

Abstract
Objectives: MicroRNA-26b (miR-26b) has been reported to be down-regulated in a wide range of malignant tumors, However, the mechanism by which miR-26b is implicated in breast cancer tumorigenesis is incompletely understood. This study was undertaken to evaluate the expression pattern of miR-26b and characterize its biological role in human breast cancer. Methods: Reverse transcription-polymerase chain reaction (RT-PCR) was used to quantify the expression levels of miR-26b in breast cancer and adjacent non-cancerous breast tissues. MTT, colony formation assay and cell cycle assay were carried out to characterize the miR-26b function. Finally, to validate the target gene of miR-26b, luciferase reporter assay was employed, followed by RT-PCR and Western blot confirmation. Results: Here, we found that miR-26b expression was relatively downregulated in breast cancer specimens (P<0.01). Overexpression of miR-26b dramatically suppressed cell proliferation, colony formation and induced G0/G1 cell cycle arrest of MDA-MB-231 and Mcf-7 cells. Luciferase assays revealed that miR-26b directly targeted the 3’UTR of CDK8. Overexpression of miR-26b led to the downregulation of CDK8 and β-catenin expression. Similarly, CDK8 knockdown by siRNA suppressed cell growth and subsequent β-catenin expression. Conclusions: These findings suggest that miR-26b exerts a tumor suppressive role in breast cancer and the miR-26b-mediated growth inhibition is achieved through suppression of a new target gene CDK8
Keywords: MiR-26b, proliferation, cell cycle, CDK8, breast cancer
Breast cancer as one of the most common female cancers is a leading cause of cancer mortality worldwide [1]. microRNAs, a class of small non-coding RNAs, can modulate gene expression post-transcriptionally through either protein translation repression or mRNA degradation of target genes [2]. Over the past decade, miRNAs are emerging as critical regulators in carcinogenesis and tumor progression by acting either as oncogenes or tumor suppressor genes [3-6]. miR-26b is consistently down-regulated in a wide range of malignant tumors, such as hepatocellular carcinoma [7], nasopharyngeal carcinomas [8], primary squamous cell lung carcinomas [9], squamous cell carcinoma of tongue [10] and breast cancer [11]. Overexpression of miR-26b promotes the apoptosis of MCF-7 breast cancer cells by targeting SLC7A11 [11]. However, the mechanism by which miR-26b is implicated in breast cancer tumorigenesis is uncompletely understood.
Cyclin-dependent kinase (CDK) 8 is located on chromosome 13q12.13 and is found to be frequently dysregulated in various human cancers, including colorectal cancers [12,13], gastric cancer [14], esophageal squamous cell carcinoma [15], bladder cancers [16] and melanoma [17]. CDK8 was identified as an oncogene involved in the proliferation and cell cycle regulation of colorectal cancer cells [12,18]. Moreover, CDK8 activation is essential for the β-catenin-dependent oncogenesis and progression in a variety of cancers [12,14,19]. On the one hand, CDK8 may directly modulate β-catenin-activated transcription. For instance, β-catenin target genes are regulated by the homologs of CDK8 module subunits MED12 and MED13 [20-22]. On the another, CDK8 has been demonstrated to repress E2F1 that suppresses β-catenin and thus enhances β-catenin indirectly [11,19].
In this study, we investigated the potential involvement of miR-26b in breast cancer and explored the possible mechanism. First of all, we examined the expression of miR-26b in human breast cancer specimens and tested its effects on cell proliferation, colony formation, and cell-cycle distribution. Finally, we searched for miR-26b targets using gene prediction-based and systematic experimental approaches.
Materials and methods
Specimens
In this study, 38 pairs of fresh breast cancer and normal specimens (>2 cm from tumor tissues) were obtained from the Department of Breast and Thyroid Surgery of Shanghai Tenth People’s Hospital. All the samples were confirmed as invasive ductal breast cancer, and no patients had received any chemotherapy or radiotherapy ahead of surgery.
Cell culture
The MDA-MB-231 and Mcf-7 breast cancer cell lines were obtained from the Chinese Science Institute and cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, USA) supplemented with 10% fetal bovine serum (FBS, Gibco), as well as 100 units of penicillin/ml and 100 μg of streptomycin/ml (Enpromise, China). Cells were incubated at 37°C in a humidified chamber containing 5% CO2.
