

Abstract
Aims
Long-QT syndromes (LQTS) are mostly autosomal-dominant congenital disorders associated with a 1:1000 mutation frequency, cardiac arrest, and sudden death. We sought to use cardiomyocytes derived from human-induced pluripotency stem cells (hiPSCs) as an in vitro model to develop and evaluate gene-based therapeutics for the treatment of LQTS.
Methods and results
We produced LQTS-type 2 (LQT2) hiPSC cardiomyocytes carrying a KCNH2 c.G1681A mutation in a IKr ion-channel pore, which caused impaired glycosylation and channel transport to cell surface. Allele-specific RNA interference (RNAi) directed towards the mutated KCNH2 mRNA caused knockdown, while leaving the wild-type mRNA unaffected. Electrophysiological analysis of patient-derived LQT2 hiPSC cardiomyocytes treated with mutation-specific siRNAs showed normalized action potential durations (APDs) and K+ currents with the concurrent rescue of spontaneous and drug-induced arrhythmias (presented as early-afterdepolarizations).
Conclusions
These findings provide in vitro evidence that allele-specific RNAi can rescue diseased phenotype in LQTS cardiomyocytes. This is a potentially novel route for the treatment of many autosomal-dominant-negative disorders, including those of the heart.
Keywords: iPS cells, Long-QT syndrome, Arrhythmia, Electrophysiology, Gene therapy
See page 1019 for the editorial comment on this article (doi:10.1093/eurheartj/eht130)
Introduction
With the prevalence of 1:1000 individuals, inherited long-QT syndromes (LQTS) are life-threatening disorders caused by abnormal ventricular repolarization1 due to mutations in at least 13 genes encoding cardiac ion-channel proteins.2 Patients with LQTS have an increased risk of Torsades de Pointes (polymorphic ventricular tachycardia), which can present as palpitations, syncope (fainting), seizures, cardiac arrest, and sudden death.3 Of LQTS cases, 45% are classified as LQTS-type 2 (LQT2), caused by mutations in KCNH2 (also known as hERG), which encodes the α-subunit of a tetrameric complex forming part of the rapid-acting inward rectifying potassium (Ikr) channel.4 Over 500 hERG mutations are identified, which primarily show autosomal-dominant-negative inheritance. Function of the wild-type (WT) protein is compromised by mutated (Mut) protein, thus resulting in reduced Ikr function due to altered gating properties5 or impaired protein trafficking sometimes linked to glycosylation defects, inappropriate protein maturation and proteasomal degradation.6
Treatments for LQT2 include the administration of β-adrenoceptor antagonists (β-blockers) or, in patients at a high risk of sudden death, surgery for implantation of pacemakers, cardioverter defibrillators, and left cardiac sympathetic denervation.7 However, limitations in current therapies have prompted investigations to further understand disease pathology, and develop and safety screen novel pharmacological treatments. Such research has been based on the use of in vitro transgenic or immortalized cell lines (e.g. HEK293) and animal models (e.g. guinea-pig myocytes, arterially perfused canine and rabbit left ventricular wedge preparations), as well as in vivo toxicity studies in monkeys, dogs, and mouse.8 However, a particular limitation of the mouse is the reliance on different ion channels relative to human (mouse, Ito, IK,slow1, IK,slow2, ISS; human, IKs, IKr) during the cardiac action potential.9 Lack of predictive translation of such models towards human behaviour4 has led to the development of a multitude of functionally relevant, humanized in vitro models for LQTS via use of induced pluripotency technologies.10 These have successfully shown that patient's clinical profiles and response to pharmacology are faithfully reflected in vitro, including for LQT2.11
Here, we sought to explore the disease-causing biophysical mechanisms of LQT2-associated c.G1681A (p.Ala561Thr) missense mutation in KCNH2, with view to developing novel patient-specific treatments. By coupling induced pluripotency technologies to RNA interference (RNAi)-based therapy, we demonstrated that allele-specific mRNA knockdown of Mut hERG is feasible in human cardiomyocytes. This treatment rescued electrophysiological characteristics of LQT2 human-induced pluripotency stem cell (hiPSC)-derived cardiomyocytes, as evidenced by normalized action potential durations and increased K+ current. Moreover, treated cells did not develop early-afterdepolarizations (EADs) in response to adrenergic stimulation or potassium blockage, drug treatments that induce arrhythmias in LQT2 patients. These data suggest that allele-specific RNAi warrants further in vivo investigation as a treatment modality for LQTS and other autosomal-dominant-negative genetic diseases.
Methods
Details on hiPSCs, including isolation of patient tissue, generation, culture, and characterization, as well as generation of high titre lentivirus are previously published.11,12 In vitro differentiation to cardiomyocytes was via embryoid body (EB) formation, based on previous publication.13
Electrophysiology analysis
For multi-electrode array (MEA) analysis, beating clusters between Days 12 and 16 of differentiation were mounted on MEAs (Multi-channel Systems), and extracellular field potential measurements performed according to previous guidelines.14 For whole-cell recordings of action potentials by patch-clamp, cardiomyocyte clusters were disaggregated to single cells using published method and buffers,15 and recordings were obtained in the current mode using an ECP-10 amplifier (HEKA). During recordings cells were maintained at 37°C in normal Tyrode's buffer, while patch pipettes and buffer were as previously described.11 Data were recorded using the Pulse software (HEKA) and analysed using Clampfit v9.0 (Molecular Devices). Cardiomyocyte subtypes were determined as previously described.11 Briefly, APD90/50 values <1.4 designated ventricular cells, 1.4–1.7 designated pacemaker cells, and >1.7 designated atrial cells.
