

SUMMARY
Diabetes is a major risk factor for atherosclerosis. Although atherosclerosis is initiated by deposition of cholesterol-rich lipoproteins in the artery wall, the entry of inflammatory leukocytes into lesions fuels disease progression and impairs resolution. We show that diabetic mice have increased numbers of circulating neutrophils and Ly6-Chi monocytes, reflecting hyperglycemia-induced proliferation and expansion of bone marrow myeloid progenitors and release of monocytes into the circulation. Increased neutrophil production of S100A8/A9, via an interaction with the receptor for advanced glycation end products on common myeloid progenitor cells, leads to enhanced myelopoiesis. Treatment of hyperglycemia reduces monocytosis, entry of monocytes into atherosclerotic lesions and promotes regression. In patients with type I diabetes plasma S100A8/A9 levels correlate with leukocyte counts and coronary artery disease. Thus, hyperglycemia drives myelopoiesis and thus promotes atherogenesis in diabetes.
INTRODUCTION
Type 1 (T1DM) and type 2 diabetes mellitus (T2DM) are associated with an increased risk of coronary artery disease (CAD). Elevated white blood cell (WBC) counts are also an independent risk factor for CAD (Coller, 2005; Danesh et al., 1998). Increased leukocyte levels are seen in diabetics (Ford, 2002; Persson et al., 1998; Schmidt et al., 1999; Vuckovic et al., 2007; Woo et al., 2011) and are associated with a higher prevalence of CAD (Orchard et al., 2003). Moreover, there is evidence that increased levels of circulating monocytes, especially inflammatory Ly6C+ CCR2+ monocytes (Swirski et al., 2007; Tacke et al., 2007), and neutrophils (Drechsler et al., 2010) leads to increased entry into plaques and drives lesion progression. Diabetes leads to a variety of metabolic changes including altered insulin signaling, greater adipose lipolysis, hyperlipidemia and hyperglycemia. However, which of these mechanisms might be responsible for leukocytosis in diabetes is unknown.
Because of the recent introduction of potent cholesterol-reduction therapies, it is now clear that markedly reducing plasma LDL cholesterol levels can promote atherosclerotic lesion regression in humans (Nicholls et al., 2011). However, diabetics with CAD appear to have impaired regression of atherosclerosis when their plasma lipid levels are controlled (Duff and Payne, 1950; Hiro et al., 2010; Parathath et al., 2011). We have recently used mouse models in which plasma levels of atherogenic lipoproteins were acutely lowered to carry out mechanistic studies of atherosclerosis regression (Feig et al., 2010; Parathath et al., 2011; Trogan et al., 2006). In this model, diabetic mice show markedly impaired regression compared to controls, despite similar plasma lipid lowering (Parathath et al., 2011); however, the underlying mechanisms were not defined.
The goals of this study were threefold: 1) to determine the mechanisms responsible for monocytosis and neutrophilia in diabetes; 2) to assess the relevance of hyperglycemia to atherosclerosis and specifically its potential role in the impaired regression of atherosclerosis in diabetes; 3) to determine if similar mechanisms might be at work in a human population with Type 1 diabetes.
RESULTS
Leukocytosis is hyperglycemia-dependent
To investigate if hyperglycemia promotes monocytosis, we studied two mouse models of insulin deficient diabetes on chow diet: chemical (streptozotocin, STZ) and genetic (C57BL/6J-Ins2Akita) islet cell destruction. In both diabetic models, we observed a significant increase in the number of circulating monocytes (predominantly the Ly6-Chi pro-inflammatory subset) and neutrophils (Fig 1A-D) but not lymphocytes (Fig S1A,B). To determine if these changes were induced by hyperglycemia, plasma glucose levels were reduced using a sodium glucose co-transporter 2 inhibitor (SGLT2i); insulin and cholesterol levels were not changed (Fig S1C-J). Lowering glucose levels reduced the levels of monocytes and neutrophils, suggesting a direct role for hyperglycemia in promoting leukocytosis. Leukocytosis was not due to decreased leukocyte apoptosis (Fig S1K). Moreover, an EdU pulse chase experiment revealed an increase in newly formed (EdU+) blood monocytes in diabetic mice (Fig S1L), suggesting an underlying proliferative defect.
Figure 1.

Leukocytosis develops in response to hyperglycemia via expansion and proliferation of BM progenitor cells. Chow fed non-diabetic WT (C57BL/6J), STZ-diabetic and Akita-diabetic mice were treated with a SGLT2i (5mg/kg; ISIS) in the drinking water for 4 wks. Representative flow cytometry plots of blood leukocyte subsets from (A) STZ-diabetic and (B) Akita-diabetic mice.
(C and D) Quantification of monocyte subsets and neutrophils in (C) STZ-diabetic and (D) Akita-diabetic mice.
(E-H) HSPC, CMP and GMP analysis in the BM: The percentage of the respective populations in (E) STZ-diabetic and (F) Akita-diabetic mice, and cell cycle (G2M phase) analysis in (G) STZ-diabetic and (H) Akita-diabetic mice was performed by flow cytometry. All experiments n=10-12/group. *P<0.05 vs. all groups and ^P<0.05 vs. WT+STZ or Akita respectively.
All values are means ± SEM.
Leukocytosis is due to enhanced myelopoiesis
Leukocytosis was associated with enhanced myelopoiesis in the bone marrow (BM) (Fig 1E-H) but not spleen (Fig S2A). The number and proliferation of granulocyte-macrophage progenitors (GMPs) and common myeloid progenitors (CMPs) (Fig 1E-H) increased with diabetes, and BM cells had a higher potential to form granulocyte/macrophage colony-forming units (GM-CFUs) (Fig S2B,C). Expansion of myeloid progenitor cells was not due to decreased apoptosis (Fig S2D). Surprisingly, there was no change in the number or proliferation of hematopoietic stem and multipotent progenitor cells (HSPCs) (Fig 1E-H). Normoglycemia attenuated the expansion and proliferation of CMP/GMPs in both STZ and Akita-diabetic mice, an effect that was reproduced with a different SGLT2i, dapagliflozin (Meng et al., 2008) (Fig S3).
Myelopoiesis is driven by S100A8/A9
Initially, we examined plasma levels of classical hematopoietic cytokines such as M-CSF, GM-CSF, G-CSF and IL-1β as potential mediators of myelopoiesis. We found only plasma G-CSF levels to be increased in diabetes and reversed by lowering blood glucose (WT: 77±7, STZ: 121±7, STZ+SGLT2i: 98±5 pg/ml). The changes in G-CSF levels may explain neutrophilia and could also contribute in part to monocytosis (Christopher et al., 2011; Lieschke et al., 1994; Roberts, 2005; Semerad et al., 1999). However, some studies suggest impaired effects of G-CSF in diabetes (Ferraro et al., 2011). We therefore assessed additional potential mechanisms. Transcriptional (mRNA) profiling analysis of circulating and BM myeloid cells from diabetic mice (Fig S4A-C) revealed increased expression of certain DAMPs, notably S100a8, S100a9 and Hmgb1 (high mobility group box1) that have been previously linked to sterile inflammatory responses. Plasma S100A8/A9 levels were increased in both STZ- (Fig 2A) and Akita-diabetic mice (Fig S3D) and were decreased by lowering blood glucose. Neutrophils were the predominant source of S100a8, S100a9 and Hmgb1 (Fig S4A-C). Expression of S100a8 and S100a9, but not Hmgb1, was decreased when glucose levels were lowered (Fig 2B). We observed an increase in reactive oxygen species (ROS) and the expression levels of the transcription factors Klf-5 and C/ebp-α in neutrophils from STZ mice (Fig S4D,E), both of which promote S100a8/a9 expression (Fujiu et al., 2011; Yao and Brownlee, 2010). Lowering glucose levels corrected the increases in ROS and expression of Klf-5 and C/ebp-α.