Real-time polymerase chain reaction (PCR) analyses
To quantify miR-26b expression, we followed the protocol of Chen et al for primer design and real-time reverse transcription (RT)-PCR [23]. The miR-26b was harvested according to the instruction of the miRcute miRNA isolation kit (Tiangen, China). The following primers were used: 5’-GTCGTATCCAGTGCAGGGTCCGA GGTATTCGCAC TGGATACGACGAGCCA-3’ (stem-loop primer), 5’-CGCCCTGTTCTCCATTACTT-3’ (sense) and 5’-CCAGTGCAGGG TCCGAGGT-3’ (antisense) for miR-26b; 5’-GTCCTAT CCAGTGCAGGGTCCGAGGTGCACTGGATACGACAAAATATGGAAC-3’ (stem-loop primer), 5’-TGCGGGTGCTCGCTTCGCAGC-3’ (sense) and 5’-CCAGTGCAGGGTCCGAGGT-3’ (antisense) for the U6 small nuclear RNA. cDNA was generated by reverse transcription according to the instructions of the PrimeScript™ RT-PCR kit (Takara, Japan). Quantitative real-time PCR was performed on a 7900HT fast RT-PCR instrument (Applied Biosystems, Singapore). PCR parameters for miRNA quantification were as follows: 2 min at 95°C, and then 40 cycles of 30 sec at 95°C, and 45 sec at 60°C.
For CDK8 mRNA expression quantification, total RNA was isolated using TRIzol (Invitrogen, USA), and cDNA was generated by reverse transcription following the protocol of the PrimeScript™ RT-PCR kit (Takara). Quantitative real-time PCR was performed on a 7900HT fast RT-PCR instrument (Applied Biosystems, Singapore). All the primers used were as follows: 5’-TCACCTTTGAAGCCTTTAGC-3’ (sense) and 5’-CTGATGTAGGAAGTGGGTCT-3’ (antisense) for CDK8; 5’-AGCGAGCATCCCCC AAAGTT-3’ (sense) and 5’-GGGCAC GAAGGCTCATCATT-3’ (antisense) for β-actin. The PCR parameters for relative quantification were as follows: 2 min at 95°C, followed by 40 cycles of 15 sec at 95°C and 30 sec at 60°C. The relative expression was calculated using the equation relative quantification (RQ) = 2-ΔΔCT [24].
Western blot analysis
48 hours after miR-26b mimics transfection, cell proteins were extracted and then quantified by BCA protein assay (Beyotime, China), equal amounts of protein samples (20 μg) were separated with 12% SDS-polyacrylamide gel (SDS-PAGE) and transferred onto PVDF membranes (Beyotime, China). Immune complexes were formed by incubation of the proteins with primary antibodies CDK8 and β-catenin (Cell signalling, USA) at the dilution of 1:1000 overnight at 4°C. Blots were washed and incubated for 1 h with anti-rabbit secondary antibody at 1:1000 dilution. Immunoreactive protein bands were detected with an odyssey scanning system.
MTT assay
MDA-MB-231 and Mcf-7 cells (5000/well) were plated in 96-well plates (BD Biosciences, USA) and incubated at 37°C overnight, when the confluence of MDA-MB-231 and Mcf-7 cells reached 50-60%, 100 nmol/L, 150 nmol/L miR-26b mimics or 50 nmol/L CDK8 siRNA (Genepharma, China) were transfected into cells according to the protocol of lipofectamine 2000 (Invitrogen, USA), The experiment used one negative control transfected with NC or siRNA control. Cell proliferation was assessed at 24, 48, 72, 96 and 120 h, following the instructions of the MTT proliferation assay kit (Sigma, USA).
Colony formation assay
MiR-26b treatment groups and control are the same with MTT assay, 24 h after miR-26b mimics transfection. MDA-MB-231 and Mcf-7 Cells were digested with trypsin and suspended into a single cell status. 500 cells from each group were cultured in the 6-well plate for 25 days. The colonies were fixed and stained with 0.5% crystal violet for 15 min. Colonies less than 2 mm in diameter and faintly stained were ignored. The number of colonies in 10 random view fields was counted under a microscope.
Cell cycle assay
After the transfection of miR-26b mimics at the concentration of 100 nmol/L for 48 h, Cells were collected and fixed in 70% ethanol at 4°C overnight. After removal of 70% ethanol, the cells were incubated with 1 mg/ml RNase A in PBS for 30 min. Then, a total of 250 μl 0.05 g/L PI staining solution was added into each sample and incubated for 30 min at room temperature and then analyzed by a flow cytometer (FACSCanto™ II, BD Biosciences).