Transduction and transfection with KCNH2 constructs
Following sterilization of plasmid DNA (Supplementary material online Methods), at 65°C for 10 min, cells were transfected using Lipofectamine™ 2000 (Invitrogen) as per previous publications,16 or transduced with lentivirus at MOI 10, generated as previously described.17 For cardiomyocyte transduction, lentivirus was pre-incubated with Polybrene® (Sigma-Aldrich) for 5 min before addition to the cells, as published before.18 This was performed to increase transduction efficiency since Polybrene® is known to neutralize charge repulsion between viral particles and sialic acid on the cell surface.19
siRNA transfection
siRNAs against WT or Mut hERG were designed based on optimum discrimination parameters,20 and ordered (Invitrogen) with dTdT 3′ modification. LacZ-siRNA was included as a non-targeting control (F: 5′-CUACACAAAUCAGCGAUUU, R: 5′-AAAUCGCUGAUUUGUGUAG) and had fluorescein 5′ modification to allow transfection efficiency quantification. For transgene knockdown, 20 µM double-stranded siRNAs (5 µL) mixed with ExGen500 lipid (7 µL; Fermentas; 2:2 siRNA:lipid ratio) were used to transfect 10 000 fibroblasts. For endogenous gene knockdown, 2.5 µL siRNA were mixed with 3.5 µL ExGen500 lipid (1:1 siRNA:lipid ratio) and used to transfect 10 000 cardiomyocytes. Transfections were performed in OptiMEM (Invitrogen) for 4 h, according to lipid manufacturer's instructions.
shRNA gateway cloning
shRNA oligos (Invitrogen) were annealed by heating to 94°C for 4 min, then 70°C for 10 min, and cooled-down at room temperature for 1 h. Double-stranded shRNAs were ligated, for 30 min, into linearized pENTR/U6 expression vector according to manufacturer's instructions (BLOCK-iT™ U6 RNAi Entry Vector Kit; Invitrogen). For transient over-expression, vectors were transfected as for pSIN-EF2 vectors above. For stable over-expression, shRNA/U6 cassette was recombined into pLenti X2 Neo DEST vector (Addgene) using Gateway® LR Clonase® II Enzyme (Invitrogen) according to manufacturer's instructions, lentivirus generated as above, and cells infected at MOI 25.
Allele-specific gene expression
RT–PCR performed as previously described11 with 2 µL cDNA, and the addition of 1.5 mM MgCl2 (QIAGEN) for WT cDNA or 2.5 mM MgCl2 for Mut cDNA. Common forward primer was used for both cDNAs (F: 5′-ACTTCAAGGGCTGGTTCCTC). Reverse primer for WT cDNA was R: 5′-ATGCAGGCTAGCCAGTGCTC and for Mut allele R: 5′-ATGCAGGCTAGCCAGTGCTT. PCR conditions were 95°C for 15 min then 37 cycles (95°C, 1 min; 59°C, 30 s; 72°C, 1 min) with the final extension at 72°C for 5 min. SYBR® Green Real-time PCR performed with 2× JumpStart™ Taq ReadyMix™ (10 µL; Sigma-Aldrich), reference dye 0.02 µL, 2.5 mM MgCl2, and same primers as above (2 µL). PCR conditions were 50°C for 20 s, 95°C for 10 min then 37 cycles (95°C, 15 s; 59°C, 1 min) with data collection after the 59°C, 1 min step. Amplification was followed by continuous melt-curve analysis.
Statistical analysis
Data presentation and significance scoring were as previously described.11 Within-group data were reported as mean values of experimental replicates ± standard deviation. Wild-type and Mut hERG monomer incorporation into tetramers before and after Mut mRNA knockdown was statistically modelled using binomial distribution. Choice of the statistical model was based on the apparent random assembly of WT and Mut hERG alleles into tetramers and their statistical independence. Therefore, the probability of a tetramer containing WT: Mut alleles at a ratio of k:(n − k) for k = 0,1, … ,n were calculated using: prob(k:n − k) = C(n, k) pk (1 − p)n−k, where n = 4, C(n, k) is the binomial coefficient, and p is the fraction of WT monomers in the total hERG population. The formula for prob(k:n− k) can be understood as follows: for a tetramer to contain WT:Mut alleles at a ratio of k:(4 − k), we need k monomers of WT giving a probability of pk, and (4 − k) monomers of Mut giving a probability of (1 − p)4 − k. However, the successful choice of the WT monomer can occur anywhere among the n = 4 trials. Namely, there are C(n, k) different ways of distributing k successes in a sequence of n trials thus giving the resulting formula.
Results
hERG c.G1681A mutation causes electrophysiological abnormalities in Long-QT syndromes-type 2 cardiomyocytes
Human-induced pluripotency stem cells were generated from a patient with LQT2-related mutation c.G1681A in KCNH2, and from genetically matched controls including the patient's father (genetically normal) and mother (asymptomatic carrier of same mutation, not under β-blocker treatment). Full 12-lead electrocardiograms (ECGs) for all family members have been previously published.11 Differentiation of hiPSCs produced beating clusters within 8–16 days,13 which contained cardiomyocytes as evidenced by characteristic cardiac muscle striations upon immunostaining (Figure 1A). Electrophysiological analysis by whole-cell patch-clamp of individual human pluripotent stem cell (hPSC) cardiomyocytes showed cultures contained ventricular (32%), atrial (41%), and pacemaker-like (27%) cell subtypes (Figure 1B). Multi-electrode array analysis showed that compared with cardiomyocytes generated from HUES7 human embryonic stem cells (hESCs; 845 ± 78 ms, n = 11), and the patient's father (LQT2-PAT; 836 ± 78 ms, n = 6) and mother (LQT2-MAT; 795 ± 174 ms, n = 6), LQT2 cardiomyocytes had significantly prolonged field potential duration (FPD: 1084 ± 1.65, P = 0.01, n = 10; Figure 1C), which correlates to the patient's ECG indications.11 The same was true when LQT2 ventricular (APD: 929 ± 167 ms, P = 0.01, n = 13), atrial (APD: 1126 ± 260 ms; P = 0.004, n = 14), and pacemaker-like (APD: 882 ± 222 ms, P = 0.02, n = 9) cardiomyocytes were compared with controls (combined data from hESCs, LQT2-PAT, and LQT2-MAT) by patch-clamp electrophysiology (Figure 1D). Treatment with E4031 (1 µM), a selective IKr channel blocker, caused APD prolongation in control cardiomyocytes (378 ± 229.0 ms, n = 6), and arrhythmogenesis appearing as EADs only in LQT2 cardiomyocytes (occurrence rate 1:2, n = 6), indicating increased sensitivity to IKr blockage (Figure 1E). All analysis was performed in two independent hiPSC-lines per patient to avoid side-effects form line-to-line variation. Deep-sequencing of the LQT2-genome also confirmed absence of additional known disease-associated mutations in cardiac arrhythmia-related genes (Table SI), therefore leaving differences between mother-daughter phenotype-to-genotype correlations unanswered.
