

Significance
How well a protein conducts electrical current depends on both the chemical nature of the protein and its contacts to the electrodes between which currents are carried. Investigating conduction via protein monolayers, we find that covalent binding to electrodes doubles room temperature conduction and halves its thermal activation energy. At low temperatures, where transport is by tunneling, covalent binding increases conduction up to 10-fold. To examine the electrical conduction across the protein, we used seven different cytochrome c mutants with surface-exposed cysteine, providing distinct electrode–heme orientations and distances. Remarkably, currents do not depend on the electrodes’ separation distance as set by a given protein binding orientation but rather on the distance between the heme and one of the electrodes.
Keywords: bioelectronics, temperature dependence, protein conduction
Abstract
Electronic coupling to electrodes, Γ, as well as that across the examined molecules, H, is critical for solid-state electron transport (ETp) across proteins. Assessing the importance of each of these couplings helps to understand the mechanism of electron flow across molecules. We provide here experimental evidence for the importance of both couplings for solid-state ETp across the electron-mediating protein cytochrome c (CytC), measured in a monolayer configuration. Currents via CytC are temperature-independent between 30 and ∼130 K, consistent with tunneling by superexchange, and thermally activated at higher temperatures, ascribed to steady-state hopping. Covalent protein–electrode binding significantly increases Γ, as currents across CytC mutants, bound covalently to the electrode via a cysteine thiolate, are higher than those through electrostatically adsorbed CytC. Covalent binding also reduces the thermal activation energy, Ea, of the ETp by more than a factor of two. The importance of H was examined by using a series of seven CytC mutants with cysteine residues at different surface positions, yielding distinct electrode–protein(–heme) orientations and separation distances. We find that, in general, mutants with electrode-proximal heme have lower Ea values (from high-temperature data) and higher conductance at low temperatures (in the temperature-independent regime) than those with a distal heme. We conclude that ETp across these mutants depends on the distance between the heme group and the top or bottom electrode, rather than on the total separation distance between electrodes (protein width).
The functional versatility that evolution imparted on proteins rationalizes the interest in their electronic charge-carrying properties for biomolecular electronics. Monolayers on conducting substrates are an appealing way to integrate proteins into possible future biomolecular electronic devices. Such devices will gain in flexibility if some proteins have current-carrying functions, explaining interest, beyond fundamental science, in understanding solid-state electron transport (ETp) characteristics of proteins. To study such ETp characteristics, the protein is sandwiched between and contacted by two electronically conducting, ion-blocking electrodes. The protein’s contacts to the electrodes and its orientation relative to them are important issues. Here we present experimental results that show how ETp via a given protein depends on its binding mode and orientation relative to the electrodes.
The examined protein is horse heart cytochrome c (CytC). Previous studies of intramolecular electron transfer (ET) in CytC used both spectroscopic (1–6) and electrochemical measurements (7–20). In the latter studies, where the rate of ET between the intrinsic redox site and a working electrode has been probed, the CytC was usually attached (covalently or electrostatically) to one electrode via a linker molecule to form a monolayer. Whereas ET processes were studied in aqueous medium, ETp via CytC was investigated in the solid state, using nanometer-scale methods such as scanning tunneling microscopy (STM) (21, 22) or conductive-probe atomic force microscopy (23). Here we used macroscopic-scale electrodes, which allow both characterizing the protein monolayer by a variety of spectroscopic methods and ETp measurements down to low temperatures.
The main goal of this study was to shed further light on the role of the protein’s coupling mode to the electrodes’ surface (Γ) and of the orientation of the bound protein, with respect to the electrodes, to the currents that flow across it. The orientation’s role was examined by using seven different mutants of CytC, where a cysteine (Cys) residue was incorporated at different loci on the protein’s surface. These mutants were bound covalently to the electrode, forming seven different orientations of CytC. By comparing ETp via these mutants with that via WT CytC, which is electrostatically adsorbed on the electrode, the importance of the protein-contact coupling was also investigated.
Results
Earlier studies resolved a large (1–1.5 orders of magnitude) difference in ETp via a given molecule with a single covalent bond compared with having only mechanical contacts with the electrodes as well as between having a single and two covalent bonds (cf. review in ref. 24). The latter difference was also shown recently to hold for ETp via a protein (25). Thus, to probe the contribution of the electronic coupling between the protein and the electrode, Γ, to the ETp process via the protein, we first compared ETp via an electrostatically adsorbed protein layer with that via a covalently bound one. A widely used method to produce a monolayer of CytC is based on using its positive surface charges at neutral pH (isoelectric point ∼10.0) (26) to electrostatically adsorb it onto a carboxylate-terminated linker that, in turn, is bound covalently to the electrode (9, 14–20). For measuring the ETp via adsorbed CytC, we functionalized the surface of high-doped Si with a carboxylate (see Experimental Procedures for further details). The successful formation of the CytC monolayers on the carboxylate-carrying surface was confirmed by ellipsometry (from which an optical thickness of 17–18 Å is deduced, comparable to that found for monolayers of other proteins of this size) (27–29), and by atomic force microscopy. The electronic conductance via the protein at room temperature (RT) was measured with an Hg drop as the top electrode, with an ∼400-µm diameter contact area, yielding rather symmetric current–voltage (I–V) curves (Fig. 1A). The order of magnitude of the measured currents (at RT) is similar (although several-fold lower at negative bias) to what we measured via azurin (Az), a Cu-containing electron-mediating protein with a molecular weight similar to that of CytC, which forms monolayers with similar density (27, 29).