Luciferase activity assay
The 3’-untranslated region (3’UTR) of mRNA sequence of CDK8 containing predicted miR-26b binding site was amplified by PCR following the protocol of Primer star kit (Takara), The corresponding mutant constructs were created by mutating the seed regions of the miR-26b-binding sites (5’-ACUUGA-3’ to 5’-AG CCCA-3’). The primers used in the reaction were (sense 5’-TAGGCGATCGCTCGAGG ACTGTGCAGCTCTCTGCG-3’; antisense 5’-AATTCCCGGGCTCGAGACATTCACAG CTTCAACTGCTAC-3’). PCR products were cloned into the Xho I site in the 3’-UTR of Renilla luciferase of psiCHECK-2 reporter vector (Promega, USA). Reporter plasmids (200 ng psiCHECK-2 reporter vector containing CDK8 3’-UTR) and 100 nmol/L miR-26b mimics were cotransfected into 293T cells (60% confluence) using Lipofectamine 2000 (Invitrogen, USA), control groups here include cells cotransfected with psiCHECK-2/CDK8 3’-UTR (200 ng) and miR-NC (100 nmol/L), psiCHECK-2/CDK8 3’-UTR mutant (200 ng) and miR-26b mimics (100 nmol/L) as well as psiCHECK-2/CDK8 3’-UTR mutant (200 ng) and miR-NC (100 nmol/L). After 48 h cells were lysed and reporter activity was determined using Dual-luciferase report assay system (Promega, USA). Firefly luciferase values were normalized to Renilla, and the ratio of firefly/renilla was presented.
Statistical analysis
Data were presented as the means ± standard deviation from at least three separate experiments. The two independent sample t-test was used to draw a comparison between groups. All tests were two-tailed, and the significance level was set at P<0.05.
Results
miR-26b expression is downregulated in human breast cancer specimens
We first determined the expression level of miR-26b in breast cancer tissues and normal adjacent tissues (NATs) by real-time RT-PCR. Quantitative analysis indicated that miR-26b expression was significantly decreased by 7.33-fold in breast cancer tissues compared with NATs (P<0.01) (Figure 1).
Figure 1.
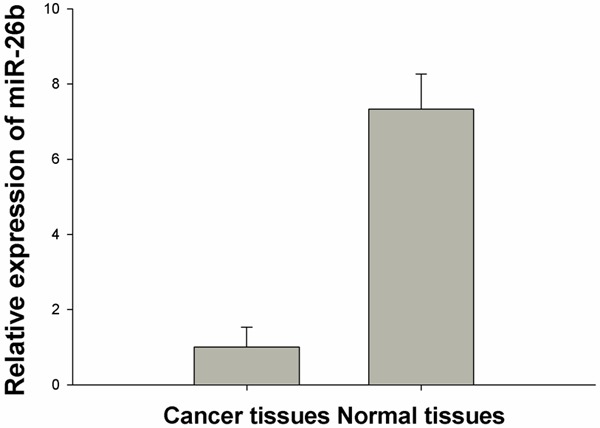
miR-26b is downregulated in breast cancer specimens.
Forced expression of miR-26b suppresses the proliferation of breast cancer cells
To determine the impact of miR-26b on the proliferation of breast cancer cells, miR-26b mimics were transfected into MDA-MB-231 and Mcf-7 cells at the final concentrations of 100 and 150 nmol/L. Figure 2 shows that cellular proliferation gradually decreased as the content of miR-26b increased. Treatment of cells with 100 nM miR-26b led to a decrease in MDA-MB-231 and Mcf-7 cell growth at 48 h (19.48% and 17.89%), 72 h (28.25% and 22.20%), 96 h (28.70% and 29.55%) and 120 h (30.15% and 33.86%) (P<0.01) compared with the negative control. This inhibitory effect was significantly enhanced following transfection with 150 nM miR-26b at 24 h (26.83% and 26.18%), 48 h (36.79% and 37.71%), 72 h (48.64% and 53.06%), 96 h (52.87% and 61.56%) and 120 h (57.58% and 60.32%) (P<0.01). Moreover, the colony formation assay showed that the colony number in miR-26b mimics-transfected groups was significantly less than that in NC (Figure 3). All these findings demonstrate that miR-26b inhibits the proliferation of MDA-MB-231 and Mcf-7 breast cancer cells. Flow cytometer analysis revealed that the percentage of G0/G1 phase MDA-MB-231 and Mcf-7 cells (45.96±2.02%, 77.24±0.60%) dramatically increased in the 100nmol/L miR-26b mimics-transfected group, which was higher than those in NC (38.21±1.86% and 64.79±0.51%), suggesting that miR-26b can initiate G0/G1 phase arrest (Figure 4). These results indicate that miR-26b may function as a growth suppressor by inducing G0/G1 phase arrest in human breast cancer.
