

Abstract
Cardiomyocyte cell division and replication in mammals proceed through embryonic development and abruptly decline soon after birth. The process governing cardiomyocyte cell cycle arrest is poorly understood. Here we carry out whole exome sequencing in an infant with evidence of persistent postnatal cardiomyocyte replication to determine the genetic risk factors. We identify compound heterozygous ALMS1 mutations in the proband, and confirm their presence in her affected sibling, one copy inherited from each heterozygous parent. Next, we recognise homozygous or compound heterozygous truncating mutations in ALMS1 in four other children with high levels of postnatal cardiomyocyte proliferation. Alms1 mRNA knockdown increases multiple markers of proliferation in cardiomyocytes, the percentage of cardiomyocytes in G2/M phases, and the number of cardiomyocytes by 10% in cultured cells. Homozygous Alms1-mutant mice have increased cardiomyocyte proliferation at two weeks postnatal compared to wild-type littermates. We conclude that deficiency of Alström protein impairs postnatal cardiomyocyte cell cycle arrest.
Mammalian cardiac development requires continuous proliferation of cardiomyocytes throughout gestation1,2. During the perinatal period, cardiomyocyte proliferation rapidly declines, and the majority of cardiomyocytes undergo cell cycle arrest with terminal differentiation3,4. Postnatal arrest of the cardiomyocyte cell cycle is a key event for maturation of the mammalian heart, but this process is poorly understood3. A recent report highlights the role of the homeodomain transcription factor, Meis1, as a critical regulator of postnatal cardiomyocyte cell cycle arrest, though this process remains incompletely understood5.
Unusual human phenotypes sometimes offer the opportunity to understand normal development or disease pathogenesis, if the cause can be determined. Mitogenic cardiomyopathy is a rare form of pediatric cardiomyopathy characterized by persistent markers of mitotic activity in cardiomyocytes6. Among five previously reported infants with this condition, there were two pairs of siblings, one of whom had parental consanguinity supporting a recessive genetic disorder. Although transgenic models of postnatal cardiomyocyte replication have been developed, no naturally inherited conditions have previously been characterized in humans that are associated with delayed postnatal cardiomyocyte cell cycle arrest7,8. Recognition and characterization of such a disorder has the potential to identify important regulators of the transition of cardiomyocytes from active proliferation to terminal differentiation.
Here we identify ALMS1 mutations in 2 siblings and 4 previously reported infants with “mitogenic cardiomyopathy.” We show that siRNA-induced knockdown of murine Alms1 increases cell cycle progression in cultured neonatal murine cardiomyocytes, and Alms1 knockdown also increases the number of induced cardiomyocytes in counting experiments. Mice with targeted mutation of Alms1 have delayed exit from active cell cycle progression, further supporting an important role for ALMS1 in regulating postnatal cardiomyocyte cell cycle arrest.
Results
Evaluation of the proband and her sibling
We identified two infant siblings with neonatal heart failure, both of whom required cardiac transplantation. The proband was normal at birth, and she had no other manifestations of Alström syndrome prior to transplantation, which was performed at three months of age. The proband’s only sibling underwent cardiac transplantation at age five months for similar cardiac dysfunction. Intracranial bleeding complicated her post-operative course, and she died one month later. Cardiac evaluations of both parents were normal. We visualized mitotic cardiomyocytes with antibodies against phosphorylated histone H3 (PH3, a marker for M-phase9) and cardiac troponin T (cTnT) (Figure 1a, b, c, and d) and as well as wheat germ agglutinin (WGA) to distinguish cell boundaries (Figure 2). Using an unbiased double-blinded approach for quantification of proliferating myocytes, we quantified PH3-positive cardiomyocytes in multiple fields from the proband and three age-matched controls with heart failure. The amount of PH3-positive cardiomyocytes was higher in the proband than in the controls (114.3 ± 31.3 per mm2 [N=4] vs. 0.28 ± 0.07 per mm2 [N=12], Student’s t-test, mean ± S.E.M., respectively). To validate the high number of proliferating cardiomyocytes in the proband, we stained with antibodies against phospho-aurora kinases (PAK) A, B, and C. PAK A/B/C are important regulators of karyokinesis and cytokinesis, and localization of phospho-aurora kinase B to the cleavage furrow is required for establishing cytokinesis10. Aurora kinases are essential for formation of the mitotic spindle, separation of centrosomes and assembly of the cleavage furrow during pro-, meta-, ana, and telophase of mitosis10–12. Immunostaining for PAK confirmed an increase in the number of positively stained cardiomyocytes in multiple fields from the proband compared to the age-matched controls with heart failure (12.2 ± 2.6 per mm2 [N=4] vs. ≤ 0.02 ± 0.01 per mm2 [N=12], Student’s t-test, mean ± S.E.M., respectively) (Figure 1e, f).
Figure 1. Increased cardiomyocyte proliferation in people with ALMS1 mutations.

(a, b) Representative confocal images of the proband heart vs. age-matched control; PH3 (green), troponin T (red), DAPI (blue). (c) Higher magnification confocal images of PH3-positive cardiomyocytes in the proband; PH3 (green), troponin T (red), DAPI (blue). (d.) Additional high-magnification confocal image of a PH3-positive cardiomyocyte in the proband; PH3 (green), troponin T (red), DAPI (blue). (e.) Phospho-aurora kinase (PAK) staining (green) is present in a dividing cardiomyocyte nucleus in the proband; PAK (green), α-sarcomeric actinin (red), DAPI (blue (f) The number of PAK-positive cardiomyocytes in the heart of the proband was compared to three age-matched controls with failing ventricles. (g) The number of PH3-positive cardiomyocytes in affected individuals was compared to three age-matched controls with failing ventricles. Error bars represent standard error of mean (SEM). All scale bars represent 10μm.
Figure 2. PH3-positive cardiomyocytes in the proband and another affected individual.
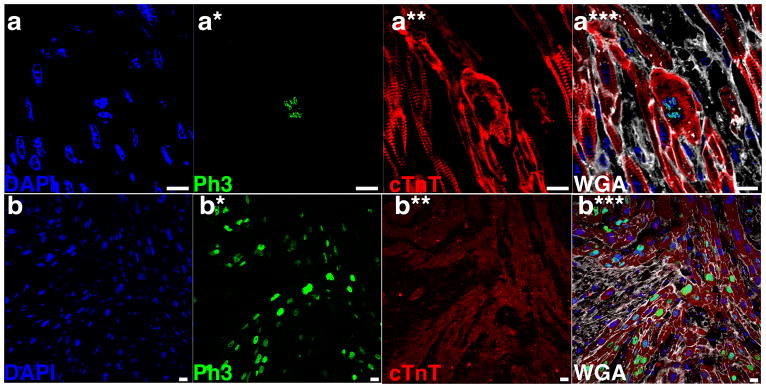
Additional representative confocal microscopic images from the heart of the proband (a) and another individual with mitogenic cardiomyopathy (b) have high levels of phosphohistone-H3 (Ph3)-positivity. Images were obtained with confocal microscopy using immunostaining for Ph3 (green), α-sarcomeric actinin or troponin T (red), wheat germ agglutinin (white) to outline cell boundaries, and DAPI (blue) to highlight nuclei. Panels a and b – DAPI; panels a* and b* – Ph3; panels a** and b** – cTnT; panels a*** and b*** – merged images with DAPI, Ph3, cTnT, and WGA. Scale bars all indicate 10 μM.