Fig. 1.

ETp via electrostatically or covalently bound CytC monolayers. (A) I–V at RT of monolayers of electrostatically bound CytC, covalently bound CytC (of the A15C and E104C mutants), along with that of WT Az. (B) Current density (measured at 0.05 V, plotted on an ln scale) as a function of inverse temperature (T) of electrostatically bound CytC. (Inset) Fit of the thermally activated regime to 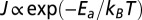 , yielding an activation energy of 105 meV. (C) Comparison between the temperature dependence of the electrostatically and covalently bound CytC monolayers (of the A15C, A51C, and E104C mutants), along with that of (covalently bound) WT Az.
, yielding an activation energy of 105 meV. (C) Comparison between the temperature dependence of the electrostatically and covalently bound CytC monolayers (of the A15C, A51C, and E104C mutants), along with that of (covalently bound) WT Az.
To gain insight into the mechanism(s) of ETp, current densities at a low bias of 50 mV were measured as a function of temperature (Fig. 1B). For the electrostatically adsorbed CytC monolayer two distinct regimes were observed, a temperature-independent (T < 165 K) regime and a thermally activated one (T > 165 K). Analysis of the Arrhenius plot of the latter indicated an activation energy, Ea, of 105 meV (Fig. 1B, Inset). A similar temperature dependence was found for CytC monolayers, formed directly on the Si–H surface (30), yet with much higher current densities due to the absence of both a separating oxide layer and linker in the latter configuration (Fig. S1).
Covalent electrode-bound CytC monolayers were prepared by using mutants with a surface-exposed Cys residue, known to have no effect on the protein’s structure. These were bound covalently to one electrode via a thiolate-terminated linker, a procedure that we used earlier to form monolayers of Az (27, 29). It has been argued that the electrostatically bound CytC monolayer has a preferred orientation, where the heme of the CytC is located proximal (<1 nm) to the carboxylate-terminated surface (15–18). Accordingly, and to focus on the effect of the covalent electrode binding on the ETp process, we first chose a CytC mutant (A15C) that binds with an orientation that can be assumed to be similar to that of the electrostatically bound CytC. The currents through such a monolayer were measured as a function of temperature at 50-mV bias and compared with those of the electrostatically bound protein. As seen in Fig. 1C, the covalent bond of the protein to the surface increases the current density, J, especially in the temperature-independent regime, and also decreases the thermal activation energy in the high-temperature regime.
The results obtained from the measurements using the CytC Cys mutants also allow a better comparison between the behavior of CytC and Az (Fig. 1A). The intrinsic Cys residues of Az allow forming monolayers by covalent binding to the surface of an Au electrode (S–Au bond) (31) or via a thiolate-carrying linker (S–S bond) (27, 29). In the produced orientation, the Az redox center, the Cu ion is very close (∼0.7–0.8 nm) to the top electrode. Thus, to compare between Az and CytC, we used a CytC mutant (E104C) that binds covalently to one (bottom) electrode in such a way that its heme is proximal to the top electrode (0.8–0.9 nm between the Fe ion and the physical top contact; 0.4 nm from the nearest heme edge to this contact). As can be seen in Fig. 1C, the current through this CytC mutant is nearly temperature-independent over the whole examined range, closely resembling the complete temperature-independent currents via Az and clearly distinct from what we observed using electrostatically bound CytC.
To further explore the impact of the separation between the electrodes dictated by the protein and the orientation of the protein relative to the electrodes on the ETp, five additional CytC mutants were used. All of the mutants used, seven in total, differ from each other only in the surface location of the introduced Cys residue. The ETp measurements of the mutants and their temperature dependencies (Fig. S2) show several differences in current density and in the activation energy in the thermally activated regime (Table 1). Comparison of the ETp via all of the mutants with that via the electrostatically adsorbed protein (Table 1) shows that covalent binding increases the current density via all of the mutants in the low temperature-independent regime and significantly lowers the activation energy in the thermally activated one.
Table 1.
Location of the heme cofactor with respect to the electrodes, the current densities at RT and 30 K, and the activation energy in the high-temperature range of the mutants and WT
| Mutant | Distance Cys–Fe, Å | Distance Fe–top electrode, Å* | Current at 297K, A/cm2† | Current at 30 K, A/cm2† | Ea, meV‡ |
| WT | <11§ | ∼19§ | 2.1 × 10−6 | 1.6 × 10−7 | 105 |
| A51C | 14.8 | 18.8 | 3.2 × 10−6 | 8.8 × 10−7 | 46 |
| G23C | 19.5 | 14.8 | 1.2 × 10−6 | 3.2 × 10−7 | 37 |
| G56C | 18.2 | 15.1 | 3.1 × 10−6 | 1.6 × 10−6 | 34 |
| A15C | 10.9 | 15.6 | 2.2 × 10−6 | 1.3 × 10−6 | 16 |
| G37C | 19.6 | 9.2 | 3.9 × 10−6 | 1.9 × 10−6 | 16 |
| V11C | 12.5 | 15.9 | 2.6 × 10−6 | 1.6 × 10−6 | 15 |
| E104C | 19.1 | 8.7 | 4.9 × 10−6 | 3.9 × 10−6 | 10 |
The distance was calculated as the average between the Fe ion and those amino acids that are most likely contacting the top electrode.
