

Significance
Structural immunology employs static, atomic-level representations to uncover the molecular mechanisms by which immune receptors sensitively discern infection and disease. Lipid membranes, particularly those exposing phosphatidylserine lipids, are now understood to be important signals in various aspects of immune regulation. However, the dynamic nature of lipid membranes makes the standard tools of structural immunology poorly suited to these systems, leaving a large gap in our understanding. Here we implement a suite of multidisciplinary tools to demonstrate, for the first time to the authors' knowledge, a mechanism for sensitivity of an immune regulatory receptor (Tim4) to the membrane context of phosphatidylserine exposure. These findings uncover new aspects of Tim4’s recognition properties and, perhaps more significantly, provide a methodology for obtaining atomic-level detail in membrane recognition.
Keywords: differential membrane recognition, PS recognition
Abstract
Recognition of phosphatidylserine (PS) lipids exposed on the extracellular leaflet of plasma membranes is implicated in both apoptotic cell removal and immune regulation. The PS receptor T cell immunoglobulin and mucin-domain-containing molecule 4 (Tim4) regulates T-cell immunity via phagocytosis of both apoptotic (high PS exposure) and nonapoptotic (intermediate PS exposure) activated T cells. The latter population must be removed at lower efficiency to sensitively control immune tolerance and memory cell population size, but the molecular basis for how Tim4 achieves this sensitivity is unknown. Using a combination of interfacial X-ray scattering, molecular dynamics simulations, and membrane binding assays, we demonstrate how Tim4 recognizes PS in the context of a lipid bilayer. Our data reveal that in addition to the known Ca2+-coordinated, single-PS binding pocket, Tim4 has four weaker sites of potential ionic interactions with PS lipids. This organization makes Tim4 sensitive to PS surface concentration in a manner capable of supporting differential recognition on the basis of PS exposure level. The structurally homologous, but functionally distinct, Tim1 and Tim3 are significantly less sensitive to PS surface density, likely reflecting the differences in immunological function between the Tim proteins. These results establish the potential for lipid membrane parameters, such as PS surface density, to play a critical role in facilitating selective recognition of PS-exposing cells. Furthermore, our multidisciplinary approach overcomes the difficulties associated with characterizing dynamic protein/membrane systems to reveal the molecular mechanisms underlying Tim4’s recognition properties, and thereby provides an approach capable of providing atomic-level detail to uncover the nuances of protein/membrane interactions.
Immunological recognition of the lipid phosphatidylserine (PS) has predominately been associated with the phagocytic removal of apoptotic cells (1). In a healthy cell, PS is typically confined to the intracellular leaflet of the plasma membrane by ATP-dependent, lipid-sorting processes (2). Apoptosis induces extracellular exposure of PS, whereupon immune scavenger cells bearing PS-specific receptors can recognize and remove the dying cells (1). This seemingly simple paradigm is complicated by the fact that extracellular PS exposure is not just a hallmark of cell death but is also functionally relevant during the activation of various immune cells, including T cells (3–7). Moreover, there exists a wide array of structurally diverse PS receptors, the expression of which is not strictly limited to immune cells capable of phagocytosis (8, 9). Accordingly, recognition of exposed PS is not always sufficient for phagocytosis (10) and can actually elicit a nonphagocytic response, such as immune cell proliferation (11).
Thus, the question remains: How does the immune system appropriately interpret apoptotic versus nonapoptotic PS exposure? Additional factors, such as the “don’t eat me” signals CD31 (12) and CD47 (13, 14), can, in some cases, modulate the immune response to PS exposure. However, it remains unclear whether all PS receptors recognize any instance of exposed PS equally or whether the physiological context of PS exposure can influence recognition. Addressing this central question has been exceedingly difficult, given that the standard tools of structural immunology are not well suited to characterizing dynamic membrane systems. Nevertheless, it appears that the ability to discern apoptotic versus nonapoptotic instances of PS exposure may indeed be a required feature of at least one PS receptor, T cell immunoglobulin and mucin-domain-containing molecule 4 (Tim4) (15, 16).