Figure 1.

Long-QT syndrome-type 2 hiPSC cardiomyocytes can recapitulate the disease phenotype in vitro. (A) Long-QT syndrome-type 2 cardiomyocytes showed characteristic cardiac muscle striations with cardiac troponin-I staining, and (B) formed ventricular (n = 11), atrial (n = 14), and pacemaker-like (n = 9) myocytes. (C) Long-QT syndrome-type 2 cardiomyocytes had prolonged field potential duration (n = 10) and (D) action potential duration compared with controls, which included cardiomyocytes derived from human embryonic stem cells, the patient's father (long-QT syndrome-type 2-PAT) and mother (long-QT syndrome-type 2-MAT). (E) E4031 (1 µM) treatment caused prolongation in action potential duration of control cardiomyocytes (n = 6), depicting the presence of functional hERG channels, and caused arrhythmias only in long-QT syndrome-type 2 cardiomyocytes (n = 6).
hERG c.G1681A mutation causes dominant-negative trafficking defect in cardiomyocytes
To explore disease-causing biophysical mechanism of KCNH2 c.G1681A mutation, we interrogated predicted hERG folding using SWISS-MODEL tools, which showed amino-acid substitution of alanine to threonine did not cause structural protein change (Figure 2A). Further studies were conducted using transgenic disease systems in fibroblasts and HEK293 cells (Supplementary material online, Figure S2), where western blot analysis confirmed the presence of glycosylation/maturation defect in Mut hERG (Figure 2C), correlating to previous publications.21 Fluorescence staining detected distinct hERG localization patterns, which for the WT overlapped to pan-cadherin and fibroblast surface markers (Figure 2Di–ii, Supplementary material online, Figure S3B), but was perinuclear for Mut hERG (Figure 2Ei–ii). This also confirmed that glycosylation defect leads to impaired protein trafficking.22 Next, clones over-expressing WT hERG-GFP were generated and showed an indistinguishable localization pattern when compared with non-tagged WT hERG clones, confirming the GFP tag did not compromise hERG localization (Supplementary material online, Figure S2E). When WT hERG-GFP was transfected into Mut transgenic lines, the GFP signal lacked surface localization (Figure 2F i and ii), indicating a dominant-negative phenotype likely due to the formation of non-functional WT/Mut tetramers with defective hERG surface transport. Finally, lentiviral transduction with paramagnetic bead-bound virus, previously proven to enhance viral uptake,12 showed that transgene over-expression can be detected in hPSC cardiomyocytes (Supplementary material online, Figure S4B) at 5–6 days post-transduction (PTD). This also enabled detection of over-expressed hERG in single cardiomyocytes demonstrating that in functionally relevant human cells, WT hERG is co-localized with surface marker pan-cadherin (Figure 2Diii) while Mut formed perinuclear aggregates (Figure 2Eiii). Defective trafficking was also seen in cardiomyocytes co-infected with WT and Mut lentivirus (Figure 2Fiii), confirming dominant-negative effect.
Figure 2.

KCNH2 mutation c.G1681A causes a hERG-dominant-negative trafficking defect. (A) SWISS-MODEL structural analysis of amino acid residues: 408 to 665 showing no effect in hERG monomer folding due to c.G1681A mutation. (B) Site-directed mutagenesis in wild-type hERG1a cDNA generated mutant form for over-expression studies. (C) Western blot showing the absence of fully glycosylated mature hERG in fibroblast lines over-expressing mutant transgene. Fluorescence staining showing over-expression of (D) wild-type and (E) Mut hERG in (i) HEK293 cells, (ii) fibroblasts, and (iii) hESC cardiomyocytes, depicting impaired surface localization of Mut hERG (red). Cell surface outlined with pan-cadherin staining (lilac; also see Supplementary material online, Figure S3). (F) Impaired surface localization of GFP-tagged wild-type hERG (green) in (i) HEK293 and (ii) fibroblast cell lines co-expressing Mut hERG, depicting the dominant-negative phenotype of the mutant form. (F) (iii) Fluorescence staining showing simultaneous over-expression of wild-type and Mut hERG in hESC-derived cardiomyocytes causes lack of surface localization. Scale-bar: 15 µm.