Currents were measured at 0.05 V.
The SD for the calculated Ea is ±6 meV.
In an electrochemical study, Bortolotti et al. (8) have shown that different orientations of three horse heart CytC mutants yield different ET rate constants, kET. In their measurements, the ET process from the heme (donor; D) to the electrode (which acts as the acceptor; A) was monitored. Their working hypothesis was that the different D–A distances, set by the different binding orientations of the protein, will affect the kET. They found a good correlation between the measured kET and the D–A separation distance of their three mutants (8). Interestingly, and in contrast to these electrochemical results, the present results show no correlation between the measured currents and the separation distance of the electrodes, either at RT or at 30 K (Fig. 2).
Fig. 2.

Current density (measured at 0.05 V) as a function of the length of the protein, separating the electrodes (sum of bottom electrode–Fe and Fe–top electrode distances), at RT and 30 K (Upper and Lower, respectively). The overlapping entries in the upper part of the figure are those of A51C and G56C.
Discussion
The use of macroscopic electrodes used here allows probing the difference in the ETp via proteins with one covalently bonded protein–electrode contact from that with no covalent contact, even at very low temperatures. The importance of this approach can be appreciated when comparing the marked impact on the ETp via cytochrome b562 (Cyt b562) achieved by STM measurements having one or both electrodes covalently bonded to the protein. Bonds to the different Cys residues in the Cyt b562 mutants produced distinct protein orientations with respect to the electrodes (25, 32), as the anisotropic structure of Cyt b562 allowed probing ETp via its long (5.2 nm) and short axis (2.4 nm). Bonding was made via Cys residues to the Au tip and/or Au substrate. Although the electrodes’ separation using the long-axis configuration is more than twice that of the short one, with two covalently bonded contacts, only a small difference in conductance was observed between the two configurations. However, no current could be measured if, for the long-axis configuration, one of the covalent bonds was removed, and one can only estimate, based on the experimental noise level, an upper bound to the current with one covalently bound contact. According to our estimates there is at least a 10- to 30-fold decrease in current, similar to the change in ETp across alkyl thiols and dithiols (one vs. two covalently bound contacts) (24). (Taking together the results of Cyt b562 and the ones in the current study, how the protein contacts the electrode in biomolecular electronics is of prime importance, especially in protein layers, which exhibit an ill-defined surface structure and relatively poor surface coverage.) The Cyt b562 results emphasize the low current detection limits of STM-based measurements caused by their nanometer-sized contact area, especially at the low-bias voltages, required for having the system close to equilibrium.
Activation Energies and I–V Asymmetry.
As seen in Table 1, the calculated activation energies, Ea, of all of the covalently bound CytC mutants are significantly lower than that of the electrostatically adsorbed WT CytC (cf. also Fig. 1C and Fig. S2). A plausible explanation is that the covalent bond to the electrode lowers the ETp barrier between the protein and the contact. This is consistent with the higher currents observed via the covalently bound CytC mutants (except for G23C and A15C at RT) than via the electrostatically adsorbed one (Table 1). It is essential to note that the barrier for ETp across WT CytC is influenced and possibly dominated by the protein’s contact to the electrode, and as such it is distinct from the Ea of ET reactions between a donor and acceptor (usually determined by the driving force and reorganization energy), as treated by Marcus theory. In this context, it is important to note that we have previously shown that in contrast to an electrochemical process, ETp across a protein does not require a redox process (33).
Based on the Ea values calculated from our results, the mutants can be roughly divided into two groups. The first, A15C, G37C, E104C, and V11C, comprises those with very low Ea values (Ea ≤ 16 meV), and the other, A51C, G23C, and G56C, includes those with ∼30–50 meV values. The differences in Ea between the two groups are rather small (only a factor of 2–3), as may be expected from ETp measurements via the same protein, differing only in orientation relative to the electrodes. These variations in Ea can be rationalized primarily by the distance and orientation of their heme groups with respect to the electrodes (Table 1 and Table S1). These distances are illustrated in Fig. 3, showing the molecular schemes of the mutants along with the calculated distances between the heme and the electrodes. Grosso modo, in the low-Ea group, the heme is located closer to either the top or the bottom electrode, whereas in the higher-Ea group it is situated approximately midway between them (cf. also Table 1).
Fig. 3.

Molecular schemes of the different CytC mutants and WT Az, bound to the bottom electrode. The calculated distances between the point of the Cys thiolate bound to the bottom electrode and the heme cofactor, and from the heme cofactor to the upper end of the protein (in contact with the top electrode), are shown. For Az the distances are those between the Cu(II) center and the thiolate (Cys3-Cys26) (bottom electrode) or to the uppermost part of the protein.