Figure 2.

Neutrophil-derived S100A8/A9 promotes leukocytosis via increased proliferation of BM progenitor cells.
(A) Plasma levels of S100A8/A9 in STZ-diabetic mice treated with SGLT2i. n=6.
(B) mRNA expression of S100a8, S100a9 and Hmgb1 in FACS isolated neutrophils. n=6, *P<0.05 vs. WT, ^P<0.05 vs. WT+STZ group.
(C) Total BM cells were isolated from WT mice and cultured in HG (25mM, 16hrs) and stimulated with S100A8/A9 complex. GMP proliferation was assessed by measuring EdU incorporation via flow cytometry. n=4 independent experiments, *P<0.05 vs. vehicle.
(D) Monocyte levels and (E) BM progenitor cells in WT mice in response to vehicle or S100A8/A9 complex (20μg/kg/mouse, i.v, twice daily, 3 days) *P<0.05 vs. vehicle.
(F-I) S100a9−/− BMT: (F) Experimental overview: WT mice were transplanted with BM from either WT or S100a9−/− mice and made diabetic with STZ. (G) Blood leukocyte levels after 4 weeks of diabetes. (H) Percentage of HSPCs, CMPs and GMPs in the BM and (I) percentage of HSPCs, CMPs and GMPs in the G2M phase of the cell cycle. D-I, n=5/group. *P<0.05 vs. all groups.
All values are means ± SEM.
Next, we tested if S100A8/A9 (the biologically active heterodimer) (Leukert et al., 2006) could induce proliferation of BM progenitor cells. Stimulation of BM cells cultured in high glucose (HG, 25 mM) with S100A8/A9 induced GMP proliferation (Fig 2C). Similarly myelopoiesis was induced by administration of S100A8/A9 to mice (Fig 2D). HMGB1, which is proposed to share receptors with S100A8/A9, did not promote myelopoiesis in vitro or in vivo (Fig S4F-I), ruling out its role in hyperglycemia-induced myelopoiesis.
Since neutrophils are the predominant source of S100A8/A9, we transplanted BM from WT and S100a9−/− mice (that also lacks S100A8 protein) into WT recipients and examined myelopoiesis in response to diabetes (Fig. 2F). Mice that received WT BM displayed enhanced myelopoiesis when rendered diabetic, whereas diabetes failed to produce this myeloproliferative phenotype in S100a9−/− transplanted mice (Fig. 2G-I). These findings confirm a central role for S100A8/A9 in diabetes-driven myelopoiesis.
S100A8/A9-induced myelopoiesis is RAGE dependent
S100A8/A9 can signal through multiple pattern recognition receptors, including RAGE and TLR2/4 (Hofmann et al., 1999; Vogl et al., 2007). To identify the specific receptor(s) involved, we cultured BM progenitor cells isolated from WT, Rage−/− and Myd88−/− mice in HG-containing medium and examined the proliferative responses to S100A8/A9. WT and Myd88−/− BM cells showed increased proliferation in response to S100A8/A9, whereas Rage−/− cells did not. Thus, the myeloproliferative effects of S100A8/A9 are mediated by RAGE (Fig 3A). The importance of RAGE in diabetic myelopoiesis was confirmed by showing that Rage−/− mice failed to develop leukocytosis in response to diabetes (Fig 3B). Since Rage−/− BM progenitor cells did not respond to S100A8/A9, we reasoned that the interaction of S100A8/A9 with RAGE on BM progenitor cells was important for their proliferation. Although hematopoietic progenitor cells from WT mice had a low level of cell surface expression of RAGE, elevated levels of RAGE were found on CMPs from diabetic mice and were reduced by glucose lowering (Fig 3C & S3E).
Figure 3.

RAGE on myeloid progenitor cells mediates S100A8/A9-stimulated leukocytosis in diabetic mice.
(A) GMP proliferation in response to S100A8/A9 (2μg/ml) as measured by EdU incorporation. n=4 independent experiments. *P<0.05 vs. vehicle in each respective genotype.
(B) Blood leukocyte levels in WT and Rage−/− mice with and without STZ-diabetes. n=5-6/group. *P<0.05 vs. all groups.
(C) Surface expression of RAGE on CMPs (histogram and quantification) in WT and STZ-diabetic mice treated with SGLT2i. n=6, *P<0.05 vs. all groups, *P<0.05 vs. WT+STZ. (D) Experimental overview: WT mice were transplanted with BM from WT or Rage−/− mice and made diabetic with STZ.
(E-G) After 4wks of diabetes, (E) blood leukocytes, (F) BM HSPC, CMP, GMP numbers and (G) proliferation measured by flow cytometry. n=5/group. *P<0.05 vs. all groups.
(H-L) Competitive BMT: (H) Experimental overview: Equally mixed portions of CD45.1 and CD45.2 BM from the respective genotypes were transplanted into WT CD45.2 recipients and made diabetic with STZ. (I) Numbers of CD45.1 and CD45.2 monocytes and neutrophils from the respective donor BM. (J) Percentage of CD45.1 and CD45.2 CMPs and GMPs and (K) percentage of CD45.1 and CD45.2 CMPs and GMPs in the cell cycle from each respective donor BM. Data are means ± SEM, n=5-6/group. Numbers in parentheses indicate ratio of CD45.1:CD45.2. *P<0.05 vs. W/W. (L) Scheme summarizing the cell extrinsic proliferative pathway induced by S100A8/A9-RAGE signaling.
(M) GMP proliferation in response to S100A8/A9 ± the NF-κB inhibitor (SN50, 20μM).
(N) mRNA of M-CSF, GM-CSF and G-CSF as quantified by qRT-PCR. n=4 independent experiments. *P<0.05 vs. all groups, ^P<0.05 vs. HG.
(O) GMP proliferation in response to S100A8/A9 ± neutralizing antibodies to M-CSF and/or GM-CSF or isotype controls (ISO) (all 30μg/ml). n=4 independent experiments.
*P<0.05 vs. ISO vehicle, ^P<0.05 vs. respective ISO control.
All values are means ± SEM.