Figure 2.
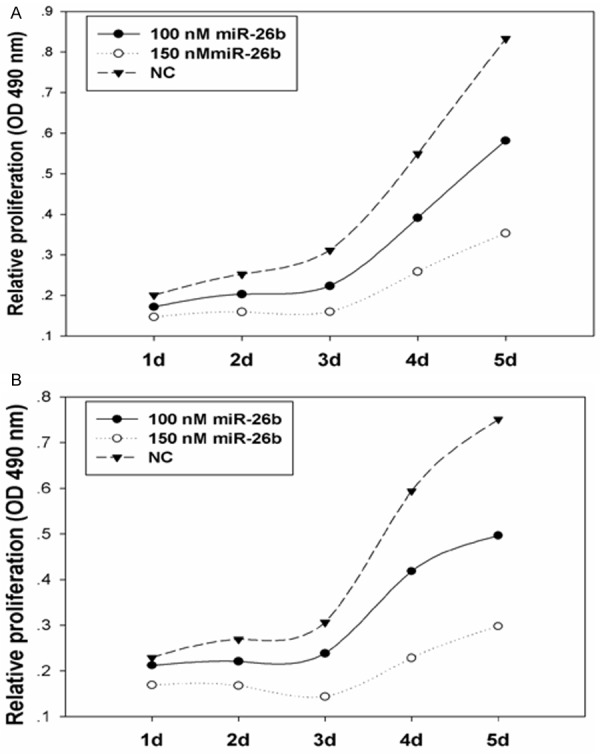
miR-26b suppresses the proliferation of MDA-MB-231 cells (A) and Mcf-7 cells (B).
Figure 3.
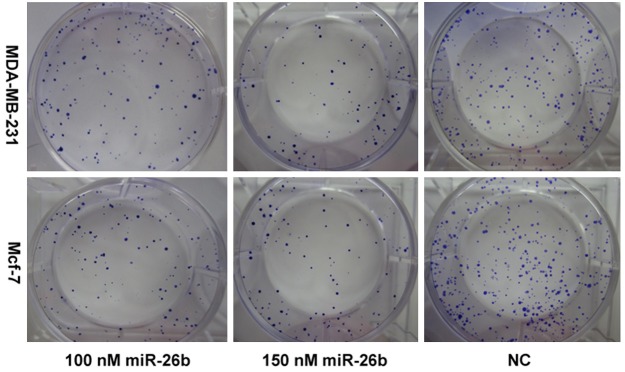
Clonogenic assay for breast cancer cells proliferative activity in different treatment groups after miR-26b transfection.
Figure 4.
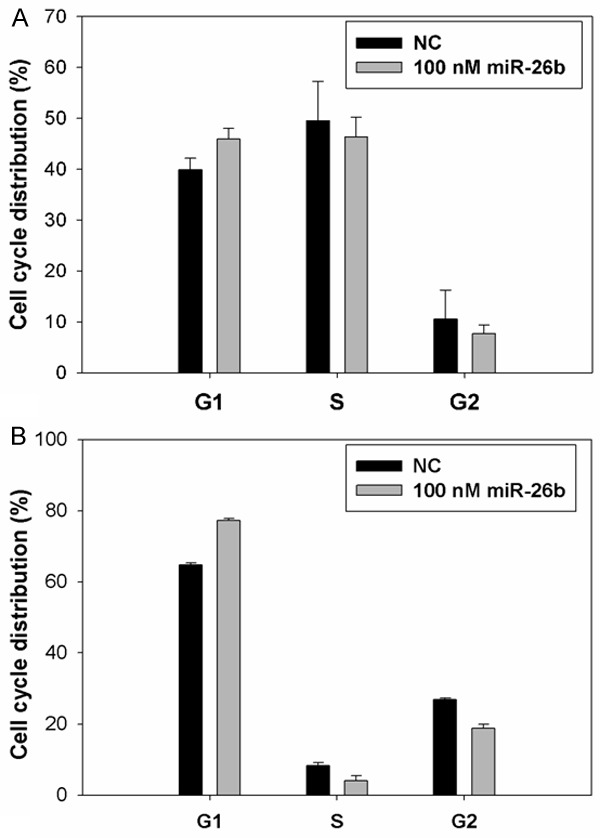
miR-26b initiates G0/G1 cell cycle arrest in MDA-MB-231 cells (A) and Mcf-7 cells (B).