An additional indication for the importance of the heme location for the ETp via the protein is given by the direction and magnitude of current rectification. Fig. S3 shows that the normalized I–V characteristics (the absolute currents were divided by the highest one for each characteristic) of the mutants are slightly rectifying, with higher currents at negative biases when the heme is closer to the top electrode (E104C and G37C; Fig. S3A). The opposite effect is seen for the mutants where the heme is closer to the bottom electrode (A15C and V11C; Fig. S3B). For the mutants where the heme is roughly midway between the two electrodes (A51C, G23C, and G56C), more symmetric I–V curves are observed (Fig. S3C).
Role of Heme–Electrode Proximity.
As noted above, ETp via Az, which is covalently bound to the electrode by its Cys thiolates, has been found to be temperature-independent (Ea = 0). In this configuration, the Cu ion is very close (∼0.7–0.8 nm) to the top electrode (Fig. 3). Among all of the CytC mutants studied, E104C shows the lowest temperature ETp dependence (cf. Fig. 1C and Fig. S2) and, hence, the lowest Ea (Table 1). Significantly, E104C is one of the two CytC mutants (the other is G37C) that has its heme closest to the top electrode (∼0.8–0.9 nm from the Fe; cf. Table 1; ∼0.4 nm from the closest heme edge; cf. Table S1). It is also the one most similar to Az in terms of rectification (Fig. S3D). This similarity suggests that temperature dependence (and currents at negative bias; cf. Fig. 1A) reflects a difference in electrode–cofactor coupling when the protein’s other end is covalently bound to the electrode. Because the contacts of both Az and E104C to the electrode, proximal to the cofactor, are not covalent but rather physical, these similarities underscore the importance of electrode–cofactor proximity for efficient ETp via proteins.
Current Dependence on Electrodes’ Separation Distance.
Assuming that the conjugated heme serves as the main charge mediator within the protein, we consider the electrodes’ separation distance as the sum of the distances between the Cys mutation and the Fe ion and the averaged distance between the Fe ion and the surface amino acid residues that are most likely to contact the top electrode (Fig. 3, Table 1, and Table S1). (We use these distances as a first approximation only, because what matters, apart from the net distance, is electronic overlap, in terms of energies and symmetries.) It was shown previously that due to the high surface tension of liquid Hg serving as the top electrode, the real contact area between Hg and a surface is limited to asperities, namely only a small fraction of the geometrical contact area (34). Thus, it is very likely that the Hg drop contacts in each case only the most exposed amino acids. The lack of correlation between the measured current densities and the calculated electrode separation distances (protein monolayer widths) is in contrast to results obtained from ETp studies via peptides (35–37) and peptide nucleic acids (38). However, none of the latter molecules contain an internal cofactor, like CytC and Az do. We also do not find a clear correlation when the distances from/to the electrodes and of the closest heme edge to the electrode are considered (see further discussion in SI Discussion, Fig. S4 and Table S1).
The ETp process can be divided into two parts, assuming the heme plays a key role in the process: from the bottom electrode, which is covalently bound to the protein via the Cys thiolate to the heme, and from the heme to the top electrode. In this case, one could argue that the rate-determining step would be the one across the longest distance. However, also in this case, no correlation was found between the measured current density and the net distance over which transport occurs (Fig. S5). A likely physical interpretation of these observations is discussed below.
Current Magnitudes.
The lack of correlation between the measured currents and the total width of the protein monolayer in the junction holds for currents both at RT and at low temperatures (<170 K). This is important because the RT ETp process is thermally activated, likely because transport is by a hopping mechanism, whereas at <170 K the temperature-independent currents suggest that ETp is by superexchange (Fig. 2). This lack of correlation between ETp and protein monolayer width (electrode separation) underscores that different orientations of the same protein can present distinct charge transport media between the two electrodes. This can also be related to the cofactor position with respect to the electrodes because, as we have shown previously, cofactors are crucial mediators in ETp, even though, in contrast to ET, they do not need to be redox-active to achieve efficient ETp (28, 33).
However, unlike the effect of the heme location on the measured Ea (as discussed above in terms of two groups of mutants), it is less straightforward to classify the mutants in terms of the current magnitudes they carry. In general, our results show that in addition to the importance of coupling to the electrodes, set by the presence/absence of chemical bonding between protein and substrate, the closer the cofactor (either the Fe or the heme edge) is to one of the electrodes, the more efficient is the ETp via the protein (Fig. S6). Nevertheless, it seems that parameters beyond simple geometric ones dictate the ETp dependence on orientation. The two observations that support this conclusion are as follows.
-
i)
The linear correlation between conductance and length of the separating protein is observed only when the heme is relatively distal from the electrode, and only at low temperature (Fig. 2), for example, when ETp is likely by superexchange (as can be observed also in Fig. S4, where the heme edges were used for the distance calculations).
-
ii)
The fact that the currents via E104C at 30 K and at RT (and also of G37C at RT) are higher than those via the other mutants, which may well be related not only to the proximity but also to the orientation of the heme group relative to the noncovalently bound electrode (Table 1 and Table S1).