Testing potential drug therapies against hERG c.G1681A dominant-negative trafficking defect
Having validated c.G1681A mutation results in a hERG-dominant-negative trafficking defect, we wished to investigate novel, effective, and patient-specific treatments for this LQT2 mutation. Studies in immortalized cell-lines have suggested thaspigargin, fexofenadine, or E4031 enhance hERG cell-surface transport.22,23 Therefore, we exposed Mut fibroblasts to above drugs (10 nM to 10 µM), and stained with anti-hERG antibody. Fluorescence microscopy did not show improved hERG trafficking with thaspigargin or fexofenadine at any concentrations tested (data not shown). Although E4031 facilitated surface localization at 1 µM (Supplementary material online, Figure S5A), our data showed this concentration also induced arrhythmogenesis in LQT2 cardiomyocytes (Figure 1E). E4031 experiments were, thus, not carried forward. To restore impaired K+ currents, LQT2 cardiomyocytes were next treated with potassium-channel openers; nicorandil (20 µM) and PD-118057 (3 µM), which enabled shortening of the otherwise prolonged APD by 293.8 ± 84.8 and 213.2 ± 26.5 ms, respectively, and by 627.4 ± 100.2 ms when used in combination (Supplementary material online, Figure S5B). Combined treatment rendered APD in LQT2 cardiomyocytes similar to controls (653 ± 100 and 613 ± 122 ms, respectively). However, the administration of potassium-channel openers and isoprenaline, to mimic stress simulation, triggered abnormalities (Supplementary material online, Figure S5B), perhaps due to excessive APD shortening requiring very fast repolarization that cells lacking functional hERG could not cope with. This suggests that LQT2 patients potentially undertaking this treatment could suffer arrhythmias during activities inducing sympathetic stimulation (e.g. exercise, stress).
Testing potential gene therapies against hERG c.G1681A dominant-negative trafficking defect
Current routes for gene therapy include targeted gene correction, transgene over-expression and RNAi for either knockdown or exon-skipping in Mut genes. Gene targeting has been achieved in vitro using hiPSC disease models,10 and in animal in vivo models using zinc-finger nucleases,24 but not yet in humans. Also, gene targeting requires proliferating cells in the S-phase of the cell-cycle,25 so it is of limited use for the treatment of predominantly quiescent heart cells, while the structurally intact state of patients' hearts (i.e. absence of scar tissue or lesions) impedes transplantation of cardiac progenitors following targeted gene correction. Figure 2Diii shows WT hERG over-expression with surface localization can be achieved in cardiomyocytes via lentiviral transduction. As a therapeutic route, transgene over-expression could be utilized to shift the ratio of WT to Mut alleles, increasing the number of functional hERG tetramers capable of surface localization. However, while lentiviral WT-hERG over-expression was achieved, onset was delayed (5–6 days PTD) and infection efficiency was low (52%). Also, since transgene over-expression levels can be uncontrolled and reach cardiotoxic levels,26 this therapeutic route could face clinical challenges.
We reasoned a more controlled way to regulate and improve formation and trafficking of functional hERG tetramers would be to use allele-specific mRNA knockdown. We designed RNAi constructs based on mismatch position guidelines previously published,20 with sequences tethered around KCNH2 c.G1681A mutation. Four siRNAs were designed against either WT or Mut KCNH2 (Figure 3A, Supplementary material online, Figure S6A), to determine the robustness of allelic knockdown, and converted to shRNA sequences cloned (Supplementary material online, Figure S6B) into pDEST/U6 Gateway lentiviral vectors (Supplementary material online, Figure S6C). Vector delivery into transgenic WT/Mut hERG fibroblasts was used to evaluate the specificity and efficiency of knockdown 72 h PTD, to allow time for VSV-G pseudo-typed vector initial integration into the host genome (can take at least 12–48 h).27 WT-G10 shRNA (75.9% knockdown) and Mut-A13-shRNA (70.5% knockdown) were most effective in reducing allele-specific transcripts without affecting the other allele (Figure 3B, Supplementary material online, Figure S6D). Flow cytometry analysis indicated hERG protein knockdown approached mRNA knockdown levels on Days 6–9 PTD (65.2 and 71.8%, respectively; Figure 3C). Delay in protein compared with mRNA knockdown was attributed to hERG protein turn-over, known to be ∼11–16 h.28,29 WT protein was unaffected by A13-shRNA, when compared with a LacZ-shRNA control (Supplementary material online, Figure S6E). Results suggested that allele-specific mRNA knockdown warranted further investigation in LQT2 cardiomyocytes.
Figure 3.
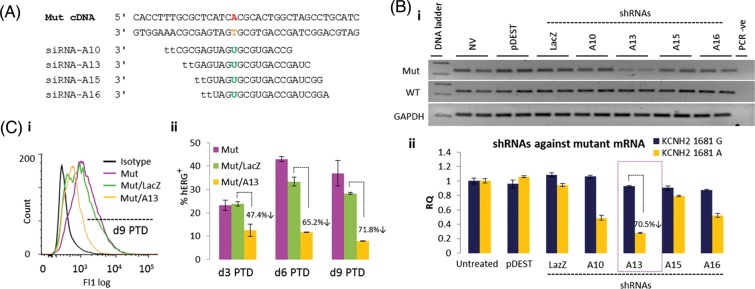
Design and validation of hERG allele-specific mRNA knockdown. (A) Rational design of siRNAs against mutated (1681A) KCNH2 mRNA. siRNA nomenclature based on distinguishing nucleotide between wild-type and mutant alleles, and its position from the siRNA 5′-end (e.g. A13). (B) (i) RT and (ii) real-time PCR showed shRNAs over-expressed through pDEST/U6 lentiviral vector enabled knockdown of Mut KCNH2 mRNA in fibroblasts carrying lentiviral over-expression of both wild-type (G) and Mut (A) mRNAs. A13-shRNA enabled the highest level knockdown (70.5%), without affecting wild-type mRNA. (C) Flow cytometry showed A13-shRNA also enabled 65–71.8% Knockdown of Mut hERG protein from Day 6 post-transduction. All mRNA and protein knockdown experiments were repeated twice with n = 2 per treatment. For further details, also see ‘Methods’ section.
