

Abstract
Background
Adenosine monophosphate-activated protein kinase (AMPK) is stimulated in embryos during diabetic pregnancy by maternal hyperglycaemia-induced embryo oxidative stress. Stimulation of AMPK disrupts embryo gene expression and causes neural tube defects. Metformin, which may be taken during early pregnancy, has been reported to stimulate AMPK activity. Thus, the benefits of improved glycaemic control could be offset by stimulated embryo AMPK activity. Here, we investigated whether metformin can stimulate AMPK activity in mouse embryos and can adversely affect embryo gene expression and neural tube defects.
Methods
Pregnant nondiabetic mice were administered metformin beginning on the first day of pregnancy. Activation of maternal and embryo AMPK [phospho-AMPK α (Thr172) relative to total AMPK], expression of Pax3, a gene required for neural tube closure, and neural tube defects were studied. Mouse embryonic stem cells were used as a cell culture model of embryonic neuroepithelium to study metformin effects on AMPK and Pax3 expression.
Results
Metformin had no effect on AMPK in embryos or maternal skeletal muscle but increased activated AMPK in maternal liver. Metformin did not inhibit Pax3 expression or increase neural tube defects. However, metformin increased activated AMPK and inhibited Pax3 expression by mouse embryonic stem cells. Mate1/Slc47a1 and Oct3/Slc22a, which encode metformin transporters, were expressed at barely detectable levels by embryos.
Conclusions
Although metformin can have effects associated with diabetic embryopathy in vitro, the lack of effects on mouse embryos in vivo may be due to lack of metformin transporters and indicates that the benefits of metformin on glycaemic control are not counteracted by stimulation of embryo AMPK activity and consequent embryopathy.
Keywords: metformin, AMP kinase, diabetic pregnancy, diabetic embryopathy, oral hypoglycaemics, Pax3
Introduction
The biguanide metformin is commonly used to treat insulin resistance, type 2 diabetes and polycystic ovary syndrome (PCOS) [1,2]. Metformin is a Pregnancy Category B drug, and women treated with metformin prior to pregnancy often continue its use during pregnancy. Nevertheless, use of metformin during early pregnancy remains controversial, and it is not clear whether metformin prevents gestational diabetes and other obstetric complications, improves fertility and improves live birth rates [1,3–8].
The main effect of metformin is to decrease hepatic glucose production, although it also increases insulin-stimulated glucose uptake by muscle and lowers plasma tri-glycerides and free fatty acids [2,9]. Metformin has been shown to stimulate adenosine monophosphate-activated protein kinase (AMPK) activity in skeletal muscle and liver by indirectly stimulating phosphorylation (Thr172) of the catalytic α subunit [9,10]. However, the precise mechanism of metformin action is unknown, as the glucose-lowering effects of metformin are maintained in mice lacking hepatic AMPK [11], and AMPK-independent effects of metformin, such as inhibition of mitochondrial complex I, modulation of several levels of the incretin, glucagon-like peptide-1, axis and stimulation of novel/conventional protein kinase C enzymes, have been reported [2,12,13].
We recently showed that embryo AMPK activity is stimulated in a mouse model of diabetic pregnancy [14]. Stimulation of AMPK resulted from hypoxic and oxidative stress caused by maternal hyperglycaemia. Stimulation of AMPK inhibited expression of Pax3, a gene that is essential for neural tube closure, and induced neural tube defects (NTD). The effects of oxidative stress and hyperglycaemia were blocked by the AMPK inhibitor, compound C, indicating that activation of AMPK mediates the embryopathic effect of maternal diabetes [14].
Whether AMPK is stimulated in embryos when metformin is administered during pregnancy has not been reported. However, if metformin can stimulate embryo AMPK activity, even when hyperglycaemia-induced oxidative stress has been suppressed, the adverse effects on embryonic development could counteract the beneficial effects on maternal glycaemic control. Here, we administered metformin to pregnant nondiabetic mice and tested whether metformin stimulates AMPK activity, inhibits Pax3 expression and increases NTD. We also used a cell culture model of embryonic neuroepithelium, murine embryonic stem cell (mESC)-derived neuronal progenitors, in which effects of adding high concentrations of metformin directly to cells on AMPK activation and Pax3 expression can be directly tested.
