tau (p-tau) protein, a microtubule assembly protein accumulating intracellularly as neurofibrillary tangles (NFTs). Additional changes include reactive microgliosis and widespread loss of neurons, white matter and synapses. The exact mechanisms leading to these changes remain to be determined.
Diagnostic criteria
Since their proposal in 1984, the key classification for the diagnosis of AD has been the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS–ADRDA) criteria.(2) These criteria combine clinical and neuropathological patterns and assign diagnoses of “possible”, “probable” and “definite AD”.(2) The AD spectrum is now recognized to be broader than was previously thought and is acknowledged to include pathological changes other than amyloid plaques and NFTs (see below). Correspondingly, the NINCDS–ADRDA criteria are again under review. The biomarkers incorporated in the updated criteria are expected to increase the diagnostic specificity. In 1999, an intermediate state between normal cognition and dementia has been defined as “mild cognitive impairment (MCI)”. In particular in clinical settings MCI has proved a useful label to define people who are at risk of developing AD.
Prevalence and incidence
By 2005, 24.2 million people worldwide had dementia and 4.6 million new cases were arising every year.(3) Approximately 70% of these cases were attributed to AD. Among regional populations of 60 year-olds, those from North America and Western Europe are believed to exhibit the highest prevalence and incidence rate of dementia, followed by those from Latin America and China and its western-Pacific neighbours (Figures 1a and 1b).(3) For all these populations, the incidence rate for dementia increases exponentially with age, with the most pronounced increase occurring through the 7th and 8th decades of life. Similar patterns are observed for the prevalence and incidence of AD. There is evidence that in western societies prevalence and increase display a cohort effect with later-born individuals having a lower risk than those born earlier in the past century.(4–7)
Figure 1a.
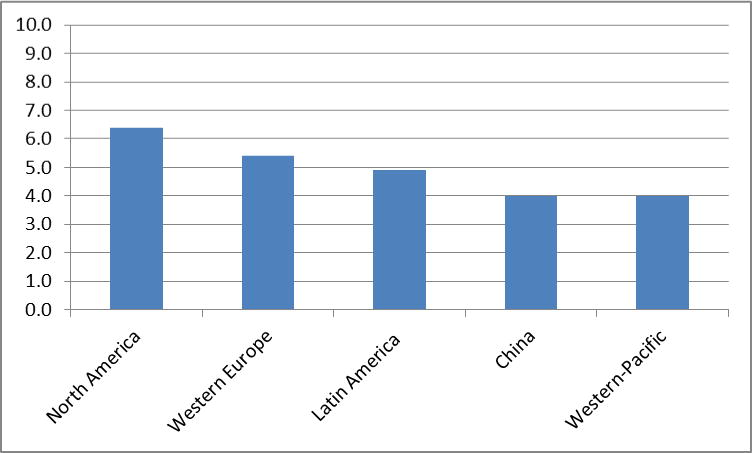
Global prevalence of dementia (%) (3)
Figure 1b.
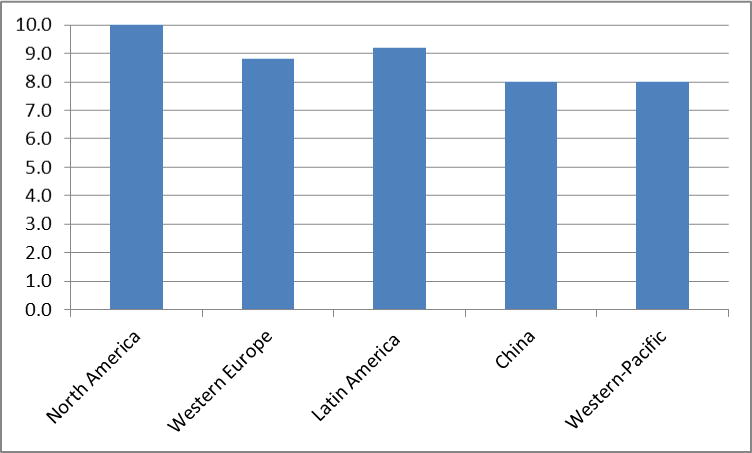
Incidence rates (per 1000 individuals in the population) (3)
Genetic epidemiology of AD
Based on its age of onset, AD is classified into early onset AD (EOAD, onset < 65 years) accounting for 1–5% of all cases, and late-onset AD (LOAD, onset ≥ 65 years) accounting for >95% of affecteds. While clinically indistinguishable from LOAD, EOAD is generally associated with a more rapid rate of progression and a Mendelian pattern of inheritance. Three genes (APP, PSEN1 and PSEN2) which all encode proteins involved in APP breakdown and Aβ generation, have been firmly implicated in the pathophysiology of EOAD. AD-linked mutations in these three genes exhibit high penetrance (>85%), are mostly autosomal dominantly inherited, and lead with certainty to Aβ aggregation and early-onset disease. Consequently, they are considered ‘diagnostic biomarkers’ of the disease.
In contrast, the genes involved in LOAD increase disease risk in a non-Mendelian fashion. First-degree relatives of patients with LOAD have twice the expected life-time risk of people without an AD-affected first-degree relative. In addition, LOAD occurs more frequently in monozygotic than in dizygotic co-twins, suggesting a substantial genetic contribution of ~60–80% to this disorder.
For more than a decade, only one genetic risk factor, the APOEε4 allele, located on chromosome 19q13, was an unequivocally established “susceptibility” gene in non-Hispanic Whites of European ancestry. APOE is a lipid-binding protein and is expressed in humans as three common isoforms coded for by three alleles, APOEε2, ε3, and ε4. A single APOE-ε4 allele is associated with a 2- to 3-fold increased risk, having two copies is associated with a five-fold or more increase.(8) In addition, each inherited APOEε4 allele lowers the age-at-onset by 6–7 years.(9–16) APOEε4 is also associated with lower cognitive performance, in particular the memory domain, is associated with mild cognitive impairment (MCI)(17–20) and with progression from MCI to dementia.(17–27) While the population attributable risk for APOEε4 is estimated at 20–50%,(28) the presence of ε4 is neither necessary nor sufficient for developing the disease.(29) In ethnic groups other than non-Hispanic Whites the association between APOE and LOAD was largely inconsistent across studies.
Findings from genome-wide association studies (GWAS)
In the beginning of the century, thousands of candidate-gene-based association studies aiming to identify additional susceptibility loci were performed but only one gene, the sortilin-related receptor (SORL1) which is implicated in intracellular trafficking of APP, could be consistently replicated in independent datasets and implicated in the disease. The main reasons for these inconsistencies between studies are sample heterogeneity with differences in linkage disequilibrium (LD) patterns and allele frequencies, and small sample sizes leading to limited power to detect small or moderate effect sizes. In the past five years, technological advances in high-throughput genome-wide arrays allowed the hypothesis-free simultaneous examination of thousands to millions of polymorphisms across the genome, and large international collaborative efforts capitalizing on this technology have significantly advanced the knowledge on the genetic underpinnings of LOAD and pathways involved by identifying more than 20 novel risk loci.
The major GWAS studies contributing to this gained knowledge are summarized in table 1. Most were performed in non-Hispanic Whites of European ancestry. The first set of studies identified CLU, PICALM, CR1 and BIN1 as susceptibility loci.(30–32) CLU, also known as apolipoprotein J (ApoJ), is a lipoprotein highly expressed in both the periphery and the brain.(33) Like ApoE, it is involved in lipid transport.(34) Clu is also hypothesized to act as an extracellular chaperone that influences Aβ-aggregation and receptor-mediated Aβ clearance by endocytosis.(33) Unlike APOE, there are no known coding variants that account for the observed genetic association to CLU, suggesting that genetic variation in expression levels may be responsible for the altered risk for LOAD.(35) BIN1 (amphiphysin II) is a member of the Bin1/amphiphysin/RVS167 (BAR) family of genes that have been involved in diverse cellular processes, including actin dynamics, membrane trafficking and clathrin-mediated endocytosis(36) which affect APP processing and Aβ production or Aβ clearance from brain. PICALM is also involved in clathrin-mediated endocytosis and recruits clathrin and adaptor protein complex 2 to sites of vesicle assembly(37). CR1 is a cell-surface receptor that is part of the complement system. It has binding sites for complement factors C3b and C4b and is involved in clearing immune-complexes containing these two proteins. Since Aβ oligomers can bind C3b, CR1 may participate in the clearance of Aβ. CR1 may also play a role in neuroinflammation, which is a prominent feature in AD.(38) Interestingly, Clu may play a role in this process as an inhibitor(39). In summary, this first set of GWAS identified loci mainly clustering in three pathways, namely immune response, APP processing and lipid metabolism and endocytosis. The second set of large GWAS studies identified additional susceptibility genes (CD33, MS4A4A/MS4A4E/MS4A6E cluster, ABCA7, CD2AP and EPHA1).(40, 41) In line with the pathways identified by the first set of GWAS, all of these five loci are likely involved in the immune system while the ABCA7 is in addition involved in lipid metabolism and APP processing. The CD33 gene encodes a protein that is a member of a family of cell surface immune receptors that bind extracellular sialylated glycans and signal via a cytoplasmic domain called the immunoreceptor tyrosine inhibitory motif.(42, 43) CD33 has primarily been studied in the peripheral immune system where it is expressed on myeloid progenitors and monocytes and also in the brain. In the periphery, CD33 appears to inhibit proliferation of myeloid cells.(44) The MS4A4A/MS4A4E/MS4A6E locus is part of a cluster of 15 MS4A genes on chromosome 11 and encodes proteins with multiple membrane-spanning domains that were initially identified by their homology to CD20, a B-lymphocyte cell surface molecule. Little is known about the function of MS4A4A gene products; however, like CD33, MS4A4A is expressed on myeloid cells and monocytes and likely has an immune-related function. EPHA1 encodes a member of the ephrin family of cell surface receptors which interact with ephrin ligands on adjacent cells to modulate cell adhesion, migration, and axon guidance and synapse formation and plasticity. While there is a substantial body of research on the function of ephrin receptors in general, little is known about the EPHA1 gene product. Like other ephrin receptors, it regulates cell morphology and motility(45) and early work implicated this receptor in regulating vascular morphogenesis and angiogenesis.(46) EPHA1 knockout in mouse results in abnormal tail and reproductive tract development,(47) but no effects on the brain. Consistent with this notion, in mouse, expression is restricted to epithelial tissue. In humans, EPHA1 is expressed by CD4-positive T lymphocytes(48), monocytes,(49) intestinal epithelium, and colon. Combined with the lack of evidence for brain expression this may suggest that, like CD33, CR1, and MS4A4/MS4A6E, the role of the EPHA1 gene product in AD may be mediated though the immune system. The CD2 associated protein gene (CD2AP) encodes a scaffolding protein that binds directly to actin(50), nephrin and other proteins involved in cytoskeletal organization. In the immune system, CD2AP is required for synapse formation(51) in a process that involves clathrin-dependent actin polymerization. ABCA7 is an integral transmembrane ATP-binding cassette transporter belonging to the ABC family proteins that mediate the biogenesis of high-density lipoprotein with cellular lipid and helical apolipoproteins.(52) It binds APOA-I and functions in apolipoprotein-mediated phospholipid and cholesterol efflux from cells.(53) In addition, ABCA7 affects the transport of other important proteins, including amyloid precursor protein,(53) through the cell membrane and is involved in host defense through effects on phagocytosis by macrophages of apoptotic cells.(52)
Table 1.