miR-26b directly targets CDK8 in breast cancer cells
Next, we wanted to investigate the mechanism of miR-26b inhibition of proliferation of MDA-MB-231 cells. Predicted miR-26b targets were identified using the algorithms of TargetScan and microRNA.org among hundreds of potential target genes, we specifically focused on CDK8, as it has been consistently reported to be oncogene in various cancers. We identified a binding site for miR-26b in the 3’-UTR of CDK8 mRNA. To confirm that miR-26b can bind to the predicted site, we performed a luciferase reporter assay in the 293T cell line. Figure 5A shows that the luciferase activity significantly decreased after co-transfection with psiCHECK-2/CDK8 3’-UTR and miR-26b mimics in comparison with control cells. The results demonstrated that miR-26b specifically binds to the 3’-UTR of CDK8 mRNA. The effect of miR-26b transfection on CDK8 mRNA and protein expression was respectively assessed using RT-PCR and Western blot in MDA-MB-231 cell line. as shown in Figure 5B and 5C, the qRT–PCR and western blotting analysis found that CDK8 expression at the mRNA and protein levels decreased in the miR-26b mimic-treatment group relative to NC. The results support the hypothesis.
Figure 5.
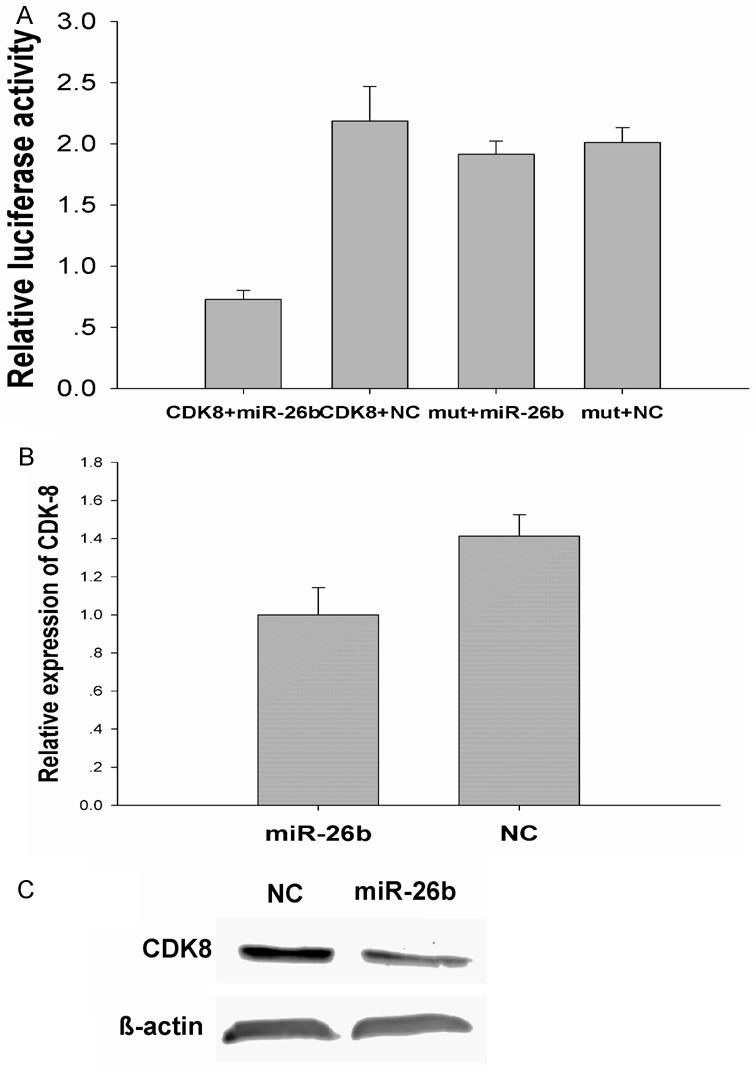
CDK8 is a direct target of miR-26b.