Materials and methods
Animal procedures
ICR mice (Taconic, Germantown, NY, USA) were employed. Animal procedures for mating, stimulating AMPK activity with 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR, Sigma, St. Louis, MO, USA) and recovering embryos were as previously described [14]. AICAR [50 mg/kg dissolved in phosphate-buffered saline (PBS)] was administered subcutaneously at noon on embryonic day (E) 7.5, with PBS administered as a control, as described [14]. Metformin (Sigma), dissolved in water, was administered at 40 mg/kg by gavage at 9:00, 12:00 and 15:00 hours, beginning on E 0.5 until the day of sacrifice. Mice used for recovery of maternal gastrocnemius muscle and liver and embryos were sacrificed on E 7.5, and tissues were frozen in liquid nitrogen and saved for assay of activated AMPK or mRNA encoding of the metformin transporters, Mate1/Slc47a1 and Oct3/Slc22. Mice were sacrificed on E 8.5 for recovery of embryos for assay of Pax3 mRNA or on E 10.5 to score for NTD as described [14]. E 7.5 is the day on which maternal hyperglycaemia or stimulation of AMPK causes decreased Pax3 expression, E 8.5 is the day that Pax3 expression begins and NTD are apparent by E 10.5 [14,15]. Maternal blood glucose was sampled from the tail vein and was measured using a Glucometer Elite (Bayer, Tanytown, NY, USA). All procedures using animals were approved by the Institutional Animal Care and Use Committee of the Joslin Diabetes Center.
Embryonic stem cell culture
Murine embryonic stem cells (D3 from ATCC, Manassas, VA, USA) were cultured on gelatin-coated 60-mm culture dishes as described [14]. Nestin-positive neuronal progenitors were induced by forming embryoid bodies and selection in media containing fibronectin (Becton Dickenson, Franklin Lakes, NJ, USA), insulin, selenium and transferrin (Sigma, St. Louis, MO, USA), as described [14,16,17]. Metformin (1 mmol/L in PBS) or AICAR (1 mmol/L in PBS) was added to cultures for 1 h for assay of activated AMPK or from days 1 to 3 during selection of neuronal progenitors to study effects on induction of Pax3 expression.
Real-time RT-PCR
Total RNA from individual culture dishes or pooled embryos from individual pregnancies was extracted using Ultraspec reagent (Biotecx Laboratories, Houston, TX, USA). About 200 ng RNA was reverse transcribed (RT) and assayed for Pax3 mRNA using a FAM-labelled probe and primers as reported [18] or for Mate1/Slc47a1 and Oct3/Slc22 mRNA using primer and probe sets from Life Technologies, by real-time RT-PCR, and expressed relative to rRNA as described [18].
Activated AMPK assays
Phosphorylated AMPK (activated), AMPK [phospho-AMPK α (Thr172)] and total AMPK were assayed by immunoblot as described [14,19] using 50 μg protein from pooled embryos from individual pregnancies, tissues from individual pregnant mice or cells from individual 60-mm culture dishes. Total AMPK was detected using an antibody against the α 1 and α 2 isoforms of the catalytic subunit (α-pan) that was obtained from EMD Millipore (Billerica, MA, USA), and phospho-AMPK α (Thr172) was detected using an antibody against Thr172-AMPK obtained from Cell Signaling Technology (Danvers, MA, USA). Bound antibodies were detected with anti-rabbit IgG (horseradish peroxidase coupled) from GE Healthcare Biosciences (Piscataway, NJ, USA). Band intensities of autoradiographs exposed in a linear range were measured using Adobe Photoshop. Activated AMPK was expressed as the intensity of phospho-AMPK α (Thr172) bands relative to the intensity of total AMPK bands.
Statistical analyses
Numbers of animals or embryonic stem cell culture dishes used are indicated in the figure legends. Data were analysed by one-way analysis of variance followed by Newman–Keuls post-test using GraphPad Prism software v. 4.0 (La Jolla, CA, USA). Data are presented as the mean ± standard error of the mean. p <0.05 was determined to be statistically significant.
Results
Activation of maternal liver AMPK is not associated with activation of embryo AMPK
Although metformin is normally administered to diabetic or insulin-resistant women, nondiabetic mice were used in this study. Because maternal hyperglycaemia-induced oxidative stress stimulates AMPK activity [14], in order to test the hypothesis that metformin stimulates AMPK activity independent of hyperglycaemia and oxidative stress, it is necessary to use a model in which AMPK is not already stimulated. Furthermore, if there were any differences between embryos from diabetic, metformin-treated pregnancies and embryos of nondiabetic pregnancies, it would not be possible to distinguish between effects of inadequate control of maternal hyperglycaemia and effects of metformin.