Major GWAS studies performed
| Study | Ethnic group | Sample size | Genes identified outside APOE region |
|---|---|---|---|
| Lambert et al. (2009) | Caucasian | Stage 1: 2,032 AD cases; 5,328 controls Stage 2: 3,978 AD cases; 3,297 controls |
CLU, CR1 |
| Harold et al. (2009) | Caucasian | Stage 1: 3,941 AD cases; 7,848 controls Stage 2: 2,023 AD cases; 2,340 controls |
CLU, PICALM |
| Seshadri et al. (2010) | Caucasian | Stage 1: 3,006 AD cases; 4,642 controls Stage 2: 2,032 AD cases; 5,328 controls Stage 3: 3,333 AD cases; 6,995 controls |
BIN1, XOC3L2/BLOC1S3/MARK4, CLU, PICALM |
| Naj et al. (2011) | Caucasian | Stage 1: 8,309 AD cases; 7,366 controls Stage 2: 3,531 AD cases; 3,565 controls |
MS4A4A, CD2AP, CD33 and EPHA1, CR1, CLU, BIN1, PICALM |
| Hollingworth et al (2011) | Caucasian | Stage 1: 6,688 AD cases; 13,685 controls Stage 2: 4,896 AD cases; 4,903 controls Stage 3: 8,286 AD cases; 21,258 controls |
ABCA7, MS4A6A/MS4A4E, EPHA1, CD33, CD2AP |
| Lambert et al. (2013) | Caucasian | Stage 1: 17,008 AD cases; 37,646 controls Stage 2: 8,572 AD cases; 11,312 controls |
CR1, CD33, BIN1, CD2AP, CLU, EPHA1, PICALM, MS4, ABCA7HLA-DRB5/HLA-DRB1, PTK2B, SORL1, SLC24A4/RIN3,DSG2 |
| Lee et al. (2011) | Caribbean Hispanic |
549 AD cases; 544 controls | CLU, PICALM, BIN1, CUGBP2, loci on 2p25.1; 3q25.2; 7p21.1; 10q23.1 |
| Reitz et al. (2013) | African American |
1,968 AD cases; 3,928 controls | ABCA7, intergenic locus on 5q35.2 |
Finally, the largest GWAS to date, performed by the International Genomics of Alzheimer’s Project (IGAP), combined all non-Hispanic White datasets from the four individual consortia and performed a mega-metaanalysis that included 74,046 subjects.(54) In addition to APOE, CR1, BIN1, CD2AP, EPHA1, CLU, MS4A6A, PICALM, ABCA7 and CD33 this GWAS identified 12 additional susceptibility loci at genome-wide significance (HLA-DRB5/HLA-DRB1, PTK2B, SORL1, SLC24A4/RIN3, DSG2, INPP5D, MEF2C, NME8, ZCWPW1, CELF1, FERMT2, CASS4) and an additional 13 candidate loci with p-values between p=7.4×10−7 and p=6.6×10−8 (intergenic locus, HS3ST1, SQSTM1, TREML2, NDUFAF6, ECHDC3, AP2A2, ADAMTS20, IGH, SPPL2A, TRIP4, SCIMP, ACE). As described above, the SORL1 (sortilin-related receptor, L(DLR class) 1) had previously been demonstrated to modulate trafficking and processing of APP in a candidate gene approach.(55, 56) With the exception of CD33 and DSG2, all loci could be validated in a replication stage. Failure to replicate DSG2 encoding desmoglein2 – a calcium-binding transmembrane glycoprotein component of desmosomes in vertebrate epithelial cells-was expected as evidence for this locus in the discovery stage was based on a single SNP and was not supported by any SNP in LD. Out of the 12 novel loci reaching genome-wide significance, most cluster in the specific pathways identified by the earlier GWAS, ie. immune response (HLA-DRB5/DRB1, INPP5D, MEF2C), APP processing (SORL1, CASS4), Tau pathology (CASS4, FERMT2), cell migration (PTK2B) and lipid transport and endocytosis (SORL1) strongly reinforcing the importance of these pathways in LOAD etiology. In addition, consistent with the notion of a complex disease, the findings of this study further suggest the existence of additional pathways. One of these may be hippocampal synaptic function: MEF2C limits excessive synapse formation during activity-dependent refinement of synaptic connectivity and thus may facilitate hippocampal-dependent learning and memory(57). Mutations and deletions at this locus have also been are associated with mental retardation, stereotypic movements, epilepsy, and cerebral malformation. The protein encoded by PTK2B is involved in induction of long-term potentiation in the hippocampal CA1(cornu ammonis 1) region, a central process in the formation of memories. PTK2B is only ~130 kb away from CLU. However, the fact that the two most strongly associated SNPs at these loci are not in LD and that there is a recombination hotspot between these loci suggests that both associations are independent. Another pathway suggested by the signals in CELF1, NME8 and CASS4 may be cytoskeletal function and axonal transport.
In these GWAS performed in non-Hispanic Whites of European ancestry, the most strongly associated SNPs at each locus other than APOE demonstrated population attributable fractions (PAFs) between 1.0%–8.0% with effect sizes ranging from ORs of 1.16 to 1.20, ie. much smaller than for APOE.(58) In the largest GWAS performed to date in Caribbean Hispanics(59) associations in CLU, PICALM, and BIN1 were replicated and several additional loci on 2p25.1, 3q25.2, 7p21.1 and 10q23.1 – which could be replicated in an independent cohort of non-Hispanic Whites of European ancestry from the National Institute on Aging Late-Onset Alzheimer’s Disease Family Study (NIA-LOAD) – were observed. Finally, in the largest GWAS of African Americans performed, Reitz et al.(60) identified ABCA7 as a major susceptibility locus in this ethnic group. Interestingly, in contrast to all GWAS loci identified in Caucasians, the ABCA7 locus had in African Americans an effect size as strong as that of APOEe4 (ie. a 70–80% increase in risk compared to a 10–20% increase in risk through the GWAS loci observed in Whites). Although this finding may represent a winner’s curse (ie. inflation of the estimated effect in a discovery set in relation to follow-up studies) and needs to be confirmed by independent studies in African Americans and functional methods, this finding may have major implications for developing targets for genetic testing, prevention and treatment in this ethnic group if proven true. In addition, this study confirmed APOE as a susceptibility gene in this ethnic group, which had been prior to this study inconsistent across studies.
Non-genetic Risk and protective factors
Cerebrovascular disease
Cerebrovascular changes such as hemorrhagic infarcts, small and large ischemic cortical infarcts, vasculopathies and white matter changes increase the risk of dementia but the specific underlying mecahnisms remain unclear. Infarcts or white matter hyperintensies may lead directly to the damage of brain regions that are important in memory function, such as the thalamus and the thalamo-cortical projections. However, they may also increase the deposition of Aβ, which in turn can lead to cognitive decline or could induce inflammatory responses impairing cognitive function. Finally, hypoperfusion may lead to overexpression of cyclin-dependent kinase 5 (CDK5), a serine–threonine kinase critical to synapse formation and synaptic plasticity.(61) Aberrant CDK5 activation is associated with neuronal apoptosis and death.(62) This kinase may also be involved in the abnormal phosphorylation of tau, contributing to the formation of NFTs,(63) and might be a key protein linking NFT pathology to amyloid plaques.
Blood pressure
Data from cross-sectional studies and longitudinal studies relating blood pressure levels measured in late life with cognitive decline and dementia remain inconsistent.(64–67) To some extent, these controveries can be attributed to differences in study design, specifically variation in the time between measurement of blood pressure and assessment of cognitive abilities and in the age at which these parameters were measured. However, data from observational studies exploring the association between elevated levels of blood pressure in mid-life (40–60 years of age) and late-life cognitive impairment have proved to be relatively consistent across cohorts suggesting that elevated blood pressure in mid-life does increase the risk of later-life cognitive impairment, dementia and AD. (68–71) Hypertension may increase the risk of AD through an effect on the vascular integrity of the blood–brain barrier (BBB), resulting in protein extravasation into brain tissue.(72) In turn, protein extravasation can lead to cell damage, a reduction in neuronal or synaptic function, apoptosis, and an increase in Aβ accumulation resulting in cognitive impairment.(73) With increasing age, the effect of elevated blood pressure on AD risk diminishes and may even become inverted, with an increase in blood pressure showing a protective effect. This observation may be explained by the fact that following the onset of AD, blood pressure begins to decrease, possibly as a result of vessel stiffening, weight loss and changes in the autonomic regulation of blood flow. The randomized, placebo-controlled trials (RCTs) evaluating the benefit of antihypertensive treatments in patients with cognitive impairment were inconsistent.(74–80)
Type 2 diabetes
In observational studies, type 2 diabetes (T2D) has been found to nearly double the risk of AD.(81, 82) The mechanisms linking T2D and LOAD are not clear but may include cerebrovascular and noncerebrovascular mechanisms.(83) T2D is a risk factor for stroke and is accompanied by other vascular risk factors including hypertension and dyslipidemia.(84) The observation from pathology studies that T2D is associated with infarcts but not AD pathology in persons with clinical LOAD(85) suggests that the presence of infacrtcs – which in turn decreases the thresehold of amyloid necessary to cause cognitive impairment- may represent the main mechanism linking T2D to LOAD.
Noncerebrovascular mechanisms potentially linking T2D and LOAD include hyperinsulinemia and advanced products of glycosylation. Hyperinsulinemia precedes and may accompany T2D.(86) Insulin can cross the blood-brain barrier, and peripheral insulin infusion in the elderly increases 42-amino-acid β-amyloid (Aβ42) levels in the CSF,(87) a surrogate marker of Aβ clearance in the brain and an indirect marker of LOAD risk. There are insulin receptors in the brain including the hippocampus and entorhinal cortex, structures affected early in LOAD. Insulin-degrading enzyme (IDE) has been linked to clearance of Aβ in the brain, and insulin and Aβ are both competing substrates for IDE. (88) Insulin in the brain can increase the deposition of Aβ and tau protein phosphorylation, which are central to the pathogenesis of LOAD.(89) Peripheral hyperinsulinemia may downregulate insulin uptake in the blood-brain barrier due to saturation over physiological levels. This may result in reduction of insulin levels in the brain and downregulation of expression of IDE and reduction in IDE-mediated amyloid reduction. This observation has been used to support the use of rosiglitazone, an insulin sensitizer, and intranasal insulin in the treatment of LOAD. In a T2D environment, diabetic animal and human tissues contain increased advanced products of glycosylation and upregulation of their receptor. Increased expression of this receptor is observed in LOAD.
Body weight
Prospective studies have linked both low and high body weight to an increased risk of cognitive impairment and AD and suggesting a U-shaped relationship(90–92) that is dependent on the age at which body weight is measured and seems to be driven by central obesity.(93) In addition, there is evidence for reverse causation in the years preceding dementia onset caused by loss of body weight due to malnutrition during the prodromal phase of dementia.(90)
Plasma lipid levels
While most cross-sectional studies and observational studies relating dyslipidemia in late life with cognitive impairment or AD are inconsistent,(94–96) studies that explored the association between lipids measured in mid-life and risk of incident AD mostly indicate a harmful effect. The latter findings are supported by genetic linkage and association studies that have clearly identified several genes involved in cholesterol metabolism or transport as AD susceptibility genes, including apolipoprotein E (APOE), apolipoprotein J (APOJ, CLU), ATP-binding cassette subfamily A member 7(ABCA7), and sortilin-related receptor (SORL1). Functional cell biology studies further support a critical involvement of lipid raft cholesterol in the modulation of Aβ precursor protein processing by β-secretase and γ-secretase resulting in altered Aβ production. However, conflicting evidence comes from epidemiological studies showing no or controversial association between dyslipidemia and AD risk, randomized clinical trials observing no beneficial effect of statin therapy, and cell biology studies suggesting that there is little exchange between circulating and brain cholesterol, that increased membrane cholesterol level is protective by inhibiting loss of membrane integrity through amyloid cytotoxicity, and that cellular cholesterol inhibits colocalization of β-secretase 1 and Aβ precursor protein in nonraft membrane domains, thereby increasing generation of plasmin, an Aβ-degrading enzyme.
Metabolic syndrome
Various studies have assessed the relationship between metabolic syndrome as a whole and the risk of AD or cognitive decline. Most of these investigations demonstrated a positive association between the presence of this syndrome and cognitive dysfunction.(97–99)
Smoking
Case–control studies have largely suggested that smoking lowers the risk of AD,(100, 101) whereas prospective studies have shown that smoking increases this risk(102–104) or has no effect on the probability of developing this disease.(105, 106) A meta-analysis that examined the relationship between smoking and AD while accounting for tobacco-industry affiliation found that the combined results of 18 crossectional studies without industry affiliation yielded no association.(107) By contrast, data from eight crossectional studies with tobacco-industry affiliation suggested that smoking protected against AD. 14 cohort studies without tobacco-industry affiliation yielded a significant increase in the risk of AD with smoking. Smoking may affect the risk of AD via several mechanisms. It may increase the generation of free radicals, leading to high oxidative stress, or affect the inflammatory immune system, leading to activation of phagocytes and further oxidative damage.(108) Second, it may affect AD risk by promoting cerebrovascular disease. However, there is also evidence for a protective effect, suggesting that nicotine induces increases in nicotinic acetylcholine receptors (nAChR) thereby counterbalancing the loss of nAChR observed in AD that leads to cholinergic deficits.
Traumatic brain injury
Retrospective studies(109–111) suggested that individuals with a history of traumatic brain injury (TBI) had a higher risk of dementia than individuals with no history of such injury. Two meta-analyses(112, 113) demonstrated that among patients with TBI, the risk of dementia was higher in men than in women. While prospective studies of the relationship between TBI and AD have proved inconsistent,(114–116) postmortem and experimental studies support a link between these conditions.(117) Evidence also exists that after human brain injury, the extent of Aβ pathology and tau pathology increases in brain tissue, cerebrospinal fluid (CSF) Aβ levels are elevated and APP is overproduced.(118)
Protective non-genetic factors
Diet
There is evidence that consumption of a Mediterranean diet, which is characterized by a high intake of plant foods and fish, with olive oil as the primary source of monounsaturated fat, a low intake of red meat and poultry and a moderate intake of wine, is associated with a reduced incidence of AD and MCI(119, 120) independent of the levels of physical activity(121) and vascular comorbidity.(122) Diets high in fish, fruit and vegetables are high in antioxidants and polyunsaturated fatty acids (PUFAs). Reactive oxygen species are clearly associated with neuronal damage in AD but wheter this association is a primary or secondary event in the neurotoxic process remains unclear. Aβ deposition leads to a decrease in cerebral iron and copper concentrations, resulting in oxidative stress and neuronal damage.(123) In vitro studies suggest that vitamin E reduces Aβ-associated lipid peroxidation and apoptosis.(124) In addition, carotenes and vitamin C protect against lipid peroxidation,(125) and vitamin C reduces the formation of nitrosamines and may affect catecholamine synthesis.(126, 127) It is also possible that intake of antioxidants reduces AD risk through decreasing the risk of cerebrovascular disease.(128) Besides reducing oxidative stress, PUFAs have favorable effects on neuronal and vascular functions and inflammatory processes.(129, 130) Observational population-based studies individually assessing the impact of vitamins E and C or PUFAs were inconclusive,(131–141) and most RCTs examining the effects of antioxidant or PUFA supplementation have found no association with cognitive performance.(142–147)
Physical activity
Epidemiological and experimental data suggest that physical exercise may promote brain health. Conflicting results have, however, emerged from cross-sectional and longitudinal observational studies that have examined the relationship between exercise levels and cognitive decline or dementia: while some studies have indicated that physical activity has a beneficial effect on brain health, other studies have shown no association between these variables.(148–152) RCTs exploring the effect of exercise on cognitive function in healthy elderly individuals have yielded conflicting results.(153–156) Physical activity could affect cognition through an increase in cerebral blood flow, oxygen extraction and glucose utilization, as well as activation of growth factors promoting structural brain changes, such as an increase in capillary density. In addition, rodent studies suggest that physical activity decreases the rate of amyloid plaque formation.(157)
Intellectual activity
Several subsequent prospective studies and RCTs suggest that people, at both young(158) and old(149) ages, who engage in cognitively stimulating activities, such as learning, reading or playing games, are less likely to develop dementia than individuals who did not engage in these activities. The benefit of cognitive training seems to be domain specific and more pronounced in persons without memory impairment, however.