To further assess the role of hematopoietic-expressed RAGE we performed a BM transplant (BMT) study using WT mice transplanted with WT, Myd88−/− and Rage−/− BM. Only mice receiving Rage−/− BM failed to show enhanced myelopoiesis when made diabetic with STZ (Fig 3D-G & S5A-C). Further, quenching RAGE ligands in diabetic mice with soluble RAGE attenuated hyperglycemia-induced leukocytosis and reduced the proliferative capacity of diabetic serum (Fig S5D-G). These data confirm that the interaction of S100A8/A9 with RAGE on myeloid cells drives myelopoiesis in diabetes.
RAGE-dependent myelopoiesis is not cell autonomous
Binding of S100A8/A9 to RAGE could directly stimulate proliferation of CMPs in a cell autonomous fashion, or alternatively induce a signaling cascade leading to the production of proliferative cytokines that act on other myeloid progenitor cells (e.g. GMPs) in a non-cell autonomous fashion. To distinguish between these two possibilities, we performed a competitive BMT study where equal portions of BM from A) WT CD45.1 and WT CD45.2 or B) WT CD45.1 and Rage−/− CD45.2 mice were transplanted into WT recipients and made diabetic with STZ (Fig 3H). In group B recipients (Rage−/− CD45.2 BM), there was an overall decrease in the number of blood monocytes and neutrophils (both CD45.1 and CD45.2 cells), compared to group A recipients. Notably however, there was no change in the ratio of CD45.1 to CD45.2 cells in group B versus group A (Fig 3I). These findings were mirrored in the BM progenitor cells (Fig 3J,K). The decrease in the total number of monocytes, neutrophils, CMPs and GMPs without a preference for CD45.1 over CD45.2 cells, indicated that RAGE stimulated proliferation of CMPs/GMPs in a non-cell autonomous fashion. This would be consistent with a model (Fig 3L) in which S100A8/A9 induces an inflammatory cytokine response on diabetic CMPs where RAGE is highly expressed (Fig. 3C), resulting in the secretion of cytokines which in turn induce proliferation and expansion of GMPs. Consistent with this, pharmacological inhibition of NF-κB, which is central to RAGE signaling (Yao and Brownlee, 2010), attenuated GMP proliferation in response to S100A8/A9 (Fig 3M).
RAGE signals via NF-κB to stimulate cytokine production
Exploring the potential mediators of GMP proliferation, we found that S100A8/A9 dramatically elevated M-CSF, GM-CSF and G-CSF expression in BM cells via NF-κB (Fig 3N). Further, antibody neutralization of M-CSF or GM-CSF reduced S100A8/9-induced GMP proliferation, while combined neutralization abolished the proliferative effects of S100A8/A9 (Fig 3O). To determine the source of the colony stimulating factors, we isolated and cultured HSPCs, CMPs and GMPs (from WT BM) in HG ± S100A8/A9. M-CSF was mainly produced from CMPs (Fig S5H,I). BM macrophages appeared to be the main source of GM-CSF as determined by flow cytometry (Fig S5J).
We next tested whether RAGE was required for M-CSF and GM-CSF induction by S100A8/A9. BM deletion of either S100A8/A9 or RAGE prevented hyperglycemia-induced expression of these CSFs (Fig S5K,L). Together these data are consistent with a model in which neutrophil-derived S100A8/A9 interacts with RAGE on BM cells including CMPs to induce M-CSF and GM-CSF, leading to GMP proliferation and increased production of monocytes and neutrophils (Fig 3L).
Neutrophils drive myelopoiesis in diabetes
To directly test whether neutrophil-derived S100A8/A9 causes hyperglycemia-induced myelopoiesis, we performed a neutrophil depletion study using the neutrophil-specific Ly6-G antibody (clone 1A8; that does not deplete monocytes) (Daley et al., 2008) (Fig 4A). Neutrophil deletion normalized monocyte levels in diabetic mice (Fig 4B). Neutrophil-depleted diabetic mice also had lower levels of S100A8/A9 in the plasma (Fig 4C) and decreased numbers and proliferation of CMPs and GMPs (Fig 4D,E). Diabetes-induced RAGE expression on CMPs was unaltered by neutrophil depletion (Fig 4F). These findings confirm that neutrophil-derived ligands (likely S100A8/A9) are required to induce myelopoiesis and imply that non-neutrophil derived RAGE-ligands are insufficient to drive myelopoiesis.
Figure 4.
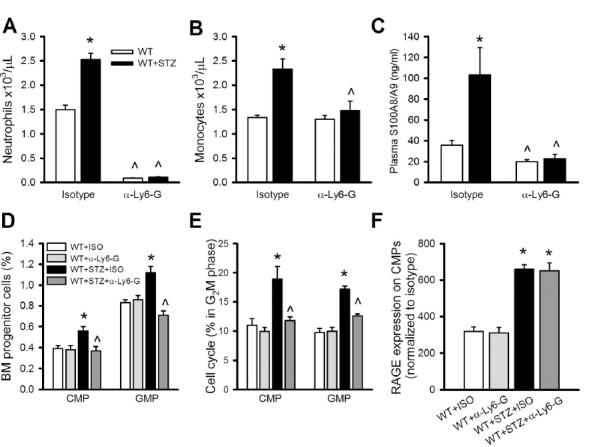
Neutrophils drive hyperglycemia-mediated leukocytosis in diabetes.
(A-F) Neutrophils in WT and STZ-diabetic mice were depleted by injecting anti-Ly6G antibody (clone 1A8, 1mg/mouse, i.p injection) every 3 days for 3 weeks.
(A) Neutrophil and (B) monocyte levels in WT and STZ mice treated with anti-Ly6-G or an isotype control.
(C) Plasma levels of S100A8/A9.
(D) CMP and GMP cell populations in the BM and (E) CMP and GMP cell proliferation assessed by DAPI staining and represented as percentage of cells in the G2M phase of the cell cycle.
(F) Surface expression of RAGE on CMPs.
All experiments n=5/group. *P<0.05 indicated diabetes effect, ^P<0.05 indicated anti-Ly6G effect. All values are means ± SEM.
Glucose reduction improves atherosclerotic lesion regression
To test the hypothesis that glucose-induced monocytosis is responsible for defective lesion regression, we performed an atherosclerosis regression study (experimental design: Fig 5A). Regression in Ldlr−/− mice with established but uncomplicated lesions was initiated by switching from a high cholesterol diet (0.15%) to a chow diet (Reg). Some mice were made diabetic (Reg + STZ) and a subgroup was treated with the SGLT2i (Reg + STZ + SGLT2i). After 6 weeks on chow diet, all mice had similarly reduced plasma cholesterol levels and importantly there was no difference in cholesterol levels between the Reg + STZ and Reg + STZ + SGLT2i mice (Fig 5B). As expected, Reg + STZ mice had monocytosis (Fig 5C), which was accompanied by increased plasma levels of S100A8/A9, enhanced expression of RAGE on CMPs and expansion and proliferation of CMPs and GMPs (Fig S6A-D). Reducing blood glucose reversed these effects, ultimately reducing the number of circulating Ly6-Chi monocytes.
Figure 5.

Defective lesion regression in diabetic mice is improved by normalizing plasma glucose.