Pregnant dams were treated with three daily doses of 40 mg/kg of metformin, beginning on E 0.5 of pregnancy. Previous studies have shown that a single daily dose of 120 mg/kg stimulates hepatic AMPK activity in rats [20], and dividing the daily dose into three treatments should more closely resemble human administration. The AMPK-activating agent, AICAR, was administered at 50 mg/kg subcutaneously at noon on E 7.5, as a control for the effects of maternal hyperglycaemia on embryo AMPK activity, Pax3 expression and NTD [14]. There were no significant effects of PBS, AICAR or metformin on maternal blood glucose levels (Table 1), indicating that any embryonic effects were due to the action of the drug, rather than indirectly due to changes in maternal glycaemia. Embryos and maternal tissues from all treatment groups were obtained on E 7.5, 90 min after AICAR or metformin administration, for assay of activated AMPK [phospho-AMPK α (Thr172) relative to total AMPK]. There were no differences in litter sizes from PBS-treated (11.67 ± 0.42 embryos/litter), AICAR-treated (12.17 ± 0.31 embryos/litter) or metformin-treated (12.50 ± 0.22 embryos/litter) pregnancies. As expected, AICAR and metformin increased activated AMPK in maternal liver, but only AICAR increased activated AMPK in maternal skeletal muscle (Figure 1A–C). Although AICAR increased activated AMPK, as observed previously [14], metformin did not increase activated AMPK in embryos (Figure 1A, D).
Table 1.
Blood glucose levels (mmol/L) at 0, 1 and 3 h after AICAR or metformin administration
| Time post-treatment (h) | PBS (n = 9) | AICAR (n = 6) | Metformin (n = 9) |
|---|---|---|---|
| 0 | 7.73 ± 0.23 | 8.57 ± 0.18 | 8.02 ± 0.2 |
| 1 | 8.82 ± 0.4 | 6.96 ± 0.64 | 9.02 ± 0.35 |
| 3 | 8.3 ± 0.31 | 7.20 ± 0.92 | 8.23 ± 0.17 |
AICAR, 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside; PBS, phosphate-buffered saline.
Figure 1.
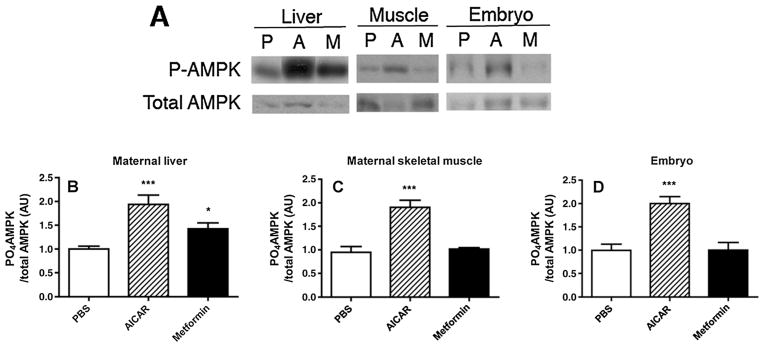
(A) Representative immunoblots of phospho-adenosine monophosphate-activated protein kinase (AMPK) α (Thr172) (P-AMPK) and total AMPK using protein extracted from maternal liver, maternal gastrocnemius muscle or embryos following administration of phosphate-buffered saline (PBS) (P), 50 mg/kg 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) (A) given at noon on E 7.5 or 40 mg/kg metformin (M) given three times/day from E 0.5 to 7.5. Embryos or maternal tissues were recovered for assay of activated AMPK 90 min after PBS or AICAR injections. (B) Quantitation of P-AMPK/total AMPK of liver of pregnant mice on E 7.5 treated as in panel (A). n = 6 for each treatment group. ***p <0.001 versus PBS; *p <0.05 versus PBS, AICAR. (C) Quantitation of PO4-AMPK/total AMPK of gastrocnemius muscle of pregnant mice on E 7.5 treated as in panel (A). n = 5 for each treatment group. ***p <0.001 versus PBS, metformin. (D) Quantitation of P-AMPK/total AMPK of E 7.5 embryos of mice treated as in panel (A). n = 6 for each treatment group. Band intensity is expressed as arbitrary units (AU) relative to PBS. ***p <0.001 versus PBS, metformin
AMPK-associated embryopathy is not induced by metformin administered to pregnant mice
To investigate whether metformin could elicit effects on embryo gene expression and malformation that occur when AMPK is stimulated, embryos were obtained on E 8.5 to assay expression of Pax3 by RT-PCR or on E 10.5 to score NTD. AICAR administered to mothers on E 7.5 inhibited Pax3 expression in embryos by more than twofold, but there was no effect of metformin administered beginning on E 0.5 on Pax3 expression (Figure 2A). AICAR increased NTD by more than fourfold, but there was no significant effect of metformin on NTD (Figure 2B).
