

Abstract
Hyperglycemia, a commonly exhibited metabolic disorder in critically ill patients, activates the body’s inflammatory defense mechanism, causing the waterfall release of numerous inflammatory mediators and cytokines, and eventually leads to organ damage. As the only glucose-lowering hormone in the body, insulin not only alleviates the detrimental effects of hyperglycemia through its metabolic regulation, but also directly modulates inflammatory mediators and acts upon immune cells to enhance immunocompetence. In this sense, hyperglycemia is pro-inflammatory whereas insulin is anti-inflammatory. Therefore, during the past 50 years, insulin has not only been used in the treatment of diabetes, but has also been put into practical use in dealing with cardiovascular diseases and critical illnesses. This review summarizes the recent advances regarding the anti-inflammatory effects of insulin in both basic research and clinical trials, with the hope of aiding in the design of further experimental research and promoting effective insulin administration in clinical practice.
Keywords: Insulin, Inflammation, Hyperglycemia
Core tip: Hyperglycemia is closely correlated with poor outcomes of morbidity and mortality in critically ill patients. As the only glucose-lowering hormone in the body, insulin not only alleviates the detrimental effects of hyperglycemia through its metabolic regulation, but also directly modulates inflammatory mediators and acts upon immune cells to enhance immunocompetence. This review summarizes the recent advances regarding the anti-inflammatory effects of insulin from our laboratory as well as others, in the hope of leading to a better understanding of this old, classic and wonder hormone and its wider and effective applications in clinical practice.
INTRODUCTION
Since its discovery in 1921, the importance of insulin in glucose homeostasis has been established, and it is universally used as a therapeutic agent for diabetes mellitus. Thousands of lives have been saved and many scientists were drawn into the study of this wonder drug. Under continuous intensive research, the mechanisms underlying the effect of insulin in its metabolic modulation, mainly glucose homeostasis, has become clearer, but there remains much interest in the elucidation of further effects of insulin.
Glucose-insulin-potassium (GIK) has been used as an adjunctive therapy in patients with acute myocardial infarction (AMI) since its introduction in 1962. However, the mechanism underlying GIK’s cardioprotection has remained largely speculative and controversial during the past 50 years. It was not until early in this century that we provided convincing in vivo evidence that insulin, rather than glucose or potassium, is the predominant protective component of GIK, and demonstrated for the first time that insulin exerted anti-apoptotic and pro-survival effects in the ischemic/reperfused (I/R) myocardium through the PI3K-Akt-eNOS-NO signaling pathway[1]. This prompted us to conceive the notion of the “survival signal”, a new mechanism of cell protection which is totally independent of the metabolic effects of insulin, and explained its cardioprotective effects. In 2001, the classical landmark clinical trial by van den Berghe[2] revealed that maintaining blood glucose at or below 110 mg/dL with low-dose insulin infusion, significantly reduced mortality and morbidity resulting from multi-organ failure among critically ill patients in the surgical intensive care unit (ICU). A further study reported that markers of inflammation, such as intercellular cell adhesion molecular-1 (ICAM-1) and E-selectin were suppressed in the liver of these patients as was inducible NO synthase (iNOS) expression, which is mainly in monocyte/macrophage cells[3], suggesting an anti-inflammatory role for insulin. This article will summarize the relationship between insulin, glucose and inflammation, and discuss the implications for the management of patients with AMI and critical illness.
GLUCOSE, OXIDATIVE STRESS AND INFLAMMATION
Hyperglycemia is common in critical illness, and may lead to severe complications. It has been reported that pronounced hyperglycemia is associated with poor outcomes of morbidity and mortality in patients with AMI, stroke and coronary artery bypass grafting[4-6]. Glucose is pro-inflammatory, and hyperglycemia is even detrimental to these patients. A total of 75 g glucose intake causes acute oxidative and inflammatory stress, as reflected in increased superoxide radical O2. generation by polymorphonuclear leukocytes, mononuclear cells and the enzyme nicotinamide adenine dinucleotide phosphate[7]. Free radical O2. generation, on the one hand, reduces NO bioavailability, as it combines with NO to form peroxynitrite ONOO-; on other hand, it activates a number of redox-sensitive major pro-inflammatory transcription factors such as nuclear factor kappa B (NFκB), activator protein-1 (AP-1), hypoxia induced factor-α (HIF-α) and early growth response-1 (Egr-1), leading to increased transcription of the pro-inflammatory genes and thus inflammation[8-10]. Meanwhile, glucose increases the expression of tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6) and monocyte chemoattractant protein-1 (MCP-1) in mononuclear cells. Moreover, it has led to increased TNF-α and IL-6 concentrations in plasma in a steady state of hyperglycemia with intravenous insulin secretion with somatostatin[11]. To sum up, glucose, oxidative stress and inflammation are inter-related, with reciprocal causation. As the only glucose-lowering hormone in the body, insulin therapy alleviates the detrimental effects of hyperglycemia through metabolic regulation, therefore hyperglycemia is pro-inflammatory whereas insulin is anti-inflammatory.
INSULIN MODULATES INFLAMMATORY MEDIATORS
The discovery of the anti-inflammatory effect of insulin can be traced back to the observation that insulin exerts a vasodilatory effect through endothelial NO release in arteries, veins and capillaries[12,13]. By inducing vasodilatation, it reduces leukocyte adhesion to the endothelium and subsequent infiltration. Furthermore, it has inhibitory effects on platelet adhesion and aggregation.