Biomarkers
Biomarkers are useful for the determination of disease risk but are also invaluable in establishing a diagnosis. While the autosomal dominantly inherited mutations are definite markers of the disease, the additional biomarkers, that have been identified and include various measurements from CSF, blood and neuroimaging, only contribute to increasing the specificity of diagnosis.
Plasma biomarkers
Plasma biomarkers of AD comprise only small or lipophilic proteins and proteins carried by transporters able to cross the BBB. Under physiological conditions there is a steady-state level of brain Aβ that is balanced by the production and deposition of Aβ in the brain and the peripheral production by platelets. As a consequence, in cognitively healthy individuals, brain Aβ levels are reflected by plasma Aβ concentrations. In patients with dementia, in who Aβ is deposited in amyloid plaques, the relationship between brain and plasma Aβ levels is not clear. In familial AD(159, 160) and Down syndrome with APP triplication,(161) total Aβ levels and Aβ1–42 levels in plasma are elevated. In sporadic AD, studies exploring the usefulness of plasma Aβ as a risk biomarker are controversial(162, 163) likely due to variability in the timing of sample collection across studies, the use of different antibodies to detect Aβ, and a lack of validation of plasma Aβ as a risk biomarker for AD. There is evidence that elevated plasma Aβ1–42 is an antecedent risk factor for sporadic AD, while decreasing levels or a decline in the Aβ1–42/Aβ1–40 ratio indicate disease onset. Several other molecules that have been investigated as plasma biomarkers for AD risk such as cholesterol, homocysteine or inflammation-related proteins including C-reactive protein, IL-1β, TNF, IL-6 and transformating growth factor β were inconsistent across studies.
CSF biomarkers
Due to the free transport of proteins between the brain and the CSF, levels of Aβ1–42, total tau (t-tau) and p-tau in CSF reflect the metabolic processes in the brain and can be used to aid the accurate diagnosis of AD at an early stage of disease.(164) In MCI or AD CSF levels of Aβ1–42 are decreased while t-tau or p-tau are increased compared to cognitively normal individuals.(165) The combined evaluation of Aβ1–37, Aβ1–38, Aβ1–39, Aβ1–40 and Aβ1–42 may further increase sensitivity and specificity in predicting progression from MCI to AD.(166) The association between CSF biomarkers and the concentrations of deposited amyloid (in plaques) and NFTs in the brain remains unclear. It has been proposed that Aβ deposition in amyloid plaques may lead to a reduction of soluble Aβ in the brain and CSF. (167) Some studies suggested a positive correlation between p-tau CSF levels and NFT concentration in people with AD(168) but other studies did not observe such correlations.(169, 170) Reasons for inconsistencies between studies may be differences in factors influencing CSF tau or Aβ levels. For example, carrying an APOE ε4 allele or higher age accelerate the deposition of Aβ1–42 in brain and lower Aβ1–42 levels in CSF.(171) Moreover, CSF APOE levels positively correlate with t-tau and 24S-hydroxycholesterol (24S-OHC) CSF levels in patients with cognitive impairment.(172) A large-scale meta-analysis of 14 studies exploring biomarkers for preclinical AD suggested that the sensitivity of Aβ1–42, t-tau and p-tau is modest (effect sizes: 0.91–1.11).(173) Several additional CSF biomarkers have been explored but were inconsistent across studies (table 2) leaving their predictive value unclear.
Table 2.
Additional CSF biomarkers assessed for prediction of Alzheimer’s disease and cognitive impairment
| Biomarker |
|---|
| 24S-Hydroxycholesterol |
| Albumin |
| Amyloid-β |
| Angiotensinogen |
| Apolipoprotein AI |
| Apolipoprotein AII |
| Apolipoprotein E |
| Complement component C3a |
| Complement component C4a |
| Cystatin C |
| Cystatin C, 8 amino acid amino-terminal truncation |
| Immunoglobulin heavy chain |
| N-acetyllactosamine |
| Neuronal pentraxin-1 |
| Prostaglandin-H2 D-isomerase |
| Retinol-binding protein |
| Thioredoxin |
| Transthyretin |
| Vascular growth factor |
| α-1-Antitrypsin |
| α-1β Glycoprotein |
| β-2-Microglobulin |
| β-Fibrinogen |
| β-Secretase |
Imaging biomarkers
a) Structural MRI
On structural MRI, LOAD is characterized by atropy in the medial temporal lobe in particlar in the (para)hippocampus and the amygdala. In EOAD, brain atrophy may be spread more posteriorly and may involve the posterior cortex,(174) occipital lobes, posterior cingulate and precuneus.(175) Atrophy in the hippocampus and entorhinal cortex is associated with a decline in memory function, progression of memory impairment(176) and an increased risk of AD.(177) In addition, white matter changes may appear. However, none of these changes are specific to AD occurring also in various other neurodegenerative disorders as well as normal aging. Nevertheless, several studies have suggested certain structural MRI biomarkers possess some degree of discriminative diagnostic power. There is evidence that among patients with amnestic MCI, those converting to LOAD show greater atrophy in the hippocampus and the inferior and middle temporal gyri than those who do not convert to AD.(178) Evidence also exists that atrophy in the corpus callosum (particularly the anterior region) may help to distinguish AD from frontotemporal dementia, in which the posterior area shows greater atrophy.(179) Diffusion tensor imaging (DTI) allows to identify reductions in white matter integrity in several brain areas of persons with AD(180–182) and MCI(182–184) compared to non-demented individuals suggesting that these changes occur early in the disease process. In line with this notion, preclinical and presymptomatic carriers of familial AD mutations display enhanced white matter degradation compared to non-carriers.(185) Arterial spin labeling (ASL)-MRI allows examination of functional cerebral perfusion deficits using MRI. ASL-MRI studies have revealed reduced cerebral blood flow in AD patients compared to non-demented persons,(186–188) can discriminate MCI from AD,(188) and can predict cognitive decline and progression from MCI to AD.(189)
Functional MRI
Functional MRI (fMRI) visualizes neuronal activity during rest or a task activating specific brain regions. The most common method used measures alterations in blood flow based on changes in deoxyhemoglobin concentration (BOLD- fMRI).(190) Several studies have demonstrated a decreased BOLD signal in the medial temporal lobe, parietal lobe and hippocampal areas of persons with AD compared to controls during a cognitive task.(191–193) In addition, some studies have observed different task-associated neuronal activity patterns in patients with MCI and healthy controls.(174)
As there is disrupted connectivity in the default mode network of AD brains compared to healthy controls,(194, 195) resting state fMRI can been used to investigate functional connectivity deficits in AD. In line with this notion, studies have shown that also resting fMRI may differentiate AD from MCI and MCI from controls.(196) In a direct comparison of resting state versus task-associated fMRI in individuals at risk for AD, the resting default network analyses provided more robust and sensitive data.(197) BOLD signals are dependent on several anatomical, physiological and imaging parameters, and can be interpreted qualitatively or semiquantitatively. As a result, interindividual and intra-individual variability limits the use in the differential diagnosis of dementia-causing disorders. Nevertheless, recent advances in fMRI have allowed intrinsic functional networks in the human brain to be defined. The study of cognitive–behavioral function in the early stages of neurodegenerative disorders may allow the identification of the neuroanatomical networks affected by these diseases, and may assist in the differential diagnosis of the various disorders that underlie dementia. BOLD levels may also have utility for assessing treatment effects in pharmacological fMRI studies. Several investigations have demonstrated increased brain activation during task performance following the administration of acetylcholinesterase inhibitors (AChEIs)(198, 199) A recent resting state fMRI study revealed AChEI-induced improvement of functional connectivity in the hippocampus which correlated with cognitive improvement.(200) Increased BOLD activation with novel therapeutics may signal potential efficacy.
Positron emission tomography (PET) and single photon emission computed tomography (SPECT)
PET and single-photon emission CT (SPECT) have been extensively evaluated as diagnostic tools for dementia, and both techniques have shown good diagnostic and prognostic capabilities. Several positron emission tomography (PET) ligands targeting amyloid, tau, or metabolic activity have been investigated. An early radiotracer shown to bind both to amyloid plaques and NFT(201) is 2-(1-{6-[(2-[F-18]fluoroethyl)(methyl)amino]-2-naphthyl}ethylidene)malononitrile (FDDNP). FDDNP visualizes protein aggregates in limbic regions including the hippocampus and amygdala, correlates with cognitive deficits in AD,(201, 202) and differentiates between persons with AD, MCI and without cognitive impairment.(203) Small signal differences in the cortex, however, may reduce its utility.(202) The first amyloid specific imaging probe that was developed is the Pittsburgh compound B (PIB). PIB binds selectively to cortical and striatal Aβ plaques, shows a strong positive correlation with AD diagnosis, fibrillary amyloid plaques at autopsy(204–206) and is inversely correlated with CSF Aβ42 levels in the presence of clinical AD.(207) However, there is also substantial PIB retention in some non-demented individuals(206) and longitudinal studies need to clarify whether this retention represents preclinical AD. Florbetaben (18F-BAY94-9172) and Florbetapir (18F AV-45) are novel amyloid imaging agents with similar binding profiles but longer half-life than PIB that can also differentiate AD from controls and other dementias(206, 208) with strong sensitivity and specificity.(197, 209)
Cerebral metabolism, as measured by 18F-fluorodeoxyglucose (FDG)-PET imaging is decreased in AD. Several studies have demonstrated reductions in regional (e.g. parietal) cerebral glucose metabolism in MCI and AD compared to healthy controls(210, 211) FDG-PET measures are strongly related to cognitive deficits(207, 211) and may further be able to predict progression from MCI to AD.(212, 213) It has a high sensitivity (94%) but a low specificity (73–78%) for the diagnosis of dementia.(214) Combining analyses of FDG-PET and ASL-MRI data differentiates AD patients from controls more effectively than either technique alone.(215)
Finally, molecular imaging probes indicative of microglial activation have been developed for use in PET imaging. Most of these radioligands bind to the 18 kDa translocator protein (TSPO), also known as the peripheral benzodiazepine receptor, which reflects neuroinflammatory processes(216) The most frequently employed PET probe, 11C-(R)-PK11195, displays increased retention in AD(217) and MCI patients(218) versus aged non-demented individuals.
Many SPECT tracers target dopamine transporters or receptors.(219) These ligands discriminate AD from DLB with high sensitivity and specificity and have also been employed to demonstrate that reduced striatal uptake corresponds to reduced nigral neuronal number but not Aβ, tau, or α-synuclein pathology.(220) Other radiotracers developed for SPECT imaging, such as 123I-quinuclidinyl benzilate (123I-QNB), target muscarinic acetylcholine (ACh) receptors and reflect cholinergic system integrity. AD patients display reduced 123I-QNB uptake compared to controls, indicating decreased cholinergic function. Probes that target vesicular ACh transporters (123I-IBVM) and nicotinic ACh receptors (123I-5IA-85380) display similar reductions in cholinergic activity in AD compared to age-matched controls(221) These tracers may be able to monitor disease progression during clinical trials of symptomatic drugs that target cholinergic neurons and may have a role in documenting neuronal preservation in trials of neuroprotective agents.
A recent study(222) has estimated the diagnostic and prognostic accuracy of different imaging markers and pertinent metrics, and the amount and source of variance among them. This study suggests that the diagnostic and prognostic accuracy of imaging AD biomarkers is at least as dependent on how the biomarker is measured as on the type of biomarker itself. While acknowledging that imaging biomarkers capture different neurobiological constructs (brain amyloidosis, neuronal injury at the molecular level, and neuronal injury at the gross structural level), this observation provides empirical support to current efforts aimed at developing standard operating procedures (SOPs) for AD biomarkers in the diagnostic routine and in clinical trials.
Conclusions
Substantial progress has been made over the past few decades in understanding AD. In particlaur findings from genetic studies have pointed to specific mechanistic pathways including APP metabolism, immune response, inflammation, lipid metabolism and intracellular trafficking/endocytosis. If confirmed, these findings have the potential to open new avenues for genetic testing, prevention and treatment. However, before this gained knowledge can applied in clinical settings several issues must be addressed. First, the specific causative variants in the known susceptibility genes as well as genes not yet identified must be mapped. Currently, large-scale whole exome and whole genome sequencing efforts are under way aiming to identify causative common and rare variants associated with LOAD. These efforts include whites and Caribbean Hispanics, and although variants identified by this effort will need to be functionally confirmed, these studies hold the promise to lead to a more accurate understanding of the genetic risk factors in these ethnic groups and refinement of risk estimates and diagnostic and predictive testing protocols specific for individual ethnic groups. Similar efforts are needed for African Americans and additional ethnic groups that have a high prevalence of LOAD but have been widely neglected by genomic LOAD research. Second, it has to be clarified through which exact meachanism these and other -not yet identified- pathways lead to the disease and how pathways interact. Monotherapy is not likely to be sufficiently effective in a complex disease, and a more detailed risk profile would provide clues for a better multitargeted interventional strategy. Third, to achieve effective prevention and treatment strategies the early identification of the disease process (before its clinical expression) must be improved; this includes the development of standard operating procedures for AD biomarkers in the diagnostic routine. Effectively screening of those at clearly definable risk in whom progression from cognitively normal function or MCI to AD would be likely and where an intervention would stop a specific pathology progressing is essential.