We can now speculate on how proximity (and, so one assumes, electronic coupling) between the heme and one of the electrodes diminishes the contribution of the remainder of the transport path, because the longer heme–electrode distance does not seem to dictate ETp efficiency in the mutants of the first group.
A possible explanation can be that when tunneling from one of the contacts to the heme group is very efficient, compared with the ETp from the heme to the other contact, the heme does not have time to relax electronically to the energy state (energy well), expected for steady-state carrier hopping (39), known from solid-state electronics (40). Then, as only close electrode–heme proximity, combined with good electronic coupling, allows efficient tunneling, this step will affect the second one, because the heme state, associated with transient hopping, will have a higher electronic energy than that of its relaxed state, associated with steady-state hopping (39). This speculation is supported by the results of Albrecht et al. (41) using electrochemical STM, and might be relevant for other molecular electronic results (42, 43).
Conclusions
The temperature dependence of solid-state ETp via CytC, measured as covalently bound or electrostatically adsorbed to the surface of one of the two electrodes, exhibits two regimes. One is a thermally activated one (at T > 185 K for the covalent and T > 165 K for the electrostatically adsorbed proteins), which can be ascribed to steady-state hopping ETp. The second is temperature-independent at lower temperatures, consistent with tunneling by superexchange. Binding the protein covalently to one electrode increases the current density in the temperature-independent regime and lowers the activation energy of the thermally activated one, showing the importance of the coupling between the protein and electrodes. To examine ETp across the protein and the role of the heme in the process, a series of seven different CytC mutants with surface Cys residues was used, providing distinct electrode–protein(–heme) orientations. These mutants were found to exhibit different ETp patterns in terms of current densities and activation energies. Surprisingly, no general correlation was observed between the measured currents via the mutants and their monolayer width, namely the electrodes’ separation distance, dictated for each mutant by its unique Cys–electrode attachment point, which also determines its orientation relative to the electrodes. Also, no simple correlation is seen between currents and the electrode–heme separation distance although, in general, ETp is more efficient for mutants having the heme closest to an electrode. This suggests that cofactor–electrode proximity (and, likely, cofactor–electrode coupling, which can also occur without a covalent bond) is important for determining solid-state ETp efficiency via the protein junction. The importance of the latter notion was illustrated by the fact that the mutant that has its cofactor closest to an electrode has the lowest temperature dependence of the ETp, in analogy to Az, which has its Cu site <1 nm from one of the electrodes and shows temperature-independent ETp.
Experimental Procedures
Preparation of CytC Mutants.
The horse heart genes for the CytC mutants were engineered using site-specific mutagenesis. The QuikChange Site-Directed Mutagenesis Kit (Stratagene) was used for the experiments. The mutagenesis procedure included synthesis of the full-length single-stranded DNA plasmid by means of the thermostable DNA polymerase Pfu, using two complementary primers (∼30–35 nt) that contained the necessary substitutions. The substitutions were localized in the middle part of the sequence, flanked by ∼15 nt. All of the mutant genes were sequenced to confirm their primary structure. The mutated genes were cloned into expression system pBP(CYC1) (44) for yeast CytC and modified for horse CytC (45). The system includes coexpression of the CytC gene and the yeast heme lyase gene. The mutant CytC expression vectors were expressed in Escherichia coli strain JM 109 for 20–22 h at 37 °C in rich super broth media. The mutant proteins were purified from the supernatant obtained after cell disruption by a French press and centrifugation (100,000 × g for 20 min at 4 °C) using two steps of liquid chromatography: cation exchange on a Mono-S column and adsorption on a hydroxyapatite column (45). Final purity of the obtained cytochrome c mutants was >95% according to SDS/PAGE. The expression efficiency of the CytC reached up to 20 mg of the heme-containing cytochrome c per L of culture.
Before the deposition on the surface, and to reduce the exposed Cys residue, 15 μL of Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) from a stock solution of 1 M TCEP (pH 7.5), was added to ∼1.5 mL of the CytC mutant. Following ∼20 min of incubation, the excess TCEP was removed by eluting the protein with a Sephadex G-50 column by using 0.05 M phosphate buffer (pH 8). The final concentration of the CytC mutant was ∼1 mg/mL.
CytC Monolayers on Si.