Figure 2.
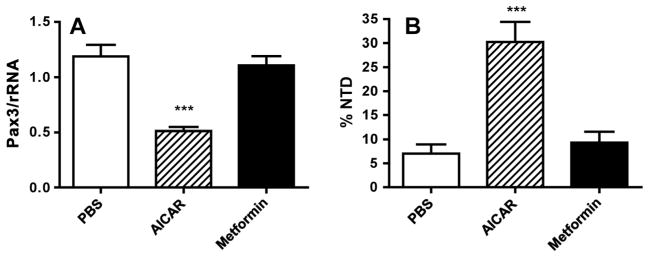
(A) Real-time RT-PCR analysis of Pax3 mRNA from E 8.5 embryos of mice treated with phosphate-buffered saline (PBS) or 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside (AICAR) on E 7.5 or metformin from E 0.5 to 8.5. Pax3 mRNA was normalized to rRNA and expressed relative to Pax3 mRNA of embryos from PBS-treated pregnancies. ***p <0.001 versus PBS, metformin. (B) Neural tube defects (NTD) of E 10.5 embryos of mice treated with PBS or AICAR on E 7.5 or metformin from E 0.5 to 10.5. NTD are expressed as the per cent of embryos in each litter with an NTD. ***p <0.001 versus PBS, metformin. n = 9 replicate litters for each treatment
Metformin stimulates AMPK and inhibits Pax3 expression by mESC
The preceding experiments indicated that metformin administration that was sufficient to increase activated AMPK in maternal liver could not increase activated AMPK in the embryo. This suggests that either the concentration of metformin delivered to the embryo was not sufficient to stimulate AMPK (because of the pharmacokinetics of metformin absorption and renal excretion in the mouse or lack of transporters to deliver metformin from maternal circulation to the embryo) or metformin does not stimulate AMPK in embryo cells. To test whether metformin could stimulate AMPK activity when directly applied to embryo cells, a cell culture model was employed. mESC can be induced to differentiate into neurons [16], and neuronal progenitors derived from mESC exhibit a gene expression profile similar to that of neuroepithelium, including expression of Pax3 [14,17,21,22]. We previously showed that AICAR stimulates AMPK activity and inhibits differentiation-induced expression of Pax3 by mESC-derived neuronal progenitors [14]. To test whether metformin can activate AMPK by mESC, cultures were treated with 1 mmol/L metformin for 1 h, conditions that stimulate AMPK by rat hepatocytes [10]. As shown in Figure 3A and B, metformin, as well as AICAR, stimulated AMPK activity by mESC by at least twofold. To test whether metformin can inhibit Pax3 expression by mESC, cultures were treated for 2 days during the induction of neural progenitors and Pax3 expression. As shown in Figure 3C, metformin, as well as AICAR, significantly inhibited Pax3 expression by differentiating mESC.
Figure 3.

(A) Representative immunoblot of phospho-adenosine monophosphate-activated protein kinase (P-AMPK) and total AMPK using protein extracted from murine embryonic stem cells (mESC) that were untreated or treated for 1 h with 1 mmol/L AICAR (A) or 1 mmol/L metformin (M) compared with time 0 (t0). (B) Quantitation of P-AMPK/total AMPK of three replicate culture dishes of mESC that were treated as in panel (A). Band intensity is expressed as arbitrary units (AU) relative to time 0. *p <0.05 versus t0. (C) Real-time RT-PCR of Pax3 in undifferentiated (UD) mESC, differentiating neuronal progenitors derived from mESC (D), differentiating neuronal progenitors treated with AICAR for 48 h (D + AICAR) or differentiating neuronal progenitors treated with metformin for 48 h (D + metformin). Pax3 mRNA was normalized to rRNA and expressed relative to that of differentiating neuronal progenitors. ***p <0.001 versus UD; *p <0.001 versus D. n = 4 replicate tissue culture dishes for each treatment
Lack of metformin transporter expression by E 7.5 embryos
The lack of a metformin effect on the embryo in vivo despite an effect on mESC in vitro indicates that the concentration entering embryo cells was insufficient. The dose of metformin that we used (120 mg/kg daily) stimulates liver AMPK activity in rats [20] and in the mice used in our study. This dose is almost four times higher than the maximum recommended dose for a woman (assuming a pre-pregnancy weight of approximately 77 kg) of 2550 mg. It is possible that higher doses, which are up to 5.5-fold higher than were used here, that stimulate AMPK activity in skeletal muscle as well as in liver [10] could have stimulated AMPK in embryos, but as these doses are approximately 15-fold higher than doses that would be administered to women, they are not likely to occur in a clinical setting. Metformin is not metabolized, but it is eliminated by renal excretion [23]. Therefore, even recognizing differences in metformin pharmacokinetics between rodents and humans [23,24], which are primarily due to differential rates of intestinal absorption and renal clearance, it is likely that mice in our study were exposed to metformin at least as high as humans taking the maximum recommended dose. However, mESC would be constantly exposed to metformin in culture media, as it could be neither metabolized nor excreted.