Figure 2.
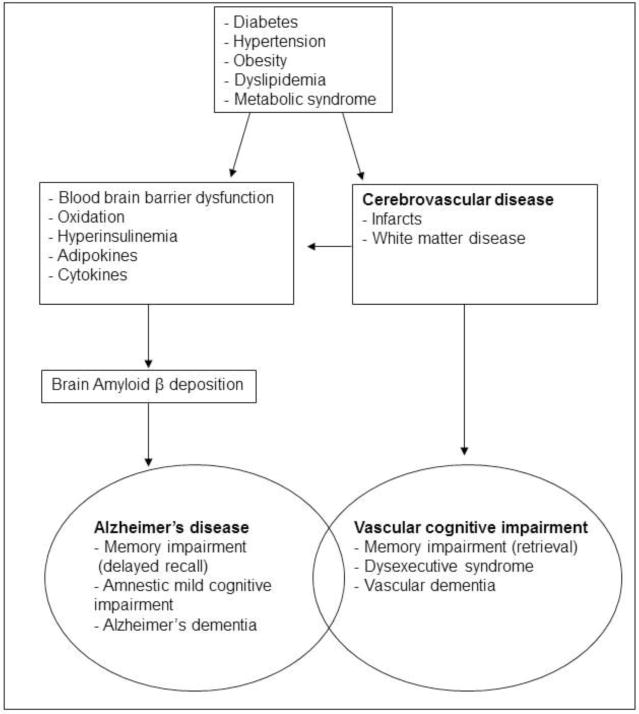
Potential mechanisms linking vascular risk factors and cognitive impairment
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.2010 Alzheimer’s disease facts and figures. Alzheimers Dement. 2010 Mar;6(2):158–94. doi: 10.1016/j.jalz.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 2.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology. 1984 Jul;34(7):939–44. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 3.Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, et al. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005 Dec 17;366(9503):2112–7. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthews FE, Arthur A, Barnes LE, Bond J, Jagger C, Robinson L, et al. A two-decade comparison of prevalence of dementia in individuals aged 65 years and older from three geographical areas of England: results of the Cognitive Function and Ageing Study I and II. Lancet. 2013 Oct 26;382(9902):1405–12. doi: 10.1016/S0140-6736(13)61570-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christensen K, Thinggaard M, Oksuzyan A, Steenstrup T, Andersen-Ranberg K, Jeune B, et al. Physical and cognitive functioning of people older than 90 years: a comparison of two Danish cohorts born 10 years apart. Lancet. 2013 Nov 2;382(9903):1507–13. doi: 10.1016/S0140-6736(13)60777-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schrijvers EM, Verhaaren BF, Koudstaal PJ, Hofman A, Ikram MA, Breteler MM. Is dementia incidence declining?: Trends in dementia incidence since 1990 in the Rotterdam Study. Neurology. 2012 May 8;78(19):1456–63. doi: 10.1212/WNL.0b013e3182553be6. [DOI] [PubMed] [Google Scholar]
- 7.Rocca WA, Petersen RC, Knopman DS, Hebert LE, Evans DA, Hall KS, et al. Trends in the incidence and prevalence of Alzheimer’s disease, dementia, and cognitive impairment in the United States. Alzheimers Dement. 2011 Jan;7(1):80–93. doi: 10.1016/j.jalz.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuusisto J, Koivisto K, Kervinen K, Mykkanen L, Helkala EL, Vanhanen M, et al. Association of apolipoprotein E phenotypes with late onset Alzheimer’s disease: population based study. BMJ. 1994 Sep 10;309(6955):636–8. doi: 10.1136/bmj.309.6955.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breitner JC, Wyse BW, Anthony JC, Welsh-Bohmer KA, Steffens DC, Norton MC, et al. APOE-epsilon4 count predicts age when prevalence of AD increases, then declines: the Cache County Study. Neurology. 1999 Jul 22;53(2):321–31. doi: 10.1212/wnl.53.2.321. [DOI] [PubMed] [Google Scholar]
- 10.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993 Aug 13;261(5123):921–3. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 11.Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locascio JJ, et al. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer’s disease. Ann Neurol. 1996 Jan;39(1):62–70. doi: 10.1002/ana.410390110. [DOI] [PubMed] [Google Scholar]
- 12.Holmes C, Levy R, McLoughlin DM, Powell JF, Lovestone S. Apolipoprotein E: non-cognitive symptoms and cognitive decline in late onset Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1996 Dec;61(6):580–3. doi: 10.1136/jnnp.61.6.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurz A, Altland K, Lautenschlager N, Zimmer R, Busch R, Gerundt I, et al. Apolipoprotein E type 4 allele and Alzheimer’s disease: effect on age at onset and relative risk in different age groups. J Neurol. 1996 Jun;243(6):452–6. doi: 10.1007/BF00900498. [DOI] [PubMed] [Google Scholar]
- 14.Murman DL, Foster NL, Kilgore SP, McDonagh CA, Fink JK. Apolipoprotein E and Alzheimer’s disease: strength of association is related to age at onset. Dementia. 1996 Sep-Oct;7(5):251–5. doi: 10.1159/000106888. [DOI] [PubMed] [Google Scholar]
- 15.Poirier J, Davignon J, Bouthillier D, Kogan S, Bertrand P, Gauthier S. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993 Sep 18;342(8873):697–9. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- 16.Roses AD. Alzheimer’s disease: the genetics of risk. Hosp Pract (Minneap) 1997 Jul 15;32(7):51–5. 8–63, 7–9. doi: 10.1080/21548331.1997.11443525. [DOI] [PubMed] [Google Scholar]
- 17.Barabash A, Marcos A, Ancin I, Vazquez-Alvarez B, de Ugarte C, Gil P, et al. APOE, ACT and CHRNA7 genes in the conversion from amnestic mild cognitive impairment to Alzheimer’s disease. Neurobiology of aging. 2007 Dec 8; doi: 10.1016/j.neurobiolaging.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 18.Petersen RC, Smith GE, Ivnik RJ, Tangalos EG, Schaid DJ, Thibodeau SN, et al. Apolipoprotein E status as a predictor of the development of Alzheimer’s disease in memory-impaired individuals. Jama. 1995 Apr 26;273(16):1274–8. [PubMed] [Google Scholar]
- 19.Sasaki M, Kodama C, Hidaka S, Yamashita F, Kinoshita T, Nemoto K, et al. Prevalence of four subtypes of mild cognitive impairment and APOE in a Japanese community. International journal of geriatric psychiatry. 2009 May 18; doi: 10.1002/gps.2234. [DOI] [PubMed] [Google Scholar]
- 20.Tyas SL, Salazar JC, Snowdon DA, Desrosiers MF, Riley KP, Mendiondo MS, et al. Transitions to mild cognitive impairments, dementia, and death: findings from the Nun Study. American journal of epidemiology. 2007 Jun 1;165(11):1231–8. doi: 10.1093/aje/kwm085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Blom ES, Giedraitis V, Zetterberg H, Fukumoto H, Blennow K, Hyman BT, et al. Rapid progression from mild cognitive impairment to Alzheimer’s disease in subjects with elevated levels of tau in cerebrospinal fluid and the APOE epsilon4/epsilon4 genotype. Dementia and geriatric cognitive disorders. 2009;27(5):458–64. doi: 10.1159/000216841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Devanand DP, Pelton GH, Zamora D, Liu X, Tabert MH, Goodkind M, et al. Predictive utility of apolipoprotein E genotype for Alzheimer disease in outpatients with mild cognitive impairment. Archives of neurology. 2005 Jun;62(6):975–80. doi: 10.1001/archneur.62.6.975. [DOI] [PubMed] [Google Scholar]
- 23.Hamalainen A, Grau-Olivares M, Tervo S, Niskanen E, Pennanen C, Huuskonen J, et al. Apolipoprotein E epsilon 4 allele is associated with increased atrophy in progressive mild cognitive impairment: a voxel-based morphometric study. Neuro-degenerative diseases. 2008;5(3–4):186–9. doi: 10.1159/000113698. [DOI] [PubMed] [Google Scholar]
- 24.Hsiung GY, Sadovnick AD, Feldman H. Apolipoprotein E epsilon4 genotype as a risk factor for cognitive decline and dementia: data from the Canadian Study of Health and Aging. Cmaj. 2004 Oct 12;171(8):863–7. doi: 10.1503/cmaj.1031789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jack CR, Jr, Petersen RC, Xu YC, O’Brien PC, Smith GE, Ivnik RJ, et al. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology. 1999 Apr 22;52(7):1397–403. doi: 10.1212/wnl.52.7.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramakers IH, Visser PJ, Aalten P, Bekers O, Sleegers K, van Broeckhoven CL, et al. The association between APOE genotype and memory dysfunction in subjects with mild cognitive impairment is related to age and Alzheimer pathology. Dementia and geriatric cognitive disorders. 2008;26(2):101–8. doi: 10.1159/000144072. [DOI] [PubMed] [Google Scholar]
- 27.Tierney MC, Szalai JP, Snow WG, Fisher RH, Tsuda T, Chi H, et al. A prospective study of the clinical utility of ApoE genotype in the prediction of outcome in patients with memory impairment. Neurology. 1996 Jan;46(1):149–54. doi: 10.1212/wnl.46.1.149. [DOI] [PubMed] [Google Scholar]
- 28.Slooter AJ, Cruts M, Kalmijn S, Hofman A, Breteler MM, Van Broeckhoven C, et al. Risk estimates of dementia by apolipoprotein E genotypes from a population-based incidence study: the Rotterdam Study. Archives of neurology. 1998 Jul;55(7):964–8. doi: 10.1001/archneur.55.7.964. [DOI] [PubMed] [Google Scholar]
- 29.Myers RH, Schaefer EJ, Wilson PW, D’Agostino R, Ordovas JM, Espino A, et al. Apolipoprotein E epsilon4 association with dementia in a population-based study: The Framingham study. Neurology. 1996 Mar;46(3):673–7. doi: 10.1212/wnl.46.3.673. [DOI] [PubMed] [Google Scholar]
- 30.Harold D, Abraham R, Hollingworth P, Sims R, Gerrish A, Hamshere ML, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet. 2009 Oct;41(10):1088–93. doi: 10.1038/ng.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet. 2009 Oct;41(10):1094–9. doi: 10.1038/ng.439. [DOI] [PubMed] [Google Scholar]
- 32.Seshadri S, Fitzpatrick AL, Ikram MA, DeStefano AL, Gudnason V, Boada M, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. Jama. 2010 May 12;303(18):1832–40. doi: 10.1001/jama.2010.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nuutinen T, Suuronen T, Kauppinen A, Salminen A. Clusterin: a forgotten player in Alzheimer’s disease. Brain Res Rev. 2009 Oct;61(2):89–104. doi: 10.1016/j.brainresrev.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 34.Wollmer MA, Sleegers K, Ingelsson M, Zekanowski C, Brouwers N, Maruszak A, et al. Association study of cholesterol-related genes in Alzheimer’s disease. Neurogenetics. 2007 Aug;8(3):179–88. doi: 10.1007/s10048-007-0087-z. [DOI] [PubMed] [Google Scholar]
- 35.DeMattos RB, Cirrito JR, Parsadanian M, May PC, O’Dell MA, Taylor JW, et al. ApoE and clusterin cooperatively suppress Abeta levels and deposition: evidence that ApoE regulates extracellular Abeta metabolism in vivo. Neuron. 2004 Jan 22;41(2):193–202. doi: 10.1016/s0896-6273(03)00850-x. [DOI] [PubMed] [Google Scholar]
- 36.Pant S, Sharma M, Patel K, Caplan S, Carr CM, Grant BD. AMPH-1/Amphiphysin/Bin1 functions with RME-1/Ehd1 in endocytic recycling. Nat Cell Biol. 2009 Dec;11(12):1399–410. doi: 10.1038/ncb1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tebar F, Bohlander SK, Sorkin A. Clathrin assembly lymphoid myeloid leukemia (CALM) protein: localization in endocytic-coated pits, interactions with clathrin, and the impact of overexpression on clathrin-mediated traffic. Mol Biol Cell. 1999 Aug;10(8):2687–702. doi: 10.1091/mbc.10.8.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crehan H, Holton P, Wray S, Pocock J, Guerreiro R, Hardy J. Complement receptor 1 (CR1) and Alzheimer’s disease. Immunobiology. 2012 Feb;217(2):244–50. doi: 10.1016/j.imbio.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 39.McGeer PL, Rogers J. Anti-inflammatory agents as a therapeutic approach to Alzheimer’s disease. Neurology. 1992 Feb;42(2):447–9. doi: 10.1212/wnl.42.2.447. [DOI] [PubMed] [Google Scholar]
- 40.Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011 May;43(5):429–35. doi: 10.1038/ng.803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011 May;43(5):436–41. doi: 10.1038/ng.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cao H, Crocker PR. Evolution of CD33-related siglecs: regulating host immune functions and escaping pathogen exploitation? Immunology. 2011 Jan;132(1):18–26. doi: 10.1111/j.1365-2567.2010.03368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.von Gunten S, Bochner BS. Basic and clinical immunology of Siglecs. Ann N Y Acad Sci. 2008 Nov;1143:61–82. doi: 10.1196/annals.1443.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vitale C, Romagnani C, Falco M, Ponte M, Vitale M, Moretta A, et al. Engagement of p75/AIRM1 or CD33 inhibits the proliferation of normal or leukemic myeloid cells. Proc Natl Acad Sci U S A. 1999 Dec 21;96(26):15091–6. doi: 10.1073/pnas.96.26.15091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamazaki T, Masuda J, Omori T, Usui R, Akiyama H, Maru Y. EphA1 interacts with integrin-linked kinase and regulates cell morphology and motility. J Cell Sci. 2009 Jan 15;122(Pt 2):243–55. doi: 10.1242/jcs.036467. [DOI] [PubMed] [Google Scholar]