Tim4 is a member of the Tim family of proteins that has collectively been associated with a variety of immune regulatory disorders including allergy, asthma, autoimmunity, and transplant intolerance (9, 17, 18). Correspondingly, each of the Tim proteins, which includes Tim1–4 in mouse and the homologous Tim1, 3, and 4 in humans (Tim2 is not expressed in humans), are associated with regulating T-cell immunity, although with distinctly different roles (9). Tim1, for example, can provide a costimulatory signal leading to T-cell proliferation (19), whereas Tim4 mediates the recognition of PS-exposing T cells for the purposes of phagocytic removal (15, 16, 20–22). The crystal structure of Tim4 in complex with a single-PS head group has provided invaluable insight into how Tim4 specifically recognizes a single-PS molecule (23). However, the molecular details of how Tim4 recognizes PS within the context of a lipid membrane remain unknown.
It is now apparent that these details may be critical to understanding the immunological function of Tim4. Tim4 has recently been shown to regulate T-cell populations during the resolution of an immune response by mediating the PS-dependent phagocytosis of both apoptotic and nonapoptotic antigen-specific T cells (15, 16). In one capacity, Tim4 initiates the highly selective and thorough clearance of apoptotic T cells by virtue of the exposure of high levels of PS on their surfaces (15, 20, 21). Equally important, however, is that Tim4 is also instrumental in pruning the population size of activated, but nonapoptotic, T cells. This activated T-cell removal is important for both immune tolerance in the lungs (16) and the maintenance of an appropriately sized pool of memory T cells after viral infection (15). Although nonapoptotic T-cell removal is believed to occur via the same PS-dependent mechanisms as apoptotic cell phagocytosis, it must necessarily be done with less vigor to ensure the appropriate level of controlled regulation (15, 16).
Activated, nonapoptotic T cells have been shown to expose PS at lower whole-cell surface densities than apoptotic T cells (3, 16, 24), but the role of these differences in level of PS surface exposure is unknown. Importantly, Tim4 ignores healthy, resting T cells, presumably because of their low basal levels of PS exposure. We therefore hypothesized that Tim4 may discriminate among these three states of T cells by virtue of their different levels of cell-surface PS exposure. Given the diversity in immunological function of the Tim proteins, we further hypothesized that Tim4 might have evolved a specific sensitivity to PS surface density distinct from that of other Tim proteins.
In this study, we develop a multidisciplinary approach to overcome the significant technological barriers associated with studying the molecular basis of membrane PS recognition by Tim4. Using a combination of interfacial X-ray scattering, molecular dynamics simulations, and membrane binding assays, we show that in addition to the known Ca2+-dependent, single-PS binding pocket (23), Tim4 also possesses four peripheral basic residues, each capable of relatively weaker ionic interactions with membrane PS lipids. Unlike other multi-PS binding proteins, such as Annexin V, which has a sharply sigmoidal, all-or-none response (25, 26), the structural characteristics of Tim4 produce a gently sigmoidal binding response. The nature of this response confers a functional sensitivity to PS surface density that is well-suited to mediating a differentiated phagocytic response to diverse populations of PS-exposing T cells. The distinctly different sensitivity of both Tim1 and Tim3 support the conclusion that Tim4 has evolved a specific sensitivity to variations in PS surface density to facilitate its unique function. These results offer a new paradigm for understanding immune recognition of PS lipid membranes by demonstrating that key characteristics of the lipid bilayer, such as PS surface density, have the potential to play an integral role in determining the appropriate immunological response.
Results
Tim4 Binding Is Sensitive to PS Surface Density.