Because the dose of metformin was sufficient to stimulate liver AMPK activity, we considered that metformin may not be transported to the embryo. Metformin is freely transported across the placenta [25,26], and the rat placenta expresses the metformin transporters, Mate1/Slc47a1 and Oct3/Slc22, at least as of E 12, with increasing expression as gestation progresses, and first-trimester human placentas express metformin transporters [27]. On the other hand, it has recently been reported that metformin transport is bidirectional in term rat placenta and may even be transported against a concentration gradient from the foetal to maternal compartments [28]. However, the placenta has not yet formed in E 7.5 embryos, and the only tissues between maternal circulation and the embryo are the trophoblast layers of the deciduum and the yolk sac. Therefore, if metformin is transported to the embryo, metformin transporters would need to be expressed by the yolk sac and the embryo.
To investigate expression of metformin transporters by E 7.5 embryos, Mate1/Slc47a1 and Oct3/Slc22 mRNAs were assayed using whole yolk sac-enclosed embryos by RT-PCR. Maternal liver was used as a positive control. As shown in Figure 4A, Mate1/Slc47a1 mRNA barely reached detectable levels in embryos and was approximately 200-fold less than that expressed by livers. We find it interesting that Mate1/Slc47a1 mRNA expression by mESC was higher than that by embryos, although this did not reach statistical significance, and it was significantly less than expression by livers. Oct3/Slc22 was barely detectable in embryos, although expression by maternal livers was also very low (i.e. CT was >33 during a 40-cycle reaction). There were no significant differences in Oct3/Slc22 mRNA between liver, embryo and mESC samples.
Figure 4.
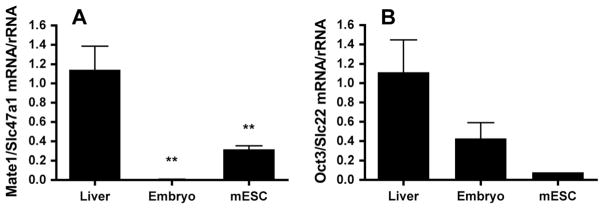
(A) Real-time RT-PCR of Mate1/Slc47a1 mRNA from maternal livers, E 7.5 embryos and murine embryonic stem cells (mESC). Mate1/Slc47a1 mRNA was normalized to rRNA and expressed relative to Mate1/Slc47a1 mRNA of liver samples. **p <0.01 versus liver. (B) Real-time RT-PCR of Oct3/Slc22 mRNA from maternal livers, E 7.5 embryos and mESC. Oct3/Slc22 mRNA was normalized to rRNA and expressed relative to Oct3/Slc22 mRNA of liver samples. n = 4 livers or pooled embryos from four replicate litters
Discussion
The US Diabetes Prevention Program has supported the administration of metformin to prevent type 2 diabetes in subjects with impaired glucose tolerance, and metformin is the drug of first choice for the treatment of type 2 diabetes [2,29,30]. Furthermore, there are recent suggestions that metformin may aid in the treatment or prevention of cancer in individuals who are obese or have type 2 diabetes [2,31]. Thus, metformin currently has widespread use, and its use may further increase in the future, including by women of childbearing age.
As a Pregnancy Category B drug, ‘Animal reproduction studies have failed to demonstrate a risk to the fetus, and there are no adequate and well-controlled studies in pregnant women OR animal studies have shown an adverse effect, but adequate and well-controlled studies in pregnant women have failed to demonstrate a risk to the fetus in any trimester’. Therefore, experiments such as the ones reported here are necessary to determine whether there is risk for metformin administration during early pregnancy.