The proband underwent clinical genetic testing to determine the cause of her cardiomyopathy with a resequencing array13. Analysis of MYH7, MYBPC3, TNNT2, TNNI3, TPM1, ACTC, LMNA, SGCD, EMD, DES, LDB3, ACTN2, CSRP3, TCAP, VCL, TAZ, PLN, ABCC9, and CTF1 showed no apparent cause for cardiomyopathy. Next, we performed whole exome sequencing using DNA obtained from the affected proband and both of her parents. With the presumption of a recessive disorder, we focused on missense, nonsense, or splice site variants that were likely to be compound heterozygote mutations in the affected offspring, since the ethnicity of the parents was geographically distant (northern European and Southeast Asian). Variants present in dbSNP131 or the 1000 genomes pilot April 2010 dataset were filtered out. After this level of filtering, six genes with compound missense variants and one gene with compound heterozygote frameshift insertion/deletion remained (Table 1). The 1000 genomes November 2010 data were subsequently available, and three of these genes with previously novel compound heterozygous missense variants were filtered. Sanger sequencing confirmed novel DNA variants in each of the remaining four genes. The absence of one or both variants in the affected sibling led to filtering of two additional genes. FERMT1 had two novel missense alleles (p.Arg98Cys and p.Val519Leu) that were present in each of the affected individuals. These novel variants in FERMT1 were prioritized lower than ALMS1 because Arg98 is not highly conserved (Cys in Danio rerio), and because valine and leucine are both neutral nonpolar amino acids (Grantham score 32); both variants are present at low levels in the NHLBI Exome Variant Server. We identified and confirmed two heterozygous ALMS1 mutations in the proband and her sibling, both of which result in frameshift and premature termination (c.1794_1801dup8 in exon 8 and c.11116_11134del19 in exon 16). Mutations in ALMS1 are known to cause Alström syndrome, a recessive systemic disorder14. The c.11116_11134del19 mutation was previously reported in a patient with Alström syndrome15. Each parent harbored one of the mutant ALMS1 alleles. Alström syndrome (OMIM #203800) is a recessive ciliopathy caused by ALMS1 mutations and characterized by childhood truncal obesity, insulin-resistant diabetes mellitus, sensorineural hearing loss, retinal degeneration, and systemic fibrosis affecting multiple organs (kidney, liver, lung, and heart)16,17. Cardiomyopathy manifests in approximately two-thirds of affected individuals, and it can precede all other manifestations, obscuring the diagnosis of Alström18,19. We hypothesized that the mutations in ALMS1 caused the delayed terminal differentiation of cardiomyocytes.
Table 1.
Filtered Exome Sequencing Results
| Gene | Variant cDNA | Variant protein | Transcript Accession number | Allele frequency in 1000 Genomes Nov 2010 | Present in Affected Sibling |
|---|---|---|---|---|---|
| ALMS1 | c.1794_1801dup8 | p.Lys601Arg_fsX3 | NM_015120 | - | Yes |
| c.11116_11134del19 | p.Arg3706Leu_fsX11 | NM_015120 | - | Yes | |
| CD248 | c.683C>G | p.Pro228Leu | NM_020404 | - | No |
| c.1235C>G | p.Pro412Arg | NM_020404 | - | No | |
| FERMT1 | c.292C>T | p.Arg98Cys | NM_017671 | - | Yes |
| c.1555G>T | p.Val519Leu | NM_017671 | - | Yes | |
| FRMD4B | c.1424C>T | p.Pro475Leu | NM_015123 | 0.6 | Not tested |
| c.2309A>C | p.Asn770Thr | NM_015123 | - | Yes | |
| RGS3 | c.231G>T | p.Gln77His | NM_144489 | 1.2 | Not tested |
| c.1327G>T | p.Ala443Ser | NM_021106 | 0.1 | Not tested | |
| RYR3 | c.4529T>C | p.Val1510Ala | NM_001036 | - | Yes |
| c.14128G>A | p.Asp4710Asn | NM_001036 | - | No | |
| SNX19 | c.842C>T | p.Ala281Val | NM_014758 | 0.5 | Not tested |
| c.2683C>T | p.Arg895Trp | NM_014758 | - | No |
Genes in which two rare variants are present in the proband, inherited in a recessive manner from each parent. Variants present in the 1000 Genomes dataset by 11/2010 were prioritized lower. Those in which one or both variants was not present in the affected sibling were excluded.
ALMS1 mutations in similarly affected infants
To extend these findings, we sequenced ALMS1 in four additional infants (two sibling pairs) with mitogenic cardiomyopathy, from whom DNA was available6. We identified homozygous or compound heterozygous mutations in each of them (Table 2). For all cellular and immunohistochemical quantifications, unpaired two-tailed Student’s t-test, type II, was used for data analysis, and P≤0.5 was considered significant. Using the same double-blinded quantification method described above, we quantified PH3-positive cardiomyocytes in three affected individuals (one from each sib-pair) and compared to three age-matched controls with heart failure, and found it to be higher in the affected children (67.3 ± 8.6 per mm2 compared to 0.28 + 0.07 per mm2, respectively, N=3, mean ±S.E.M., P<0.01; Figure 1g). Accordingly, DNA content in dividing cardiomyocytes was 1.9-fold greater in PH3-positive myocytes than in non-dividing cells, confirming that PH3-positive myocytes are undergoing mitosis (Figure 3).
Table 2.
Mutations in ALMS1 in people with mitogenic cardiomyopathy:
| Individual | Exon | cDNA | protein |
|---|---|---|---|
| 1a | 8 | 1794_1801dup8 | Lys601Arg_fsX3 |
| 16 | 11116_11134del19 | Arg3706Leu_fsX11 | |
| 1b | 8 | 1794_1801dup8 | Lys601Arg_fsX3 |
| 16 | 11116_11134del19 | Arg3706Leu_fsX11 | |
| 2a | 8 | 4296_4299delCACA | His1432Gln_fsX40 |
| 8 | 5926delG | Glu1976Ser_fsX8 | |
| 2b | 8 | 4296_4299delCACA | His1432Gln_fsX40 |
| 8 | 5926delG | Glu1976Ser_fsX8 | |
| 3a | 8 | 1894C>T | Gln632Ter |
| 8 | 1894C>T | Gln632Ter | |
| 3b | 8 | 1894C>T | Gln632Ter |
| 8 | 1894C>T | Gln632Ter |
The proband and her sibling are 1a and 1b. Individuals 2a, 2b, 3a, and 3b are affected sibling pairs that were previously reported with this phenotype. Parental testing in 2a and 2b demonstrated compound heterozygosity, because both parents are heterozygous for a single ALMS1 mutation.