Studies have further confirmed that insulin suppressed three important inflammatory mediators: intercellular cell adhesion molecular-1 (ICAM-1), MCP-1 expression and NFκB binding in human aortic endothelial cells in vitro[14,15]. These suppressive effects can be blocked by the NOS inhibitor N(G)-nitro-L-arginine, indicating the effects are mediated by NO release. Among all the pro-inflammatory cytokines, TNF-α is the most active one in triggering the production of other cytokines such as IL-6 and other expression molecules[16]. We provided direct evidence in myocardial ischemia/reperfusion (I/R) rats that insulin inhibits TNF-α induction locally and systemically, and demonstrated for the first time that in vitro treatment with insulin attenuated I/R-induced TNF-α production in cardiomyocytes via the Akt-eNOS-NO signaling pathway[17]. Polymorphonuclear neutrophils (PMN) are the first defense line against infection and invasive microorganisms. Adherence of PMN to endothelial cells is an early requisite event in I/R-induced inflammatory injury. Thus we performed in vivo and in vitro experiments in a rabbit model to investigate whether insulin inhibits PMN-mediated adherence[18]. It was found that insulin reduced P-selectin and ICAM-1 expression in endothelium which mediates the initial interaction between PMNs and the endothelial cell surface, thus insulin attenuated PMN adherence and I/R-induced inflammatory injury. The Akt-eNOS-NO signaling pathway was involved in these effects. Moreover, insulin has been reported to ameliorate the endotoxin-induced systemic inflammatory response by decreasing IL-6, TNF-α expression and increasing the anti-inflammatory cascade in the context of normoglycemia in rat[19] and porcine models[20]. All these data indicate that insulin alleviates inflammation through suppression of pro-inflammatory cytokines and immune mediators, pointing strongly to its role as an anti-inflammatory agent.
INSULIN SUPPRESSES TOLL-LIKE RECEPTOR EXPRESSION
Toll-like receptors (TLRs) are a variety of conserved pattern recognition receptors that have been implicated in innate immune responses. Accumulating evidence suggests that TRLs play an essential role in tissue inflammation and damage such as cardiac I/R, post-ischemic remodeling and atherosclerosis[21-23]. TLR signaling and its critical roles in inflammatory cardiac conditions has been intensively studied, especially TRL2, TRL4’ role with myocardial infarction and reperfusion injury. TRL2 aggravated myocardial tissue injury in I/R-based experimental animal models and its deletion was associated with a smaller MI size compared with control[24]. The TLR-deficient model, TLR2-/- mice, exhibited improved left ventricular dysfunction following I/R[25]. Besides, administration of anti-TLR2 antibody prior to reperfusion reduced MI sites and preserved cardiac function. TLR4 is the specific receptor of endotoxin, thus it mediates inflammatory changes induced by endotoxins. Oyama et al[26] first demonstrated that TLR4-deficient mice had more than 50% reduction in MI area, which was associated with attenuated myocardial inflammation, as evidenced by less neutrophil infiltration and fewer lipid peroxides. Inhibited by eritoran, a specific TLR4 antagonist, resulted in a 40% reduction in MI and decrease in TNF-α, IL-1β, IL-6 and MCP-1 expression[27,28]. Moreover, TRL4 has been found to act as a determinant of neutrophil infiltration after global MI through mediating KC and MCP-1 expression[29]. Suppression of TRL signaling is associated with smaller MI size and is beneficial in I/R-based animal models. It has been reported that insulin infusion (2 U/h) with type 2 diabetes (T2D) patients within 2 h has significantly suppressed TLR1, -2, -4, -7 and -9 mRNA expressions in MNCs, and this prompt suppression may be mediated by the suppression of PU.1 binding and subsequent activation of TLRs[30]. Thus, insulin suppresses the expression of several TLRs at the transcriptional level and alleviates TRL-mediated inflammatory injury.
INSULIN ACTS UPON IMMUNE CELLS
Peripheral blood mononuclear cells (PBMCs) is a critical component in the immune system, and mainly comprised of lymphocytes and monocytes. Investigations have been conducted to study the effects of insulin upon mononuclear cells in obese non-diabetic subjects[31]. The results showed that insulin reduced activation of the pro-inflammatory transcription factor NFκB, with downregulation of plasma soluble intercellular adhesion molecular-1, which facilitates the attachment of monocytes to endothelial cells and chemotactic factor MCP-1, which encourages monocyte migration into the subintimal space. This suppressive effect on NFκB in PBMC has also been reported in critically ill patients with intensive insulin therapy[32]. Similarly, Egr-1, another important pro-inflammatory transcription factor, was notably reduced in mononuclear cells with insulin treatment, resulting in decreased plasma concentrations of tissue factor and plasminogen activator inhibitor-1 (PAI-1)[33]. Taken together, insulin suppresses pro-inflammatory transcription factors in mononuclear cells and the subsequent inflammatory mediators regulated by them, thus ameliorating MNC-mediated inflammation.