- 46.Adams RH, Klein R. Eph receptors and ephrin ligands. essential mediators of vascular development. Trends Cardiovasc Med. 2000 Jul;10(5):183–8. doi: 10.1016/s1050-1738(00)00046-3. [DOI] [PubMed] [Google Scholar]
- 47.Duffy SL, Coulthard MG, Spanevello MD, Herath NI, Yeadon TM, McCarron JK, et al. Generation and characterization of EphA1 receptor tyrosine kinase reporter knockout mice. Genesis. 2008 Oct;46(10):553–61. doi: 10.1002/dvg.20434. [DOI] [PubMed] [Google Scholar]
- 48.Holen HL, Nustad K, Aasheim HC. Activation of EphA receptors on CD4+CD45RO+ memory cells stimulates migration. J Leukoc Biol. 2010 Jun;87(6):1059–68. doi: 10.1189/jlb.0709497. [DOI] [PubMed] [Google Scholar]
- 49.Sakamoto A, Sugamoto Y, Tokunaga Y, Yoshimuta T, Hayashi K, Konno T, et al. Expression profiling of the ephrin (EFN) and Eph receptor (EPH) family of genes in atherosclerosis-related human cells. J Int Med Res. 2011;39(2):522–7. doi: 10.1177/147323001103900220. [DOI] [PubMed] [Google Scholar]
- 50.Lehtonen S, Zhao F, Lehtonen E. CD2-associated protein directly interacts with the actin cytoskeleton. Am J Physiol Renal Physiol. 2002 Oct;283(4):F734–43. doi: 10.1152/ajprenal.00312.2001. [DOI] [PubMed] [Google Scholar]
- 51.Dustin ML, Olszowy MW, Holdorf AD, Li J, Bromley S, Desai N, et al. A novel adaptor protein orchestrates receptor patterning and cytoskeletal polarity in T-cell contacts. Cell. 1998 Sep 4;94(5):667–77. doi: 10.1016/s0092-8674(00)81608-6. [DOI] [PubMed] [Google Scholar]
- 52.Tanaka N, Abe-Dohmae S, Iwamoto N, Yokoyama S. Roles of ATP-binding cassette transporter A7 in cholesterol homeostasis and host defense system. J Atheroscler Thromb. 2011;18(4):274–81. doi: 10.5551/jat.6726. [DOI] [PubMed] [Google Scholar]
- 53.Chan SL, Kim WS, Kwok JB, Hill AF, Cappai R, Rye KA, et al. ATP-binding cassette transporter A7 regulates processing of amyloid precursor protein in vitro. J Neurochem. 2008 Jul;106(2):793–804. doi: 10.1111/j.1471-4159.2008.05433.x. [DOI] [PubMed] [Google Scholar]
- 54.Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013 Dec;45(12):1452–8. doi: 10.1038/ng.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rogaeva E, Meng Y, Lee JH, Gu Y, Kawarai T, Zou F, et al. The neuronal sortilin-related receptor SORL1 is genetically associated with Alzheimer disease. Nat Genet. 2007 Feb;39(2):168–77. doi: 10.1038/ng1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reitz C, Cheng R, Rogaeva E, Lee J, Tokuhiro S, Zou F, et al. Meta-analysis of the association between variants in SORL1 and Alzheimer’s disease Archives of neurology. 2010 doi: 10.1001/archneurol.2010.346. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akhtar MW, Kim MS, Adachi M, Morris MJ, Qi X, Richardson JA, et al. In vivo analysis of MEF2 transcription factors in synapse regulation and neuronal survival. PLoS One. 2012;7(4):e34863. doi: 10.1371/journal.pone.0034863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. Jama. 1997 Oct 22–29;278(16):1349–56. [PubMed] [Google Scholar]
- 59.Lee JH, Cheng R, Barral S, Reitz C, Medrano M, Lantigua R, et al. Identification of novel loci for Alzheimer disease and replication of CLU, PICALM, and BIN1 in Caribbean Hispanic individuals. Archives of neurology. 2011 Mar;68(3):320–8. doi: 10.1001/archneurol.2010.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reitz C, Jun G, Naj A, Rajbhandary R, Vardarajan BN, Wang LS, et al. Variants in the ATP-binding cassette transporter (ABCA7), apolipoprotein E 4,and the risk of late-onset Alzheimer disease in African Americans. Jama. 2013 Apr 10;309(14):1483–92. doi: 10.1001/jama.2013.2973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheung ZH, Gong K, Ip NY. Cyclin-dependent kinase 5 supports neuronal survival through phosphorylation of Bcl-2. J Neurosci. 2008 May 7;28(19):4872–7. doi: 10.1523/JNEUROSCI.0689-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Weishaupt JH, Kussmaul L, Grotsch P, Heckel A, Rohde G, Romig H, et al. Inhibition of CDK5 is protective in necrotic and apoptotic paradigms of neuronal cell death and prevents mitochondrial dysfunction. Mol Cell Neurosci. 2003 Oct;24(2):489–502. doi: 10.1016/s1044-7431(03)00221-5. [DOI] [PubMed] [Google Scholar]
- 63.Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, et al. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochimica et biophysica acta. 2007 Apr;1772(4):473–83. doi: 10.1016/j.bbadis.2006.10.011. [DOI] [PubMed] [Google Scholar]
- 64.Glynn RJ, Beckett LA, Hebert LE, Morris MC, Scherr PA, Evans DA. Current and remote blood pressure and cognitive decline. Jama. 1999 Feb 3;281(5):438–45. doi: 10.1001/jama.281.5.438. [DOI] [PubMed] [Google Scholar]
- 65.Knopman D, Boland LL, Mosley T, Howard G, Liao D, Szklo M, et al. Cardiovascular risk factors and cognitive decline in middle-aged adults. Neurology. 2001 Jan 9;56(1):42–8. doi: 10.1212/wnl.56.1.42. [DOI] [PubMed] [Google Scholar]
- 66.Posner HB, Tang MX, Luchsinger J, Lantigua R, Stern Y, Mayeux R. The relationship of hypertension in the elderly to AD, vascular dementia, and cognitive function. Neurology. 2002 Apr 23;58(8):1175–81. doi: 10.1212/wnl.58.8.1175. [DOI] [PubMed] [Google Scholar]
- 67.Skoog I, Lernfelt B, Landahl S, Palmertz B, Andreasson LA, Nilsson L, et al. 15-year longitudinal study of blood pressure and dementia. Lancet. 1996 Apr 27;347(9009):1141–5. doi: 10.1016/s0140-6736(96)90608-x. [DOI] [PubMed] [Google Scholar]
- 68.Kivipelto M, Helkala EL, Hanninen T, Laakso MP, Hallikainen M, Alhainen K, et al. Midlife vascular risk factors and late-life mild cognitive impairment: A population-based study. Neurology. 2001 Jun 26;56(12):1683–9. doi: 10.1212/wnl.56.12.1683. [DOI] [PubMed] [Google Scholar]
- 69.Launer LJ, Ross GW, Petrovitch H, Masaki K, Foley D, White LR, et al. Midlife blood pressure and dementia: the Honolulu-Asia aging study. Neurobiology of aging. 2000 Jan-Feb;21(1):49–55. doi: 10.1016/s0197-4580(00)00096-8. [DOI] [PubMed] [Google Scholar]
- 70.Swan GE, DeCarli C, Miller BL, Reed T, Wolf PA, Jack LM, et al. Association of midlife blood pressure to late-life cognitive decline and brain morphology. Neurology. 1998 Oct;51(4):986–93. doi: 10.1212/wnl.51.4.986. [DOI] [PubMed] [Google Scholar]
- 71.Whitmer RA, Sidney S, Selby J, Johnston SC, Yaffe K. Midlife cardiovascular risk factors and risk of dementia in late life. Neurology. 2005 Jan 25;64(2):277–81. doi: 10.1212/01.WNL.0000149519.47454.F2. [DOI] [PubMed] [Google Scholar]
- 72.Kalaria RN. Vascular basis for brain degeneration: faltering controls and risk factors for dementia. Nutr Rev. 2010 Dec;68(Suppl 2):S74–87. doi: 10.1111/j.1753-4887.2010.00352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deane R, Wu Z, Zlokovic BV. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004 Nov;35(11 Suppl 1):2628–31. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- 74.Prevention of stroke by antihypertensive drug treatment in older persons with isolated systolic hypertension. Final results of the Systolic Hypertension in the Elderly Program (SHEP). SHEP Cooperative Research Group. JAMA. 1991 Jun 26;265(24):3255–64. [PubMed] [Google Scholar]
- 75.Forette F, Seux ML, Staessen JA, Thijs L, Babarskiene MR, Babeanu S, et al. The prevention of dementia with antihypertensive treatment: new evidence from the Systolic Hypertension in Europe (Syst-Eur) study. Archives of internal medicine. 2002 Oct 14;162(18):2046–52. doi: 10.1001/archinte.162.18.2046. [DOI] [PubMed] [Google Scholar]
- 76.Lithell H, Hansson L, Skoog I, Elmfeldt D, Hofman A, Olofsson B, et al. The Study on Cognition and Prognosis in the Elderly (SCOPE): principal results of a randomized double-blind intervention trial. Journal of hypertension. 2003 May;21(5):875–86. doi: 10.1097/00004872-200305000-00011. [DOI] [PubMed] [Google Scholar]
- 77.Peters R, Pinto E, Beckett N, Swift C, Potter J, McCormack T, et al. Association of depression with subsequent mortality, cardiovascular morbidity and incident dementia in people aged 80 and over and suffering from hypertension. Data from the Hypertension in the Very Elderly Trial (HYVET) Age and ageing. Jul;39(4):439–45. doi: 10.1093/ageing/afq042. [DOI] [PubMed] [Google Scholar]
- 78.Prince MJ, Bird AS, Blizard RA, Mann AH. Is the cognitive function of older patients affected by antihypertensive treatment? Results from 54 months of the Medical Research Council’s trial of hypertension in older adults. BMJ. 1996 Mar 30;312(7034):801–5. doi: 10.1136/bmj.312.7034.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tzourio C, Anderson C, Chapman N, Woodward M, Neal B, MacMahon S, et al. Effects of blood pressure lowering with perindopril and indapamide therapy on dementia and cognitive decline in patients with cerebrovascular disease. Archives of internal medicine. 2003 May 12;163(9):1069–75. doi: 10.1001/archinte.163.9.1069. [DOI] [PubMed] [Google Scholar]
- 80.Starr JM, Whalley LJ, Deary IJ. The effects of antihypertensive treatment on cognitive function: results from the HOPE study. J Am Geriatr Soc. 1996 Apr;44(4):411–5. doi: 10.1111/j.1532-5415.1996.tb06412.x. [DOI] [PubMed] [Google Scholar]
- 81.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, et al. The risk of dementia among persons with diabetes mellitus: a population-based cohort study. Ann N Y Acad Sci. 1997 Sep 26;826:422–7. doi: 10.1111/j.1749-6632.1997.tb48496.x. [DOI] [PubMed] [Google Scholar]
- 82.Luchsinger JA, Tang MX, Stern Y, Shea S, Mayeux R. Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort. American journal of epidemiology. 2001 Oct 1;154(7):635–41. doi: 10.1093/aje/154.7.635. [DOI] [PubMed] [Google Scholar]
- 83.Luchsinger JA. Adiposity, hyperinsulinemia, diabetes and Alzheimer’s disease: an epidemiological perspective. Eur J Pharmacol. 2008 May 6;585(1):119–29. doi: 10.1016/j.ejphar.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sacco RL, Benjamin EJ, Broderick JP, Dyken M, Easton JD, Feinberg WM, et al. American Heart Association Prevention Conference. IV. Prevention and Rehabilitation of Stroke. Risk factors. Stroke. 1997 Jul;28(7):1507–17. doi: 10.1161/01.str.28.7.1507. [DOI] [PubMed] [Google Scholar]
- 85.Arvanitakis Z, Schneider JA, Wilson RS, Li Y, Arnold SE, Wang Z, et al. Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology. 2006 Dec 12;67(11):1960–5. doi: 10.1212/01.wnl.0000247053.45483.4e. [DOI] [PubMed] [Google Scholar]
- 86.Festa A, Williams K, D’Agostino R, Jr, Wagenknecht LE, Haffner SM. The natural course of beta-cell function in nondiabetic and diabetic individuals: the Insulin Resistance Atherosclerosis Study. Diabetes. 2006 Apr;55(4):1114–20. doi: 10.2337/diabetes.55.04.06.db05-1100. [DOI] [PubMed] [Google Scholar]
- 87.Watson GS, Peskind ER, Asthana S, Purganan K, Wait C, Chapman D, et al. Insulin increases CSF Abeta42 levels in normal older adults. Neurology. 2003 Jun 24;60(12):1899–903. doi: 10.1212/01.wnl.0000065916.25128.25. [DOI] [PubMed] [Google Scholar]
- 88.Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, et al. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A. 2003 Apr 1;100(7):4162–7. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park CR. Cognitive effects of insulin in the central nervous system. Neurosci Biobehav Rev. 2001 Jun;25(4):311–23. doi: 10.1016/s0149-7634(01)00016-1. [DOI] [PubMed] [Google Scholar]