The highly doped (<0.001 Ω·cm) <100> oriented p-type Si surface was cleaned by bath sonication by placing the Si surface in a vial containing ethyl acetate/acetone/ethanol (2 min in each), followed by 30 min of piranha treatment [7/3 (vol/vol) H2SO4/H2O2] at 80 °C. The Si surface was then thoroughly rinsed in Milli-Q (18 MΩ) water (Millipore) and dipped in 2% (vol/vol) HF solution for 90 s to etch the Si surface (leaving an Si–H surface). For controlled growth of a thin oxide layer (9–10 Å), the etched Si surface was put in a fresh piranha solution for 4–5 s and then immediately rinsed thoroughly with Milli-Q water and dried under a nitrogen stream. For preparation of the electrostatically adsorbed CytC monolayers, a monolayer of 3-aminopropyl trimethoxysilane (3-APTMS; NH2-terminated linker, 97%; Sigma-Aldrich) was prepared by immersing the above SiO2 substrate for ∼4 h in a 10% 3-APTMS in methanol solution, followed by 3 min of bath sonication in methanol, yielding a monolayer thickness of ∼5–6 Å. The 3-APTMS decorations were extended to exhibit a carboxylate-terminated surface by placing the surfaces in saturated succinic anhydride (≥99%; Sigma-Aldrich) in dimethylformamide (DMF) containing 1 mg/mL of triethylamine for 2 h followed by 3 min of bath sonication in DMF, yielding an overall monolayer thickness of 8–9 Å. The latter surfaces were immersed in a sealed vial containing ∼1 mg/mL wild-type horse heart CytC in 0.05 M phosphate buffer (pH 8). For preparation of the covalently bound CytC surfaces, a monolayer of 3-mercaptopropyl trimethoxysilane (3-MPTMS; SH-terminated linker, 95%; Sigma-Aldrich) was prepared by immersing the SiO2 substrate in 10 mM 3-MPTMS in bicyclohexyl for 1 h, followed by 3 min of bath sonication in acetone and 10 s in hot ethanol, yielding a monolayer thickness of ∼7 Å. The latter surfaces were immersed in a sealed vial containing ∼1 mg/mL reduced CytC mutant.
Ellipsometry Measurements.
Ellipsometry measurements were performed with a Woollam M-2000 V multiple-wavelength ellipsometer at an angle of incidence of 70°. The Cauchy model was used to estimate the thickness of the organic layers (the linker and the protein).
Current–Voltage Measurements.
The top contact was made by placing a drop of Hg by capillary on top of the protein monolayer. InGa was used as a back contact by scratching the back side of the Si surface and rubbing an InGa eutectic paste onto it. The top Hg contact was biased, and the back contact was grounded. For temperature-controlled measurements, the sample was placed in a low-vacuum (0.1 mbar) chamber in a TTPX cryogenic four-probe electrical measurement system (Lake Shore), and both sample holder and probes were cooled. The temperature was monitored and controlled ±0.2 K.
Supplementary Material
Acknowledgments
We are very grateful to A. Kotlyar (Tel Aviv University) for his help with the mutants; Y. Barak for help with FPLC; D. Marchak, S. Mukhopadhyay, L. Sepunaru, and S. Raichlin for stimulating discussions; and the reviewers for constructive comments. We are grateful to the Minerva Foundation (Munich), The Nancy and Stephen Grand Centre for Sensors and Security, and The Kimmelman Center for Biomolecular Structure and Assembly for partial support. M.S. holds the Katzir-Makineni Chair in Chemistry. D.C. holds the Schaefer Chair in Energy Research.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1319351111/-/DCSupplemental.
References
- 1.Nocera DG, Winkler JR, Yocom KM, Bordignon E, Gray HB. Kinetics of intermolecular and intramolecular electron transfer from ruthenium(II) complexes to ferricytochrome c. J Am Chem Soc. 1984;106(18):5145–5150. [Google Scholar]
- 2.Therien MJ, Selman M, Gray HB, Chang IJ, Winkler JR. Long-range electron transfer in ruthenium-modified cytochrome c: Evaluation of porphyrin-ruthenium electronic couplings in the Candida krusei and horse heart proteins. J Am Chem Soc. 1990;112(6):2420–2422. [Google Scholar]
- 3.Meade TJ, Gray HB, Winkler JR. Driving-force effects on the rate of long-range electron transfer in ruthenium-modified cytochrome c. J Am Chem Soc. 1989;111(12):4353–4356. [Google Scholar]
- 4.Chance B. The interaction of energy and electron transfer reactions in mitochondria. II. General properties of adenosine triphosphate-linked oxidation of cytochrome and reduction of pyridine nucleotide. J Biol Chem. 1961;236(5):1544–1554. [PubMed] [Google Scholar]