Allele-specific RNAi can knockdown mutated hERG in long-QT syndrome-type 2 cardiomyocytes
Having validated allele-specific knockdown at the gene and protein levels, we synthesized siRNAs corresponding to A13 sequences. siRNA transfection efficiencies were optimized using fluorescently tagged oligos in transgenic fibroblast lines (98.8–99.8%, Supplementary material online, Figure S7) and LQT2 hiPSC cardiomyocytes (84%, Figure 4B, Supplementary material online, Figure S8). In disaggregated single cardiomyocytes or small beating clusters of five to six cells, siRNA transfection was not significantly cytotoxic as it did not compromise spontaneous contraction or structural integrity when the 1:1 siRNA:lipid ratio was used (see section ‘Methods’, Figure 4B, Supplementary material online, Figure S8B). siRNA treatment at the 2:2 ratio (or higher), enabled ∼100% cardiomyocyte transfection but was detrimental to structural integrity as shown by cardiac troponin-I staining (Supplementary material online, Figure S8A) and was not used in knockdown experiments.
Figure 4.

siRNA-mediated knockdown of mutated hERG in long-QT syndrome-type 2 cardiomyocytes. (A) In fibroblasts carrying lentiviral over-expression of both wild-type and mutant alleles. (i) A13-siRNAs against mutant KCNH2 enabled allele-specific mRNA knockdown for up to 48 h. (ii) Repeat-transfection with siRNAs every 48 h (red arrows) sustained knockdown up to Day 6 post-initial transfection. All mRNA knockdown experiments repeated twice with n = 2 per treatment. (B) long-QT syndrome-type 2 cardiomyocytes were successfully transfected with fluorescein-tagged siRNAs (green, n = 42), while maintaining (i) morphology, spontaneous contraction and (ii) structural integrity, as shown by staining with cardiac troponin-I (cardiac troponin-I = red; blue = DAPI nuclear stain). (C) A13-siRNA-mediated 61.8% knockdown of mutant KCNH2 in long-QT syndrome-type 2 cardiomyocytes. (D) Statistical analysis of the likelihood for functional hERG tetramer formation following mutant knockdown showed that the formation of functional hERG tetramers can increase by 4.5-fold, from 6 to 27%. (E) Fluorescence staining showing co-expression of (i) wild-type hERG, Mut hERG, and LacZ-shRNA (n = 10), or (ii) wild-type hERG, Mut hERG, and A13-shRNA (n = 10) in hESC cardiomyocytes, depicting impaired hERG (red) surface localization in (i) and considerably improved localization in (ii). Cell surface outlined with pan-cadherin staining (lilac). Scale-bar: 15 µm.
Allele-specific mRNA knockdown by A13-siRNA was confirmed in fibroblast transgenic lines (Figure 4Ai), and repeated transfection with siRNA every 48 h for 6 days sustained knockdown of Mut KCNH2 (Figure 4Aii). In LQT2 cardiomyocytes, A13-siRNA enabled 61.8% knockdown of endogenous Mut KCNH2 mRNA compared with controls (untreated and treated with LacZ-siRNA), without affecting WT mRNA (Figure 4C). This is the first report documenting allele-specific knockdown of an endogenous mRNA in heart cells, or indeed hiPSCs and their differentiated derivatives. Based on observations that gene knockdown levels correlate to protein knockdown (Figure 3C), statistical modelling fitted to the binomial distribution was used to predict the likelihood of functional hERG tetramer formation following Mut hERG knockdown. Results indicated that this level knockdown of endogenous Mut hERG could enhance the likelihood of functional protein tetramer formation from 0.0625 (6.25%) to 0.2757 (27.57%; Figure 4D), meaning a potential 4.5-fold increase in IKr current. This could shift the phenotype of LQT2 cardiomyocytes towards that of an asymptomatic mutation carrier or normal individual. Indeed, simultaneous transduction of cardiomyocytes with hERG WT, Mut, and A13-shRNA lentiviral constructs co-incubated onto paramagnetic beads (can bind hundreds of virions/bead), indicated considerable improvement in surface localization for hERG (Figure 4E). This demonstrated specific knockdown of Mut transgene could alleviate dominant-negative phenotype and allow corrected WT localization.
Electrophysiological properties of siRNA-treated long-QT syndrome-type 2 cardiomyocytes
We next investigated the effect of siRNA knockdown on LQT2 hiPSC-cardiomyocyte function. Patch-clamp electrophysiology showed the otherwise prolonged APD of ventricular, atrial, and pacemaker-like myocytes was shortened by 279 ± 148 ms (P = 0.35, n = 8), 438 ± 142 ms (P = 0.01, n = 15), and 212 ± 66 ms (P = 0.03, n = 17), respectively, following A13 treatment when compared with control LQT2 cardiomyocytes (Figure 5A and B). APD50 and APD9011 values were also shortened (% decrease in Figure 5A). Spontaneous arrhythmias observed in control LQT2 myocytes (occurrence rate 1:3, n = 21) were never detected in A13-treated cells (Figure 5C), indicating rescue of LQT2 arrhythmogenic phenotype. Voltage-clamp recordings demonstrated increased K+ current in A13-treated LQT2 cardiomyocytes (n = 5), suggesting APD shortening could be due to improved repolarization ability from A13 treatment.
Figure 5.

Electrophysiology analysis in siRNA-treated long-QT syndrome-type 2 cardiomyocytes. (A) Patch-clamp electrophysiology showing A13-siRNA treatment decreased action potential duration, APD50 and APD90, in long-QT syndrome-type 2 cardiomyocytes (ventricular: n = 8, atrial: n = 15, pacemaker: n = 17). (B) Overlays of representative traces from control and A13-treated long-QT syndrome-type 2 ventricular, atrial, and pacemaker cardiomyocytes. (C) Averaged and raw data of spontaneous arrhythmias, in the form of early-afterdepolarizations, observed only in control long-QT syndrome-type 2 cardiomyocytes (n = 21). (D) Voltage-clamp showing increased K+ currents with A13 treatment (n = 5).