- 90.Gustafson DR, Backman K, Waern M, Ostling S, Guo X, Zandi P, et al. Adiposity indicators and dementia over 32 years in Sweden. Neurology. 2009 Nov 10;73(19):1559–66. doi: 10.1212/WNL.0b013e3181c0d4b6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Razay G, Vreugdenhil A. Obesity in middle age and future risk of dementia: midlife obesity increases risk of future dementia. BMJ. 2005 Aug 20;331(7514):455. doi: 10.1136/bmj.331.7514.455. author reply. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stewart R, Masaki K, Xue QL, Peila R, Petrovitch H, White LR, et al. A 32-year prospective study of change in body weight and incident dementia: the Honolulu-Asia Aging Study. Archives of neurology. 2005 Jan;62(1):55–60. doi: 10.1001/archneur.62.1.55. [DOI] [PubMed] [Google Scholar]
- 93.Whitmer RA, Gustafson DR, Barrett-Connor E, Haan MN, Gunderson EP, Yaffe K. Central obesity and increased risk of dementia more than three decades later. Neurology. 2008 Sep 30;71(14):1057–64. doi: 10.1212/01.wnl.0000306313.89165.ef. [DOI] [PubMed] [Google Scholar]
- 94.Kuo YM, Emmerling MR, Bisgaier CL, Essenburg AD, Lampert HC, Drumm D, et al. Elevated low-density lipoprotein in Alzheimer’s disease correlates with brain abeta 1–42 levels. Biochem Biophys Res Commun. 1998 Nov 27;252(3):711–5. doi: 10.1006/bbrc.1998.9652. [DOI] [PubMed] [Google Scholar]
- 95.Michikawa M. Cholesterol paradox: is high total or low HDL cholesterol level a risk for Alzheimer’s disease? J Neurosci Res. 2003 Apr 15;72(2):141–6. doi: 10.1002/jnr.10585. [DOI] [PubMed] [Google Scholar]
- 96.Muckle TJ, Roy JR. High-density lipoprotein cholesterol in differential diagnosis of senile dementia. Lancet. 1985 May 25;1(8439):1191–3. doi: 10.1016/s0140-6736(85)92866-1. [DOI] [PubMed] [Google Scholar]
- 97.Raffaitin C, Gin H, Empana JP, Helmer C, Berr C, Tzourio C, et al. Metabolic syndrome and risk for incident Alzheimer’s disease or vascular dementia: the Three-City Study. Diabetes care. 2009 Jan;32(1):169–74. doi: 10.2337/dc08-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Solfrizzi V, Scafato E, Capurso C, D’Introno A, Colacicco AM, Frisardi V, et al. Metabolic syndrome and the risk of vascular dementia: the Italian Longitudinal Study on Ageing. J Neurol Neurosurg Psychiatry. Apr;81(4):433–40. doi: 10.1136/jnnp.2009.181743. [DOI] [PubMed] [Google Scholar]
- 99.Yaffe K, Weston AL, Blackwell T, Krueger KA. The metabolic syndrome and development of cognitive impairment among older women. Archives of neurology. 2009 Mar;66(3):324–8. doi: 10.1001/archneurol.2008.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ferini-Strambi L, Smirne S, Garancini P, Pinto P, Franceschi M. Clinical and epidemiological aspects of Alzheimer’s disease with presenile onset: a case control study. Neuroepidemiology. 1990;9(1):39–49. doi: 10.1159/000110750. [DOI] [PubMed] [Google Scholar]
- 101.Tyas SL. Are tobacco and alcohol use related to Alzheimer’s disease? A critical assessment of the evidence and its implications. AddictBiol. 1996;1(3):237. doi: 10.1080/1355621961000124856. [DOI] [PubMed] [Google Scholar]
- 102.Launer LJ, Feskens EJ, Kalmijn S, Kromhout D. Smoking, drinking, and thinking. The Zutphen Elderly Study. AmJEpidemiol. 1996;143(3):219. doi: 10.1093/oxfordjournals.aje.a008732. [DOI] [PubMed] [Google Scholar]
- 103.Merchant C, Tang MX, Albert S, Manly J, Stern Y, Mayeux R. The influence of smoking on the risk of Alzheimer’s disease. Neurology. 1999;52(7):1408. doi: 10.1212/wnl.52.7.1408. [DOI] [PubMed] [Google Scholar]
- 104.Ott A, Slooter AJ, Hofman A, van Harskamp F, Witteman JC, Van Broeckhoven C, et al. Smoking and risk of dementia and Alzheimer’s disease in a population-based cohort study: the Rotterdam Study. Lancet. 1998;351(9119):1840. doi: 10.1016/s0140-6736(97)07541-7. [DOI] [PubMed] [Google Scholar]
- 105.Elias PK, Elias MF, Robbins MA, Budge MM. Blood pressure-related cognitive decline: does age make a difference? Hypertension. 2004 Nov;44(5):631–6. doi: 10.1161/01.HYP.0000145858.07252.99. [DOI] [PubMed] [Google Scholar]
- 106.Hebert LE, Scherr PA, Beckett LA, Funkenstein HH, Albert MS, Chown MJ, et al. Relation of smoking and alcohol consumption to incident Alzheimer’s disease. American journal of epidemiology. 1992 Feb 15;135(4):347–55. doi: 10.1093/oxfordjournals.aje.a116296. [DOI] [PubMed] [Google Scholar]
- 107.Cataldo JK, Prochaska JJ, Glantz SA. Cigarette smoking is a risk factor for Alzheimer’s Disease: an analysis controlling for tobacco industry affiliation. J Alzheimers Dis. 19(2):465–80. doi: 10.3233/JAD-2010-1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Traber MG, van der Vliet A, Reznick AZ, Cross CE. Tobacco-related diseases. Is there a role for antioxidant micronutrient supplementation? Clin Chest Med. 2000 Mar;21(1):173–87. x. doi: 10.1016/s0272-5231(05)70016-2. [DOI] [PubMed] [Google Scholar]
- 109.Mayeux R, Ottman R, Maestre G, Ngai C, Tang MX, Ginsberg H, et al. Synergistic effects of traumatic head injury and apolipoprotein-epsilon 4 in patients with Alzheimer’s disease. Neurology. 1995 Mar;45(3 Pt 1):555–7. doi: 10.1212/wnl.45.3.555. [DOI] [PubMed] [Google Scholar]
- 110.Rasmusson DX, Brandt J, Martin DB, Folstein MF. Head injury as a risk factor in Alzheimer’s disease. Brain Inj. 1995 Apr;9(3):213–9. doi: 10.3109/02699059509008194. [DOI] [PubMed] [Google Scholar]
- 111.Schofield PW, Tang M, Marder K, Bell K, Dooneief G, Chun M, et al. Alzheimer’s disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry. 1997 Feb;62(2):119–24. doi: 10.1136/jnnp.62.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003 Jul;74(7):857–62. doi: 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mortimer JA, van Duijn CM, Chandra V, Fratiglioni L, Graves AB, Heyman A, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case-control studies. EURODEM Risk Factors Research Group. International journal of epidemiology. 1991;20(Suppl 2):S28–35. doi: 10.1093/ije/20.supplement_2.s28. [DOI] [PubMed] [Google Scholar]
- 114.Guo Z, Cupples LA, Kurz A, Auerbach SH, Volicer L, Chui H, et al. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000 Mar 28;54(6):1316–23. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- 115.Mehta KM, Ott A, Kalmijn S, Slooter AJ, van Duijn CM, Hofman A, et al. Head trauma and risk of dementia and Alzheimer’s disease: The Rotterdam Study. Neurology. 1999 Dec 10;53(9):1959–62. doi: 10.1212/wnl.53.9.1959. [DOI] [PubMed] [Google Scholar]
- 116.Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000 Oct 24;55(8):1158–66. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 117.Hartman RE, Laurer H, Longhi L, Bales KR, Paul SM, McIntosh TK, et al. Apolipoprotein E4 influences amyloid deposition but not cell loss after traumatic brain injury in a mouse model of Alzheimer’s disease. J Neurosci. 2002 Dec 1;22(23):10083–7. doi: 10.1523/JNEUROSCI.22-23-10083.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Franz G, Beer R, Kampfl A, Engelhardt K, Schmutzhard E, Ulmer H, et al. Amyloid beta 1-42 and tau in cerebrospinal fluid after severe traumatic brain injury. Neurology. 2003 May 13;60(9):1457–61. doi: 10.1212/01.wnl.0000063313.57292.00. [DOI] [PubMed] [Google Scholar]
- 119.Scarmeas N, Stern Y, Mayeux R, Manly JJ, Schupf N, Luchsinger JA. Mediterranean diet and mild cognitive impairment. Archives of neurology. 2009 Feb;66(2):216–25. doi: 10.1001/archneurol.2008.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gu Y, Nieves JW, Stern Y, Luchsinger JA, Scarmeas N. Food combination and Alzheimer disease risk: a protective diet. Archives of neurology. 2010 Jun;67(6):699–706. doi: 10.1001/archneurol.2010.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Scarmeas N, Luchsinger JA, Schupf N, Brickman AM, Cosentino S, Tang MX, et al. Physical activity, diet, and risk of Alzheimer disease. JAMA. 2009 Aug 12;302(6):627–37. doi: 10.1001/jama.2009.1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Scarmeas N, Stern Y, Mayeux R, Luchsinger JA. Mediterranean diet, Alzheimer disease, and vascular mediation. Archives of neurology. 2006 Dec;63(12):1709–17. doi: 10.1001/archneur.63.12.noc60109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nagano S, Huang X, Moir RD, Payton SM, Tanzi RE, Bush AI. Peroxidase activity of cyclooxygenase-2 (COX-2) cross-links beta-amyloid (Abeta) and generates Abeta-COX-2 hetero-oligomers that are increased in Alzheimer’s disease. The Journal of biological chemistry. 2004 Apr 9;279(15):14673–8. doi: 10.1074/jbc.M313003200. [DOI] [PubMed] [Google Scholar]
- 124.Butterfield DA, Castegna A, Drake J, Scapagnini G, Calabrese V. Vitamin E and neurodegenerative disorders associated with oxidative stress. Nutritional neuroscience. 2002 Sep;5(4):229–39. doi: 10.1080/10284150290028954. [DOI] [PubMed] [Google Scholar]
- 125.Pitchumoni SS, Doraiswamy PM. Current status of antioxidant therapy for Alzheimer’s Disease. J Am Geriatr Soc. 1998 Dec;46(12):1566–72. doi: 10.1111/j.1532-5415.1998.tb01544.x. [DOI] [PubMed] [Google Scholar]
- 126.Weisburger JH. Vitamin C and prevention of nitrosamine formation. Lancet. 1977 Sep 17;2(8038):607. doi: 10.1016/s0140-6736(77)91451-9. [DOI] [PubMed] [Google Scholar]
- 127.Pardo B, Mena MA, Fahn S, Garcia de Yebenes J. Ascorbic acid protects against levodopa-induced neurotoxicity on a catecholamine-rich human neuroblastoma cell line. Mov Disord. 1993 Jul;8(3):278–84. doi: 10.1002/mds.870080305. [DOI] [PubMed] [Google Scholar]
- 128.Voko Z, Hollander M, Hofman A, Koudstaal PJ, Breteler MM. Dietary antioxidants and the risk of ischemic stroke: the Rotterdam Study. Neurology. 2003 Nov 11;61(9):1273–5. doi: 10.1212/01.wnl.0000090458.67821.a3. [DOI] [PubMed] [Google Scholar]
- 129.Calder PC. Polyunsaturated fatty acids, inflammation, and immunity. Lipids. 2001 Sep;36(9):1007–24. doi: 10.1007/s11745-001-0812-7. [DOI] [PubMed] [Google Scholar]
- 130.Yehuda S, Rabinovitz S, Carasso RL, Mostofsky DI. The role of polyunsaturated fatty acids in restoring the aging neuronal membrane. Neurobiology of aging. 2002 Sep-Oct;23(5):843–53. doi: 10.1016/s0197-4580(02)00074-x. [DOI] [PubMed] [Google Scholar]
- 131.Engelhart MJ, Geerlings MI, Ruitenberg A, Van Swieten JC, Hofman A, Witteman JC, et al. Diet and risk of dementia: Does fat matter?: The Rotterdam Study. Neurology. 2002 Dec 24;59(12):1915–21. doi: 10.1212/01.wnl.0000038345.77753.46. [DOI] [PubMed] [Google Scholar]
- 132.Engelhart MJ, Geerlings MI, Ruitenberg A, van Swieten JC, Hofman A, Witteman JC, et al. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA. 2002 Jun 26;287(24):3223–9. doi: 10.1001/jama.287.24.3223. [DOI] [PubMed] [Google Scholar]
- 133.Masaki KH, Losonczy KG, Izmirlian G, Foley DJ, Ross GW, Petrovitch H, et al. Association of vitamin E and C supplement use with cognitive function and dementia in elderly men. Neurology. 2000 Mar 28;54(6):1265–72. doi: 10.1212/wnl.54.6.1265. [DOI] [PubMed] [Google Scholar]
- 134.Morris MC, Evans DA, Tangney CC, Bienias JL, Wilson RS. Fish consumption and cognitive decline with age in a large community study. Archives of neurology. 2005 Dec;62(12):1849–53. doi: 10.1001/archneur.62.12.noc50161. [DOI] [PubMed] [Google Scholar]
- 135.Huang TL, Zandi PP, Tucker KL, Fitzpatrick AL, Kuller LH, Fried LP, et al. Benefits of fatty fish on dementia risk are stronger for those without APOE epsilon4. Neurology. 2005 Nov 8;65(9):1409–14. doi: 10.1212/01.wnl.0000183148.34197.2e. [DOI] [PubMed] [Google Scholar]
- 136.Laurin D, Masaki KH, Foley DJ, White LR, Launer LJ. Midlife dietary intake of antioxidants and risk of late-life incident dementia: the Honolulu-Asia Aging Study. American journal of epidemiology. 2004 May 15;159(10):959–67. doi: 10.1093/aje/kwh124. [DOI] [PubMed] [Google Scholar]