Figure 3. Human cells analyzed for ploidy status.

(a.) The DNA content in dividing cardiomyocyte nuclei was compared to non-dividing nuclei (both cardiomyocytes and non-cardiomyocytes) in the proband using quantitative confocal laser cytometry with DAPI nuclear staining. The ratio of DNA in nuclei from proliferating cardiomyocytes compared to non-proliferating (cardiomyocyte and lymphocyte) nuclei in the proband is 1.9, indicating predominantly 4N chromosomal content in these cells. (b.) Summary of the ploidy analysis using centromeric probes for 4 affected individuals and 2 unaffected controls. Error bars indicate S.E.M.. (c.) An example of ploidy analysis using one of the centromere FISH probes for chromosome 8 (CEP8) in an affected individual. Color staining is shown in the bottom left. Scale bars indicate 10 μM; These five panels are the same field with: C) DAPI; C*) CEP8; C**) DAPI and CEP8; C***) DAPI, CEP8 and sarcomeric actinin; and C****) DAPI, CEP8, sarcomeric actinin, and WGA.
Cardiomyocytes can undergo DNA replication without completing the cell cycle. Polyploidization occurs during early postnatal development and in response to myocardial stress4,20,21. We considered the possibility that ALMS1-deficiency could result in increased polyploidy. We applied centromeric FISH probes to determine the ploidy status in these infants with mitogenic cardiomyopathy. The percentage of 4N cardiomyocytes was 31.1+7.2% among affected individuals (N=4) and 15.5+8.3% in the controls (N=12), Student’s t-test, mean±S.E.M., P=0.26. Although we found cardiomyocytes that were polyploid (>4N), there was no difference between affected individuals (0.4%, N=4) and failing heart age matched controls (0.6%, N=2) (Figure 3). Taken together, these results indicate that ALMS1 deficiency in cardiomyocytes does not lead to increased polyploidy.
Alms1 knockdown increases cardiomyocyte cell cycling
To confirm that ALMS1 loss is sufficient for extending the postnatal proliferative window of cardiomyocytes, we treated cultured neonatal mouse cardiomyocytes with Alms1 siRNA (Supplementary Fig. 1). After 48 hours, cells were stained with antibodies against Ki67 (a wide-ranging marker of proliferation) or Vybrant DyeCycle Ruby Stain (a marker of DNA content for determination of cell cycle; Invitrogen), together with cTnT, and analyzed by flow cytometry. In ALMS1-deficient cardiomyocytes, Ki67-positive cardiomyocytes were 2.5-fold higher compared to control (4.2% vs. 1.7% respectively; N=8, Student’s t-test, P<0.05) (Supplementary Fig. 2). Likewise, we observed more cells in G2/M phases after Alms1 knockdown compared to the control (13.3+1.8% vs. 9.6+1.3% mean ± S.E.M., N=4, respectively; Student’s t-test, P<0.05) (Figure 4). We also analyzed Ki67 expression in cells stained with the fibroblast marker Thy-122 and found no significant difference between ALMS1-deficient cardiac fibroblasts compared to control (3.5±0.5% vs. 2.9±0.8% mean ± S.E.M., N=4, respectively; Student’s t-test, P=0.55), indicating that the observed increase of cells in G2/M may be in cardiomyocytes. To determine whether the observed increase in G2/M was mediated by cardiomyocytes, we cultured neonatal mouse cardiomyocytes obtained from transgenic mice in which the αMHC promoter drives expression of green fluorescent protein (GFP), and thus only cardiomyocytes produce GFP23. After Alms1 knockdown, the number of GFP-positive cells in phases G2/M was increased compared to the control (11.3±1.0% vs. 7.0±0.9% mean ± S.E.M., N=3, respectively, Student’s t-test, P<0.05) (Figure 5a), indicating that ALMS1-deficiency leads to impaired cell cycle arrest in cardiomyocytes.
Figure 4. Knockdown of Alms1 increases the number of cells in G2/M phases in cardiomyocyte enriched cultures.

Comparison of the % of cells in different phases of cell cycle between control transfection and Alms1 knockdown (KD) using siRNA; red = G0 or G1 phase, white = S phase, blue = G2 or M phase. (*) refers to P< 0.05 using unpaired Student’s t-test.
Figure 5. Increased cardiomyocyte proliferation in cultured cells after Alms1 knockdown.

(a.) Comparison of α-MHC-GFP positive cardiomyocytes in different phases of cell cycle after control siRNA transfection compared with Alms1 knockdown (KD) by siRNA; red = G0 or G1 phase, white = S phase, blue = G2 or M phase; N=3. Alms1 KD increases the proportion of cells in G2/M phase (blue). (b., c., d., e.) PAK-positive cardiomyocytes in different phases of mitosis; cTnT (red), PAK (green), and DAPI (blue); b is prophase, c is metaphase, d is anaphase, e is telophase. (f.) Flow cytometry of puromycin-selected cardiomyocytes (cTnT+ cells) shows that after Alms1 knockdown (KD), there is increased PAK expression compared to controls; N=3. (g.) Scatter plot showing increased incorporation of EdU (Y-axis) and increased DNA content by DAPI (X-axis) in puromycin-selected (cTnT+) cardiomyocytes with ALMS1 KD; N=15. (h.) Total number of puromycin-selected cardiomyocytes after Alms1 knockdown (KD) is increased compared to controls; N=4. In all Figures, (*) refers to P<0.05 using Student’s t-test. Error bars represent standard error of mean (SEM), and all scale bars represent 5 μM.
To confirm that ALMS1-deficient cardiomyocytes proliferate with impaired cell cycle arrest, we differentiated cardiomyocytes from mouse ESCs that carry a cardiomyocyte-specific promoter (Ncx1), which drives puromycin resistance, allowing efficient purification of cardiomyocytes (>98%)24. (Supplementary Fig. 3) By immunohistochemistry, we confirmed the presence of cardiomyocytes in pro-, meta-, ana- and telophase of mitosis after knockdown of Alms1 (Figure 5b, c, d, e), demonstrating that these cardiomyocytes complete the cell cycle in vitro. In accordance with this observation, the amount PAK-positive cardiomyocytes was higher in ALMS1-deficient cardiomyocytes compared to control (0.41±0.09% vs. 0.15±0.03%; mean ± S.E.M., N=3, respectively, Student’s t-test, P<0.05; Figure 5f).