Monocytes/macrophages (MO/Mϕ) initiate immune and inflammatory responses. Insulin administration (10-7 mmol/L) retarded macrophage apoptosis and enhances BclXL mRNA expression by activating phosphatidylinositol 3’-kinase (PI3K) in a dose-dependent manner, thus improving macrophage survival[34]. Use of wortmannin, a specific inhibitor of PI-3K, has further confirmed its position in the anti-apoptotic effect of insulin in lipopolysaccharide-challenged THP-1 cells[35]. HLA-DR is a cluster of membrane molecules of MO/Mϕ which are involved in the MO antigen presentation to T cells. The intensity of HLA-DR expression is associated with immunocompetency of MO/Mϕ[36]. Insulin treatment with blood glucose maintained between 4.4-6.1 mmol/L increased HLA-DR expressions of peripheral MO cells. This upregulation means enhanced antigen presentation of MO cells, indicative of improved immune function. Moreover, the phagocytosis, chemotaxis, and oxidative burst capacity of MO have also been assessed in a burn-injured rabbit model, suggesting that insulin improved the capacity for phagocytosis and oxidative burst, but had no effect on chemotaxis[37].
T cell differentiation is important in the immune response. A single naïve T cell under cell differentiation is able to generate multiple subsets of memory T cells with different phenotypic and functional properties in response to infections, resulting in acquisition of immune functions required for pathogen clearance. Insulin was first confirmed to induce a shift in Th cell differentiation toward Th2 cells which is involved in secretion of inflammatory mediators (IL-4, IL-10, IL-13, etc.) and enhanced antibody-mediated responses[38]. Myocarditis is a severe disease of myocardial inflammation and often results from an autoimmune reaction. Significant T cell reduction was observed in cardiac myocarditis[39]. Thus, we investigated the effect of insulin on myocardial inflammation in experimental myocarditis in mice and its potential role in T cell regulation. The results showed that insulin promoted T cell recovery, particularly CD3+ T cells without changing the naïve-to-memory T-cell ratio and had a direct effect on T cell proliferation, thus alleviating myocarditis[40]. It is possible that insulin may promote T cell recovery in myocarditis, especially in diabetic or hyperglycemic patients.
ANTI-INFLAMMATORY EFFECTS OF INSULIN IN HUMAN STUDIES
Cardiovascular disease (CVD) is the leading cause of death worldwide, and remains a great challenge in healthcare. Various risk factors of CVD, including hypertension, diabetes and smoking, can initiate a chronic inflammatory reaction. Accumulating epidemiological and clinical studies have found strong and consistent relationships between markers of inflammation and the risk of future cardiovascular events[41]. Thus, inflammation is established as a definitive cardiovascular risk factor.
Hyperglycemia is pro-inflammatory and damaging, especially in critically ill patients. Pronounced hyperglycemia at hospital admission is associated with poor outcomes of morbidity and mortality in patients with AMI, thus effective glucose management is a necessary therapeutic intervention. It has been shown in large pilot studies, Diabetes and Insulin-Glucose Infusion in Acute Myocardial Infarction (DIGAMI)[42] and the Estudios Cardiologicos Latinoamerica (ECLA) study[43], that small doses of intravenously delivered insulin markedly improved clinical outcomes in patients with AMI. There was a marked 29% reduction in 1-year mortality in the insulin-glucose infusion group in the 1995 DIGAMI study, and a statistically significant reduction in mortality and a consistent trend toward fewer in-hospital events in the GIK group in the 1998 ECLA pilot trial, possibly as a result of rigorous glycemic control. The anti-inflammatory effect of insulin have been applied clinically. Plasma C-reactive protein (CRP) and serum amyloid A (SAA) concentrations are the two accepted markers of systemic inflammation which were impressively reduced to 40% in patients with AMI when treated with low-dose insulin infusion[44]. As the CRP concentration is correlated with the size of the infarct in AMI, a reduction is indicative of insulin’s cardioprotective effects. Moreover, intensive insulin therapy has been given to critically ill patients in surgical and medical ICUs with improved outcomes[2,45]. In 1548 critically ill patients undergoing surgery, insulin infusion which maintained fasting blood glucose concentrations under 110 mg/dL dramatically improved the clinical outcomes with a reduction in total mortality by 48%, the incidence of bacteremia by 46%, acute renal failure requiring dialysis by 41%, ICU poly-neuropathy by 44%, and the need for red cell transfusion by 50% when compared with controls[2]. Mortality and morbidity in the surgical ICUs was dramatically reduced, as was morbidity in medical ICUs. No other agent has been shown to reduce mortality and morbidity by this magnitude in so many diverse ways in the ICU setting. Glucose control seems crucial, but several potential mechanisms may add to the benefits, including reduction of systemic inflammation[46], prevention of immune dysfunction[37], and protection of the endothelium[3,47]. The exact mechanisms underlying this simple and cost-effective intervention need further investigations.
INSULIN RESISTANCE AND INFLAMMATION
Insulin resistance (IR) is a pathological condition wherein insulin-stimulated glucose uptake and clearance in targeted organs are decreased. A few studies suggested that obesity, inflammation and IR are inextricably linked through the actions of specific inflammatory immune cells. The development of IR is thought to occur in response to increased production of pro-inflammatory cytokines by adipose tissue in obesity, that then have an inhibitory effect on insulin signaling pathways in multiple tissues. TNF-α was first found to be increased in adipose tissue of obese mice and able to induce IR[48]. In animal studies, administration of exogenous TNF-α induces IR, whereas neutralization of TNF-α improves insulin sensitivity. IL-1β, another key inflammatory cytokine, interferes with insulin signaling which leads to IR. TNF-α, and more generally, inflammation, activates and increases the expression of several proteins that suppress and impair specific pathways of insulin signaling, making the human body less responsive to insulin and increasing the risk of IR. In turn, IR states are pro-inflammatory. Increased levels of markers and mediators of inflammation such as fibrinogen, CRP, IL-6, PAI-1 and white cell count were shown to correlate with T2D[49-53]. These inflammatory mediators perpetuate and promote the progression of IR. Polycystic ovary syndrome, another IR state, was found to have chronic low-grade inflammation[54]. In other words, inflammation causes IR, and IR is inflammatory. Thus, anti-inflammatory treatment could be proposed as a therapeutic strategy in the treatment of IR.