The crystal structure of Tim4 in complex with a PS head group analog revealed only a single, Ca2+-dependent PS binding site (23). If indeed Tim4 binds as a monomeric protein to a single, membrane–PS molecule, then the binding affinity should not be sensitive to the surface density of PS, so long as the total amount of PS in solution remains constant. To assess whether Tim4 binding affinity is sensitive to PS surface density, we used tryptophan spectral shift as an assay for binding. The PS recognition domain of murine Tim4 (hereafter simply referred to as Tim4) was assayed for binding on titrations of large unilamellar vesicles (LUVs) comprised of 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS; Fig. 1A) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; Fig. 1A) lipids at a PS mole percentage of either 10% or 30%.
Fig. 1.

Tim4 binding is sensitive to PS surface density. (A) POPS and POPC lipids. (B) Comparison of 170 nM Tim4 binding titration for lipid vesicle systems of 7:3 (circles) and 9:1 (squares) POPC:POPS. Total lipid concentration at any given point is 3× higher for 10% versus 30%, so that the total PS concentration is matched. Lines through data points are the fits to a single-site binding model. Addition of 10 mM EGTA to 90 μM [PS] in the 30% system is shown as x. (C) Surface titration of PS mole fraction at constant total lipid concentration of 600 μM for 170 nM Tim4. Line through points is fit to a Hill binding model. For all data points, error bars denote 1 SD for measurements done at least in triplicate.
To match the total amount of PS in solution between the 10% and 30% systems, each titration point for the 10% PS systems used a threefold higher total lipid concentration relative to that of the 30%. Despite the equivalence in the total amount of PS available for binding between the two systems, binding to 30% PS vesicles had an equilibrium disassociation constant (Kd) ∼sixfold stronger than that of the 10% PS vesicles (Fig. 1B). Addition of 10 mM EGTA to chelate all available calcium significantly reduced binding of Tim4 to 30% PS vesicles from ∼80% to ∼7% bound at 300 μM.
The sensitivity of Tim4 to PS surface density suggests interactions with multiple PS molecules. Moreover, the fact that some weak binding does occur in the absence of Ca2+ (i.e., without binding of PS in the primary, Ca2+-coordinated pocket) further suggests the presence of secondary PS interactions that are independent of Ca2+.
To probe this possibility further, we next assayed Tim4 binding as a function of increasing PS surface density. Here, the total lipid concentration was held constant, and the PS mole percentage was increased from 0% to 30%. The fraction of total protein bound was measured as a function of PS surface density, and the data were fit to the Hill equation (Fig. 1C). Typically, the Hill equation is used when measuring the fractional saturation of binding sites as a function of ligand concentration in order to parameterize, via the Hill coefficient, the degree of cooperativity in the system (i.e., how much the binding of one ligand enhances the binding of additional ligands) and provide a minimum value of stoichiometry. However, when measuring total fraction of protein bound to membrane (as done here), the Hill coefficient instead describes an “apparent cooperativity” that is not necessarily an indication of allosteric cooperativity, but nevertheless does provide a minimum value of stoichiometry (26). Indeed, Tim4 binding showed a sigmoidal dependence on PS surface density with a Hill coefficient of 2.40 ± 0.08, suggesting the presence of multi-PS interactions (Fig. 1C). This value further predicts a minimum stoichiometry of at least 3 PS per protein, as the stoichiometry, by definition, must exceed the Hill coefficient (27).
Previous characterizations of other multi-PS binding proteins demonstrate that dimensionality effects can increase apparent cooperativity even in the absence of allosteric (i.e., conformational) changes in the protein (26, 28). That is, the binding of one PS necessarily confines the protein to the lipid membrane surface, and thereby enhances the effective concentration of other membrane PS. Thus, the initial interaction can affect an apparent increase in PS affinity at the remaining sites, leading to a more strongly sigmoidal binding response. Accordingly, even strong sigmoidicity in the binding of multivalent proteins to membrane surfaces does not require an allosteric change in the protein.