MiR-26b inhibits proliferation via regulation of CDK8
Given that CDK8 is a direct target of miR-26b, we hypothesized that the inhibitory effect of miR-26b on breast cancer cell viability might be achieved via targeting CDK8. To investigate this, we knocked down CDK8 expression and checked its impact on the growth of MDA-MB-231 cells. Treatment of cells with 50 nmol/L CDK8 siRNA markedly suppressed cell viability by 19%, 23%, 37% and 27% at 24 h, 48 h, 72 h and 96 h, respectively, compared with control siRNA (P<0.01) (Figure 6A). These results demonstrate that downregulation of CDK8 expression by miR-26b contributes, at least partially, to the suppression of the growth of breast cancer cells. It has been reported that CKD8 activates the β-catenin-dependent oncogenesis in various cancers, therefore, miR-26b might be implicated in the Wnt/β-catenin-dependent oncogenesis via regulation of CDK8. As shown in Figure 6B, CDK8 siRNA remarkably silenced the expression of CDK8, and β-catenin expression was relatively decreased as compared with siRNA control, In agreement with this finding, Overexpression of miR-26b led to the downregulation of CDK8 and β-catenin expression (Figure 6C). All these findings support the involvement of miR-26b in the proliferation of breast cancer cells via targeting CDK8.
Figure 6.
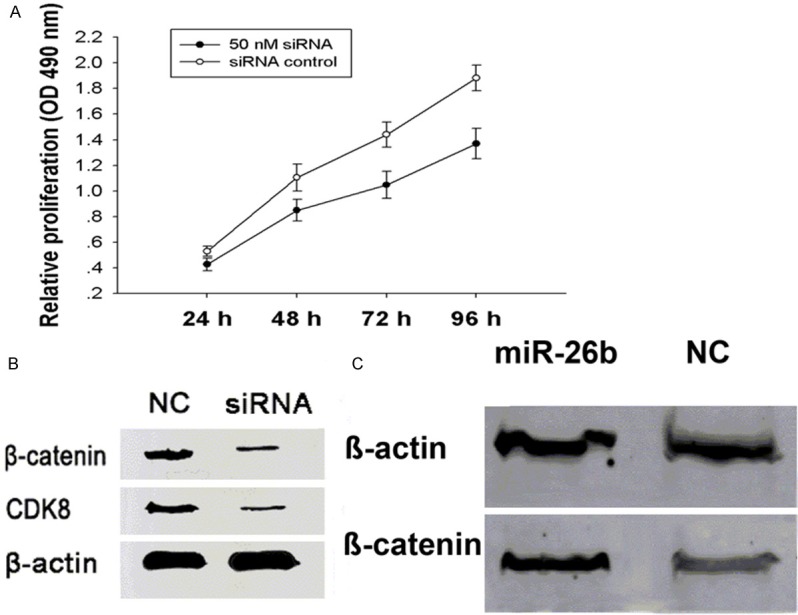
MiR-26b inhibits proliferation via regulation of CDK8.
Discussion
The discovery of the first miRNA, lin-4, in Caenorhabditis elegans initiated a new era of miRNA biology. Since then, thousands of miRNAs have been identified and annotated. Furthermore, an increasing number of evidences indicate that miRNAs are differentially expressed between normal and malignant tumor tissues, miRNA deregulation is a critical cause of cancer formation. In this study, we performed RT-PCR to determine the expression pattern of miR-26b in breast cancer. Compared to the normal breast tissues, miR-26b expression was significantly down-regulated in the breast cancer specimens. Similar finding has been revealed in a number of cancers [7-11]. Xiao-Xiao Liu et al reported miR-26b expression is relatively downregulated in breast cancer cell lines MCF7, HCC1937, MDA-MB-231, MDA-MB-468, MDA-MB-453, BT-549 and BT-474 as comparing with the normal breast skin cells CCD-1095Sk [11]. These results indicate that miR-26b is downregulated in human breast cancer specimens and cell lines.
miR-26b has been reported as a critical regulator in carcinogenesis and tumor progression by acting as tumor suppressor gene in various cancers [8,11]. In this study, we found that transfection of miR-26b mimics into MDA-MB-231 and Mcf-7 cells significantly suppressed cellular proliferation, which is in consistence with previously published studies in glioma and desferrioxamine (DFOM)-treated carcinoma of nasopharyngeal epithelial (CNE) cells [8]. Furthermore, overexpression of miR-26b increased the proportion of breast cancer cells in G0/G1 phase. Similar to this finding, miR-26b and its host genes CTDSP1/2/L could cooperate to block G1/S-phase progression by activating pRb protein in hepatocellular carcinoma [25].