A13-treated LQT2-myocytes did not develop isoprenaline-induced arrhythmias (n = 10; Figure 6B), suggesting patients under siRNA therapy could have increased tolerance to exercise or stress. This was in contrast to control LQT2 cardiomyocytes (n = 10, Figure 6A), and cardiomyocytes under combined Nicorandil and PD-118057 treatment which had enabled comparable APD decrease to siRNA treatment (Supplementary material online, Figure S5B). In addition, siRNA-treated myocytes showed no arrhythmias with E4031 treatment (n = 5, Figure 6B), which indicates that patients potentially undertaking this therapy could be less susceptible to drug-induced cardiac episodes when administered drugs with cardio-toxic QT-elongation side-effects.30 In summary, although focusing on one specific LQT2 mutation, the results presented here are evidence that allele-specific RNAi can rescue LQTS phenotype, in a humanized functionally relevant in vitro disease model.
Figure 6.
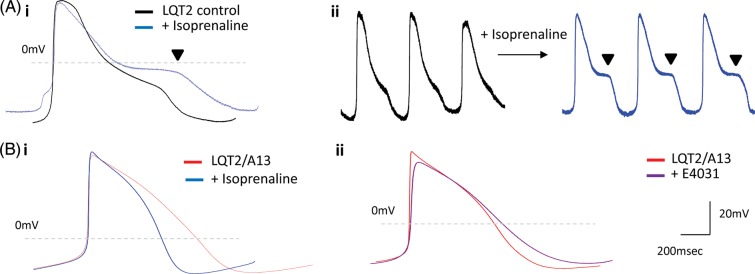
Isoprenaline and E4031 drug treatment in siRNA-treated long-QT syndrome-type 2 cardiomyocytes. (A) (i) Averaged and (ii) raw data of isoprenaline-induced arrhythmias in the form of early-afterdepolarizations observed only in control long-QT syndrome-type 2 cardiomyocytes (n = 10). (B). (i) Isoprenaline (100 nM, n = 10) or (ii) E4031 (1 µM, n = 5) treatment in A13-treated long-QT syndrome-type 2 cardiomyocytes did not cause early-afterdepolarizations, depicting rescue of long-QT syndrome-type 2-phenotype by RNAi-based therapy.
Discussion
Biophysical disease mechanisms of LQT2-associated mutation c.G1681A were analysed in this paper and exploited to validate a novel therapeutic treatment in patient-specific hiPSC cardiomyocytes. Treatment was based on RNAi technologies to specifically knockdown Mut hERG alleles, while leaving the WT unaffected, and maintaining contractibility and structural integrity in cardiomyocytes. The quiescent state of human cardiomyocytes poses a major obstacle for other in vivo cardiac therapies, such as a targeted gene correction by zinc-finger nucleases where cells need to enter the S-phase of the cell-cycle.25 In contrast, we show that human cardiomyocytes can readily uptake and express siRNA molecules. In LQT2 cardiomyocytes, Mut mRNA was knocked-down by 61.8%, increasing likelihood of functional hERG tetramer formation by 4.5-fold. Consequently, APD in siRNA-treated LQT2 cardiomyocytes was shortened, K+ currents increased, and spontaneous as well as isoprenaline and E4031-induced arrhythmias abolished. These are significant findings since they demonstrate that adrenergic stimulation and potassium current blockage, which often evoke arrhythmic episodes in LQT2 patients,30,31 are prevented by RNAi treatment.
In vivo specificity and efficacy of allele-specific RNAi in animal models of LQTS, delivery methods, safety, dosage, and timing issues would need to be addressed before this therapy for LQTS was transferred to the clinic. Other RNAi-based animal and clinical trials for the treatment of disorders such as cancer, age-related macular degeneration, respiratory syncytical virus pulmonary infections, and Duchenne muscular dystrophy use targeted systemic nanoparticle delivery for siRNA administration to patients,32 delivery via intravitreal and i.v. injection of naked siRNA33,34 or administration in the form of nasal sprays.35 In vivo siRNA delivery to human cardiomyocytes has not been reported to date, but mouse studies show rapid and efficient siRNA biodistribution in several internal organs, including the heart, via i.v. injection.36 Injected macroscopic biomaterial scaffolds or conjugation to peptides could also allow siRNA delivery specifically to human hearts.37 Tissue-specific delivery would alleviate potential off-target effects of siRNAs in non-cardiac cells.
Allele-specific mRNA knockdown is a largely unexploited novel concept with wide applications the future treatment of human diseases. It has previously been tested against triplet repeat expansions in the Huntingtin gene with knockdown validation in HeLa cells,38 and SNP mutation in the procollagen type III gene with validation in skin fibroblasts form a rare (prevalence 1:100 000) Ehlers-Danlos syndrome patient.39 Study of allele-specific mRNA knockdown has never been shown for common conditions and where isolation of affected human tissue is challenging, such as in cardiac disorders. Here, hiPSC reprogramming technologies enabled generation of patient-specific cardiomyocytes, faithfully recapitulating disease pathologies in vitro, compared with genetically matched controls, where benefits of allele-specific knockdown were assessed in functional human cells. This would pave the way towards further in vivo testing in animal models of LQTS and ultimately clinical trials that could personalize clinical treatment for not only LQTS mutations (prevalence 1:1000), but also other congenital cardiovascular and non-cardiovascular disorders caused by autosomal-dominant-negative mutations.
Supplementary material
Supplementary material is available at European Heart Journal online.
Acknowledgements
The authors would like to thank Kati Kemppainen and Dr Kid Törnquist (Åbo Akademi University, Finland) as well as Jennifer Abbruzzese and Prof Michael Sanguinetti (The University of Utah, USA) for wild-type hERG1a cDNA constructs, Dr Paul Burridge (Stanford University School of Medicine, USA) for advice on high-efficiency differentiation of hESCs to cardiomyocytes, and João Bigares (Cardiff University, UK) for advice on cardiomyocyte electrophysiology analysis.