In the case of Tim4, the 13-kDal PS recognition domain is a single, IgV-like fold made rigid by three pairs of disulfide bonds (23). This structure would be considered unlikely to produce significant conformational changes to expose extra binding sites following membrane binding. In addition, multiple previous binding studies have effectively ruled out the existence of direct Tim4–Tim4 interactions (9, 21, 29); thus, it is also not likely that a trimeric (or higher oligomeric) Tim4 state mediates the minimal stoichiometry of 3 PSs per protein binding unit required by a Hill coefficient of 2.40. We therefore applied a combination of structural and biochemical approaches to determine how a monomeric Tim4 protein can mediate direct contacts with multiple PS lipids to yield a sigmoidal dependence of protein binding on increasing PS surface density.
X-Ray Reflectivity Reveals Membrane-Bound Tim4 Orientation.
The central positioning of the Ca2+-coordinated, single-PS binding pocket on Tim4 suggested the possibility that additional peripheral residues might also form membrane contacts (23). Previous binding studies with alanine mutants have validated the importance of a select few of these residues (20, 23, 30). However, the current structural information was derived from a complex with a single, soluble PS molecule, and therefore cannot adequately account for the observed multi-PS interactions. To generate a comprehensive structural model, we used interfacial X-ray reflectivity on Tim4 bound to a lipid monolayer. This technique measures the reflected X-ray intensity from an interfacial surface as a function of incident angle. This intensity versus incident angle relationship is essentially an interference pattern produced by variations in the 1D electron density profile along the dimension perpendicular to the reflecting interface, averaged over the in-plane dimension parallel to the interface (31). Given that we know the general molecular structure of both the lipid monolayer and the protein (from the crystal structure), this 1D electron density profile is, in principle, sufficient to determine a precise orientation of a protein bound to a membrane, presuming the electron density of the protein is not symmetrically distributed (32, 33).
To determine the membrane-bound orientation of Tim4, we performed X-ray reflectivity experiments on lipid monolayers of 7:3 1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine (SOPC) to 1-stearoyl-2-oleoyl-sn-glycero-3-phosphoserine (SOPS) in the presence and absence of 1 μM Tim4 (subphase concentration). The lipids were deposited to an area/molecule roughly equivalent to that of a SOPC bilayer (∼65 Å2/molecule) (34). The SO lipids were used in keeping with previous studies to help minimize oxidative damage of the unsaturated tail by X-ray radiation (32, 33). The reflected intensity as a function of momentum transfer vector [Qz = 4π sin(α)/λ, where α is the incident angle and λ is the X-ray wavelength] for lipid monolayers with or without 1 μM Tim4 is shown normalized by Fresnel reflectivity from an ideal air/buffer interface (Fig. 2A). Fig. 2B depicts the corresponding 1D electron density profiles determined by fits of the data in Fig. 2A. The lipid-only data yielded mean values of 13.4 ± 0.1 Å and 10.5 ± 0.4 Å for the thickness of the lipid tail and head regions, respectively; the electron density values were 0.231 ± 0.004 electrons/Å3 for the tail region and 0.453 ± 0.004 electrons/Å3 for the head group region. These values are in general agreement with previously measured values of similar systems, allowing for small variations resulting from different buffer conditions (32, 33). Table S1 provides the specific parameter values associated with the best fits to the lipid-only and +1 μM Tim4 data.
Fig. 2.