To understand the mechanisms by which miR-26b suppresses tumor cell proliferation in breast cancer, we identified CDK8 as one of potential target genes using TargetScan and microRNA.org. Our data showed that miR-26b directly interacted with the CDK8 3’UTR and downregulated CDK8 mRNA and protein levels in BC cells. These results demonstrate that CDK8 is a direct target gene of miR-26b in breast cancer. CDK8 kinase activity is necessary for β-catenin-driven transformation and for expression of several β-catenin transcriptional targets in a variety of cancers. Our study found that miR-26b was able to inhibit CDK8 and β-catenin expression, knockdown of CDK8 caused significant decrease of cellular proliferation and β-catenin expression. Therefore, downregulation of CDK8 by miR-26b contributes to miR-26b mediated proliferation suppression in MDA-MB-231 cells.
Collectively, Given the downregulation of miR-26b and its ability to suppress proliferation and cell-cycle progression, restoration of miR-26b could be a promising therapeutic target for the treatment of breast cancer in the future.
Acknowledgements
This research was made possible with financial support from National Natural Sciences Foundation of China, for the project 81272240, Shanghai Science Committee Foundation (to Lin FANG) (No. STCSM 10411964700).
Disclosure of conflict of interest
The authors declare no conflict of interest.
Abbreviations
- miRNAs
MicroRNAs
- RT-PCR
Reverse transcription-polymerase chain reaction
- CDK8
Cyclin-dependent kinase 8
- FBS
Fetal bovine serum
- DMEM
Dulbecco’s modified Eagle’s medium
- SDS-PAGE
SDS-polyacrylamide gel
- NATs
Normal adjacent tissues
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nature Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 3.Guttilla IK, White BA. Coordinate regulation of FOXO1 by miR-27a, miR-96, and miR-182 in breast cancer cells. J Biol Chem. 2009;284:23204–23216. doi: 10.1074/jbc.M109.031427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsu PY, Deatherage DE, Rodriguez BA, Liyanarachchi S, Weng YI, Zuo T, Liu J, Cheng AS, Huang TH. Xenoestrogen-induced epigenetic repression of microRNA-9-3 in breast epithelial cells. Cancer Res. 2009;69:5936–5945. doi: 10.1158/0008-5472.CAN-08-4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu HL, Zhu SM, Mo YY. Suppression of cell growth and invasion by miR-205 in breast cancer. Cell Res. 2009;19:439–448. doi: 10.1038/cr.2009.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liang ZX, Wu H, Xia J, Li YH, Zhang YW, Huang K, Wagar N, Yoon Y, Cho HT, Scala S, Shim H. Involvement of miR-326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance-associated protein 1. Biochem Pharmacol. 2010;79:817–824. doi: 10.1016/j.bcp.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 7.Ji J, Shi J, Budhu A, Yu Z, Forgues M, Roessler S, Ambs S, Chen Y, Meltzer PS, Croce CM, Qin LX, Man K, Lo CM, Lee J, Ng IO, Fan J, Tang ZY, Sun HC, Wang XW. MicroRNA expression, survival, and response to interferon in liver cancer. N Engl J Med. 2009;361:1437–1447. doi: 10.1056/NEJMoa0901282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ji Y, He Y, Liu L, Zhong X. MiRNA-26b regulates the expression of cyclooxygenase-2 in desferrioxamine-treated CNE cells. FEBS Lett. 2010;584:961–967. doi: 10.1016/j.febslet.2010.01.036. [DOI] [PubMed] [Google Scholar]
- 9.Gao W, Shen H, Liu L, Xu J, Xu J, Shu Y. MiR-21 overexpression in human primary squamous cell lung carcinoma is associated with poor patient prognosis. J Cancer Res Clin Oncol. 2011;137:557–566. doi: 10.1007/s00432-010-0918-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong TS, Liu XB, Wong BY, Ng RW, Yuen AP, Wei WI. Mature miR-184 as potential oncogenic microRNA of squamous cell carcinoma of tongue. Clin Cancer Res. 2008;14:2588–2592. doi: 10.1158/1078-0432.CCR-07-0666. [DOI] [PubMed] [Google Scholar]
- 11.Liu XX, Li XJ, Zhang B, Liang YJ, Zhou CX, Cao DX, He M, Chen GQ, He JR, Zhao Q. MicroRNA-26b is underexpressed in human breast cancer and induces cell apoptosis by targetingSLC7A11. FEBS Lett. 2011;585:1363–1367. doi: 10.1016/j.febslet.2011.04.018. [DOI] [PubMed] [Google Scholar]