Funding
This work was supported by the British Heart Foundation (BHF), the Medical Research Council (MRC), and the Biotechnology and Biological Sciences Research Council (BBSRC).
Conflict of interest: none declared.
References
- 1.Morita H, Wu J, Zipes DP. The QT syndromes: long and short. Lancet. 2008;372:750–763. doi: 10.1016/S0140-6736(08)61307-0. [DOI] [PubMed] [Google Scholar]
- 2.Bokil NJ, Baisden JM, Radford DJ, Summers KM. Molecular genetics of long QT syndrome. Mol Genet Metab. 2011;101:1–8. doi: 10.1016/j.ymgme.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 3.Tester DJ, Ackerman MJ. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation. 2011;123:1021–1037. doi: 10.1161/CIRCULATIONAHA.109.914838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehnart SE, Ackerman MJ, Benson DW, Jr, Brugada R, Clancy CE, Donahue JK, George AL, Jr, Grant AO, Groft SC, January CT, Lathrop DA, Lederer WJ, Makielski JC, Mohler PJ, Moss A, Nerbonne JM, Olson TM, Przywara DA, Towbin JA, Wang LH, Marks AR. Inherited arrhythmias: a National Heart, Lung, and Blood Institute and Office of Rare Diseases workshop consensus report about the diagnosis, phenotyping, molecular mechanisms, and therapeutic approaches for primary cardiomyopathies of gene mutations affecting ion channel function. Circulation. 2007;116:2325–2345. doi: 10.1161/CIRCULATIONAHA.107.711689. [DOI] [PubMed] [Google Scholar]
- 5.Yang HT, Sun CF, Cui CC, Xue XL, Zhang AF, Li HB, Wang DQ, Shu J. HERG-F463L potassium channels linked to long QT syndrome reduce I(Kr) current by a trafficking-deficient mechanism. Clin Exp Pharmacol Physiol. 2009;36:822–827. doi: 10.1111/j.1440-1681.2009.05150.x. [DOI] [PubMed] [Google Scholar]
- 6.Mihic A, Chauhan VS, Gao X, Oudit GY, Tsushima RG. Trafficking defect and proteasomal degradation contribute to the phenotype of a novel KCNH2 long QT syndrome mutation. PLoS One. 2011;6:e18273. doi: 10.1371/journal.pone.0018273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schimpf R, Veltmann C, Wolpert C, Borggrefe M. Arrhythmogenic hereditary syndromes: Brugada Syndrome, long QT syndrome, short QT syndrome and CPVT. Minerva Cardioangiol. 2010;58:623–636. [PubMed] [Google Scholar]
- 8.Medeiros-Domingo A, Iturralde-Torres P, Ackerman MJ. Clinical and genetic characteristics of long QT syndrome. Rev Esp Cardiol. 2007;60:739–752. [PubMed] [Google Scholar]
- 9.Davis RP, van den Berg CW, Casini S, Braam SR, Mummery CL. Pluripotent stem cell models of cardiac disease and their implication for drug discovery and development. Trends Mol Med. 2011;17:475–484. doi: 10.1016/j.molmed.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Matsa E, Denning C. In vitro uses of human pluripotent stem cell-derived cardiomyocytes. J Cardiovasc Trans Res. 2012;5:581–592. doi: 10.1007/s12265-012-9376-5. [DOI] [PubMed] [Google Scholar]
- 11.Matsa E, Rajamohan D, Dick E, Young L, Mellor I, Staniforth A, Denning C. Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J. 2011;32:952–962. doi: 10.1093/eurheartj/ehr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dick E, Matsa E, Bispham J, Reza M, Guglieri M, Staniforth A, Watson S, Kumari R, Lochmuller H, Young L, Darling D, Denning C. Two new protocols to enhance the production and isolation of human induced pluripotent stem cell lines. Stem Cell Res. 2011;6:158–167. doi: 10.1016/j.scr.2010.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Burridge PW, Thompson S, Millrod MA, Weinberg S, Yuan X, Peters A, Mahairaki V, Koliatsos VE, Tung L, Zambidis ET. A universal system for highly efficient cardiac differentiation of human induced pluripotent stem cells that eliminates interline variability. PLoS One. 2011;6:e18293. doi: 10.1371/journal.pone.0018293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mahlstedt MM, Anderson D, Sharp JS, McGilvray R, Barbadillo Munoz MD, Buttery LD, Alexander MR, Rose FR, Denning C. Maintenance of pluripotency in human embryonic stem cells cultured on a synthetic substrate in conditioned medium. Biotechnol Bioeng. 2010;105:130–140. doi: 10.1002/bit.22520. [DOI] [PubMed] [Google Scholar]
- 15.Moore JC, van Laake LW, Braam SR, Xue T, Tsang SY, Ward D, Passier R, Tertoolen LL, Li RA, Mummery CL. Human embryonic stem cells: genetic manipulation on the way to cardiac cell therapies. Reprod Toxicol. 2005;20:377–391. doi: 10.1016/j.reprotox.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 16.Braam SR, Denning C, Matsa E, Young LE, Passier R, Mummery CL. Feeder-free culture of human embryonic stem cells in conditioned medium for efficient genetic modification. Nat Protoc. 2008;3:1435–1443. doi: 10.1038/nprot.2008.140. [DOI] [PubMed] [Google Scholar]
- 17.Dick E, Matsa E, Young LE, Darling D, Denning C. Faster generation of hiPSCs by coupling high-titer lentivirus and column-based positive selection. Nat Protoc. 2011;6:701–714. doi: 10.1038/nprot.2011.320. [DOI] [PubMed] [Google Scholar]