(A) Experimental overview: Ldlr−/− mice were fed a HCD (0.15%) for 16 weeks to develop atherosclerotic lesions. At this time point a group of mice was sacrificed to determine baseline (pre-regression) lesion characteristics. The remaining mice were divided into 3 groups; Reg, Reg+STZ and Reg+STZ+SGLT2i and placed on chow diet for 6 weeks (n=10-11/group).
(B) Blood glucose and total cholesterol levels at baseline and 6 weeks of regression. *P<0.05 vs. all groups.
(C) Blood leukocyte levels at baseline and after 6 weeks of regression as assessed by flow cytometry. n=9-11/group. *P<0.05 vs. all groups, ^P<0.05 vs. Reg+STZ.
(D) Quantification of mean lesion areas.
(E) Representative ORO stained lesions and quantification of ORO stain as percent of lesion area.
(F) Representative CD68+ stained lesions and quantification as CD68+ area/lesion. *P<0.05 vs. Reg and Reg+STZ+SGLT2i, ^P<0.05 vs Reg+STZ.
(G-H) FACS isolated Ly6-Chi monocytes were (G) allowed to adhere to cultured human aortic endothelial cells under static conditions or (H) assessed for their ability to migrate towards CCL2 (in a transwell chamber). *P<0.05 vs. all groups, ^P<0.05 vs. Reg+STZ n=4-6/group.
(I) Representative images and quantification of lesions stained with an anti-Ly6-C antibody (FITC; green) and Hoechst dye (blue; nuclei) using confocal microscopy. Arrows indicate Ly6-C+ cells (Ly6-Chi monocytes). *P<0.05 vs. all groups, ^P<0.05 vs. Reg+STZ. All values are means ± SEM.
Reg mice showed a marked reduction in lesion area; lipid (Oil Red O) and macrophage content (CD68+ cells) were decreased compared to baseline. Diabetes severely impaired the regression process. However, lowering plasma glucose significantly improved lesion regression as shown by decreased lesion area, lipid accumulation and macrophage content (Fig 5D,E & S6E).
mRNA analysis of lesional CD68+ cells revealed that the macrophages in diabetic mice had increased production of inflammatory molecules (Fig S7A). This could be due to a number of factors including recruitment of inflammatory Ly6-Chi monocytes. Consistent with increased Ly6-Chi monocyte recruitment, we observed changes in a number of parameters of the cell adhesion cascade including an increase in the number (Fig 5C), activation (CD11b; Fig 5G), adhesion (Fig 5H) and migration (Fig 5I) of Ly6-Chi monocytes from diabetic (Reg+STZ) mice. There was a non-significant trend for increased expression of ICAM-1 and VCAM-1 on the endothelial cells covering the lesion (data not shown). Increased lesional Ly6-C staining (Fig 5J) and Ccr2 mRNA was consistent with greater recruitment of Ly6-Chi (CCR2+) monocytes (Swirski et al., 2007; Tacke et al., 2007) (Fig S7B). We also observed an increase in the expression of a number of chemokines in lesional macrophages (Fig S7B), consistent with enhanced monocyte recruitment. These key processes in the cell adhesion cascade along with the Ly6-C staining and expression of chemotactic genes in the lesion were all decreased by lowering blood glucose levels.
Defective lesion regression in diabetes reflects persistent monocyte recruitment
The results from our regression study (Fig 5) pointed towards changes in monocyte recruitment as the mechanism for defective lesion regression in diabetes. Defects in lesion regression can result from changes in monocyte entry (Potteaux et al., 2011), macrophage egress (Trogan et al., 2006), or both. To assess monocyte entry vs. egress on macrophage burden in diabetic mice, we performed fate mapping studies using two different models; aortic transplantation (Trogan et al., 2006) and diet-induced regression. To assess monocyte entry, atherosclerotic segments of the aorta were transplanted into Akita mice and they were injected with fluorescent beads to label their monocytes 48 hrs before sacrifice (Outline: Fig 6A). Compared to untreated mice, there was a significant reduction in the uptake of beads in the lesions of SGTL2i treated Akita mice (Fig 6B), indicating reduce monocyte entry/recruitment. To determine the effect of glucose reduction on egress of monocytes/macrophages from lesions, in the same experiment we had labeled Ly6-Chi monocytes in the blood of transplant donors using EdU. A group of donor mice were killed to assess the amount of EdU+ labeling in aortic arch lesions and this served as a baseline measurement to calculate egress. The was a similar reduction of EdU+ cells in lesions of the transplanted aortas in both groups of Akita mice (Fig 6C), suggesting that blood glucose did not affect the emigration of macrophages from lesions.
Figure 6.

Impaired lesion regression in diabetic mice is due to increased monocyte recruitment.
(A) Ldlr−/− mice were fed a HCD for 16wks to develop atherosclerotic lesions. Aortic transplantation model overview: All donor Ldlr−/− mice were injected with EdU (1 mg, i.v) to label newly formed monocytes. 48 hrs post-injection, aortas were dissected from these mice and either processed for baseline measurement of EdU+ cells or transplanted into chow fed Akita mice ± SGLT2i. 48hrs prior to termination, mice were injected with green fluorescent microspheres to determine monocyte entry.
(B) Monocyte entry determined by fluorescent beads.
(C-D) Monocyte egress: (C) Representative images showing EdU stain (red) in the aortic arch sections of donor mice (baseline) and grafts from Akita and Akita+SGLT2i.
(D) Quantification of monocyte egress as represented by the number of EdU+ cells per section. *P<0.05 vs. baseline, n=5/group.
(E) Diet induced regression model overview: Ldlr−/− mice fed with a HCD for 16 wks were divided into 3 groups; regression (reg), reg+STZ and reg+STZ+SGLT2i and placed on chow diet. At the end of 4 weeks, all mice were injected (i.v) with clodronate liposomes (CLO, 250μl) to deplete the circulating monocytes. 48 hrs later they were injected with green fluorescent microspheres. 4 days later, a portion for mice from each group were sacrificed to determine the baseline measurement of beads in the atheroma and to assess Ly6-Chi monocyte entry. A second group of mice was assessed 14 days later (6 weeks of regression) for quantification of labeled macrophages remaining in plaques. 48 hrs prior to sacrificing the final group of mice they were injected with EdU to also assess Ly6-Chi monocyte entry.
(F) Ly6-Chi monocyte entry as determined by EdU+ cells in the lesion.
(G) Ly6-Chi monocyte entry and monocyte/macrophage retention in the lesion as assessed by number of beads/section.
(H) Percentage of monocyte/macrophage egress compared to baseline. *P<0.05 vs. all groups, ^P<0.05 final vs. baseline, n=6/group.
All values are means ± SEM.
