

1. DISEASE CHARACTERISTICS
1.1 Name of the disease (synonyms)
Cystinosis.
1.2 OMIM# of the disease
Nephropathic infantile form (MIM #219800), nephropathic juvenile form (MIM #219900) and non-nephropathic adult form (MIM #219750).
1.3 Name of the analysed genes or DNA/chromosome segments
CTNS, 17p13.2.
1.4 OMIM# of the gene(s)
606272.
1.5 Mutational spectrum
Multi-exon 57-kb deletion, small insertions, deletions, duplications, point mutations (missense, nonsense), splice-site mutations, promoter mutations, genomic rearrangements.1, 2, 3, 4, 5, 6
The 57-kb deletion is detected in up to 76% of affected northern European alleles and is due to a founder effect arising around the middle of the first millennium AD.1, 2
Currently, approximately 100 mutations in the CTNS are described by HGMD (http://www.hgmd.cf.ac.uk/ac/index.php), however, novel mutations are still being reported, especially when genetically different populations are tested.7, 8
The standard reference sequence indicating reported variants (ENSG00000040531) and a reference for exon numbering (ENST00000046640) can be found at www.ensembl.org.
1.6 Analytical methods
The 57-kb deletion: rapid PCR assay with the 57-kb deletion breakpoint primer sets, FISH analysis, and MLPA typing. Sanger sequencing of the total coding region, the exon-intron boundaries, and promoter region of the CTNS gene. Analysis of microsatellite markers for detection of uniparental heterodisomy of the chromosome 17.6
1.7 Analytical validation
The segregation of the mutations should be confirmed in the parents. Pathogenicity of novel missense variants has to be verified by testing a set of at least 100 control chromosomes of the same ethnic origin and by in-silico prediction methods (like SIFT, PolyPhen and/or Align). CTNS gene transcripts should be analysed in patients with exon-skipping or splice-site mutations.9 Analysis of functional consequences of the CTNS mutation on protein level in cell models is available in a few laboratories in the world.
1.8 Estimated frequency of the disease (Incidence at birth (‘birth prevalence') or population prevalence. If known to be variable between ethnic groups, please report)
Birth prevalence: 1:100 000 to 1:200 000. Increased incidence in some populations: for example Brittany, France: 1:26 000.
1.9 If applicable, prevalence in the ethnic group of the investigated person
Not applicable.
1.10 Diagnostic setting

Comment:
The common 57-kb deletion encompasses the first nine exons and a part of exon 10 of the CTNS gene and the upstream 5′ region that encodes for the CARKL gene and the first two non-coding alternative exons of the transient receptor potential vanilloid 1 receptor (TRPV1) gene.
So far, there is no evidence confirming pathological involvement of the CARKL and the TRPV1 genes in the clinical phenotype of cystinosis patients.
The CARKL gene encodes for the enzyme sedoheptulose kinase responsible for the phosphorylation of sedoheptulose into sedoheptulose-7-phosphate within the pentose phosphate pathway. Patients carrying the common 57-kb deletion accumulate sedoheptulose. Increased concentration of sedoheptulose in blood or urine can be used for identifying patients with the homozygous 57-kb deletion.10, 11
The TRPV1 gene encodes for the TRPV1 expressed in the primary sensory neurons and activated by heat and various chemical compounds, including capsaicin.12 Two first non-coding exons of the TRPV1 are removed by the 57-kb deletion. Hypothetically, the reduced sweating and crafting for spicy food described in cystinosis patients might be attributed to a decreased function of the TRPPV1 receptor.
2. TEST CHARACTERISTICS
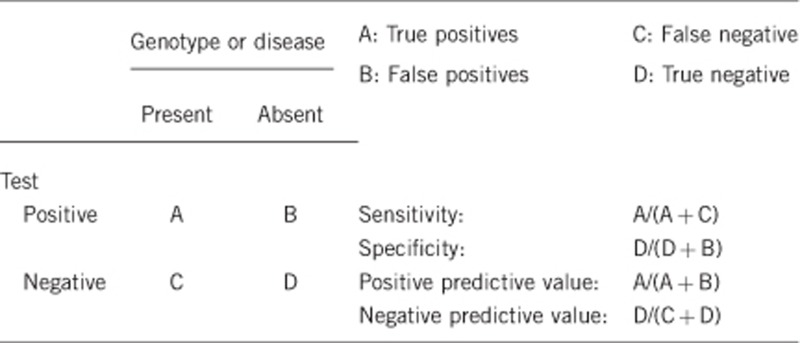
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Close to 100%.
Not all found variants have been tested functionally,13 and therefore a theoretical possibility exists that some of them might be rare polymorphisms. The pathogenicity of most mutants has been confirmed by in-silico prediction methods.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
Close to 100%.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)The clinical sensitivity can be dependent on variable factors, such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Close to 100%.
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)The clinical specificity can be dependent on variable factors, such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
Close to 100%.