- 5.DeVault D, Chance B. Studies of photosynthesis using a pulsed laser. I. Temperature dependence of cytochrome oxidation rate in chromatium. Evidence for tunneling. Biophys J. 1966;6(6):825–847. doi: 10.1016/s0006-3495(66)86698-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimura M, Roy SB, Schleyer H, Chance B. Studies on the electron-transfer systems in photosynthetic bacteria. IV. Kinetics of light-induced cytochrome reactions and analysis of electron-transfer paths. Biochim Biophys Acta. 1964;88(2):251–266. doi: 10.1016/0926-6577(64)90181-0. [DOI] [PubMed] [Google Scholar]
- 7.Tarlov MJ, Bowden EF. Electron-transfer reaction of cytochrome-c adsorbed on carboxylic-acid terminated alkanethiol monolayer electrodes. J Am Chem Soc. 1991;113(5):1847–1849. [Google Scholar]
- 8.Bortolotti CA, et al. Orientation-dependent kinetics of heterogeneous electron transfer for cytochrome c immobilized on gold: Electrochemical determination and theoretical prediction. J Phys Chem C. 2007;111(32):12100–12105. [Google Scholar]
- 9.Avila A, Gregory BW, Niki K, Cotton TM. An electrochemical approach to investigate gated electron transfer using a physiological model system: Cytochrome c immobilized on carboxylic acid-terminated alkanethiol self-assembled monolayers on gold electrodes. J Phys Chem B. 2000;104(12):2759–2766. [Google Scholar]
- 10.Cooper JM, Greenough KR, McNeil CJ. Direct electron transfer reactions between immobilized cytochrome c and modified gold electrodes. J Electroanal Chem. 1993;347(1–2):267–275. [Google Scholar]
- 11.El Kasmi A, et al. Adsorptive immobilization of cytochrome c on indium/tin oxide (ITO): Electrochemical evidence for electron transfer-induced conformational changes. Electrochem Commun. 2002;4(2):177–181. [Google Scholar]
- 12.El Kasmi A, Wallace JM, Bowden EF, Binet SM, Linderman RJ. Controlling interfacial electron-transfer kinetics of cytochrome c with mixed self-assembled monolayers. J Am Chem Soc. 1998;120(1):225–226. [Google Scholar]
- 13.Ji XP, Jin BK, Jin JY, Nakamura T. Voltammetry of immobilized cytochrome c on novel binary self-assembled monolayers of thioctic acid and thioctic amide modified gold electrodes. J Electroanal Chem. 2006;590(2):173–180. [Google Scholar]
- 14.Song S, Clark RA, Bowden EF, Tarlov MJ. Characterization of cytochrome-c alkanethiolate structures prepared by self-assembly on gold. J Phys Chem. 1993;97(24):6564–6572. [Google Scholar]
- 15.Wei JJ, et al. Electron-transfer dynamics of cytochrome c: A change in the reaction mechanism with distance. Angew Chem Int Ed Engl. 2002;41(24):4700–4703. doi: 10.1002/anie.200290021. [DOI] [PubMed] [Google Scholar]
- 16.Yue H, et al. On the electron transfer mechanism between cytochrome c and metal electrodes. Evidence for dynamic control at short distances. J Phys Chem B. 2006;110(40):19906–19913. doi: 10.1021/jp0620670. [DOI] [PubMed] [Google Scholar]
- 17.Nakano K, Yoshitake T, Yamashita Y, Bowden EF. Cytochrome c self-assembly on alkanethiol monolayer electrodes as characterized by AFM, IR, QCM, and direct electrochemistry. Langmuir. 2007;23(11):6270–6275. doi: 10.1021/la063697w. [DOI] [PubMed] [Google Scholar]
- 18.Loftus AF, Reighard KP, Kapourales SA, Leopold MC. Monolayer-protected nanoparticle film assemblies as platforms for controlling interfacial and adsorption properties in protein monolayer electrochemistry. J Am Chem Soc. 2008;130(5):1649–1661. doi: 10.1021/ja076312k. [DOI] [PubMed] [Google Scholar]
- 19.Yue H, et al. Multiple sites for electron tunneling between cytochrome c and mixed self-assembled monolayers. J Phys Chem C. 2008;112(7):2514–2521. [Google Scholar]
- 20.Lee JB, Kim DJ, Choi JW, Koo KK. Preparation of a self-assembled cytochrome c monolayer on a gold substrate for biomolecular device architecture. Mater Sci Eng C. 2004;24(1–2):79–81. [Google Scholar]
- 21.Khomutov GB, et al. STM study of morphology and electron transport features in cytochrome c and nanocluster molecule monolayers. Bioelectrochemistry. 2002;55(1–2):177–181. doi: 10.1016/s1567-5394(01)00135-9. [DOI] [PubMed] [Google Scholar]
- 22.Morimoto J, Tanaka H, Kawai T. Direct measurement of electron transport features in cytochrome c via V–I characteristics of STM currents. Surf Sci. 2005;580(1–3):L103–L108. [Google Scholar]
- 23.Davis JJ, Peters B, Xi W. Force modulation and electrochemical gating of conductance in a cytochrome. J Phys Condens Matter. 2008;20(37):374123. doi: 10.1088/0953-8984/20/37/374123. [DOI] [PubMed] [Google Scholar]
- 24.Akkerman HB, de Boer B. Electrical conduction through single molecules and self-assembled monolayers. J Phys Condens Matter. 2008;20(1):013001. [Google Scholar]
- 25.Della Pia EA, Elliott M, Jones DD, Macdonald JE. Orientation-dependent electron transport in a single redox protein. ACS Nano. 2012;6(1):355–361. doi: 10.1021/nn2036818. [DOI] [PubMed] [Google Scholar]