- 12.Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, Freed E, Ligon AH, Vena N, Ogino S, Chheda MG, Tamayo P, Finn S, Shrestha Y, Boehm JS, Jain S, Bojarski E, Mermel C, Barretina J, Chan JA, Baselga J, Tabernero J, Root DE, Fuchs CS, Loda M, Shivdasani RA, Meyerson M, Hahn WC. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature. 2008;455:547–551. doi: 10.1038/nature07179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheffer M, Bacolod MD, Zuk O, Giardina SF, Pincas H, Barany F, Paty PB, Gerald WL, Notterman DA, Domany E. Association of survival and disease progression with chromosomal instability: a genomic exploration ofcolorectal cancer. Proc Natl Acad Sci U S A. 2009;106:7131–7136. doi: 10.1073/pnas.0902232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim MY, Han SI, Lim SC. Roles of cyclin-dependent kinase 8 and β-catenin in the oncogenesis and progression of gastric adenocarcinoma. Int J Oncol. 2011;38:1375–1383. doi: 10.3892/ijo.2011.948. [DOI] [PubMed] [Google Scholar]
- 15.Chattopadhyay I, Singh A, Phukan R, Purkayastha J, Kataki A, Mahanta J, Saxena S, Kapur S. Genome-wide analysis of chromosomal alterations in patients with esophageal squamous cell carcinoma exposed to tobacco and betel quid from high-risk area in India. Mutat Res. 2010;696:130–138. doi: 10.1016/j.mrgentox.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Mitra AP, Almal AA, George B, Fry DW, Lenehan PF, Pagliarulo V, Cote RJ, Datar RH, Worzel WP. The use of genetic programming in the analysis of quantitative gene expression profiles for identification of nodal status in bladder cancer. BMC Cancer. 2006;6:159. doi: 10.1186/1471-2407-6-159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapoor A, Goldberg MS, Cumberland LK, Ratnakumar K, Segura MF, Emanuel PO, Menendez S, Vardabasso C, Leroy G, Vidal CI, Polsky D, Osman I, Garcia BA, Hernando E, Bernstein E. The histone variant macroH2A suppresses melanoma progression through regulation of CDK8. Nature. 2010;468:1105–1109. doi: 10.1038/nature09590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.He SB, Yuan Y, Wang L, Yu MJ, Zhu YB, Zhu XG. Effects of cyclin-dependent kinase 8 specific siRNA on the proliferation and apoptosis of colon cancer cells. J Exp Clin Cancer Res. 2011;30:109. doi: 10.1186/1756-9966-30-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris EJ, Ji JY, Yang F, Di Stefano L, Herr A, Moon NS, Kwon EJ, Haigis KM, Näär AM, Dyson NJ. E2F1 represses beta-catenin transcription and is antagonized by both pRB and CDK8. Nature. 2008;455:552–556. doi: 10.1038/nature07310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim S, Xu X, Hecht A, Boyer TG. Mediator is a transducer of Wnt/beta-catenin signaling. J Biol Chem. 2006;281:14066–14075. doi: 10.1074/jbc.M602696200. [DOI] [PubMed] [Google Scholar]
- 21.Carrera I, Janody F, Leeds N, Duveau F, Treisman JE. Pygopus activates Wingless target gene transcription through the mediator complex subunits Med12 and Med13. Proc Natl Acad Sci U S A. 2008;105:6644–6649. doi: 10.1073/pnas.0709749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoda A, Kouike H, Okano H, Sawa H. Components of the transcriptional mediator complex are required for asymmetric cell division in C. elegans. Development. 2005;132:1885–1893. doi: 10.1242/dev.01776. [DOI] [PubMed] [Google Scholar]
- 23.Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, Nguyen JT, Barbisin M, Xu NL, Mahuvakar VR, Andersen MR, Lao KQ, Livak KJ, Guegler KJ. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using Real-time quantitative PCR and the 2-ΔΔct method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 25.Zhu Y, Lu Y, Zhang Q, Liu JJ, Li TJ, Yang JR, Zeng C, Zhuang SM. MicroRNA-26a/b and their host genes cooperate to inhibit the G1/S transition by activating the pRb protein. Nucleic Acids Res. 2012;40:4615–4625. doi: 10.1093/nar/gkr1278. [DOI] [PMC free article] [PubMed] [Google Scholar]