ANTI-INFLAMMATION THERAPY FOR INSULIN RESISTANCE
Inflammation is hallmark of diabetes and a main cause of its long-term complications. Particularly in obese conditions in humans and animals, it contributes to the pathogenesis of T2D through IR. Therefore, anti-inflammation therapy may be proposed as a strategy for the improvement of IR.
TNF-α is a critical mediator of inflammation, and its increased expression was found to be associated with IR in the adipose tissue of obese mice[48]. In vitro studies demonstrated that TNF-α had a direct inhibitory effect on insulin signaling and impaired insulin-stimulated glucose uptake and metabolism in human subjects[55]. Clinically, neutralization of TNF-α with infliximab in patients with rheumatoid arthritis has significantly improved IR as reflected by the significant reduction in the Homeostasis Model Assessment Index[56]. Peroxisome proliferator-activated receptors (PPAR)γ inactivation leads to suppression of IRS-2, which is a signaling molecular in insulin pathways, thus further promotes IR. The anti-diabetic thiazolidinediones (TZDs), which include pioglitazone, rosiglitazone and troglitazone, are clinically used to improve insulin sensitivity in patients with T2D by lowering free fatty acids (FFA) in blood by activating PPARγ. Aspirin, another therapeutic agent, inhibits the activity of multiple kinases induced by TNF-α, and thus enhances insulin sensitivity by protecting proteins from serine phosphorylation[57]. Statins, as a class of anti-inflammatory drugs, have been shown to downregulate transcriptional activities of NFκB, AP-1 and HIF-1α, with reductions in inflammatory cytokines[58]. Despite these modest anti-inflammatory properties, the statins do not appear to significantly influence either IR or glycemic status. In contrast, high-dose salicylates directly suppress inflammation by targeting NFκB, which improves insulin sensitivity and reduces blood glucose in patients with diabetes[59-61]. The anti-inflammatory properties of TZDs and statins have associated side effects apart from their primary modes of action, thus they may not be safe in the long term. It is necessary to investigate new classes of drugs.
Histone deacetylases (HDACs) are key enzymes that regulate gene expression. Inhibition of histone deacetylase activity has been reported as a new approach to treat diabetes mellitus. Butyrate or trichostatin A, which are histone deacetylase inhibitors, prevented high fat-induced obesity and improved IR in mice[62]. The multiple beneficial effects included: reduced systemic chronic inflammation[63-66], reduced lipid toxicity[67,68], promotion of beta-cell development, proliferation, differentiation and function[69]. Thus HDAC inhibitors may represent a novel drug in the treatment of IR.
CONCLUSION
Hyperglycemia, a commonly exhibited metabolic disorder in critically ill patients, activates the body’s inflammatory defense system, causing the cascade release of numerous inflammatory mediators and cytokines, and eventually leads to organ damage. Insulin inhibits hypermetabolism, such as hyperglycemia and lipid degradation, thus could attenuate glucose and FFA-mediated inflammation and improve immunocompetence. More importantly, insulin directly suppresses pro-inflammatory cytokines and induces anti-inflammatory mediators through non-metabolic pathways (Figure 1). Currently, the effects are dependent upon its suppression of innate immune mechanisms and the suppression of transcription factors such as NFκB and Egr-1. With further investigation, the discovery and understanding of the mechanisms underlying the anti-inflammatory effects of insulin opens up the possibility that insulin therapy could be used in multiple clinical practices.
Figure 1.
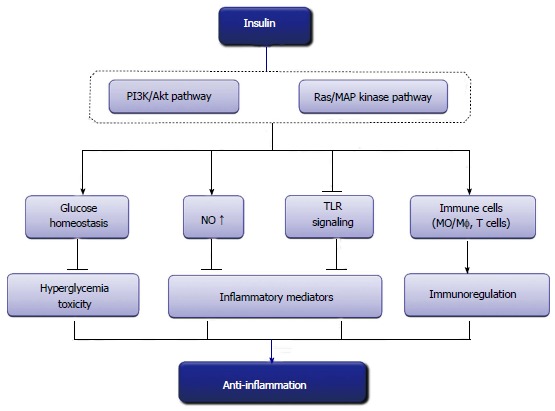
Anti-inflammatory effects of insulin.TLR: Toll-like receptor.
Hyperglycemia, inflammation and IR are inter-related and of reciprocal causation. The relationships between the three entities are far from being elucidated. Hyperglycemia leads to oxidative stress, which further results in inflammation. IR, commonly as a manifestation of hyperglycemia, is pro-inflammatory. Reactive oxygen species is believed to be an important cause of many pathological conditions, including inflammation and IR. It has been established that hyperglycemia is inflammatory whereas insulin is anti-inflammatory. From simple glucose maintenance to the discovery of cardiovascular protection, the knowledge and understanding about insulin is increasing. The pleiotropic effects of insulin including glucose control, and reduction in apoptosis, oxidative/nitrative stress and inflammation, contribute to cardiovascular protection and are beneficial in critical illness. It is not a single effect that mediates the important role of insulin, but it is the whole scenario that promotes its myriad effects. With consistent research, we will gain a better understanding of these working mechanisms, and in doing so, are likely to find more therapeutic targets and wider applications for this wonder drug.