To confirm that ALMS1 loss extends the proliferative window of cardiomyocytes, we treated puromycin-selected cardiomyocytes with Alms1 siRNA or control siRNA. After 48 hours, we pulse-labeled cardiomyocytes with 5-ethynyl-2′-deoxyuridine (EdU) for 12h. We observed an increase in EdU-positive cardiomyocytes after Alms1 knockdown compared to controls (13.6 ± 1.0% vs. 10.2 ± 0.6%, mean ± S.E.M., N=15, respectively, Student’s t-test, P<0.05)(Figure 5g). During maturation, cardiomyocytes undergo karyokinesis (nuclear division) but not cytokinesis (cell division) and become terminally differentiated25. To exclude the possibility that the observed increase in EdU-positive uptake is due to karyokinesis and not cytokinesis, we counted cardiomyocytes 72 hours after siRNA treatment and found that the number of cardiomyocytes was higher after Alms1 knockdown compared to control (51,915±1821/cm2 vs. 46,696±1311/cm2, mean ± S.E.M., N= 4, respectively; Student’s t-test, P<0.05) (Figure 5h). Taken together, these results confirm that ALMS1-deficiency increases cardiomyocyte proliferation in vitro.
Impaired cardiomyocyte cell cycle arrest in Alms1Gt/Gt mice
To investigate the role of ALMS1 in vivo, we utilized Alms1Gt/Gt mice with truncated Alms1 mRNA and many characteristics of human Alström syndrome26. In mice, the maturation process from mono- to binucleate state occurs during postnatal day 5–10, resulting in 95–99% cardiomyocytes being binucleated and terminally differentiated at 10 days of age21,25. We therefore analyzed mouse hearts at 15.5 days of age for persistent cardiomyocyte proliferation. To do this, we treated 15-day old mice with a pulse-dose of EdU, and 12 hours later, their hearts were isolated and EdU uptake in cardiomyocytes was analyzed using the double-blinded quantification method described above. In the Alms1Gt/Gt mutant mice, more than 8% of cardiomyocytes were EdU-positive, compared to very low levels in the wild-type littermate controls (8.3 ± 0.91% vs. 0.3 ± 0.04%, mean ± S.E.M., N=4, respectively; Student’s t-test, P<0.001, Figure 6a, b, and c). Accordingly, we also observed a higher number of PH3-positive cardiomyocytes in homozygous Alms1Gt/Gt mutant mice compared to their wild-type littermates (13.2 ± 2.6 per mm2 compared to less than 1 per mm2; mean ± S.E.M., N= 3, Student’s t-test, P<0.01)(Figure 6d, e, and f). Additional staining demonstrated increased PAK-positive cardiomyocytes in the Alms1Gt/Gt mice compared to wild-type controls (1.36 ± 0.1 vs. 0.11 ± 0.1 per mm2; mean ± S.E.M., N= 3, Student’s t-test, respectively; P<0.001)(Figure 6g, h, and i). Importantly, we did not observe any difference in apoptosis (caspase3-positive or TUNEL-positive cells) between Alms1Gt/Gt mutant mice and their littermate controls (Supplementary Fig. 4, 5).
Figure 6. Increased cardiomyocyte proliferation in homozygous Alms1Gt/Gt mutant mice.

(a, b.) Representative confocal images demonstrating EdU (green) incorporation in Alms1Gt/Gt vs. wild-type littermate control mouse cardiac myocytes; α-sarcomeric actinin (red), wheat germ agglutinin (WGA; white), and DAPI (blue). (c.) Bar graph comparing cardiomyocyte EdU incorporation in Alms1Gt/Gt mutant (KO) vs. wild-type littermate control mice (N=4 each). (d, e.) Representative confocal images demonstrating PH3 (green) in a cardiomyocyte nucleus in a Alms1Gt/Gt mutant vs. wild-type littermate control mouse, with cTnT (red), wheat germ agglutinin (white), and DAPI (blue). (f.) Comparison of the number of PH3-positive cardiomyocytes in Alms1Gt/Gt (KO) vs. wild-type littermate control mice (N=3 each). (g, h.) Representative confocal images demonstrating PAK staining (green) is present in a dividing cardiomyocyte nucleus in an Alms1Gt/Gt mouse (KO); DAPI (blue) highlights the nucleus, which is surrounded by α-sarcomeric actinin (red) to highlight cardiomyocytes and WGA (white) to identify cell boundaries. (i.) Comparison of the number of PAK-positive cardiomyocytes in Alms1Gt/Gt (KO) vs. wild-type controls. All scale bars represent 10μm. Error bars represent S.E.M.. N=3 each.
Phenotypic characterization of Alms1Gt/Gt at postnatal day 15.5 demonstrated that ALMS1-mutant mouse heart/body ratio was larger in Alms1Gt/Gt mice (N=7) compared to wild-type littermates (N=16) (7.2 ± 0.3 vs. 6.4 ± 0.2; mean ± S.E.M., Student’s t-test, P<0.05; Figure 7). In addition, Alms1Gt/Gt cardiomyocytes were smaller compared to wild-type littermate controls (128.2 ± 10.6 vs. 176.1 ± 8.5 μM2, mean ± S.E.M., N= 4, Student’s t-test, P < 0.0.001) (Figure 7), implying that cardiomyocyte number is increased in Alms1Gt/Gt hearts. Together, these findings indicate that terminal differentiation is impaired in ALMS1-deficient cardiomyocytes and that ALMS1-deficient cardiomyocytes remain proliferative beyond the normal window of postnatal cardiomyocyte cell cycle arrest. Alms1 levels in dividing cardiomyocytes obtained from proliferative mouse hearts (embryonic day 15.5) are relatively low, compared to levels at the beginning of the maturation process (postnatal day 6) when cytokinesis rapidly declines, suggesting that ALMS1 may be transcriptionally regulated during perinatal development (Figure 8).
Figure 7. Increased cardiomyocyte density and normalized heart size in homozygous Alms1Gt/Gt mutant mice.

Representative images of (A.) wild-type and (B.) Alms1Gt/Gt hearts stained with wheat germ agglutinin (white) and cTnT (red); scale bars = 50 uM. (C) Measured cardiomyocyte cross sectional area, N=12 each. (D) Heart/body weight ratios, N=16 for WT and N=7 for Alms1−/−; error bars represent S.E.M., (*) indicates P<0.05 using Student’s t-test.
Figure 8. Perinatal expression of Alms1 in murine cardiomyocytes.

Murine GFP-positive cardiomyocytes were isolated from α-MHC-GFP transgenic mice by FACS. Alms1 mRNA levels were normalized to Gapdh. Relative Alms1 mRNA levels at embryonic day 15.5 (ED15.5; N=4) and postnatal day 0 (P0; N=4) were compared by student’s T-Test to postnatal days 5.5/6.5 (P5/6; N=10), p=0.02 and p=0.013, respectively. (*) denotes P<0.05. There was no significant difference in Alms1 expression between days E15.5 and P0; error bars indicate S.E.M.