Recent literature analysis found no evidence for teratogenic effects of metformin in animal models [32], although an earlier study showed that culture of yolk sac-enclosed neurulating mouse embryos with therapeutic concentrations of metformin only caused a delay in neural tube closure in about 10% of embryos [33]. However, reports of in vivo administration of metformin to pregnant animals during organogenesis are limited. With regard to human studies, evaluation of the risk of metformin during early pregnancy is complicated because subjects taking metformin are already at high risk for obstetric complications. Indeed, metformin improves hyperglycaemia, which is known to be teratogenic, associated with PCOS, and hyperinsulinemia, which increases risk for early pregnancy loss [8]. Moreover, women with PCOS may fail to ovulate and conceive altogether without metformin [7]. Some studies have observed an increase in live birth rate of metformin-treated pregnancies [4], although other studies have failed to see such effect [7]. One study found no increased risk for congenital malformations in the offspring of women with PCOS treated with metformin in the first trimester [34], although the study may not have been sufficiently powered to observe an effect.
None of the previous studies using animal models investigated whether metformin, at doses that would stimulate AMPK activity in maternal tissues, could stimulate AMPK activity in embryos. This study was undertaken because we had shown that AMPK activity is stimulated in embryos during murine diabetic pregnancy and that increased AMPK activity inhibits expression of Pax3 and causes NTD [14]. Although recent evidence indicates that stimulation of AMPK may not be the main mechanism by which metformin improves glycaemic control [2], if metformin does stimulate AMPK activity in embryos, this could counteract its beneficial effects on maternal glycaemic control.
As reported here, metformin did not increase activated AMPK in embryos and did not elicit the adverse effects on Pax3 expression or NTD that occur upon AMPK activation during diabetic pregnancy, despite activation of maternal liver AMPK. However, metformin did increase activated AMPK and inhibited Pax3 expression in mESC-derived neural progenitors in vitro. This demonstrated that metformin can stimulate AMPK in embryonic cells and can elicit a downstream effect under some conditions.
The lack of effect of metformin on the embryo maybe due to insufficient expression of metformin transporters, especially Mate1/Slc47a1, compared with metformin-responsive maternal liver. However, the expression of Mate1/Slc47a1 by mESC, which was not statistically significantly different from that by E 7.5 embryos, was sufficient for activation of AMPK and inhibition of Pax3 expression. We also detected Mate1/Slc47a1 mRNA in four additional independently isolated mESC lines, and expression ranged from being fivefold higher than embryos to being the same as that of maternal liver (data not shown), although we did not test these lines for metformin responsiveness. These differences between Mate1/Slc47a1 expression by embryonic cells in culture and by the intact embryo could be due to adaptation to the culture environment, or it could be due to a difference between cells derived from the inner cell mass (which are homogeneous) and the distribution of metformin transporters in whole embryos that included the yolk sac.
In summary, we found no evidence for activation of AMPK signalling in embryos by metformin administered to pregnant mice, and subsequent embryopathic effects, although metformin added directly to a cell culture model of neuroepithelium had an effect. The lack of metformin responsiveness by embryos may be explained by insufficient expression of metformin transporters during neurulation. However, our studies focused specifically on activation of AMPK and its downstream effects during neural tube formation. Additional studies are necessary to determine whether metformin is safe and has no adverse effects on overall embryo or foetal development or long-term effects on the health and metabolic control of the offspring. Thus, our experiments do not resolve the issue of whether metformin should be a Category B drug and did not provide evidence that metformin has adverse effects during early embryonic development.
Acknowledgments
We are grateful to Dr Florence Brown (Joslin Diabetes Center) for the helpful discussion and critical review of the manuscript. We appreciate the help of Eric B. Taylor from the Goodyear lab (Joslin Diabetes Center) for the advice in the assay of AMPK. We are thankful to Kaitlyn Sanders for assistance with mouse mating and to Jin Hyuk Jung for assistance with real-time PCR (members of the Loeken lab, Joslin Diabetes Center). This project was supported by RO1 DK58300 (to MRL) and was assisted by core facilities supported by a Diabetes Endocrine Research Center grant, P30DK036836, from the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Conflict of interest
The authors have no conflicts of interest to report.