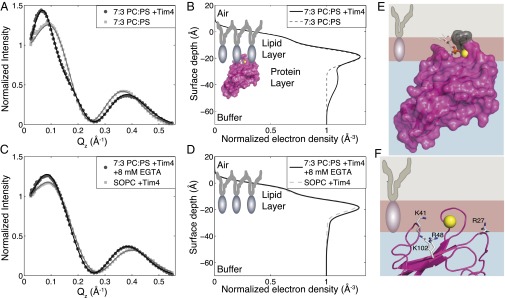
X-ray reflectivity reveals membrane-bound Tim4 orientation. (A) Representative X-ray reflectivity data sets and fits for 7:3 SOPC:SOPS alone (light gray squares) or with 1 μM Tim4 (dark gray circles). Lines through data are fits based on a model-dependent, iterative fitting procedure. Data are normalized by Fresnel reflectivity of an ideal air–water interface. (B) Best fit, 1D electron density profiles corresponding to fits of reflectivity data in A (dashed light gray: 7:3 SOPC:SOPS alone; solid black: with 1μM Tim4). The cartoon overlay shows the approximate orientation of Tim4 that was used to calculate the profile. (C) Representative X-ray reflectivity data sets and fits for 7:3 SOPC:SOPS with 1 μM Tim4 after injection of EGTA to 8 mM final concentration to remove bound protein layer (dark gray circles). Also shown is pure SOPC with 1 μM Tim4 (light gray squares). Lines through points are model-dependent fits, as in A. (D) The 1D electron density profiles corresponding to the best fits from C showing the absence of a significant protein layer either after EGTA injection (solid black) or with pure SOPC + 1 μM Tim4 (dashed light gray). (E) Scale model of Tim4 best-fit orientation from X-ray reflectivity measurements. Lipid is shown as a two-box model with tail group in gray and head group in burgundy. Hydrophobic residues W97 and F98 (gray) are shown penetrating the tail region. The PS head group analog from the Tim4 crystal structure [3BIB (23)] is shown as sticks, with the coordinating Ca2+ shown as a yellow sphere. (F) Structural diagram of four peripheral basic residues within close proximity of the membrane surface.
The best-fit orientation shown by the cartoon in Fig. 2B was generated using recently developed methods for incorporating protein crystal structures to fit reflectivity data (32, 33). Under this approach, a reduced χ2 map depicting the fit quality as a function of protein orientation was generated. The protein orientation is varied along two rotational degrees of freedom (θ and Φ) depicted in Fig. S1. The reduced χ2 map reveals two equally well-fit regions of essentially identical 1D electron density profiles (Fig. S1). Given that we have collapsed a 3D structure down to a 1D profile, it is not unexpected that electron density symmetries may arise depending on the underlying structure of the protein studied. However, in this case, only one of the two orientations places the known Ca2+-coordinated PS pocket proximal to the membrane surface and would therefore be the physiologically relevant orientation. The second orientation is reversed with the Ca2+ pocket far from the membrane surface (Fig. S1). Fig. 2 C and D show that the presence of a significant Tim4 interfacial layer is dependent on both the presence of PS and, most important, is removable by the addition of 8 mM EGTA to chelate all of the Ca2+ in solution. The EGTA control confirms that the Ca2+-coordinated, PS-binding pocket, and hence the protein, is properly oriented.
Finally, Fig. 2E depicts the most probable orientation of Tim4 bound to a PS membrane based on the parameters extracted from the X-ray reflectivity data. This orientation verifies two predictions from the Tim4/single-PS crystal structure (23). First, two solvent-exposed hydrophobic residues (W97 and F98) penetrate the hydrophobic lipid tail region (W97 is presumably the residue that yields the spectral shift we used in membrane binding assays). In addition, the PS head group analog and coordinating Ca2+ ion that were cocrystalized with Tim4 are well aligned with the lipid monolayer head group region. These details support the validity of this membrane binding orientation.
We next examined this orientation to determine how Tim4 mediates interactions with multiple PS. A ubiquitous feature of many PS-membrane binding proteins is the interfacial presence of basic lysine (K) and arginine (R) residues (35). Our observation of weak Tim4 binding in the absence of Ca2+ (Fig. 1A), suggested the possibility that the additional contacts with negatively charged PS lipids might involve ionic interactions via positively charged K or R residues. Fig. 2F reveals that the bound orientation of Tim4 positions two R and two K residues in close proximity to the membrane; no other positively charged residues were within ∼10 Å of the model membrane interface. Notably, these four residues are positioned essentially equidistant from one another along two orthogonal axes, with R27 opposing K41 on one axis and K102 opposing R48 along a nearly perpendicular axis. The separated positioning of these residues makes it unlikely that any two could simultaneously coordinate a single PS but, rather, that they individually interact with PS lipids. The static structure also suggests that R27 and K41 deeply penetrate the lipid head group region, whereas R48 and K102 merely graze the lipid head groups at the buffer interface.