We considered whether our findings could be due to an increased number of cardiac stem cells. However we observed no difference in the number of c-kit+ or Sca-1+ cardiac stem cell markers in Alms1Gt/Gt compared to littermates (both markers <1 cell per 225 μM2). As cardiac stem cells differentiate, they may lose c-kit or Sca-1 markers. Thus, we cannot exclude that the increase in proliferating cardiomyocytes may arise in part from resident cardiac stem cells.
Discussion
Our data show that ALMS1 is a key molecule for cell cycle regulation in perinatal cardiomyocytes. ALMS1 is a component of the non-motile primary cilium, a subcellular organelle that projects from most cell types. Its presence is temporally associated with cellular quiescence, with resorption during mitosis27. Induction of a longer cilium causes delay in G1/S transition, and ciliary disassembly increases S-phase28,29. Prior reports show that certain components of the primary cilium restrain canonical Wnt/β-catenin signaling, which plays an important role in cardiomyogenesis30,31. Deficiency of ALMS1 may activate Wnt/β-catenin signaling, thereby activating the transcription factor TCF/LEF (T-cell factor/lymphoid enhancer factor) and inducing transcription of genes that promote cell cycle proliferation32,33.
Definitive proof of cardiomyocyte cytokinesis can be challenging. Proliferation markers, such as PH3, Ki67, and EdU, cannot discern karyokinesis from cytokinesis. Increased PAK staining in humans and mice with ALMS1 deficiency is consistent with increased cytokinesis, but it is insufficient. The increased density of cardiomyocytes in Alms1-mutant mice and the increased number of cardiomyocytes with cell counting studies after Alms1 knockdown are further supportive of amplified cardiomyocyte cytokinesis. However, the evidence for cytokinesis, and therefore the completion of mitosis, is inconclusive and warrants further investigation.
Mitogenic cardiomyopathy is a very rare human phenotype, previously of unknown etiology. Our finding of homozygous or compound heterozygous mutations in ALMS1 among all six affected infants from whom DNA was available suggests that this is the main cause. However, in each of these individuals, hearts were removed either at the time of transplantation due to end-stage heart failure or after death from heart failure6. Cardiomyopathy is found in approximately two-thirds of affected individuals, which suggests that other factors contribute to its development besides ALMS1 mutations18. When cardiomyopathy develops in infancy, it can precede other manifestations, at times obscuring the diagnosis of Alström syndrome, as among these 6 infants and as previously reported19. The cardiac function of the ALMS1-mutant mice is normal26. However, our findings do not rule out an injury response that contributes to myocyte proliferation in the context of an ALMS1 mutation.
ALMS1 localizes to the centrosome, which plays a pivotal role in regulation of the cell cycle34,35. At postnatal day 15 (1 week beyond the normal window of postnatal cardiomyocyte cell cycle arrest), ALMS1-deficient mice display persistent cardiomyocyte proliferation, indicating that ALMS1-deficient cardiomyocytes may have an impaired ability to undergo cell cycle arrest. A similar finding was previously reported for the transcription factor MEIS1, although no direct link between MEIS1 and ALMS1 is currently known5. Throughout mitosis, ALMS1 localizes to the centrosomal spindle poles, and during late mitosis, ALMS1 localizes to the contractile ring and the cleavage furrow36. ALMS1 retains the centrosome cohesion protein C-NAP1, an important regulator of centrosome organization during mitosis. Depletion of ALMS1 reduces centrosomal levels of C-NAP1 and increases centrosome splitting, a key event for chromosomal division during anaphase34. In addition, ALMS1 interacts with several cytoskeleton-associated components that are necessary for the recycling of receptors to the plasma membrane, also referred to as endocytic trafficking36. During mitosis endocytic trafficking is repressed, as continued endocytosis may interfere with accurate chromosome segregation37–39. Interestingly, endocytic trafficking is significantly reduced in ALMS1-deficient cells36. Whether the impairment of terminal differentiation of cardiomyocyte proliferation is partly due to diminished endocytic trafficking remains unclear. Further investigation of this role of ALMS1 may identify novel and therapeutically important avenues to alter cardiomyocyte replication.
Methods
Exome sequencing
All human subjects (or their legal representative) signed informed consent, and the research was approved by the Johns Hopkins University Institutional Review Board. Genomic DNA was obtained from the proband’s dermal fibroblasts and from blood from each of the parents by the Gentra Puregene Tissue/Blood Kit (Qiagen). Three μg of genomic DNA was sheared using the Covaris S-2 instrument (Covaris) using the recommended settings according to the Agilent protocol (SureSelectXT Target enrichment System for Illumina Paired End and Multiplexed Sequencing Library v1.0) for a 150–200bp fragment size. Libraries were prepared for targeted enrichment using the SPRIworks Fragment Library System I (200–400bp size selection) according to Beckman-Coulter’s protocol (version 2.0). Adapter ligated fragments were amplified according to the Agilent protocol for 6 cycles; 500ng of amplified library was used in a whole exome enrichment reaction with the SureSelect Human All Exon, 50 Mb product (Agilent WE). Samples were clustered one per flow cell lane using the Illumina cBot Paired End Cluster Generation Kit with v1 HiSeq flow cell (Illumina). Seventy-six bp paired-end sequencing was performed on the HiSeq2000 with TruSeq SBS v1 chemistry (Illumina).
Intensity analysis and base calling were performed through the Illumina Real Time Analysis (RTA) software (version 1.7.48.0). Average sequence yield for the samples was 9.2 Gb having a quality value of Q30 or greater. Basecall files were converted from a binary format (BCL) to flat file format (qseq.txt) using the Illumina BCL Converter software (version 1.7.1).
Basecall files were converted to fastq format, aligned with BWA40 version 0.5.7 to the GRCh37 human genome reference. Duplicate molecules were flagged with Picard version 1.26. SNVs and indels were called using SAMtools41 version 0.1.7. Only variant calls from the reference genome having a minimum depth of 10x were considered for downstream analysis. In addition, for SNVs, only those calls where the root mean square of their read mapping qualities (RMS) was greater than 20 were considered while for indels only those with an RMS greater than 15 were considered.
Samples were processed on the Illumina Infinium HumanOmniExpress beadchip (OE) to confirm family and gender relationships and provide sample identity confirmation against the sequencing data. The mean on bait coverage (regions covered by probes in the Agilent WE) was greater than 77x for each sequencing experiment and greater than 92% of on bait bases had a depth greater than 10x. Overall concordance to genotypes for each sample was greater than 99.5%. Greater than 93% of the OE heterozygote genotypes for each sample within the baited regions were called as variants against reference in the sequencing experiments.