- 137.Luchsinger JA, Tang MX, Shea S, Mayeux R. Antioxidant vitamin intake and risk of Alzheimer disease. Archives of neurology. 2003 Feb;60(2):203–8. doi: 10.1001/archneur.60.2.203. [DOI] [PubMed] [Google Scholar]
- 138.Kalmijn S, Launer LJ, Ott A, Witteman JC, Hofman A, Breteler MM. Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol. 1997 Nov;42(5):776–82. doi: 10.1002/ana.410420514. [DOI] [PubMed] [Google Scholar]
- 139.Roberts RO, Cerhan JR, Geda YE, Knopman DS, Cha RH, Christianson TJ, et al. Polyunsaturated fatty acids and reduced odds of MCI: the Mayo Clinic Study of Aging. J Alzheimers Dis. 21(3):853–65. doi: 10.3233/JAD-2010-091597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schaefer EJ, Bongard V, Beiser AS, Lamon-Fava S, Robins SJ, Au R, et al. Plasma phosphatidylcholine docosahexaenoic acid content and risk of dementia and Alzheimer disease: the Framingham Heart Study. Archives of neurology. 2006 Nov;63(11):1545–50. doi: 10.1001/archneur.63.11.1545. [DOI] [PubMed] [Google Scholar]
- 141.Solfrizzi V, Colacicco AM, D’Introno A, Capurso C, Torres F, Rizzo C, et al. Dietary intake of unsaturated fatty acids and age-related cognitive decline: a 8.5-year follow-up of the Italian Longitudinal Study on Aging. Neurobiology of aging. 2006 Nov;27(11):1694–704. doi: 10.1016/j.neurobiolaging.2005.09.026. [DOI] [PubMed] [Google Scholar]
- 142.Kang JH, Cook N, Manson J, Buring JE, Grodstein F. A randomized trial of vitamin E supplementation and cognitive function in women. Archives of internal medicine. 2006 Dec 11–25;166(22):2462–8. doi: 10.1001/archinte.166.22.2462. [DOI] [PubMed] [Google Scholar]
- 143.Petersen RC, Thomas RG, Grundman M, Bennett D, Doody R, Ferris S, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. The New England journal of medicine. 2005 Jun 9;352(23):2379–88. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 144.Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K, Grundman M, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. The Alzheimer’s Disease Cooperative Study. The New England journal of medicine. 1997 Apr 24;336(17):1216–22. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 145.Yaffe K, Clemons TE, McBee WL, Lindblad AS. Impact of antioxidants, zinc, and copper on cognition in the elderly: a randomized, controlled trial. Neurology. 2004 Nov 9;63(9):1705–7. doi: 10.1212/01.wnl.0000142969.19465.8f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chiu CC, Su KP, Cheng TC, Liu HC, Chang CJ, Dewey ME, et al. The effects of omega-3 fatty acids monotherapy in Alzheimer’s disease and mild cognitive impairment: a preliminary randomized double-blind placebo-controlled study. Progress in neuro-psychopharmacology & biological psychiatry. 2008 Aug 1;32(6):1538–44. doi: 10.1016/j.pnpbp.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 147.Freund-Levi Y, Eriksdotter-Jonhagen M, Cederholm T, Basun H, Faxen-Irving G, Garlind A, et al. Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: a randomized double-blind trial. Archives of neurology. 2006 Oct;63(10):1402–8. doi: 10.1001/archneur.63.10.1402. [DOI] [PubMed] [Google Scholar]
- 148.Abbott RD, White LR, Ross GW, Masaki KH, Curb JD, Petrovitch H. Walking and dementia in physically capable elderly men. JAMA. 2004 Sep 22;292(12):1447–53. doi: 10.1001/jama.292.12.1447. [DOI] [PubMed] [Google Scholar]
- 149.Fratiglioni L, Paillard-Borg S, Winblad B. An active and socially integrated lifestyle in late life might protect against dementia. Lancet neurology. 2004 Jun;3(6):343–53. doi: 10.1016/S1474-4422(04)00767-7. [DOI] [PubMed] [Google Scholar]
- 150.Rovio S, Kareholt I, Helkala EL, Viitanen M, Winblad B, Tuomilehto J, et al. Leisure-time physical activity at midlife and the risk of dementia and Alzheimer’s disease. Lancet neurology. 2005 Nov;4(11):705–11. doi: 10.1016/S1474-4422(05)70198-8. [DOI] [PubMed] [Google Scholar]
- 151.Scarmeas N, Levy G, Tang MX, Manly J, Stern Y. Influence of leisure activity on the incidence of Alzheimer’s disease. Neurology. 2001 Dec 26;57(12):2236–42. doi: 10.1212/wnl.57.12.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Verghese J, Lipton RB, Katz MJ, Hall CB, Derby CA, Kuslansky G, et al. Leisure activities and the risk of dementia in the elderly. The New England journal of medicine. 2003 Jun 19;348(25):2508–16. doi: 10.1056/NEJMoa022252. [DOI] [PubMed] [Google Scholar]
- 153.Emery CF, Schein RL, Hauck ER, MacIntyre NR. Psychological and cognitive outcomes of a randomized trial of exercise among patients with chronic obstructive pulmonary disease. Health Psychol. 1998 May;17(3):232–40. doi: 10.1037//0278-6133.17.3.232. [DOI] [PubMed] [Google Scholar]
- 154.Fabre C, Chamari K, Mucci P, Masse-Biron J, Prefaut C. Improvement of cognitive function by mental and/or individualized aerobic training in healthy elderly subjects. International journal of sports medicine. 2002 Aug;23(6):415–21. doi: 10.1055/s-2002-33735. [DOI] [PubMed] [Google Scholar]
- 155.Kramer AF, Erickson KI, Colcombe SJ. Exercise, cognition, and the aging brain. J Appl Physiol. 2006 Oct;101(4):1237–42. doi: 10.1152/japplphysiol.00500.2006. [DOI] [PubMed] [Google Scholar]
- 156.Lautenschlager NT, Cox KL, Flicker L, Foster JK, van Bockxmeer FM, Xiao J, et al. Effect of physical activity on cognitive function in older adults at risk for Alzheimer disease: a randomized trial. JAMA. 2008 Sep 3;300(9):1027–37. doi: 10.1001/jama.300.9.1027. [DOI] [PubMed] [Google Scholar]
- 157.Dishman RK, Berthoud HR, Booth FW, Cotman CW, Edgerton VR, Fleshner MR, et al. Neurobiology of exercise. Obesity (Silver Spring) 2006 Mar;14(3):345–56. doi: 10.1038/oby.2006.46. [DOI] [PubMed] [Google Scholar]
- 158.Carlson MC, Helms MJ, Steffens DC, Burke JR, Potter GG, Plassman BL. Midlife activity predicts risk of dementia in older male twin pairs. Alzheimers Dement. 2008 Sep;4(5):324–31. doi: 10.1016/j.jalz.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature medicine. 1996 Aug;2(8):864–70. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 160.Kosaka T, Imagawa M, Seki K, Arai H, Sasaki H, Tsuji S, et al. The beta APP717 Alzheimer mutation increases the percentage of plasma amyloid-beta protein ending at A beta42(43) Neurology. 1997 Mar;48(3):741–5. doi: 10.1212/wnl.48.3.741. [DOI] [PubMed] [Google Scholar]
- 161.Schupf N, Patel B, Silverman W, Zigman WB, Zhong N, Tycko B, et al. Elevated plasma amyloid beta-peptide 1-42 and onset of dementia in adults with Down syndrome. Neuroscience letters. 2001 Apr 6;301(3):199–203. doi: 10.1016/s0304-3940(01)01657-3. [DOI] [PubMed] [Google Scholar]
- 162.Mayeux R, Tang MX, Jacobs DM, Manly J, Bell K, Merchant C, et al. Plasma amyloid beta-peptide 1-42 and incipient Alzheimer’s disease. Ann Neurol. 1999 Sep;46(3):412–6. doi: 10.1002/1531-8249(199909)46:3<412::aid-ana19>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 163.van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM. Plasma Abeta(1-40) and Abeta(1-42) and the risk of dementia: a prospective case-cohort study. Lancet neurology. 2006 Aug;5(8):655–60. doi: 10.1016/S1474-4422(06)70501-4. [DOI] [PubMed] [Google Scholar]
- 164.Hansson O, Zetterberg H, Buchhave P, Andreasson U, Londos E, Minthon L, et al. Prediction of Alzheimer’s disease using the CSF Abeta42/Abeta40 ratio in patients with mild cognitive impairment. Dementia and geriatric cognitive disorders. 2007;23(5):316–20. doi: 10.1159/000100926. [DOI] [PubMed] [Google Scholar]
- 165.Ewers M, Zhong Z, Burger K, Wallin A, Blennow K, Teipel SJ, et al. Increased CSF-BACE 1 activity is associated with ApoE-epsilon 4 genotype in subjects with mild cognitive impairment and Alzheimer’s disease. Brain. 2008 May;131(Pt 5):1252–8. doi: 10.1093/brain/awn034. [DOI] [PubMed] [Google Scholar]
- 166.Hoglund K, Hansson O, Buchhave P, Zetterberg H, Lewczuk P, Londos E, et al. Prediction of Alzheimer’s disease using a cerebrospinal fluid pattern of C-terminally truncated beta-amyloid peptides. Neuro-degenerative diseases. 2008;5(5):268–76. doi: 10.1159/000119457. [DOI] [PubMed] [Google Scholar]
- 167.Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006 Mar;59(3):512–9. doi: 10.1002/ana.20730. [DOI] [PubMed] [Google Scholar]
- 168.Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, et al. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain. 2006 Nov;129(Pt 11):3035–41. doi: 10.1093/brain/awl269. [DOI] [PubMed] [Google Scholar]
- 169.Buerger K, Alafuzoff I, Ewers M, Pirttila T, Zinkowski R, Hampel H. No correlation between CSF tau protein phosphorylated at threonine 181 with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain. 2007 Oct;130(Pt 10):e82. doi: 10.1093/brain/awm140. [DOI] [PubMed] [Google Scholar]
- 170.Engelborghs S, Sleegers K, Cras P, Brouwers N, Serneels S, De Leenheir E, et al. No association of CSF biomarkers with APOEepsilon4, plaque and tangle burden in definite Alzheimer’s disease. Brain. 2007 Sep;130(Pt 9):2320–6. doi: 10.1093/brain/awm136. [DOI] [PubMed] [Google Scholar]
- 171.Fukumoto H, Tennis M, Locascio JJ, Hyman BT, Growdon JH, Irizarry MC. Age but not diagnosis is the main predictor of plasma amyloid beta-protein levels. Archives of neurology. 2003 Jul;60(7):958–64. doi: 10.1001/archneur.60.7.958. [DOI] [PubMed] [Google Scholar]
- 172.Shafaati M, Solomon A, Kivipelto M, Bjorkhem I, Leoni V. Levels of ApoE in cerebrospinal fluid are correlated with Tau and 24S-hydroxycholesterol in patients with cognitive disorders. Neuroscience letters. 2007 Sep 25;425(2):78–82. doi: 10.1016/j.neulet.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 173.Schmand B, Huizenga HM, van Gool WA. Meta-analysis of CSF and MRI biomarkers for detecting preclinical Alzheimer’s disease. Psychol Med. 2010 Jan;40(1):135–45. doi: 10.1017/S0033291709991516. [DOI] [PubMed] [Google Scholar]
- 174.Teipel SJ, Schapiro MB, Alexander GE, Krasuski JS, Horwitz B, Hoehne C, et al. Relation of corpus callosum and hippocampal size to age in nondemented adults with Down’s syndrome. The American journal of psychiatry. 2003 Oct;160(10):1870–8. doi: 10.1176/appi.ajp.160.10.1870. [DOI] [PubMed] [Google Scholar]
- 175.Karas G, Scheltens P, Rombouts S, van Schijndel R, Klein M, Jones B, et al. Precuneus atrophy in early-onset Alzheimer’s disease: a morphometric structural MRI study. Neuroradiology. 2007 Dec;49(12):967–76. doi: 10.1007/s00234-007-0269-2. [DOI] [PubMed] [Google Scholar]
- 176.Mungas D, Harvey D, Reed BR, Jagust WJ, DeCarli C, Beckett L, et al. Longitudinal volumetric MRI change and rate of cognitive decline. Neurology. 2005 Aug 23;65(4):565–71. doi: 10.1212/01.wnl.0000172913.88973.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Apostolova LG, Dutton RA, Dinov ID, Hayashi KM, Toga AW, Cummings JL, et al. Conversion of mild cognitive impairment to Alzheimer disease predicted by hippocampal atrophy maps. Archives of neurology. 2006 May;63(5):693–9. doi: 10.1001/archneur.63.5.693. [DOI] [PubMed] [Google Scholar]
- 178.Chetelat G, Landeau B, Eustache F, Mezenge F, Viader F, de la Sayette V, et al. Using voxel-based morphometry to map the structural changes associated with rapid conversion in MCI: a longitudinal MRI study. NeuroImage. 2005 Oct 1;27(4):934–46. doi: 10.1016/j.neuroimage.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 179.Likeman M, Anderson VM, Stevens JM, Waldman AD, Godbolt AK, Frost C, et al. Visual assessment of atrophy on magnetic resonance imaging in the diagnosis of pathologically confirmed young-onset dementias. Archives of neurology. 2005 Sep;62(9):1410–5. doi: 10.1001/archneur.62.9.1410. [DOI] [PubMed] [Google Scholar]