- 18.Li S, Kimura E, Fall BM, Reyes M, Angello JC, Welikson R, Hauschka SD, Chamberlain JS. Stable transduction of myogenic cells with lentiviral vectors expressing a minidystrophin. Gene Ther. 2005;12:1099–1108. doi: 10.1038/sj.gt.3302505. [DOI] [PubMed] [Google Scholar]
- 19.Davis HE, Rosinski M, Morgan JR, Yarmush ML. Charged polymers modulate retrovirus transduction via membrane charge neutralization and virus aggregation. Biophys J. 2004;86:1234–1242. doi: 10.1016/S0006-3495(04)74197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang H, Qiao R, Zhao D, Zhang T, Li Y, Yi F, Lai F, Hong J, Ding X, Yang Z, Zhang L, Du Q, Liang Z. Profiling of mismatch discrimination in RNAi enabled rational design of allele-specific siRNAs. Nucleic Acids Res. 2009;37:7560–7569. doi: 10.1093/nar/gkp835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chugh SS, Senashova O, Watts A, Tran PT, Zhou Z, Gong Q, Titus JL, Hayflick SJ. Postmortem molecular screening in unexplained sudden death. J Am Coll Cardiol. 2004;43:1625–1629. doi: 10.1016/j.jacc.2003.11.052. [DOI] [PubMed] [Google Scholar]
- 22.Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ, January CT. Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking-deficient) mechanism. Circulation. 2006;113:365–373. doi: 10.1161/CIRCULATIONAHA.105.570200. [DOI] [PubMed] [Google Scholar]
- 23.Rajamani S, Anderson CL, Anson BD, January CT. Pharmacological rescue of human K(+) channel long-QT2 mutations: human ether-a-go-go-related gene rescue without block. Circulation. 2002;105:2830–2835. doi: 10.1161/01.cir.0000019513.50928.74. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Haurigot V, Doyon Y, Li T, Wong SY, Bhagwat AS, Malani N, Anguela XM, Sharma R, Ivanciu L, Murphy SL, Finn JD, Khazi FR, Zhou S, Paschon DE, Rebar EJ, Bushman FD, Gregory PD, Holmes MC, High KA. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Carroll D. Genome Engineering With Zinc-Finger Nucleases. Genetics. 2011;188:773–782. doi: 10.1534/genetics.111.131433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salama G, London B. Mouse models of long QT syndrome. J Physiol. 2007;578(Pt 1):43–53. doi: 10.1113/jphysiol.2006.118745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Maele B, De Rijck J, De Clercq E, Debyser Z. Impact of the central polypurine tract on the kinetics of human immunodeficiency virus type 1 vector transduction. J Virol. 2003;77:4685–4694. doi: 10.1128/JVI.77.8.4685-4694.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Soltan MK, Ghonaim HM, El Sadek M, Kull MA, El-aziz LA, Blagbrough IS. Design and synthesis of N4,N9-disubstituted spermines for non-viral siRNA delivery—structure-activity relationship studies of siFection efficiency versus toxicity. Pharm Res. 2009;26:286–295. doi: 10.1007/s11095-008-9731-z. [DOI] [PubMed] [Google Scholar]
- 29.Walker VE, Atanasiu R, Lam H, Shrier A. Co-chaperone FKBP38 promotes HERG trafficking. J Biol Chem. 2007;282:23509–23516. doi: 10.1074/jbc.M701006200. [DOI] [PubMed] [Google Scholar]
- 30.Vieweg WV, Wood MA. Tricyclic antidepressants, QT interval prolongation, and torsade de pointes. Psychosomatics. 2004;45:371–377. doi: 10.1176/appi.psy.45.5.371. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103:89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 32.Davis ME, Zuckerman JE, Choi CHJ, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaiser PK, Symons RC, Shah SM, Quinlan EJ, Tabandeh H, Do DV, Reisen G, Lockridge JA, Short B, Guerciolini R, Nguyen QD. RNAi-based treatment for neovascular age-related macular degeneration by Sirna-027. Am J Ophthalmol. 2010;150:33–39. doi: 10.1016/j.ajo.2010.02.006. e2. [DOI] [PubMed] [Google Scholar]
- 34.Cirak S, Feng L, Anthony K, Arechavala-Gomeza V, Torelli S, Sewry C, Morgan JE, Muntoni F. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol Ther. 2012;20:462–467. doi: 10.1038/mt.2011.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lam JK, Liang W, Chan HK. Pulmonary delivery of therapeutic siRNA. Adv Drug Deliv Rev. 2012;64:1–15. doi: 10.1016/j.addr.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arnold AS, Tang YL, Qian K, Shen L, Valencia V, Phillips MI, Zhang YC. Specific beta1-adrenergic receptor silencing with small interfering RNA lowers high blood pressure and improves cardiac function in myocardial ischemia. J Hypertens. 2007;25:197–205. doi: 10.1097/01.hjh.0000254374.73241.ab. [DOI] [PubMed] [Google Scholar]
- 37.Krebs MD, Alsberg E. Localized, Targeted, and Sustained siRNA Delivery. Vol. 17. Chemistry; 2011. pp. 3054–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Takahashi M, Watanabe S, Murata M, Furuya H, Kanazawa I, Wada K, Hohjoh H. Tailor-made RNAi knockdown against triplet repeat disease-causing alleles. Proc Natl Acad Sci U S A. 2010;107:21731–21736. doi: 10.1073/pnas.1012153107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muller GA, Hansen U, Xu Z, Griswold B, Talan MI, McDonnell NB, Briest W. Allele-specific siRNA knockdown as a personalized treatment strategy for vascular Ehlers-Danlos syndrome in human fibroblasts. FASEB J. 2012;26:668–677. doi: 10.1096/fj.11-182162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.