To further explore the effect of glucose reduction on Ly6-Chi monocyte entry and egress, we used a combination approach involving both EdU and bead fate mapping of monocytes in diet-induced regression model (Outline: Fig 6D). We employed clodronate liposomes to deplete circulating monocytes and specifically labeled Ly6-Chi cells with fluorescent beads (Tacke et al., 2007). This was done at week 4 of the regression period, with a group of mice sacrificed 4 days post-liposome injection (48hrs post-beads). This allowed us to examine recruitment of Ly6-Chi monocytes to the lesion and this data also provided a baseline value of the number of Ly6-Chi monocytes/macrophages in order for us to determine monocyte/macrophage retention and egress rates in the respective regressing lesions. 48 hrs before the end of the regression period (T= −48hrs) we also labeled circulating Ly6-Chi monocytes with EdU as a confirmatory measure of monocyte entry into the atheroma. The number of Ly6-Chi cells that entered lesions from Reg+STZ group was significantly greater as measured by the number of EdU+ cells or beads per lesion (Fig 6F,G). While there was increased retention of macrophages as determined by the absolute number of beads in the lesion after the regression period (Fig 6G), the percentage of egress was similar in both groups (Fig 6H), consistent with our findings in the aortic transplant model (Fig 6C). Moreover, our lesional macrophage gene expression data revealed that Ccr7, a key receptor for macrophage egress (Forster et al., 1999; Trogan et al., 2006), was decreased in diabetic mice, but not reversed by glucose lowering (Fig S7B), which also supports the conclusion that egress levels are similar in both groups of mice. We acknowledge that EdU is incorporated into the DNA of dividing cells and our data could also in part be affected by macrophage proliferation or apoptosis in the lesions, however, the data obtained with EdU was consistent with that obtained with fluorescent beads. Importantly, taken together, these data support the hypothesis that persistent recruitment of Ly6-Chi monocytes into lesions is responsible for greater macrophage content in hyperglycemia and impaired lesion regression.
S100A8/A9 is associated with leukocytosis and the prevalence of CAD in T1D patients
To determine if total WBC count, leukocytes subsets, and S100A8/A9 levels correlate with CAD in T1DM, we analyzed data from the 18th year follow up of the Pittsburgh Epidemiology of Diabetes Complications (EDC) study. T1DM patients with CAD had increased numbers of WBCs, monocytes and neutrophils and serum levels of S100A8/A9 (Fig 7A). In univariate analysis, total WBC, neutrophil counts and monocyte counts were all significantly associated with CAD in the 290 participants with Odds Ratios (OR) of 1.44, 1.64 and 1.56, respectively. However, after adjusting for other established risk factors only neutrophil count remained independently associated with CAD (OR 1.65, p=0.03). In a subgroup of 49 subjects, we measured serum S100A8/A9 levels and found a borderline association with CAD (OR 1.45, p=0.06), which was a stronger correlate than total WBCs (1.20, p=0.31). Further analysis revealed a positive correlation between serum S100A8/A9 and both neutrophils and WBC (Fig 7B,C).
Figure 7.

S100A8/A9 correlates with leukocytes in T1DM patients with CAD and stimulates myelopoiesis in human CD34+ progenitor cells.
(A) Leukocyte and S100A8/A9 levels in T1DM patients with and without CAD.
(B-C) Regression analysis: (B) S100A8/A9 vs. WBCs and (C) S100A8/A9 vs. neutrophils. n=49.
(D) Proliferation of CD34+ progenitor cells to increasing doses of S100A8/A9 was measured by EdU incorporation.
(E) Production of CD14+ monocytes from CD34+ progenitor cells to increasing doses of S100A8/A9. D, E, n=4 independent experiments, data are means ± SEM, *P<0.05 vs. control.
(F) Schematic overview: In response to hyperglycemia, (1) neutrophils produce S100A8/A9, which is (2) sensed by RAGE on BM CMPs and macrophages to signal via NF-κB to induce M-CSF, GM-CSF production. M-CSF and GM-CSF in turn acts in an autocrine/paracrine fashion to stimulate CMP and GMP proliferation, producing monocytes and neutrophils. (3) Ly6-Chi monocytes in the circulation become activated, adhere to the endothelium and readily enter lesions leading to increased lesional macrophages.
To examine if S100A8/A9 promotes myelopoiesis using human cells, we stimulated cultured hematopoietic progenitor cells obtained from human cord blood (CD34+ cells) with S100A8/A9. CD34+ cells cultured in HG and varying concentrations of S100A8/A9 showed a dose-dependent increase in proliferation (Fig 7D) and in production of CD14+ monocytes (Fig 7E).
DISCUSSION
We have shown that hyperglycemia causes increased production of inflammatory cells in the bone marrow, monocytosis and neutrophilia and impairs atherosclerotic lesion regression. In response to hyperglycemia, neutrophils produce S100A8/A9, which interacts with glucose-inducible RAGE on CMPs resulting in the release of inflammatory Ly6-Chi cells. The enhanced and persistent entry of these cells into lesions impairs regression. Normalizing blood glucose facilitated resolution by dampening S100A8/A9-initiated monocytosis and causing an overall decrease in inflammatory status (summarized in Fig 7F). These observations demonstrate a previously unknown crosstalk between diabetic neutrophils and myeloid progenitor cells, leading to increased production of monocytes and neutrophils, and provide a mechanistic link between diabetes and CAD.
Diabetes is generally considered to be an inflammatory state (Wellen and Hotamisligil, 2005) but the underlying mechanisms are not well understood. Surprisingly, we discovered prominent monocytosis and neutrophilia in a two commonly used mouse models of T1DM. Mice with defective cholesterol efflux pathways due to deficiency of Apoe or Abca1/g1 also have leukocytosis; these studies have provided evidence that increased entry of circulating monocytes into lesions promotes atherogenesis (Murphy et al., 2011; Potteaux et al., 2011; Robbins et al., 2011; Swirski et al., 2007; Tacke et al., 2007; Yvan-Charvet et al., 2010). In part these changes reflected a cell autonomous proliferation and expansion of hematopoietic stem and progenitor cells in the bone marrow linked to increased levels of growth factor receptors (Murphy et al., 2011; Yvan-Charvet et al., 2010). However, our investigations in the diabetic mouse models uncovered a distinctive set of mechanisms underlying the increased production of inflammatory cells. First, monocytosis and neutrophilia could be completely dissociated from effects of hypercholesterolemia, since they were observed in normolipidemic chow fed diabetic mice. Rather, making use of a glycosuric agent, we were able to demonstrate a direct effect of hyperglycemia on myelopoiesis. Moreover, increased production of monocytes and neutrophils was not associated with expansion of the stem cell pool, but rather with increased proliferation and numbers of CMPs and GMPs, and shown to be mediated by cell extrinsic factors. These factors were found to be increased production of S100A8/9 by neutrophils, which stimulated RAGE on CMPs, leading to secretion of colony stimulating factors that induced proliferation of GMPs and increased production of monocytes and neutrophils. Although S100A8/9 and RAGE have been previously implicated in diabetic inflammation and atherogenesis, these studies have uncovered a signaling axis in which diabetes led to increased expression of RAGE in CMPs, mediating an augmented BM proliferative response to the elevated production of S100A8/9 from neutrophils.