2.5 Positive clinical predictive value
(life-time risk to develop the disease if the test is positive)
100%.
2.6 Negative clinical predictive value
(Probability of not developing the disease if the test is negative) Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
100%.
Index case in that family had not been tested:
Depending on the degree of relationship and consanguinity (25% risk for developing disease in siblings). Less than 1% chance to develop the disease in other relatives in populations with a low degree of consanguinity.
3. CLINICAL UTILITY
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.9 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2 Describe the burden of alternative diagnostic methods to the patient
In the majority of cases, the diagnosis of cystinosis is based on the high degree of clinical suspicion (presence of renal Fanconi syndrome, corneal cystine accumulation) and on the biochemical detection of elevated cystine levels in white blood cells (WBC) or preferentially in polymorphonuclear cells having a higher degree of cystine accumulation.
Biochemical methods of cystine measurement (high-performance liquid chromatography or tandem mass spectrometry) are time consuming, require rapid blood processing for cell isolation and can only be performed in specialised laboratories experienced in this measurement.
Young children may have rather low cystine accumulation in WBC and low amount of corneal cystine crystals, complicating the diagnosis of cystinosis shortly after birth and during the first months of life.
Corneal crystals may be absent until 16 months of age.16
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
The cost effectiveness of clinical eye investigation and biochemical cystine measurements is very high.
The differences in price between tests depend on the laboratory and the country where testing is performed. Clinical eye examination is always cheaper compared with biochemical or genetic testing, but it is not contributive in patients below 1–2 years of age.16
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive Setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.9 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe):
In affected persons cysteamine therapy should be started immediately after the diagnosis. Early therapy postpones renal failure and the development of extra-renal manifestations of cystinosis.
If the test result is negative (please describe)
No further medical follow-up will be required in subjects from families with a known genetic defect. In case of an unknown mutation in the family, at-risk individuals should be followed medically.
3.2.2 Which options in view of lifestyle and prevention does a person at risk have if no genetic test has been done (please describe)?
No life-style adaptations or other type of prevention can be applied. No medical follow-up will be required in persons with a negative genetic test within a family with identified mutations.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.9 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
Yes. If the index case has known mutations, siblings, parents and other family members can be screened for disease or carriership.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes. Genetic test results in the index case allow prenatal diagnosis in the next siblings. In populations with a high degree of consanguinity, carriership in partners can be tested as well.
3.4 Prenatal diagnosis
(To be answered if in 1.9 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Yes.
4. IF APPLICABLE, FURTHER CONSEQUENCES OF TESTING
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Yes, for performing genetic counselling in a family.
In general, there is no consensus on genetic testing of partners of affected female patients planning pregnancies. Some genetic laboratories recommend to screen partners for the mutations common in their population (for example: for 57-kb deletion in the Northern European population). Male cystinosis patients are infertile.23
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics. E Levtchenko is supported by the Fund for Scientific Research, Flanders (Belgium) (FWO Vlaanderen) (Grant Agreement 1801110N). E Levtchenko, L van den Heuvel, F Emma and C Antignac are supported by the Cystinosis Research Foundation, USA.
The authors declare no conflict of interest.
References
- Town M, Jean G, Cherqui S, et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat Genet. 1998;18:319–324. doi: 10.1038/ng0498-319. [DOI] [PubMed] [Google Scholar]
- Forestier L, Jean G, Attard M, et al. Molecular characterization of CTNS deletions in nephropathic cystinosis: development of a PCR-based detection assay. Am J Hum Genet. 1999;65:353–359. doi: 10.1086/302509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalatzis V, Cohen-Solal L, Cordier B, et al. Identification of 14 novel CTNS mutations and characterization of seven splice site mutations associated with cystinosis. Hum Mutat. 2002;20:439–446. doi: 10.1002/humu.10141. [DOI] [PubMed] [Google Scholar]