- 26.Lvov Y, Ariga K, Ichinose I, Kunitake T. Assembly of multicomponent protein films by means of electrostatic layer-by-layer adsorption. J Am Chem Soc. 1995;117(22):6117–6123. [Google Scholar]
- 27.Ron I, et al. Proteins as electronic materials: Electron transport through solid-state protein monolayer junctions. J Am Chem Soc. 2010;132(12):4131–4140. doi: 10.1021/ja907328r. [DOI] [PubMed] [Google Scholar]
- 28.Amdursky N, Pecht I, Sheves M, Cahen D. Doping human serum albumin with retinoate markedly enhances electron transport across the protein. J Am Chem Soc. 2012;134(44):18221–18224. doi: 10.1021/ja308953q. [DOI] [PubMed] [Google Scholar]
- 29.Sepunaru L, Pecht I, Sheves M, Cahen D. Solid-state electron transport across azurin: From a temperature-independent to a temperature-activated mechanism. J Am Chem Soc. 2011;133(8):2421–2423. doi: 10.1021/ja109989f. [DOI] [PubMed] [Google Scholar]
- 30.Amdursky N, Pecht I, Sheves M, Cahen D. Electron transport via cytochrome c on Si-H surfaces: Roles of Fe and heme. J Am Chem Soc. 2013;135(16):6300–6306. doi: 10.1021/ja4015474. [DOI] [PubMed] [Google Scholar]
- 31.Li W, et al. Temperature and force dependence of nanoscale electron transport via the Cu protein azurin. ACS Nano. 2012;6(12):10816–10824. doi: 10.1021/nn3041705. [DOI] [PubMed] [Google Scholar]
- 32.Della Pia EA, et al. Fast electron transfer through a single molecule natively structured redox protein. Nanoscale. 2012;4(22):7106–7113. doi: 10.1039/c2nr32131a. [DOI] [PubMed] [Google Scholar]
- 33.Amdursky N, Ferber D, Pecht I, Sheves M, Cahen D. Redox activity distinguishes solid-state electron transport from solution-based electron transfer in a natural and artificial protein: Cytochrome C and hemin-doped human serum albumin. Phys Chem Chem Phys. 2013;15(40):17142–17149. doi: 10.1039/c3cp52885e. [DOI] [PubMed] [Google Scholar]
- 34.Timsit RS. The true area of contact at a liquid metal-solid interface. Appl Phys Lett. 1982;40(5):379–381. [Google Scholar]
- 35.Juhaniewicz J, Sek S. Peptide molecular junctions: Distance dependent electron transmission through oligoprolines. Bioelectrochemistry. 2012;87:21–27. doi: 10.1016/j.bioelechem.2011.11.013. [DOI] [PubMed] [Google Scholar]
- 36.Pawlowski J, Juhaniewicz J, Tymecka D, Sek S. Electron transfer across α-helical peptide monolayers: Importance of interchain coupling. Langmuir. 2012;28(50):17287–17294. doi: 10.1021/la302716n. [DOI] [PubMed] [Google Scholar]
- 37.Sek S, Misicka A, Swiatek K, Maicka E. Conductance of alpha-helical peptides trapped within molecular junctions. J Phys Chem B. 2006;110(39):19671–19677. doi: 10.1021/jp063073z. [DOI] [PubMed] [Google Scholar]
- 38.Wierzbinski E, et al. The single-molecule conductance and electrochemical electron-transfer rate are related by a power law. ACS Nano. 2013;7(6):5391–5401. doi: 10.1021/nn401321k. [DOI] [PubMed] [Google Scholar]
- 39.Segal D, Nitzan A, Davis WB, Wasielewski MR, Ratner MA. Electron transfer rates in bridged molecular systems 2. A steady-state analysis of coherent tunneling and thermal transitions. J Phys Chem B. 2000;104(16):3817–3829. [Google Scholar]
- 40.Quéré Y. Physics of Materials. Amsterdam: Gordon and Breach; 1998. [Google Scholar]
- 41.Albrecht T, Guckian A, Kuznetsov AM, Vos JG, Ulstrup J. Mechanism of electrochemical charge transport in individual transition metal complexes. J Am Chem Soc. 2006;128(51):17132–17138. doi: 10.1021/ja066213r. [DOI] [PubMed] [Google Scholar]
- 42.Arielly R, Vadai M, Kardash D, Noy G, Selzer Y. Real-time detection of redox events in molecular junctions. J Am Chem Soc. 2014;136(6):2674–2680. doi: 10.1021/ja412668f. [DOI] [PubMed] [Google Scholar]
- 43.Tran E, Cohen AE, Murray RW, Rampi MA, Whitesides GM. Redox site-mediated charge transport in a Hg-SAM//Ru(NH(3))(6)(3+/2+)//SAM-Hg junction with a dynamic interelectrode separation: Compatibility with redox cycling and electron hopping mechanisms. J Am Chem Soc. 2009;131(6):2141–2150. doi: 10.1021/ja804075y. [DOI] [PubMed] [Google Scholar]
- 44.Pollock WBR, Rosell FI, Twitchett MB, Dumont ME, Mauk AG. Bacterial expression of a mitochondrial cytochrome c. Trimethylation of Lys72 in yeast iso-1-cytochrome c and the alkaline conformational transition. Biochemistry. 1998;37(17):6124–6131. doi: 10.1021/bi972188d. [DOI] [PubMed] [Google Scholar]
- 45.Abdullaev ZKh, et al. A cytochrome c mutant with high electron transfer and antioxidant activities but devoid of apoptogenic effect. Biochem J. 2002;362(Pt 3):749–754. doi: 10.1042/0264-6021:3620749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.