Previous studies have reported increased levels of S100A8/A9 in the plasma and lesions of diabetic mice (Soro-Paavonen et al., 2008). However, the specific role of these proteins in atherosclerosis has been controversial. Deletion of S100A9 reduced atherosclerosis in one study performed on the Apoe−/− background (Croce et al., 2009), while transplant of S100a9−/− BM into Ldlr−/− mice did not affect lesion size (Averill et al., 2011). These discrepancies could be due to different genetic backgrounds and disease states but importantly, neither of these studies was conducted in the setting of diabetes.
RAGE has been previously implicated in atherosclerosis but not as a receptor mediating increased production of inflammatory cells. Deletion of RAGE globally (Soro-Paavonen et al., 2008) or in hematopoietic cells (Morris-Rosenfeld et al., 2011) reduces atherosclerosis, likely by dampening the production of inflammatory mediators. Previous studies implicating RAGE in diabetic atherosclerosis have mainly suggested that RAGE was activated by advanced glycation endproducts (Bucciarelli et al., 2002; Harja et al., 2008; Park et al., 1998; Soro-Paavonen et al., 2008). However, a seminal study by Reaven and colleagues revealed that accumulation of AGEs does not influence atherosclerosis (Reaven et al., 1997). Our studies indicate that ligands other than AGEs notably S100A8/A9 are important for RAGE activation and its inflammatory effects.
Our study using murine atherosclerosis regression models shows that persistent monocytosis and increased monocyte entry into lesions likely contributes to the previously documented impaired regression in diabetes (Parathath et al., 2011). These findings provide conceptual support for the work of Potteaux et al., demonstrating that persistent monocyte entry into lesions may contribute to impaired regression, and show for the first time that excessive bone marrow production of inflammatory cells contributes to this process. While increased macrophage production of chemokines was likely an important factor in both studies, our study reveals a coordinated process in which increased production and activation of monocytes is coupled with enhanced calling of cells into the inflammatory milieu of the plaque. An important implication is that normalizing monocyte production and decreasing monocyte influx into the plaque may be an avenue to facilitate lesion regression in the setting of diabetes. Glucose reduction may also have beneficial effects on macrophages and other cells in the atheroma including, vascular smooth muscle and regulatory T-cells (Bornfeldt and Tabas, 2011).
A limitation of our study is that our untreated diabetic mice were not under any form of glucose control, unlike diabetic patients where glucose is usually controlled. Therefore it is possible that the hyperglycemia-driven leukocytosis observed in our mouse models is dampened in humans. However, intermittent spikes of hyperglycemia appear to be sufficient to develop leukocytosis (Ford, 2002; Persson et al., 1998; Schmidt et al., 1999; Vuckovic et al., 2007; Woo et al., 2011), likely by S100A8/A9-mediated mechanisms. These hyperglycemia-induced changes in WBCs could affect vascular disease.
Suggesting human relevance of our mechanistic mouse studies, we found a significant correlation among leukocytes, serum S100A8/A9 and the incidence of CAD in a subset of T1DM patients from the Pittsburg EDC study. While these data only describe correlations and not a direct role for S100A8/A9, our in vitro studies using human CD34+ cells also suggest that our findings in the mouse may be translatable to these patients. These findings are also consistent with other studies in which plasma S100A8/A9 concentration (Healy et al., 2006) or circulating leukocyte numbers (Orchard et al., 2003) independently predicts future CAD.
Although our studies largely focused on hyperglycemia in insulin deficient mouse models of diabetes, patients with either T1DM or T2DM have increased leukocyte counts (Ford, 2002; Schmidt et al., 1999; Tong et al., 2004; van Oostrom et al., 2004) and are at greater risk of developing CAD. Our study in mouse models strongly suggests that hyperglycemia per se promotes monocyte production in patients with diabetes and this may be a factor contributing to higher risk of CAD. Thus, our findings highlight the potential importance of glucose control as well as lipid lowering therapy as strategies to promote regression of atherosclerosis in diabetics, and also suggest a number of therapeutic targets, including disruption of the S100A8/A9-RAGE signaling axis.
Experimental Procedures
Mice and Treatments
8-12 weeks old male mice – wild type, Ins2Akita/J, S100a9−/−, Ldlr−/−, Rage−/−, Myd88−/− in C57BL/6J backgrounds – were used for this study. Diabetes was induced by streptozotocin (STZ; 50mg/kg. i.p. for 5 days). Sodium glucose transporter 2 inhibitors (SGLT2i) from ISIS Pharmaceuticals Inc. and Bristol Myers Squibb Company were administered in drinking water at a dose of 5 mg and 25 mg/kg (b.w), respectively. Mice in the STZ- and Akita-diabetic groups were treated for 4 weeks with the SGLT2i, while Ldlr−/− mice in the atherosclerosis regression study were treated for 6 weeks. Unless specifically indicated all diabetic mice were treated with SGLT2i from ISIS Pharmaceuticals at a dose of 5 mg/kg/day in drinking water.
For S100A8/A9 and HMGB1 in vivo studies, WT mice were injected with vehicle or human recombinant proteins at a dose of 20 μg/kg/day (i.v), twice a day for 3 days after which their blood and BM cells were analyzed by flow cytometry.
Flow Cytometry
Blood leukocytes
Leukocyte subsets were identified from whole blood as previously described (Murphy et al., 2011). EDTA anti-coagulated blood was subjected to RBC lysis. WBCs were re-suspended in flow-buffer (Hanks balanced salt solution + 0.1% BSA w/v, 5mM EDTA) and stained with a cocktail of antibodies. Monocytes were identified as CD45hiCD115hi and subsets as Ly6-Chi and Ly6-Clo; neutrophils were identified as CD45hiCD115loLy6-C/Ghi (Gr-1). CD11b mean fluorescent intensity (geometric mean) was measured as marker of monocyte activation (Murphy et al., 2008).
Hematopoietic Stem Cells
Hematopoietic stem and progenitor cells were analyzed by flow cytometry as previously described (Murphy et al., 2011). BM was harvested from femurs and tibias, RBCs lysed and the cell suspension incubation with a cocktail of antibodies against lineage-committed cells (B220, CD19, CD11b, CD3e, TER-119, CD2, CD8, CD4, Ly6-C/G: All FITC) and markers to identify; HSPCs lineage−, Sac1+ and ckit+ (LSK); CMPs were identified as lineage−, Sca1−, ckit+, CD34int, FcγRII/IIIint; GMPs as lineage−, Sca1−, ckit+, CD34int, FcγRII/IIIhi. RAGE expression was measured using a primary antibody to RAGE followed by a fluorescently conjugated secondary antibody. Cell cycle analysis was performed using DAPI. Flow cytometry was performed using an LSRII (for analysis) or FACS Aria (for sorting), both machines running FACS DiVa software. All flow cytometry data were analyzed using FlowJo software (Tree Star Inc.)
Supplementary Material
Highlights.