Dynamic Model of Membrane-Bound Tim4.
We next used all-atom molecular dynamics (MD) simulations to confirm that the Tim4 orientation derived from X-ray experiments on lipid monolayers is representative of the more physiological bilayer case. The MD simulations were also used to place the static model derived from X-ray scattering in a more realistic dynamic context. The Chemistry at Harvard Macromolecular Mechanics-Graphical User Interface (CHARMM-GUI) Membrane Builder Web site (www.charmm-gui.org/?doc=input/membrane) (36, 37) was used to build a 7:3 POPC:POPS lipid bilayer comprised of ∼70 lipids per leaflet, with Tim4 docked according to the orientational parameters derived from the X-ray scattering best fit (θ, Φ, and membrane depth). Both the coordinating Ca2+ ion and the bound PS lipid were built into the central PS-binding pocket on the basis of the Tim4 crystal structure (23). After 1 ns of equilibration, unrestrained simulation was performed for 100 ns. Fig. 3A depicts the structure of the full lipid bilayer, protein, and centrally bound PS/Ca2+ used for simulation and also defines our surface depth and rotational parameters.
Fig. 3.

Dynamic model of membrane-bound Tim4. (A) Depiction of simulated lipid bilayer system with Tim4 docked according to parameters derived from X-ray fitting. Centrally bound PS is shown as spheres, as is the coordinating Ca2+ ion (yellow). Also shown is the definition of bilayer surface depth and protein rotational angles. (B) Trajectories of θ (blue) and Φ (red) angles, as defined in A, over the course of the MD simulation. Overlaying dashed lines depict best-fit orientation (black) from X-ray reflectivity and 95% confidence window (gray). (C) Surface depth distribution of the center of mass of W97 (Left) relative to the start of the tail region (dashed light gray line). (Right) Center-of-mass, surface depth distribution of the Ca2+-bound, central PS-phosphate (horizontal orange bars) relative to the distribution of all other phosphates in the leaflet (burgundy line). All distributions are normalized to total number of sampled frames. (D) Center-of-mass, surface depth distributions of side chain nitrogens (cationic sites) for the four residues identified in Fig. 2F (horizontal blue bars). These are shown relative to distributions of the two different species of anionic binding partners: phosphates (orange line; includes both PC and PS) and carboxyl moieties of PS head groups (burgundy line includes only PS). The anion distributions are normalized to the total frames of the phosphate to show the relative abundance of phosphates (100% of lipids) versus carboxyl moieties (30% of lipids). The dashed black line denotes phosphate-dominated regions (above the line) versus carboxyl-dominated regions (below the line) of potential anionic interaction.
The protein remained stably bound to both the central PS lipid (Ca2+ coordinated) and the bulk membrane surface throughout the simulation. Fig. 3 B and C demonstrate this stability. Fig. 3B depicts values for the θ and Φ rotational angles over the course of the simulated trajectory. These values are shown relative to the mean and 95% confidence window of the experimental values from X-ray reflectivity. This result confirms that the Tim4 orientation derived from X-ray fitting is also a stable configuration in silico. Fig. 3C shows the center-of-mass, surface depth distribution for the phosphate of the PS bound in the central PS pocket over the entire simulation (all distributions are normalized to total simulated frames). This distribution is entirely contained within the broader distribution of all other membrane phosphates in that leaflet. Thus, the PS bound within the Tim4 PS pocket is in a stable configuration relative to the rest of the lipid membrane. Finally, the surface depth distribution for the benzene ring of W97 (Fig. 3C) shows consistent penetration into the lipid tail region. This further confirms that Tim4 remains stably bound to the lipid membrane. Considered in total, these features validate the simulation with respect to the experimental results and further suggest that our dynamic model is representative of an energetically stable Tim4 orientation.