References
- 1.Glueck CJ, Pranikoff J, Aregawi D, Wang P. Prevention of gestational diabetes by metformin plus diet in patients with polycystic ovary syndrome. Fertil Steril. 2008;89(3):625–634. doi: 10.1016/j.fertnstert.2007.03.036. [DOI] [PubMed] [Google Scholar]
- 2.Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond) 2012;122(6):253–270. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Johnson N. Metformin is a reasonable first-line treatment option for non-obese women with infertility related to anovulatory polycystic ovary syndrome – a meta-analysis of randomised trials. Aust N Z J Obstet Gynaecol. 2011;51(2):125–129. doi: 10.1111/j.1479-828X.2010.01274.x. [DOI] [PubMed] [Google Scholar]
- 4.Morin-Papunen L, Rantala AS, Unkila-Kallio L, et al. Metformin improves pregnancy and live-birth rates in women with polycystic ovary syndrome (PCOS): a multicenter, double-blind, placebo-controlled randomized trial. J Clin Endocrinol Metab. 2012;97(5):1492–1500. doi: 10.1210/jc.2011-3061. [DOI] [PubMed] [Google Scholar]
- 5.Hughes RC, Rowan JA. Pregnancy in women with type 2 diabetes: who takes metformin and what is the outcome? Diabet Med. 2006;23(3):318–322. doi: 10.1111/j.1464-5491.2006.01750.x. [DOI] [PubMed] [Google Scholar]
- 6.Brown FM, Wyckoff J, Rowan JA, Jovanovic L, Sacks DA, Briggs GG. Metformin in pregnancy: its time has not yet come. Diabetes Care. 2006;29(2):485–486. doi: 10.2337/diacare.29.02.06.dc05-2098. [DOI] [PubMed] [Google Scholar]
- 7.Siebert TI, Viola MI, Steyn DW, Kruger TF. Is metformin indicated as primary ovulation induction agent in women with PCOS? A systematic review and meta-analysis. Gynecol Obstet Invest. 2012;73 (4):304–313. doi: 10.1159/000335253. [DOI] [PubMed] [Google Scholar]
- 8.Ghazeeri GS, Nassar AH, Younes Z, Awwad JT. Pregnancy outcomes and the effect of metformin treatment in women with polycystic ovary syndrome: an overview. Acta Obstet Gynecol Scand. 2012;91(6):658–678. doi: 10.1111/j.1600-0412.2012.01385.x. [DOI] [PubMed] [Google Scholar]
- 9.Hawley SA, Gadalla AE, Olsen GS, Hardie DG. The antidiabetic drug metformin activates the AMP-activated protein kinase cascade via an adenine nucleotide-independent mechanism. Diabetes. 2002;51(8):2420–2425. doi: 10.2337/diabetes.51.8.2420. [DOI] [PubMed] [Google Scholar]
- 10.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foretz M, Hebrard S, Leclerc J, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120(7):2355–2369. doi: 10.1172/JCI40671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maida A, Lamont BJ, Cao X, Drucker DJ. Metformin regulates the incretin receptor axis via a pathway dependent on peroxisome proliferator-activated receptor-alpha in mice. Diabetologia. 2011;54(2):339–349. doi: 10.1007/s00125-010-1937-z. [DOI] [PubMed] [Google Scholar]
- 13.Turban S, Stretton C, Drouin O, et al. Defining the contribution of AMP-activated protein kinase (AMPK) and protein kinase C (PKC) in regulation of glucose uptake by metformin in skeletal muscle cells. J Biol Chem. 2012;287(24):20088–20099. doi: 10.1074/jbc.M111.330746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu Y, Viana M, Thirumangalathu S, Loeken MR. AMP-activated protein kinase mediates effects of oxidative stress on embryo gene expression in a mouse model of diabetic embryopathy. Diabetologia. 2012;55(1):245–254. doi: 10.1007/s00125-011-2326-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fine E, Horal M, Chang T, Fortin G, Loeken M. Evidence that hyperglycemia causes altered gene expression, apoptosis, and neural tube defects in a mouse model of diabetic pregnancy. Diabetes. 1999;48:2454–2462. doi: 10.2337/diabetes.48.12.2454. [DOI] [PubMed] [Google Scholar]
- 16.Lee SH, Lumelsky N, Studer L, Auerbach JM, McKay RD. Efficient generation of midbrain and hindbrain neurons from mouse embryonic stem cells. Nat Biotechnol. 2000;18(6):675–679. doi: 10.1038/76536. [DOI] [PubMed] [Google Scholar]