Hyperglycemia causes myeloproliferation
Neutrophil S100A8/A9 interacts with RAGE to stimulate myelopoiesis
Glucose reduction improves atherosclerosis regression
ACKNOWLEDGEMENTS
We thank ISIS Pharmaceuticals Inc. and Bristol-Myers Squibb for providing SGLT2 inhibitors. We also acknowledge the assistance from Flow Cytometry and Imaging Core Facilities at Columbia University. This project was funded by grants from NIH: U01-HL087945 & DK095684 – Dual-PI grants to IJG & EAF, P01-HL54591 (IJG) and HL87123, HL107653 (ART) and a Pilot and Feasibility Award from the Diabetes Complications Consortium (DCC to IJG). PRN was supported by a post-doctoral fellowship from the Canadian Institutes of Health Research and AJM was supported by a post-doctoral fellowship from the American Heart Association (12POST11890019).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Detailed methods are available online as a supplementary file.
References
- Averill MM, Barnhart S, Becker L, Li X, Heinecke JW, Leboeuf RC, Hamerman JA, Sorg C, Kerkhoff C, Bornfeldt KE. S100A9 differentially modifies phenotypic states of neutrophils, macrophages, and dendritic cells: implications for atherosclerosis and adipose tissue inflammation. Circulation. 2011;123:1216–1226. doi: 10.1161/CIRCULATIONAHA.110.985523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornfeldt KE, Tabas I. Insulin resistance, hyperglycemia, and atherosclerosis. Cell Metab. 2011;14:575–585. doi: 10.1016/j.cmet.2011.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, Goova MT, Moser B, Kislinger T, Lee DC, Kashyap Y, Stern DM, Schmidt AM. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 2002;106:2827–2835. doi: 10.1161/01.cir.0000039325.03698.36. [DOI] [PubMed] [Google Scholar]
- Christopher MJ, Rao M, Liu F, Woloszynek JR, Link DC. Expression of the G-CSF receptor in monocytic cells is sufficient to mediate hematopoietic progenitor mobilization by G-CSF in mice. The Journal of experimental medicine. 2011;208:251–260. doi: 10.1084/jem.20101700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller BS. Leukocytosis and ischemic vascular disease morbidity and mortality: is it time to intervene? Arterioscler Thromb Vasc Biol. 2005;25:658–670. doi: 10.1161/01.ATV.0000156877.94472.a5. [DOI] [PubMed] [Google Scholar]
- Croce K, Gao H, Wang Y, Mooroka T, Sakuma M, Shi C, Sukhova GK, Packard RR, Hogg N, Libby P, Simon DI. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation. 2009;120:427–436. doi: 10.1161/CIRCULATIONAHA.108.814582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- Danesh J, Collins R, Appleby P, Peto R. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: meta-analyses of prospective studies. JAMA. 1998;279:1477–1482. doi: 10.1001/jama.279.18.1477. [DOI] [PubMed] [Google Scholar]
- Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. 2010;122:1837–1845. doi: 10.1161/CIRCULATIONAHA.110.961714. [DOI] [PubMed] [Google Scholar]
- Duff GL, Payne TP. The effect of alloxan diabetes on experimental cholesterol atherosclerosis in the rabbit. III. The mechanism of the inhibition of experimental cholesterol atherosclerosis in alloxan-diabetic rabbits. J Exp Med. 1950;92:299–317. doi: 10.1084/jem.92.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig JE, Pineda-Torra I, Sanson M, Bradley MN, Vengrenyuk Y, Bogunovic D, Gautier EL, Rubinstein D, Hong C, Liu J, Wu C, van Rooijen N, Bhardwaj N, Garabedian M, Tontonoz P, Fisher EA. LXR promotes the maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J Clin Invest. 2010;120:4415–4424. doi: 10.1172/JCI38911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro F, Lymperi S, Mendez-Ferrer S, Saez B, Spencer JA, Yeap BY, Masselli E, Graiani G, Prezioso L, Rizzini EL, Mangoni M, Rizzoli V, Sykes SM, Lin CP, Frenette PS, Quaini F, Scadden DT. Diabetes impairs hematopoietic stem cell mobilization by altering niche function. Sci Transl Med. 2011;3:104ra101. doi: 10.1126/scitranslmed.3002191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford ES. Leukocyte Count, Erythrocyte Sedimentation Rate, and Diabetes Incidence in a National Sample of US Adults. Am J Epidemiol. 2002;155:57–64. doi: 10.1093/aje/155.1.57. [DOI] [PubMed] [Google Scholar]
- Forster R, Schubel A, Breitfeld D, Kremmer E, Renner-Muller I, Wolf E, Lipp M. CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell. 1999;99:23–33. doi: 10.1016/s0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Fujiu K, Manabe I, Nagai R. Renal collecting duct epithelial cells regulate inflammation in tubulointerstitial damage in mice. J Clin Invest. 2011;121:3425–3441. doi: 10.1172/JCI57582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, Kalea AZ, Lu Y, Rosario RH, Oruganti S, Nikolla Z, Belov D, Lalla E, Ramasamy R, Yan SF, Schmidt AM. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE−/− mice. J Clin Invest. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy AM, Pickard MD, Pradhan AD, Wang Y, Chen Z, Croce K, Sakuma M, Shi C, Zago AC, Garasic J, Damokosh AI, Dowie TL, Poisson L, Lillie J, Libby P, Ridker PM, Simon DI. Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation. 2006;113:2278–2284. doi: 10.1161/CIRCULATIONAHA.105.607333. [DOI] [PubMed] [Google Scholar]
- Hiro T, Kimura T, Morimoto T, Miyauchi K, Nakagawa Y, Yamagishi M, Ozaki Y, Kimura K, Saito S, Yamaguchi T, Daida H, Matsuzaki M. Diabetes mellitus is a major negative determinant of coronary plaque regression during statin therapy in patients with acute coronary syndrome--serial intravascular ultrasound observations from the Japan Assessment of Pitavastatin and Atorvastatin in Acute Coronary Syndrome Trial (the JAPAN-ACS Trial) Circ J. 2010;74:1165–1174. doi: 10.1253/circj.cj-09-0766. [DOI] [PubMed] [Google Scholar]
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- Leukert N, Vogl T, Strupat K, Reichelt R, Sorg C, Roth J. Calcium-dependent tetramer formation of S100A8 and S100A9 is essential for biological activity. J. Mol. Biol. 2006;359:961–972. doi: 10.1016/j.jmb.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Lieschke GJ, Grail D, Hodgson G, Metcalf D, Stanley E, Cheers C, Fowler KJ, Basu S, Zhan YF, Dunn AR. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood. 1994;84:1737–1746. [PubMed] [Google Scholar]
- Meng W, Ellsworth BA, Nirschl AA, McCann PJ, Patel M, Girotra RN, Wu G, Sher PM, Morrison EP, Biller SA, Zahler R, Deshpande PP, Pullockaran A, Hagan DL, Morgan N, Taylor JR, Obermeier MT, Humphreys WG, Khanna A, Discenza L, Robertson JG, Wang A, Han S, Wetterau JR, Janovitz EB, Flint OP, Whaley JM, Washburn WN. Discovery of dapagliflozin: a potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J Med Chem. 2008;51:1145–1149. doi: 10.1021/jm701272q. [DOI] [PubMed] [Google Scholar]