Footnotes
P- Reviewers: Collino M, Gómez-Sáez J, Kumar K S- Editor: Zhai HH L- Editor: Cant MR E- Editor: Wu HL
Supported by The State Key Program of National Natural Science Foundation of China, No. 81030005; The National High Technology Research and Development Program of China, No. 2014AA020542; and National Natural Science Foundation of China, No. 31371150
References
- 1.Gao F, Gao E, Yue TL, Ohlstein EH, Lopez BL, Christopher TA, Ma XL. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: the roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation. 2002;105:1497–1502. doi: 10.1161/01.cir.0000012529.00367.0f. [DOI] [PubMed] [Google Scholar]
- 2.van den Berghe G, Wouters P, Weekers F, Verwaest C, Bruyninckx F, Schetz M, Vlasselaers D, Ferdinande P, Lauwers P, Bouillon R. Intensive insulin therapy in critically ill patients. N Engl J Med. 2001;345:1359–1367. doi: 10.1056/NEJMoa011300. [DOI] [PubMed] [Google Scholar]
- 3.Langouche L, Vanhorebeek I, Vlasselaers D, Vander Perre S, Wouters PJ, Skogstrand K, Hansen TK, Van den Berghe G. Intensive insulin therapy protects the endothelium of critically ill patients. J Clin Invest. 2005;115:2277–2286. doi: 10.1172/JCI25385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Capes SE, Hunt D, Malmberg K, Gerstein HC. Stress hyperglycaemia and increased risk of death after myocardial infarction in patients with and without diabetes: a systematic overview. Lancet. 2000;355:773–778. doi: 10.1016/S0140-6736(99)08415-9. [DOI] [PubMed] [Google Scholar]
- 5.Capes SE, Hunt D, Malmberg K, Pathak P, Gerstein HC. Stress hyperglycemia and prognosis of stroke in nondiabetic and diabetic patients: a systematic overview. Stroke. 2001;32:2426–2432. doi: 10.1161/hs1001.096194. [DOI] [PubMed] [Google Scholar]
- 6.Furnary AP, Gao G, Grunkemeier GL, Wu Y, Zerr KJ, Bookin SO, Floten HS, Starr A. Continuous insulin infusion reduces mortality in patients with diabetes undergoing coronary artery bypass grafting. J Thorac Cardiovasc Surg. 2003;125:1007–1021. doi: 10.1067/mtc.2003.181. [DOI] [PubMed] [Google Scholar]
- 7.Mohanty P, Hamouda W, Garg R, Aljada A, Ghanim H, Dandona P. Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. J Clin Endocrinol Metab. 2000;85:2970–2973. doi: 10.1210/jcem.85.8.6854. [DOI] [PubMed] [Google Scholar]
- 8.Dandona P, Chaudhuri A, Ghanim H, Mohanty P. Proinflammatory effects of glucose and anti-inflammatory effect of insulin: relevance to cardiovascular disease. Am J Cardiol. 2007;99:15B–26B. doi: 10.1016/j.amjcard.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Dhindsa S, Tripathy D, Mohanty P, Ghanim H, Syed T, Aljada A, Dandona P. Differential effects of glucose and alcohol on reactive oxygen species generation and intranuclear nuclear factor-kappaB in mononuclear cells. Metabolism. 2004;53:330–334. doi: 10.1016/j.metabol.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 10.Aljada A, Friedman J, Ghanim H, Mohanty P, Hofmeyer D, Chaudhuri A, Dandona P. Glucose ingestion induces an increase in intranuclear nuclear factor kappaB, a fall in cellular inhibitor kappaB, and an increase in tumor necrosis factor alpha messenger RNA by mononuclear cells in healthy human subjects. Metabolism. 2006;55:1177–1185. doi: 10.1016/j.metabol.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 11.Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, Ciotola M, Quagliaro L, Ceriello A, Giugliano D. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation. 2002;106:2067–2072. doi: 10.1161/01.cir.0000034509.14906.ae. [DOI] [PubMed] [Google Scholar]
- 12.Grover A, Padginton C, Wilson MF, Sung BH, Izzo JL, Dandona P. Insulin attenuates norepinephrine-induced venoconstriction. An ultrasonographic study. Hypertension. 1995;25:779–784. doi: 10.1161/01.hyp.25.4.779. [DOI] [PubMed] [Google Scholar]
- 13.Steinberg HO, Brechtel G, Johnson A, Fineberg N, Baron AD. Insulin-mediated skeletal muscle vasodilation is nitric oxide dependent. A novel action of insulin to increase nitric oxide release. J Clin Invest. 1994;94:1172–1179. doi: 10.1172/JCI117433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aljada A, Ghanim H, Saadeh R, Dandona P. Insulin inhibits NFkappaB and MCP-1 expression in human aortic endothelial cells. J Clin Endocrinol Metab. 2001;86:450–453. doi: 10.1210/jcem.86.1.7278. [DOI] [PubMed] [Google Scholar]
- 15.Aljada A, Saadeh R, Assian E, Ghanim H, Dandona P. Insulin inhibits the expression of intercellular adhesion molecule-1 by human aortic endothelial cells through stimulation of nitric oxide. J Clin Endocrinol Metab. 2000;85:2572–2575. doi: 10.1210/jcem.85.7.6677. [DOI] [PubMed] [Google Scholar]