- Mason S, Pepe G, Dall'Amico R, et al. Mutational spectrum of the CTNS gene in Italy. Eur J Hum Genet. 2003;11:503–508. doi: 10.1038/sj.ejhg.5200993. [DOI] [PubMed] [Google Scholar]
- Phornphutkul C, Anikster Y, Huizing M, et al. The promoter of a lysosomal membrane transporter gene, CTNS, binds Sp-1, shares sequences with the promoter of an adjacent gene, CARKL, and causes cystinosis if mutated in a critical region. Am J Hum Genet. 2001;69:712–721. doi: 10.1086/323484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebre AS, Morinière V, Dunand O, Bensman A, Morichon-Delvallez N, Antignac C. Maternal uniparental heterodisomy of chromosome 17 in a patient with nephropathic cystinosis. Eur J Hum Genet. 2009;17:1019–1023. doi: 10.1038/ejhg.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topaloglu R, Vilboux T, Coskun T, et al. Genetic basis of cystinosis in Turkish patients: a single-center experience. Pediatr Nephrol. 2012;27:115–121. doi: 10.1007/s00467-011-1942-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahkarami S, Galehdari H, Ahmadzadeh A, et al. The first molecular genetics analysis of individuals suffering from nephropatic cystinosis in the Southwestern Iran. Nefrologia. 2013;33:308–315. doi: 10.3265/Nefrologia.pre2012.Sep.11558. [DOI] [PubMed] [Google Scholar]
- Taranta A, Wilmer MJ, van den Heuvel LP, et al. Analysis of CTNS gene transcripts in nephropathic cystinosis. Pediatr Nephrol. 2010;25:1263–1267. doi: 10.1007/s00467-010-1502-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamelink MM, Struys EA, Jansen EE, et al. Sedoheptulokinase deficiency due to a 57-kb deletion in cystinosis patients causes urinary accumulation of sedoheptulose: elucidation of the CARKL gene. Hum Mutat. 2008;29:532–536. doi: 10.1002/humu.20685. [DOI] [PubMed] [Google Scholar]
- Wamelink MM, Struys EA, Jansen EE, et al. Elevated concentrations of sedoheptulose in bloodspots of patients with cystinosis caused by the 57-kb deletion: implications for diagnostics and neonatal screening. Mol Genet Metab. 2011;102:339–342. doi: 10.1016/j.ymgme.2010.12.002. [DOI] [PubMed] [Google Scholar]
- Gees M, Owsianik G, Nilius B, Voets TTRP. Channels. Compr Physiol. 2012;2:563–608. doi: 10.1002/cphy.c110026. [DOI] [PubMed] [Google Scholar]
- Kalatzis V, Nevo N, Cherqui S, Gasnier B, Antignac C. Molecular pathogenesis of cystinosis: effect of CTNS mutations on the transport activity and subcellular localization of cystinosin. Hum Mol Genet. 2004;13:1361–1371. doi: 10.1093/hmg/ddh152. [DOI] [PubMed] [Google Scholar]
- Labbé A, Niaudet P, Loirat C, Charbit M, Guest G, Baudouin C. In vivo confocal microscopy and anterior segment optical coherence tomography analysis of the cornea in nephropathic cystinosis. Ophthalmology. 2009;116:870–876. doi: 10.1016/j.ophtha.2008.11.021. [DOI] [PubMed] [Google Scholar]
- Chiavérini C, Kang HY, Sillard L, et al. In vivo reflectance confocal microscopy of the skin: a noninvasive means of assessing body cystine accumulation in infantile cystinosis. J Am Acad Dermatol. 2013;68:e111–e116. doi: 10.1016/j.jaad.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Kuehl EM, Iwata F, Lindblad A, Kaiser-Kupfer MI. Corneal crystals in nephropathic cystinosis: natural history and treatment with cysteamine eyedrops. Mol Genet Metab. 2000;71:100–120. doi: 10.1006/mgme.2000.3062. [DOI] [PubMed] [Google Scholar]
- Markello TC, Bernardini IM, Gahl WA. Improved renal function in children with cystinosis treated with cysteamine. N Engl J Med. 1993;328:1157–1162. doi: 10.1056/NEJM199304223281604. [DOI] [PubMed] [Google Scholar]
- Greco M, Brugnara M, Zaffanello M, Taranta A, Pastore A, Emma F. Long-term outcome of nephropathic cystinosis: a 20-year single-center experience. Pediatr Nephrol. 2010;25:2459–2467. doi: 10.1007/s00467-010-1641-8. [DOI] [PubMed] [Google Scholar]
- Nesterova G, Gahl W. Nephropathic cystinosis: late complications of a multisystemic disease. Pediatr Nephrol. 2008;23:863–878. doi: 10.1007/s00467-007-0650-8. [DOI] [PubMed] [Google Scholar]
- Kleta R, Kaskel F, Dohil R, et al. First NIH/Office of Rare Diseases Conference on Cystinosis: past, present, and future. Pediatr Nephrol. 2005;20:452–454. doi: 10.1007/s00467-004-1777-5. [DOI] [PubMed] [Google Scholar]
- Van Stralen KJ, Emma F, Jager KJ, et al. Improvement in the renal prognosis in nephropathic cystinosis. Clin J Am Soc Nephrol. 2011;6:2485–2491. doi: 10.2215/CJN.02000311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmer MJ, Schoeber JP, van den Heuvel LP, Levtchenko EN. Cystinosis: practical tools for diagnosis and treatment. Pediatr Nephrol. 2011;26:205–215. doi: 10.1007/s00467-010-1627-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besouw MT, Kremer JA, Janssen MC, Levtchenko EN. Fertility status in male cystinosis patients treated with cysteamine. Fertil Steril. 2010;93:1880–1883. doi: 10.1016/j.fertnstert.2008.12.113. [DOI] [PubMed] [Google Scholar]