- 17.Wang XD, Morgan SC, Loeken MR. Pax3 stimulates p53 ubiquitination and degradation independent of transcription. PLoS One. 2011;6(12):e29379. doi: 10.1371/journal.pone.0029379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang TI, Horal M, Jain S, Wang F, Patel R, Loeken MR. Oxidant regulation of gene expression and neural tube development: insights gained from diabetic pregnancy on molecular causes of neural tube defects. Diabetologia. 2003;46:538–545. doi: 10.1007/s00125-003-1063-2. [DOI] [PubMed] [Google Scholar]
- 19.Musi N, Fujii N, Hirshman MF, et al. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes. 2001;50(5):921–927. doi: 10.2337/diabetes.50.5.921. [DOI] [PubMed] [Google Scholar]
- 20.Cleasby ME, Dzamko N, Hegarty BD, Cooney GJ, Kraegen EW, Ye JM. Metformin prevents the development of acute lipid-induced insulin resistance in the rat through altered hepatic signaling mechanisms. Diabetes. 2004;53(12):3258–3266. doi: 10.2337/diabetes.53.12.3258. [DOI] [PubMed] [Google Scholar]
- 21.Yamada G, Kioussi C, Schubert FR, et al. Regulated expression of Brachyury (T), NKX1. 1 and Pax genes in embryoid bodies. Biochem Biophys Res Commun. 1994;199:552–563. doi: 10.1006/bbrc.1994.1264. [DOI] [PubMed] [Google Scholar]
- 22.Perry P, Sauer S, Billon N, et al. A dynamic switch in the replication timing of key regulator genes in embryonic stem cells upon neural induction. Cell Cycle. 2004;3(12):1645–1650. [PubMed] [Google Scholar]
- 23.Scheen AJ. Clinical pharmacokinetics of metformin. Clin Pharmacokinet. 1996;30(5):359–371. doi: 10.2165/00003088-199630050-00003. [DOI] [PubMed] [Google Scholar]
- 24.Junien JL, Brohon J, Guillaume M, Sterne J. DBM mice as a pharmacological model of maturity onset diabetes. Studies with metformin. Arch Int Pharmacodyn Ther. 1979;241(1):165–176. [PubMed] [Google Scholar]
- 25.Terada T, Masuda S, Asaka J, Tsuda M, Katsura T, Inui K. Molecular cloning, functional characterization and tissue distribution of rat H+/organic cation antiporter MATE1. Pharm Res. 2006;23(8):1696–1701. doi: 10.1007/s11095-006-9016-3. [DOI] [PubMed] [Google Scholar]
- 26.Kovo M, Kogman N, Ovadia O, Nakash I, Golan A, Hoffman A. Carrier-mediated transport of metformin across the human placenta determined by using the ex vivo perfusion of the placental cotyledon model. Prenat Diagn. 2008;28(6):544–548. doi: 10.1002/pd.2026. [DOI] [PubMed] [Google Scholar]
- 27.Ahmadimoghaddam D, Zemankova L, Nachtigal P, et al. Organic cation transporter 3 (OCT3/SLC22A3) and multidrug and toxin extrusion 1 (MATE1/SLC47A1) transporter in the placenta and fetal tissues: expression profile and fetus protective role at different stages of gestation. Biol Reprod. 2013;88(3):55. doi: 10.1095/biolreprod.112.105064. [DOI] [PubMed] [Google Scholar]
- 28.Ahmadimoghaddam D, Staud F. Transfer of metformin across the rat placenta is mediated by organic cation transporter 3 (OCT3/SLC22A3) and multidrug and toxin extrusion 1 (MATE1/SLC47A1) protein. Reprod Toxicol. 2013;39:17–22. doi: 10.1016/j.reprotox.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 29.DeFronzo RA, Abdul-Ghani MA. Preservation of beta-cell function: the key to diabetes prevention. J Clin Endocrinol Metab. 2011;96(8):2354–2366. doi: 10.1210/jc.2011-0246. [DOI] [PubMed] [Google Scholar]
- 30.Association AD. Standards of medical care in diabetes – 2012. Diabetes Care. 2012;35 (Suppl 1):S11–63. doi: 10.2337/dc12-s011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suh S, Kim KW. Diabetes and cancer: is diabetes causally related to cancer? Diabetes Metab J. 2011;35(3):193–198. doi: 10.4093/dmj.2011.35.3.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feig DS, Briggs GG, Koren G. Oral antidiabetic agents in pregnancy and lactation: a paradigm shift? Ann Pharmacother. 2007;41(7):1174–1180. doi: 10.1345/aph.1K045. [DOI] [PubMed] [Google Scholar]
- 33.Denno KM, Sadler TW. Effects of the biguanide class of oral hypoglycemic agents on mouse embryogenesis. Teratology. 1994;49(4):260–266. doi: 10.1002/tera.1420490405. [DOI] [PubMed] [Google Scholar]
- 34.Bolton S, Cleary B, Walsh J, Dempsey E, Turner MJ. Continuation of metformin in the first trimester of women with polycystic ovarian syndrome is not associated with increased perinatal morbidity. Eur J Pediatr. 2009;168(2):203–206. doi: 10.1007/s00431-008-0737-7. [DOI] [PubMed] [Google Scholar]