- 16.Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996;334:1717–1725. doi: 10.1056/NEJM199606273342607. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Zhang H, Wu F, Nan Y, Ma H, Guo W, Wang H, Ren J, Das UN, Gao F. Insulin inhibits tumor necrosis factor-alpha induction in myocardial ischemia/reperfusion: role of Akt and endothelial nitric oxide synthase phosphorylation. Crit Care Med. 2008;36:1551–1558. doi: 10.1097/CCM.0b013e3181782335. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Wu F, Zhang H, Fu F, Ji L, Dong L, Li Q, Liu W, Zhang Y, Lv A, et al. Insulin inhibits leukocyte-endothelium adherence via an Akt-NO-dependent mechanism in myocardial ischemia/reperfusion. J Mol Cell Cardiol. 2009;47:512–519. doi: 10.1016/j.yjmcc.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 19.Jeschke MG, Klein D, Bolder U, Einspanier R. Insulin attenuates the systemic inflammatory response in endotoxemic rats. Endocrinology. 2004;145:4084–4093. doi: 10.1210/en.2004-0592. [DOI] [PubMed] [Google Scholar]
- 20.Brix-Christensen V, Andersen SK, Andersen R, Mengel A, Dyhr T, Andersen NT, Larsson A, Schmitz O, Ørskov H, Tønnesen E. Acute hyperinsulinemia restrains endotoxin-induced systemic inflammatory response: an experimental study in a porcine model. Anesthesiology. 2004;100:861–870. doi: 10.1097/00000542-200404000-00016. [DOI] [PubMed] [Google Scholar]
- 21.Chao W. Toll-like receptor signaling: a critical modulator of cell survival and ischemic injury in the heart. Am J Physiol Heart Circ Physiol. 2009;296:H1–12. doi: 10.1152/ajpheart.00995.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Michelsen KS, Arditi M. Toll-like receptor signaling and atherosclerosis. Curr Opin Hematol. 2006;13:163–168. doi: 10.1097/01.moh.0000219662.88409.7c. [DOI] [PubMed] [Google Scholar]
- 23.Vallejo JG. Role of toll-like receptors in cardiovascular diseases. Clin Sci (Lond) 2011;121:1–10. doi: 10.1042/CS20100539. [DOI] [PubMed] [Google Scholar]
- 24.Shishido T, Nozaki N, Takahashi H, Arimoto T, Niizeki T, Koyama Y, Abe J, Takeishi Y, Kubota I. Central role of endogenous Toll-like receptor-2 activation in regulating inflammation, reactive oxygen species production, and subsequent neointimal formation after vascular injury. Biochem Biophys Res Commun. 2006;345:1446–1453. doi: 10.1016/j.bbrc.2006.05.056. [DOI] [PubMed] [Google Scholar]
- 25.Sakata Y, Dong JW, Vallejo JG, Huang CH, Baker JS, Tracey KJ, Tacheuchi O, Akira S, Mann DL. Toll-like receptor 2 modulates left ventricular function following ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2007;292:H503–H509. doi: 10.1152/ajpheart.00642.2006. [DOI] [PubMed] [Google Scholar]
- 26.Oyama J, Blais C, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–789. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 27.Chong AJ, Shimamoto A, Hampton CR, Takayama H, Spring DJ, Rothnie CL, Yada M, Pohlman TH, Verrier ED. Toll-like receptor 4 mediates ischemia/reperfusion injury of the heart. J Thorac Cardiovasc Surg. 2004;128:170–179. doi: 10.1016/j.jtcvs.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 28.Shimamoto A, Chong AJ, Yada M, Shomura S, Takayama H, Fleisig AJ, Agnew ML, Hampton CR, Rothnie CL, Spring DJ, et al. Inhibition of Toll-like receptor 4 with eritoran attenuates myocardial ischemia-reperfusion injury. Circulation. 2006;114:I270–I274. doi: 10.1161/CIRCULATIONAHA.105.000901. [DOI] [PubMed] [Google Scholar]
- 29.Ao L, Zou N, Cleveland JC, Fullerton DA, Meng X. Myocardial TLR4 is a determinant of neutrophil infiltration after global myocardial ischemia: mediating KC and MCP-1 expression induced by extracellular HSC70. Am J Physiol Heart Circ Physiol. 2009;297:H21–H28. doi: 10.1152/ajpheart.00292.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghanim H, Mohanty P, Deopurkar R, Sia CL, Korzeniewski K, Abuaysheh S, Chaudhuri A, Dandona P. Acute modulation of toll-like receptors by insulin. Diabetes Care. 2008;31:1827–1831. doi: 10.2337/dc08-0561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dandona P, Aljada A, Mohanty P, Ghanim H, Hamouda W, Assian E, Ahmad S. Insulin inhibits intranuclear nuclear factor kappaB and stimulates IkappaB in mononuclear cells in obese subjects: evidence for an anti-inflammatory effect? J Clin Endocrinol Metab. 2001;86:3257–3265. doi: 10.1210/jcem.86.7.7623. [DOI] [PubMed] [Google Scholar]