All remaining variants were annotated using the SeattleSeq Annotation Server build 6.0342. Variants present in dbSNP131 or the 1000 genomes pilot April 2010 dataset were filtered out. Only missense, nonsense or splice site compound heterozygotes present in the offspring where one allele each was inherited from the parents were considered. After this level of filtering, 6 compound missense heterozygotes and 1 compound heterozygote frameshift indel remained.
Statistical analyses
For all cellular and immunohistochemistry quantification, the unpaired two-tailed Student’s t-test, type II, was used for data analyses. P<0.05 was considered significant.
Cell culture and analysis
Mouse neonatal cardiomyocytes were isolated from newborn mouse hearts on PD0.5. Hearts were minced and digested with collagenase type II and pancreatase myocyte digestion buffer. To enrich for cardiomyocytes, dissociated cells were pre-plated by the conventional pre-plating method22,43. Enriched cardiomyocytes were cultured in Dulbecco’s Modified Eagle Medium (GIBCO) supplemented with 20% horse serum and 2 mM GlutaMAX (Invitrogen) in 5% CO2. Cardiac fibroblasts were identified by their expression of the cardiac fibroblast marker Thy-122,23. Mouse embryonic stem cells (mESCs) carrying a puromycin resistance cassette driven by the cardiomyocyte-specific sodium-calcium exchanger 1 gene (Ncx1) promoter44 were maintained and cultured on gelatin-coated tissue culture dishes in standard maintenance medium (Glasgow minimum essential medium with 10% fetal bovine serum and 1500 Units lif/ml (millipore), Glutamax, sodium pyruvate and non-essential amino acids 45. For differentiation, ESCs were allowed to form embryoid bodies (EBs) in the IMDM/Ham-F12 (Cellgro) (3:1) supplemented with N2, B27, Penicillin/Streptomycin, 2mM GlutaMAX, 0.05% BSA, 5 ng/ml L-ascorbic acid (Sigma-aldrich), α-Monothioglycerol (MTG, Sigma-Aldrich). For mesoderm induction EBs were dissociated and reaggregated in the presence of Activin A, BMP4 and VEGF for 48 hours46. Cardiomyocytes were selected with Puromycin (2.5ug/ml) for 48h 44, dissociated and counted using an automated cell counter (Scepter 2.0, Millipore).
RNA suppression and cell cycle analysis
Cardiomyocytes were transfected with Lipofectamine RNAiMAX (Invitrogen). For Alms1 knockdown experiments, Alms1 ON-TARGETplus SMARTpool and Alms1 siRNA (Ambion) or scrambled siRNA oligonucleotides (Dharmacon) and siRNA negative control (Ambion) were used at a final concentration of 100 nM for cell transfection. Forty-eight hours after transfection, cells were fixed and stained for flow cytometry with the following antibodies: anti-Ki67 (1:200, cat# ab15580, Abcam), -phospho-aurora A/B/C kinase (1:200, cat# 2914S, Cell Signaling), -phospho histone 3 (1:400, cat# ab5176, Abcam), -cardiac troponin T (1:500, cat #13-11, Thermo scientific), -Thy1-APC (1:200, cat# 17-0902-81, eBioscience), with secondary detection using the appropriate Alexa Fluor-conjugated antibody (1:400, Invitrogen). For S-phase analysis, cardiomyocytes were treated with 100 μM EdU for 30 min prior to fixation and stained using Click-iT EdU cell proliferation kit (Invitrogen). For cell cycle analysis, dissociated cardiomyocytes were incubated for 30 min with 5 μM Vybrant Dye Cycle Ruby Stain (Invitrogen). For EdU analysis, cells were treated with 100 μM EdU for 30 minutes prior to isolation and fixation. EdU incorporation was detected using Click-it EdU Alexa Fluor Imaging Kit (Invitrogen) according to manufacturer’s protocol. Cells were analysed on an Accuri C6 flow cytometer (BD Biosciences) using FlowJo software (Treestar).
Mice
The Alms1Gt/Gt mice (B6.129P2-Alms1Gt(XH152)Byg/Pjn) used in this study were generated from gene-trapped ES cells (MMRC#008633)26,47. Mice were fed ad libitum a 4K54 diet (PMI Nutrition International, St. Louis, MO) and provided an unlimited access to water in a temperature/humidity controlled setting with a 12 hour light/dark cycle at The Jackson Laboratory Research Animal Facility. All mouse protocols used in this study were approved by the JAX institutional Animal Care and Use Committee. Mouse hearts were extracted from mice euthanized by carbon dioxide asphyxiation and allowed to contract in phosphate buffered saline for 5–10 minutes and immediately placed in 4% paraformaldeyde at 4 °C overnight. Subsequently, tissues were embedded in paraffin, sectioned, and immunostained as previously reported48. The aMHC promoter-driven EGFP-IRES-puromycin transgenic mice (alphaMHC-GFP), in which only cardiomyocytes express the green fluorescent protein (GFP) facilitate rapid and efficient isolation of cardiomyocytes23. Hearts from these mice were dissociated using collagense II/TrypLE. GFP+ cardiomyocytes were isolated by Fluorescence Activated Cell Sorting (FACS) using an SH800 sorter (Sony Biotechnology).
Histology and immunohistochemistry
Paraffin-embedded heart sections were rehydrated, followed by antigen retrieval. Immunostaining was performed on cultured cardiomyocytes and mouse cardiac tissue with primary antibodies against Ki67, cardiac troponin T, α-sarcomeric actinin, phosphohistone-H3, phosphoaurora A/B/C, or activated caspase-3 followed by secondary detection with appropriate Alexa Fluor conjugated antibodies. Additional staining was done with wheat germ agglutinin conjugated with Alexa Fluor-647 dye to show cell boundaries. Analyses using confocal microscopy were quantified in a blinded fashion by 2 independent observers. All images were acquired with a Zeiss LSM 510 Meta Confocal system and analyzed with Volocity imaging software. Puromycin selected cardiomyocytes were fixed in 4% formaldehyde. Cardiomyocytes were permabilized with 0.5% saponin/PBS and stained with antibodies against cTnT or isotype control antibodies followed the appropriate Alexa Fluor-conjugated antibody (Invitrogen).
Determination of cardiomyocyte density and size
Postnatal day 15 wild-type and Alms1Gt/Gt mouse hearts were fixed with methanol, paraffin embedded, sectioned, deparaffinized, rehydrated, and subjected to citrate-based heat-mediated antigen retrieval. Slides were incubated with 5 μg/ml Alexa Fluor 647-conjugated wheat germ agglutinin (Invitrogen) overnight at 4 °C and mounted in Vectashield containing DAPI (Vector Labs, CA). Image acquisition was performed on an EVOS epifluorescence microscope (Life Technologies). Cardiomyocyte cross sectional area was measured using an automated algorithm with NIH Image J 1.47i software analyzing 300 – 600 cells from 3–4 areas per mouse heart.