- 180.Rose SE, Chen F, Chalk JB, Zelaya FO, Strugnell WE, Benson M, et al. Loss of connectivity in Alzheimer’s disease: an evaluation of white matter tract integrity with colour coded MR diffusion tensor imaging. J Neurol Neurosurg Psychiatry. 2000 Oct;69(4):528–30. doi: 10.1136/jnnp.69.4.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Stebbins GT, Murphy CM. Diffusion tensor imaging in Alzheimer’s disease and mild cognitive impairment. Behav Neurol. 2009;21(1):39–49. doi: 10.3233/BEN-2009-0234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zhang Y, Schuff N, Jahng GH, Bayne W, Mori S, Schad L, et al. Diffusion tensor imaging of cingulum fibers in mild cognitive impairment and Alzheimer disease. Neurology. 2007 Jan 2;68(1):13–9. doi: 10.1212/01.wnl.0000250326.77323.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Medina D, DeToledo-Morrell L, Urresta F, Gabrieli JD, Moseley M, Fleischman D, et al. White matter changes in mild cognitive impairment and AD: A diffusion tensor imaging study. Neurobiology of aging. 2006 May;27(5):663–72. doi: 10.1016/j.neurobiolaging.2005.03.026. [DOI] [PubMed] [Google Scholar]
- 184.Fellgiebel A, Wille P, Muller MJ, Winterer G, Scheurich A, Vucurevic G, et al. Ultrastructural hippocampal and white matter alterations in mild cognitive impairment: a diffusion tensor imaging study. Dementia and geriatric cognitive disorders. 2004;18(1):101–8. doi: 10.1159/000077817. [DOI] [PubMed] [Google Scholar]
- 185.Ringman JM, Younkin SG, Pratico D, Seltzer W, Cole GM, Geschwind DH, et al. Biochemical markers in persons with preclinical familial Alzheimer disease. Neurology. 2008 Jul 8;71(2):85–92. doi: 10.1212/01.wnl.0000303973.71803.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Alsop DC, Detre JA, Grossman M. Assessment of cerebral blood flow in Alzheimer’s disease by spin-labeled magnetic resonance imaging. Ann Neurol. 2000 Jan;47(1):93–100. [PubMed] [Google Scholar]
- 187.Chen Y, Wolk DA, Reddin JS, Korczykowski M, Martinez PM, Musiek ES, et al. Voxel-level comparison of arterial spin-labeled perfusion MRI and FDG-PET in Alzheimer disease. Neurology. 2011 Nov 29;77(22):1977–85. doi: 10.1212/WNL.0b013e31823a0ef7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Johnson NA, Jahng GH, Weiner MW, Miller BL, Chui HC, Jagust WJ, et al. Pattern of cerebral hypoperfusion in Alzheimer disease and mild cognitive impairment measured with arterial spin-labeling MR imaging: initial experience. Radiology. 2005 Mar;234(3):851–9. doi: 10.1148/radiol.2343040197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Chao LL, Schuff N, Kramer JH, Du AT, Capizzano AA, O’Neill J, et al. Reduced medial temporal lobe N-acetylaspartate in cognitively impaired but nondemented patients. Neurology. 2005 Jan 25;64(2):282–9. doi: 10.1212/01.WNL.0000149638.45635.FF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Sperling R. Potential of functional MRI as a biomarker in early Alzheimer’s disease. Neurobiology of aging. 2011 Dec;32(Suppl 1):S37–43. doi: 10.1016/j.neurobiolaging.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Celone KA, Calhoun VD, Dickerson BC, Atri A, Chua EF, Miller SL, et al. Alterations in memory networks in mild cognitive impairment and Alzheimer’s disease: an independent component analysis. J Neurosci. 2006 Oct 4;26(40):10222–31. doi: 10.1523/JNEUROSCI.2250-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, et al. Increased hippocampal activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005 Aug 9;65(3):404–11. doi: 10.1212/01.wnl.0000171450.97464.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Golby A, Silverberg G, Race E, Gabrieli S, O’Shea J, Knierim K, et al. Memory encoding in Alzheimer’s disease: an fMRI study of explicit and implicit memory. Brain. 2005 Apr;128(Pt 4):773–87. doi: 10.1093/brain/awh400. [DOI] [PubMed] [Google Scholar]
- 194.Rombouts SA, Barkhof F, Goekoop R, Stam CJ, Scheltens P. Altered resting state networks in mild cognitive impairment and mild Alzheimer’s disease: an fMRI study. Hum Brain Mapp. 2005 Dec;26(4):231–9. doi: 10.1002/hbm.20160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer’s disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004 Mar 30;101(13):4637–42. doi: 10.1073/pnas.0308627101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sorg C, Riedl V, Muhlau M, Calhoun VD, Eichele T, Laer L, et al. Selective changes of resting-state networks in individuals at risk for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2007 Nov 20;104(47):18760–5. doi: 10.1073/pnas.0708803104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Fleisher AS, Sherzai A, Taylor C, Langbaum JB, Chen K, Buxton RB. Resting-state BOLD networks versus task-associated functional MRI for distinguishing Alzheimer’s disease risk groups. NeuroImage. 2009 Oct 1;47(4):1678–90. doi: 10.1016/j.neuroimage.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Venneri A, McGeown WJ, Shanks MF. Responders to ChEI treatment of Alzheimer’s disease show restitution of normal regional cortical activation. Curr Alzheimer Res. 2009 Apr;6(2):97–111. doi: 10.2174/156720509787602933. [DOI] [PubMed] [Google Scholar]
- 199.Bentley P, Driver J, Dolan RJ. Cholinesterase inhibition modulates visual and attentional brain responses in Alzheimer’s disease and health. Brain. 2008 Feb;131(Pt 2):409–24. doi: 10.1093/brain/awm299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Goveas JS, Xie C, Ward BD, Wu Z, Li W, Franczak M, et al. Recovery of hippocampal network connectivity correlates with cognitive improvement in mild Alzheimer’s disease patients treated with donepezil assessed by resting-state fMRI. J Magn Reson Imaging. 2011 Oct;34(4):764–73. doi: 10.1002/jmri.22662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, et al. Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002 Jan-Feb;10(1):24–35. [PubMed] [Google Scholar]
- 202.Shin J, Kepe V, Barrio JR, Small GW. The merits of FDDNP-PET imaging in Alzheimer’s disease. J Alzheimers Dis. 2011;26(Suppl 3):135–45. doi: 10.3233/JAD-2011-0008. [DOI] [PubMed] [Google Scholar]
- 203.Small GW, Kepe V, Ercoli LM, Siddarth P, Bookheimer SY, Miller KJ, et al. PET of brain amyloid and tau in mild cognitive impairment. The New England journal of medicine. 2006 Dec 21;355(25):2652–63. doi: 10.1056/NEJMoa054625. [DOI] [PubMed] [Google Scholar]
- 204.Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, et al. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008 Dec;32(3):412–9. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 205.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004 Mar;55(3):306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 206.Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007 May 15;68(20):1718–25. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 207.Jagust WJ, Landau SM, Shaw LM, Trojanowski JQ, Koeppe RA, Reiman EM, et al. Relationships between biomarkers in aging and dementia. Neurology. 2009 Oct 13;73(15):1193–9. doi: 10.1212/WNL.0b013e3181bc010c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Barthel H, Gertz HJ, Dresel S, Peters O, Bartenstein P, Buerger K, et al. Cerebral amyloid-beta PET with florbetaben (18F) in patients with Alzheimer’s disease and healthy controls: a multicentre phase 2 diagnostic study. Lancet neurology. 2011 May;10(5):424–35. doi: 10.1016/S1474-4422(11)70077-1. [DOI] [PubMed] [Google Scholar]
- 209.Camus V, Payoux P, Barre L, Desgranges B, Voisin T, Tauber C, et al. Using PET with 18F-AV-45 (florbetapir) to quantify brain amyloid load in a clinical environment. Eur J Nucl Med Mol Imaging. 2012 Apr;39(4):621–31. doi: 10.1007/s00259-011-2021-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Landau SM, Harvey D, Madison CM, Koeppe RA, Reiman EM, Foster NL, et al. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiology of aging. 2011 Jul;32(7):1207–18. doi: 10.1016/j.neurobiolaging.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Langbaum JB, Chen K, Lee W, Reschke C, Bandy D, Fleisher AS, et al. Categorical and correlational analyses of baseline fluorodeoxyglucose positron emission tomography images from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) NeuroImage. 2009 May 1;45(4):1107–16. doi: 10.1016/j.neuroimage.2008.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Arnaiz E, Jelic V, Almkvist O, Wahlund LO, Winblad B, Valind S, et al. Impaired cerebral glucose metabolism and cognitive functioning predict deterioration in mild cognitive impairment. Neuroreport. 2001 Mar 26;12(4):851–5. doi: 10.1097/00001756-200103260-00045. [DOI] [PubMed] [Google Scholar]
- 213.Drzezga A, Riemenschneider M, Strassner B, Grimmer T, Peller M, Knoll A, et al. Cerebral glucose metabolism in patients with AD and different APOE genotypes. Neurology. 2005 Jan 11;64(1):102–7. doi: 10.1212/01.WNL.0000148478.39691.D3. [DOI] [PubMed] [Google Scholar]
- 214.Silverman DH, Small GW, Chang CY, Lu CS, Kung De Aburto MA, Chen W, et al. Positron emission tomography in evaluation of dementia: Regional brain metabolism and long-term outcome. JAMA. 2001 Nov 7;286(17):2120–7. doi: 10.1001/jama.286.17.2120. [DOI] [PubMed] [Google Scholar]
- 215.Dashjamts T, Yoshiura T, Hiwatashi A, Yamashita K, Monji A, Ohyagi Y, et al. Simultaneous arterial spin labeling cerebral blood flow and morphological assessments for detection of Alzheimer’s disease. Acad Radiol. 2011 Dec;18(12):1492–9. doi: 10.1016/j.acra.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 216.Lang S. The role of peripheral benzodiazepine receptors (PBRs) in CNS pathophysiology. Curr Med Chem. 2002 Aug;9(15):1411–5. doi: 10.2174/0929867023369745. [DOI] [PubMed] [Google Scholar]
- 217.Tomasi G, Edison P, Bertoldo A, Roncaroli F, Singh P, Gerhard A, et al. Novel reference region model reveals increased microglial and reduced vascular binding of 11C-(R)-PK11195 in patients with Alzheimer’s disease. J Nucl Med. 2008 Aug;49(8):1249–56. doi: 10.2967/jnumed.108.050583. [DOI] [PubMed] [Google Scholar]
- 218.Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R, et al. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009 Jan 6;72(1):56–62. doi: 10.1212/01.wnl.0000338622.27876.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Cummings JL, Henchcliffe C, Schaier S, Simuni T, Waxman A, Kemp P. The role of dopaminergic imaging in patients with symptoms of dopaminergic system neurodegeneration. Brain. 2011 Nov;134(Pt 11):3146–66. doi: 10.1093/brain/awr177. [DOI] [PubMed] [Google Scholar]
- 220.Colloby SJ, McParland S, O’Brien JT, Attems J. Neuropathological correlates of dopaminergic imaging in Alzheimer’s disease and Lewy body dementias. Brain. 2012 Sep;135(Pt 9):2798–808. doi: 10.1093/brain/aws211. [DOI] [PubMed] [Google Scholar]
- 221.Colloby SJ, Perry EK, Pakrasi S, Pimlott SL, Wyper DJ, McKeith IG, et al. Nicotinic 123I-5IA-85380 single photon emission computed tomography as a predictor of cognitive progression in Alzheimer’s disease and dementia with Lewy bodies. Am J Geriatr Psychiatry. 2010 Jan;18(1):86–90. doi: 10.1097/JGP.0b013e3181b972aa. [DOI] [PubMed] [Google Scholar]
- 222.Frisoni GB, Bocchetta M, Chetelat G, Rabinovici GD, de Leon MJ, Kaye J, et al. Imaging markers for Alzheimer disease: Which vs how. Neurology. 2013 Jul 30;81(5):487–500. doi: 10.1212/WNL.0b013e31829d86e8. [DOI] [PMC free article] [PubMed] [Google Scholar]