- 32.Ma C, Liu WY, Cui Q, Gu CH, Dou YW, Zhao R, Chen M, Zheng X. [Effects of intensive insulin therapy on plasma nitric oxide and endothelin-1 levels in patients undergoing cardiac surgery under cardiopulmonary bypass] Zhonghua Waike Zazhi. 2008;46:443–445. [PubMed] [Google Scholar]
- 33.Aljada A, Ghanim H, Mohanty P, Kapur N, Dandona P. Insulin inhibits the pro-inflammatory transcription factor early growth response gene-1 (Egr)-1 expression in mononuclear cells (MNC) and reduces plasma tissue factor (TF) and plasminogen activator inhibitor-1 (PAI-1) concentrations. J Clin Endocrinol Metab. 2002;87:1419–1422. doi: 10.1210/jcem.87.3.8462. [DOI] [PubMed] [Google Scholar]
- 34.Iida KT, Suzuki H, Sone H, Shimano H, Toyoshima H, Yatoh S, Asano T, Okuda Y, Yamada N. Insulin inhibits apoptosis of macrophage cell line, THP-1 cells, via phosphatidylinositol-3-kinase-dependent pathway. Arterioscler Thromb Vasc Biol. 2002;22:380–386. doi: 10.1161/hq0302.105272. [DOI] [PubMed] [Google Scholar]
- 35.Leffler M, Hrach T, Stuerzl M, Horch RE, Herndon DN, Jeschke MG. Insulin attenuates apoptosis and exerts anti-inflammatory effects in endotoxemic human macrophages. J Surg Res. 2007;143:398–406. doi: 10.1016/j.jss.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 36.Yoshida S. [Monocyte HLA-DR expression as predictors of clinical outcome for patients with sepsis] Nihon Rinsho. 2004;62:2281–2284. [PubMed] [Google Scholar]
- 37.Weekers F, Giulietti AP, Michalaki M, Coopmans W, Van Herck E, Mathieu C, Van den Berghe G. Metabolic, endocrine, and immune effects of stress hyperglycemia in a rabbit model of prolonged critical illness. Endocrinology. 2003;144:5329–5338. doi: 10.1210/en.2003-0697. [DOI] [PubMed] [Google Scholar]
- 38.Viardot A, Grey ST, Mackay F, Chisholm D. Potential antiinflammatory role of insulin via the preferential polarization of effector T cells toward a T helper 2 phenotype. Endocrinology. 2007;148:346–353. doi: 10.1210/en.2006-0686. [DOI] [PubMed] [Google Scholar]
- 39.Kishimoto C, Kuribayashi K, Fukuma K, Masuda T, Tomioka N, Abelmann WH, Kawai C. Immunologic identification of lymphocyte subsets in experimental murine myocarditis with encephalomyocarditis virus. Different kinetics of lymphocyte subsets between the heart and the peripheral blood, and significance of Thy 1.2+ (pan T) and Lyt 1+, 23+ (immature T) subsets in the development of myocarditis. Circ Res. 1987;61:715–725. doi: 10.1161/01.res.61.5.715. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Zhuang R, Geng C, Cai X, Lei W, Tian N, Gao F. Insulin promotes T cell recovery in a murine model of autoimmune myocarditis. Clin Exp Immunol. 2013;171:46–53. doi: 10.1111/j.1365-2249.2012.04662.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tousoulis D, Antoniades C, Koumallos N, Stefanadis C. Pro-inflammatory cytokines in acute coronary syndromes: from bench to bedside. Cytokine Growth Factor Rev. 2006;17:225–233. doi: 10.1016/j.cytogfr.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 42.Malmberg K, Rydén L, Hamsten A, Herlitz J, Waldenström A, Wedel H. Mortality prediction in diabetic patients with myocardial infarction: experiences from the DIGAMI study. Cardiovasc Res. 1997;34:248–253. doi: 10.1016/s0008-6363(96)00263-5. [DOI] [PubMed] [Google Scholar]
- 43.Díaz R, Paolasso EA, Piegas LS, Tajer CD, Moreno MG, Corvalán R, Isea JE, Romero G. Metabolic modulation of acute myocardial infarction. The ECLA (Estudios Cardiológicos Latinoamérica) Collaborative Group. Circulation. 1998;98:2227–2234. doi: 10.1161/01.cir.98.21.2227. [DOI] [PubMed] [Google Scholar]
- 44.Chaudhuri A, Janicke D, Wilson MF, Tripathy D, Garg R, Bandyopadhyay A, Calieri J, Hoffmeyer D, Syed T, Ghanim H, et al. Anti-inflammatory and profibrinolytic effect of insulin in acute ST-segment-elevation myocardial infarction. Circulation. 2004;109:849–854. doi: 10.1161/01.CIR.0000116762.77804.FC. [DOI] [PubMed] [Google Scholar]
- 45.Aberegg SK. Intensive insulin therapy in the medical ICU. N Engl J Med. 2006;354:2069–2071; author reply 2069-2071;. [PubMed] [Google Scholar]
- 46.Hansen TK, Thiel S, Wouters PJ, Christiansen JS, Van den Berghe G. Intensive insulin therapy exerts antiinflammatory effects in critically ill patients and counteracts the adverse effect of low mannose-binding lectin levels. J Clin Endocrinol Metab. 2003;88:1082–1088. doi: 10.1210/jc.2002-021478. [DOI] [PubMed] [Google Scholar]
- 47.Van den Berghe G. How does blood glucose control with insulin save lives in intensive care? J Clin Invest. 2004;114:1187–1195. doi: 10.1172/JCI23506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt MI, Duncan BB, Sharrett AR, Lindberg G, Savage PJ, Offenbacher S, Azambuja MI, Tracy RP, Heiss G. Markers of inflammation and prediction of diabetes mellitus in adults (Atherosclerosis Risk in Communities study): a cohort study. Lancet. 1999;353:1649–1652. doi: 10.1016/s0140-6736(99)01046-6. [DOI] [PubMed] [Google Scholar]