Quantitative qRT-PCR
RNA was extracted with TRIzol (Invitrogen). Reverse transcriptase–quantitative PCR (qPCR) was performed using the Superscript III first-strand synthesis system (Invitrogen) followed by use of TaqMan probes on the ABI 7900HT (Applied Biosystems) according to the manufacturer’s protocols. Optimized primers from TaqMan Gene Expression Array were used. Expression levels were normalized to Gapdh expression. All samples were run at least intriplicate. Real-time PCR data were normalized and standardized with SDS2.2 software.
Quantitative fluorescence
To measure nuclear DNA content, 20um thick histology sections of the proband’s heart were stained with DAPI. Using a Zeiss LSM 510 Meta confocal microscope and Zen software for image acquisition, the nuclear DNA content of cardiac myocytes was assayed by measuring the total fluorescence from each selected nucleus with Volocity™ imaging software. Background fluorescence was excluded prior to image acquisition using Zen software. Nuclei in contact with the edges of the image stack or touching other nuclei were excluded. The total DAPI fluorescence signal associated with diploid status was designated 1 in non-proliferating cells (negative for PH3 staining). The DNA content of proliferating cardiomyocytes (positive for PH3 staining) was compared to non-proliferating cells and displayed as a ratio of the total quantitative fluorescence. A value of 2 would indicate cells that have a full duplication of nuclear DNA prior to karyokinesis.
Supplementary Material
Acknowledgments
We thank the families presented in this report for their participation. We thank Dr. Peter Rainer for assistance with analysis of myocyte sizes, Dr Kenneth Boheler for providing mouse embryonic stem cells with the Ncx-puromycin transgene, and Dr Deepak Srivastava for providing αMHC-GFP transgenic mice. We also thank Drs. Loren Field, Charles Steenbergen, and E. Rene Rodriguez for helpful advice and guidance. Supported by funding from the JHU Friends in Red, the Zegar Family Foundation, The Michel Mirowski Discovery Fund (DPJ), Mrs. Seena Lubcher (DPJ), R01HL111198 and 4R00HL09223 (NIH/NHLBI)(CK), Maryland Stem Cell Research Fund(CK), Magic That Matters Fund (CK), the Lundbeck Foundation (PA), and HD036878 (NIH) (JKN and GBC). The JHU Center for Inherited Disease Research is supported by funding from NIH contract HHSN268200782096C.
Footnotes
Author Contributions D.P.J. managed and supervised the project. L.T.S., P.A., and M.K.H. performed the histological analyses. P.A., L.T.S., L.F., N.A.A., Y.C., and S.C. performed the cellular studies. C.L., W.M., E.C, and K.C. characterized the Canadian samples. G.B.C and J.K.N developed, characterized, and provided the Alms1-mutant mice. R.Y. and D.A.S.B. performed the ploidy analyses. J.E.C, J.S., and L.V. identified, characterized, and referred the proband and her sibling. B.D.C, B.A.M., D.W.M, K.N.H., J.M.R., A.F.S, D.V., and K.F.D. performed the exome sequencing and bioinformatics analysis. L.T.S., P.A., C.K., and D.P.J. designed the experiments, interpreted the data, wrote and revised the manuscript.
Conflict of Interests The authors declare no competing financial interests.
Accession Codes:
Whole exome sequence data for the proband has been deposited in the GenBank dbGaP (database of Genotypes and Phenotypes) database under the accession code phs000711.v1.p1. Sequence data for ALMS1 from additional patients have been deposited in the GenBank nucleotide core database under the accession codes AB905563 to AB905565.
References
- 1.Drenckhahn JD, et al. Compensatory growth of healthy cardiac cells in the presence of diseased cells restores tissue homeostasis during heart development. Dev Cell. 2008;15:521–33. doi: 10.1016/j.devcel.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 2.Meilhac SM, et al. A retrospective clonal analysis of the myocardium reveals two phases of clonal growth in the developing mouse heart. Development. 2003;130:3877–89. doi: 10.1242/dev.00580. [DOI] [PubMed] [Google Scholar]
- 3.Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle regulation. Circ Res. 2002;90:1044–54. doi: 10.1161/01.res.0000020201.44772.67. [DOI] [PubMed] [Google Scholar]
- 4.Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15–26. doi: 10.1161/01.res.83.1.15. [DOI] [PubMed] [Google Scholar]
- 5.Mahmoud AI, et al. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013;497:249–53. doi: 10.1038/nature12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chang KT, Taylor GP, Meschino WS, Kantor PF, Cutz E. Mitogenic cardiomyopathy: a lethal neonatal familial dilated cardiomyopathy characterized by myocyte hyperplasia and proliferation. Hum Pathol. 2010;41:1002–8. doi: 10.1016/j.humpath.2009.12.008. [DOI] [PubMed] [Google Scholar]
- 7.Pasumarthi KB, Nakajima H, Nakajima HO, Soonpaa MH, Field LJ. Targeted expression of cyclin D2 results in cardiomyocyte DNA synthesis and infarct regression in transgenic mice. Circ Res. 2005;96:110–8. doi: 10.1161/01.RES.0000152326.91223.4F. [DOI] [PubMed] [Google Scholar]
- 8.Cottage CT, et al. Cardiac Progenitor Cell Cycling Stimulated by Pim-1 Kinase. Circ Res. 2010;106:891–901. doi: 10.1161/CIRCRESAHA.109.208629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei Y, Mizzen CA, Cook RG, Gorovsky MA, Allis CD. Phosphorylation of histone H3 at serine 10 is correlated with chromosome condensation during mitosis and meiosis in Tetrahymena. Proc Natl Acad Sci U S A. 1998;95:7480–4. doi: 10.1073/pnas.95.13.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim Y, Holland AJ, Lan W, Cleveland DW. Aurora kinases and protein phosphatase 1 mediate chromosome congression through regulation of CENP-E. Cell. 2010;142:444–55. doi: 10.1016/j.cell.2010.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang KT, et al. Aurora-C Kinase Deficiency Causes Cytokinesis Failure in Meiosis I and Production of Large Polyploid Oocytes in Mice. Mol Biol Cell. 2010;21:2371–2383. doi: 10.1091/mbc.E10-02-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science. 2008;320:1655–8. doi: 10.1126/science.1157425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimmerman RS, et al. A novel custom resequencing array for dilated cardiomyopathy. Genetics in Medicine. 2010;12:268–278. doi: 10.1097/GIM.0b013e3181d6f7c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collin GB, et al. Mutations in ALMS1 cause obesity, type 2 diabetes and neurosensory degeneration in Alstrom syndrome. Nat Genet. 2002;31:74–8. doi: 10.1038/ng867. [DOI] [PubMed] [Google Scholar]