- Morris-Rosenfeld S, Blessing E, Preusch MR, Albrecht C, Bierhaus A, Andrassy M, Nawroth PP, Rosenfeld ME, Katus HA, Bea F. Deletion of bone marrow-derived receptor for advanced glycation end products inhibits atherosclerotic plaque progression. Eur J Clin Invest. 2011;41:1164–1171. doi: 10.1111/j.1365-2362.2011.02514.x. [DOI] [PubMed] [Google Scholar]
- Murphy AJ, Akhtari M, Tolani S, Pagler T, Bijl N, Kuo CL, Wang M, Sanson M, Abramowicz S, Welch C, Bochem AE, Kuivenhoven JA, Yvan-Charvet L, Tall AR. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J Clin Invest. 2011;121:4138–4149. doi: 10.1172/JCI57559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AJ, Woollard KJ, Hoang A, Mukhamedova N, Stirzaker RA, McCormick SP, Remaley AT, Sviridov D, Chin-Dusting J. High-density lipoprotein reduces the human monocyte inflammatory response. Arterioscler Thromb Vasc Biol. 2008;28:2071–2077. doi: 10.1161/ATVBAHA.108.168690. [DOI] [PubMed] [Google Scholar]
- Nicholls SJ, Ballantyne CM, Barter PJ, Chapman MJ, Erbel RM, Libby P, Raichlen JS, Uno K, Borgman M, Wolski K, Nissen SE. Effect of two intensive statin regimens on progression of coronary disease. N Engl J Med. 2011;365:2078–2087. doi: 10.1056/NEJMoa1110874. [DOI] [PubMed] [Google Scholar]
- Orchard TJ, Olson JC, Erbey JR, Williams K, Forrest KY, Smithline Kinder L, Ellis D, Becker DJ. Insulin resistance-related factors, but not glycemia, predict coronary artery disease in type 1 diabetes: 10-year follow-up data from the Pittsburgh Epidemiology of Diabetes Complications Study. Diabetes Care. 2003;26:1374–1379. doi: 10.2337/diacare.26.5.1374. [DOI] [PubMed] [Google Scholar]
- Parathath S, Grauer L, Huang LS, Sanson M, Distel E, Goldberg IJ, Fisher EA. Diabetes Mellitus Adversely Affects Macrophages During Atherosclerotic Plaque Regression in Mice. Diabetes. 2011;60:1759–1769. doi: 10.2337/db10-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr., Chow WS, Stern D, Schmidt AM. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- Persson SU, Larsson H, Odeberg H. Reduced number of circulating monocytes after institution of insulin therapy--relevance for development of atherosclerosis in diabetics? Angiology. 1998;49:423–433. doi: 10.1177/000331979804900602. [DOI] [PubMed] [Google Scholar]
- Potteaux S, Gautier EL, Hutchison SB, van Rooijen N, Rader DJ, Thomas MJ, Sorci-Thomas MG, Randolph GJ. Suppressed monocyte recruitment drives macrophage removal from atherosclerotic plaques of Apoe−/− mice during disease regression. J Clin Invest. 2011;121:2025–2036. doi: 10.1172/JCI43802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaven P, Merat S, Casanada F, Sutphin M, Palinski W. Effect of streptozotocin-induced hyperglycemia on lipid profiles, formation of advanced glycation endproducts in lesions, and extent of atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 1997;17:2250–2256. doi: 10.1161/01.atv.17.10.2250. [DOI] [PubMed] [Google Scholar]
- Robbins CS, Chudnovskiy A, Rauch PJ, Figueiredo JL, Iwamoto Y, Gorbatov R, Etzrodt M, Weber GF, Ueno T, van Rooijen N, Mulligan-Kehoe MJ, Libby P, Nahrendorf M, Pittet MJ, Weissleder R, Swirski FK. Extramedullary Hematopoiesis Generates Ly-6Chigh Monocytes that Infiltrate Atherosclerotic Lesions. Circulation. 2011 doi: 10.1161/CIRCULATIONAHA.111.061986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AW. G-CSF: a key regulator of neutrophil production, but that's not all! Growth Factors. 2005;23:33–41. doi: 10.1080/08977190500055836. [DOI] [PubMed] [Google Scholar]
- Schmidt MI, Duncan BB, Sharrett AR, Lindberg G, Savage PJ, Offenbacher S, Azambuja MI, Tracy RP, Heiss G. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet. 1999;353:1649–1652. doi: 10.1016/s0140-6736(99)01046-6. [DOI] [PubMed] [Google Scholar]
- Semerad CL, Poursine-Laurent J, Liu F, Link DC. A role for G-CSF receptor signaling in the regulation of hematopoietic cell function but not lineage commitment or differentiation. Immunity. 1999;11:153–161. doi: 10.1016/s1074-7613(00)80090-4. [DOI] [PubMed] [Google Scholar]
- Soro-Paavonen A, Watson AM, Li J, Paavonen K, Koitka A, Calkin AC, Barit D, Coughlan MT, Drew BG, Lancaster GI, Thomas M, Forbes JM, Nawroth PP, Bierhaus A, Cooper ME, Jandeleit-Dahm KA. Receptor for advanced glycation end products (RAGE) deficiency attenuates the development of atherosclerosis in diabetes. Diabetes. 2008;57:2461–2469. doi: 10.2337/db07-1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M, van Rooijen N, Lira SA, Habenicht AJ, Randolph GJ. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185–194. doi: 10.1172/JCI28549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong PC, Lee KF, So WY, Ng MH, Chan WB, Lo MK, Chan NN, Chan JC. White blood cell count is associated with macro- and microvascular complications in chinese patients with type 2 diabetes. Diabetes Care. 2004;27:216–222. doi: 10.2337/diacare.27.1.216. [DOI] [PubMed] [Google Scholar]
- Trogan E, Feig JE, Dogan S, Rothblat GH, Angeli V, Tacke F, Randolph GJ, Fisher EA. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc Natl Acad Sci U S A. 2006;103:3781–3786. doi: 10.1073/pnas.0511043103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Oostrom AJ, van Wijk JP, Sijmonsma TP, Rabelink TJ, Castro Cabezas M. Increased expression of activation markers on monocytes and neutrophils in type 2 diabetes. Neth J Med. 2004;62:320–325. [PubMed] [Google Scholar]
- Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, Roth J. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- Vuckovic S, Withers G, Harris M, Khalil D, Gardiner D, Flesch I, Tepes S, Greer R, Cowley D, Cotterill A, Hart DN. Decreased blood dendritic cell counts in type 1 diabetic children. Clin. Immunol. 2007;123:281–288. doi: 10.1016/j.clim.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo SJ, Ahn SJ, Ahn J, Park KH, Lee K. Elevated systemic neutrophil count in diabetic retinopathy and diabetes: a hospital-based cross-sectional study of 30,793 Korean subjects. Invest Ophthalmol Vis Sci. 2011;52:7697–7703. doi: 10.1167/iovs.11-7784. [DOI] [PubMed] [Google Scholar]
- Yao D, Brownlee M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes. 2010;59:249–255. doi: 10.2337/db09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yvan-Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, Tall AR. ATP-binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. doi: 10.1126/science.1189731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.