- 50.Duncan BB, Schmidt MI, Pankow JS, Ballantyne CM, Couper D, Vigo A, Hoogeveen R, Folsom AR, Heiss G. Low-grade systemic inflammation and the development of type 2 diabetes: the atherosclerosis risk in communities study. Diabetes. 2003;52:1799–1805. doi: 10.2337/diabetes.52.7.1799. [DOI] [PubMed] [Google Scholar]
- 51.Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 52.Barzilay JI, Abraham L, Heckbert SR, Cushman M, Kuller LH, Resnick HE, Tracy RP. The relation of markers of inflammation to the development of glucose disorders in the elderly: the Cardiovascular Health Study. Diabetes. 2001;50:2384–2389. doi: 10.2337/diabetes.50.10.2384. [DOI] [PubMed] [Google Scholar]
- 53.Vozarova B, Weyer C, Lindsay RS, Pratley RE, Bogardus C, Tataranni PA. High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes. 2002;51:455–461. doi: 10.2337/diabetes.51.2.455. [DOI] [PubMed] [Google Scholar]
- 54.González F. Inflammation in Polycystic Ovary Syndrome: underpinning of insulin resistance and ovarian dysfunction. Steroids. 2012;77:300–305. doi: 10.1016/j.steroids.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, Pedersen BK. Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes. 2005;54:2939–2945. doi: 10.2337/diabetes.54.10.2939. [DOI] [PubMed] [Google Scholar]
- 56.Tam LS, Tomlinson B, Chu TT, Li TK, Li EK. Impact of TNF inhibition on insulin resistance and lipids levels in patients with rheumatoid arthritis. Clin Rheumatol. 2007;26:1495–1498. doi: 10.1007/s10067-007-0539-8. [DOI] [PubMed] [Google Scholar]
- 57.Gao Z, Zuberi A, Quon MJ, Dong Z, Ye J. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in tumor necrosis factor-treated cells through targeting multiple serine kinases. J Biol Chem. 2003;278:24944–24950. doi: 10.1074/jbc.M300423200. [DOI] [PubMed] [Google Scholar]
- 58.Castrillo A, Tontonoz P. Nuclear receptors in macrophage biology: at the crossroads of lipid metabolism and inflammation. Annu Rev Cell Dev Biol. 2004;20:455–480. doi: 10.1146/annurev.cellbio.20.012103.134432. [DOI] [PubMed] [Google Scholar]
- 59.Kopp E, Ghosh S. Inhibition of NF-kappa B by sodium salicylate and aspirin. Science. 1994;265:956–959. doi: 10.1126/science.8052854. [DOI] [PubMed] [Google Scholar]
- 60.Williamson RT. On the Treatment of Glycosuria and Diabetes Mellitus with Sodium Salicylate. Br Med J. 1901;1:760–762. doi: 10.1136/bmj.1.2100.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. 1998;396:77–80. doi: 10.1038/23948. [DOI] [PubMed] [Google Scholar]
- 62.Gao Z, Yin J, Zhang J, Ward RE, Martin RJ, Lefevre M, Cefalu WT, Ye J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes. 2009;58:1509–1517. doi: 10.2337/db08-1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Adcock IM, Ito K, Barnes PJ. Histone deacetylation: an important mechanism in inflammatory lung diseases. COPD. 2005;2:445–455. doi: 10.1080/15412550500346683. [DOI] [PubMed] [Google Scholar]
- 64.Blanchard F, Chipoy C. Histone deacetylase inhibitors: new drugs for the treatment of inflammatory diseases? Drug Discov Today. 2005;10:197–204. doi: 10.1016/S1359-6446(04)03309-4. [DOI] [PubMed] [Google Scholar]
- 65.Dinarello CA. Inhibitors of histone deacetylases as anti-inflammatory drugs. Ernst Schering Res Found Workshop. 2006:45–60. doi: 10.1007/3-540-37673-9_3. [DOI] [PubMed] [Google Scholar]
- 66.Zhang L, Fang H, Xu W. Strategies in developing promising histone deacetylase inhibitors. Med Res Rev. 2010;30:585–602. doi: 10.1002/med.20169. [DOI] [PubMed] [Google Scholar]
- 67.Fajas L, Egler V, Reiter R, Hansen J, Kristiansen K, Debril MB, Miard S, Auwerx J. The retinoblastoma-histone deacetylase 3 complex inhibits PPARgamma and adipocyte differentiation. Dev Cell. 2002;3:903–910. doi: 10.1016/s1534-5807(02)00360-x. [DOI] [PubMed] [Google Scholar]
- 68.Zhang J, Henagan TM, Gao Z, Ye J. Inhibition of glyceroneogenesis by histone deacetylase 3 contributes to lipodystrophy in mice with adipose tissue inflammation. Endocrinology. 2011;152:1829–1838. doi: 10.1210/en.2010-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Christensen DP, Dahllöf M, Lundh M, Rasmussen DN, Nielsen MD, Billestrup N, Grunnet LG, Mandrup-Poulsen T. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol Med. 2011;17:378–390. doi: 10.2119/molmed.2011.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]