- 15.Marshall JD, et al. Spectrum of ALMS1 variants and evaluation of genotype-phenotype correlations in Alstrom syndrome. Hum Mutat. 2007;28:1114–23. doi: 10.1002/humu.20577. [DOI] [PubMed] [Google Scholar]
- 16.Alstrom CH, Hallgren B, Nilsson LB, Asander H. Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence-Moon-Bardet-Biedl syndrome: a clinical, endocrinological and genetic examination based on a large pedigree. Acta Psychiat Neurol Scand. 1959;34:1–35. [PubMed] [Google Scholar]
- 17.Girard D, Petrovsky N. Alstrom syndrome: insights into the pathogenesis of metabolic disorders. Nat Rev Endocrinol. 2011;7:77–88. doi: 10.1038/nrendo.2010.210. [DOI] [PubMed] [Google Scholar]
- 18.Marshall JD, Maffei P, Collin GB, Naggert JK. Alstrom syndrome: genetics and clinical overview. Curr Genomics. 2011;12:225–35. doi: 10.2174/138920211795677912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bond J, et al. The importance of seeking ALMS1 mutations in infants with dilated cardiomyopathy. J Med Genet. 2005;42:10. doi: 10.1136/jmg.2004.026617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li F, Wang X, Capasso JM, Gerdes AM. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol. 1996;28:1737–46. doi: 10.1006/jmcc.1996.0163. [DOI] [PubMed] [Google Scholar]
- 21.Soonpaa MH, Kim KK, Pajak L, Franklin M, Field LJ. Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol. 1996;271:H2183–H2189. doi: 10.1152/ajpheart.1996.271.5.H2183. [DOI] [PubMed] [Google Scholar]
- 22.Ieda M, et al. Cardiac Fibroblasts Regulate Myocardial Proliferation through β Integrin Signaling. Developmental Cell. 2009;16:233–244. doi: 10.1016/j.devcel.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ieda M, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–86. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boheler KR, et al. Embryonic stem cell-derived cardiomyocyte heterogeneity and the isolation of immature and committed cells for cardiac remodeling and regeneration. Stem Cells Int. 2011;2011:214203. doi: 10.4061/2011/214203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walsh S, Ponten A, Fleischmann BK, Jovinge S. Cardiomyocyte cell cycle control and growth estimation in vivo--an analysis based on cardiomyocyte nuclei. Cardiovasc Res. 2010;86:365–73. doi: 10.1093/cvr/cvq005. [DOI] [PubMed] [Google Scholar]
- 26.Collin GB, et al. Alms1-disrupted mice recapitulate human Alstrom syndrome. Hum Mol Genet. 2005;14:2323–33. doi: 10.1093/hmg/ddi235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rash JE, Shay JW, Biesele JJ. Cilia in cardiac differentiation. J Ultrastruct Res. 1969;29:470–84. doi: 10.1016/s0022-5320(69)90067-7. [DOI] [PubMed] [Google Scholar]
- 28.Kim S, et al. Nde1-mediated inhibition of ciliogenesis affects cell cycle re-entry. Nat Cell Biol. 2011;13:351–360. doi: 10.1038/ncb2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li A, et al. Ciliary transition zone activation of phosphorylated Tctex-1 controls ciliary resorption, S-phase entry and fate of neural progenitors. Nat Cell Biol. 2011;13:402–411. doi: 10.1038/ncb2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ajima R, Hamada H. Wnt signalling escapes to cilia. Nat Cell Biol. 2011;13:636–637. doi: 10.1038/ncb0611-636. [DOI] [PubMed] [Google Scholar]
- 31.Kwon C, et al. Canonical Wnt signaling is a positive regulator of mammalian cardiac progenitors. Proc Natl Acad Sci U S A. 2007;104:10894–9. doi: 10.1073/pnas.0704044104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. doi: 10.1146/annurev.cellbio.20.010403.113126. [DOI] [PubMed] [Google Scholar]
- 33.Hayward P, Kalmar T, Arias AM. Wnt/Notch signalling and information processing during development. Development. 2008;135:411–24. doi: 10.1242/dev.000505. [DOI] [PubMed] [Google Scholar]
- 34.Knorz VJ, et al. Centriolar association of ALMS1 and likely centrosomal functions of the ALMS motif-containing proteins C10orf90 and KIAA1731. Mol Biol Cell. 2010;21:3617–29. doi: 10.1091/mbc.E10-03-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hinchcliffe EH. Cell cycle: seeking permission from the mother centriole. Current Biology. 2003;13:R646–648. doi: 10.1016/s0960-9822(03)00572-4. [DOI] [PubMed] [Google Scholar]
- 36.Collin GB, et al. The Alström Syndrome Protein, ALMS1, Interacts with α-Actinin and Components of the Endosome Recycling Pathway. PLoS One. 2012;7:e37925. doi: 10.1371/journal.pone.0037925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fielding AB, Willox AK, Okeke E, Royle SJ. Clathrin-mediated endocytosis is inhibited during mitosis. Proc Natl Acad Sci U S A. 2012;109:6572–7. doi: 10.1073/pnas.1117401109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fielding AB, Royle SJ. Mitotic inhibition of clathrin-mediated endocytosis. Cell Mol Life Sci. 2013;70:3423–33. doi: 10.1007/s00018-012-1250-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sager PR, Brown PA, Berlin RD. Analysis of transferrin recycling in mitotic and interphase HeLa cells by quantitative fluorescence microscopy. Cell. 1984;39:275–82. doi: 10.1016/0092-8674(84)90005-9. [DOI] [PubMed] [Google Scholar]
- 40.Li H, Durbin R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ng SB, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–6. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simpson P, Savion S. Differentiation of rat myocytes in single cell cultures with and without proliferating nonmyocardial cells. Cross-striations, ultrastructure, and chronotropic response to isoproterenol. Circulation Research. 1982;50:101–116. doi: 10.1161/01.res.50.1.101. [DOI] [PubMed] [Google Scholar]
- 44.Yamanaka S, Zahanich I, Wersto RP, Boheler KR. Enhanced Proliferation of Monolayer Cultures of Embryonic Stem (ES) Cell-Derived Cardiomyocytes Following Acute Loss of Retinoblastoma. PLoS One. 2008;3:e3896. doi: 10.1371/journal.pone.0003896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kwon C, et al. Notch post-translationally regulates beta-catenin protein in stem and progenitor cells. Nat Cell Biol. 2011;13:1244–51. doi: 10.1038/ncb2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kattman SJ, et al. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell. 2011;8:228–40. doi: 10.1016/j.stem.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 47.Jagger D, et al. Alstrom Syndrome protein ALMS1 localizes to basal bodies of cochlear hair cells and regulates cilium-dependent planar cell polarity. Hum Mol Genet. 2011;20:466–81. doi: 10.1093/hmg/ddq493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shenje LT, et al. Lineage tracing of cardiac explant derived cells. PLoS One. 2008;3:e1929. doi: 10.1371/journal.pone.0001929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.