Fig. 3D displays the center-of-mass, surface depth distributions of the charged moieties for the side chains of the four basic residues identified in Fig. 2F. These distributions are shown relative to the full membrane distribution of phosphates (including both PC and PS lipids) and PS head group carboxyls. The dashed black line depicts the transition between the phosphate-dominated (above the line) and PS carboxyl-dominated (below the line) regions of potential anionic binding. The individual distributions of the four cationic side chains reveal that all four residues reside either entirely (R27, K41) or significantly (R48, K102) within the region populated by lipid phosphate or carboxyl anionic binding partners.
Thus, dynamic modeling suggests that all four peripheral basic residues can act as sites of additional PS interactions. Including the Ca2+-coordinated PS bound within the central pocket, Tim4 can then be characterized as having an effective stoichiometry of five PS. Notably, the surface depth distributions of the four residues are not identical. Similar to the static model, R27 and K41 reside predominantly in the phosphate-dominated region, whereas R48 and K102 are most frequently found in the PS carboxyl region. These predicted differences in surface depth distribution suggest the possibility that these four basic residues may not all contribute equally to the Tim4 binding characteristics.
Alanine Mutants Verify Functional Relevance of Four Peripheral Basic Residues.
Alanine point mutations at each of the four peripheral basic residue sites were individually assayed for binding, as in Fig. 1 (Fig. 4 A and B). Fig. 4C demonstrates the change in free energy for each of the mutants relative to wild type (WT). Alanine substitutions at three residues (K41, K102, and R27) led to significant reductions in the free energy of binding, whereas mutation of R48 to alanine had a slightly beneficial effect on binding. Notably, the binding of WT for 90 μM PS in the absence of Ca2+ was significantly weaker than any of the individual alanine mutants at the identical titration point. This suggests that the Ca2+-coordinated PS bound within the central pocket contributes significantly more to the total binding energy than PS interactions at any of the peripheral basic residues.
Fig. 4.

Alanine mutants verify functional relevance of four peripheral basic residues. (A) Lipid vesicle titrations of 7:3 POPC:POPS for Tim4 mutants. Open shapes represent binding data, whereas filled shapes correspond to 90 μM [PS] + 10 mM EGTA for mutants (all points are overlapping at ∼0% binding), and the X is the corresponding WT EGTA control. Lines through data are fits to a single-site binding model. (B) PS mole fraction titrations at 600 μM total lipid with 170 nM protein for each alanine mutant. Lines through the data are fits to a Hill model. All data points are mean of measurements done at least in triplicate, with error bars showing 1 SD. (C) Apparent change in free energy for the Tim4 mutants calculated from binding titration data in A or Fig. S2 (for N39A). *P < 0.0001; N.S., statistically nonsignificant differences (N39A P value = 0.7228). (D) Bar plot of relative change in Hill coefficient, as determined by fitting to data in C or Fig. S2 (for N39A). *Statistically significant changes (P values relative to WT: R48A and K41A, P < 0.0001; R27A, P = 0.002; K102, P = 0.0195; and P values relative to R48A: R27A, P = 0.0013; K41A, P = 0.0044). N.S., statistically nonsignificant differences (N39A P value = 0.9628). All error bars denote 1 SD.
An uncharged polar residue (N39) near K41 was also targeted for mutation. Previously, mutation of N39 to aspartic acid (D) showed a significant reduction in binding (30) to PS-containing vesicles, suggesting that this residue lies close to the membrane after Tim4 binding. However, based on our model and previous studies documenting the importance of interfacial basic residues for membrane binding proteins (35), we hypothesized that this reduction in binding was a result of charge repulsion between the negatively charged D residue and negatively charged PS lipids, and not evidence that N39 forms significant hydrogen bond contacts with the lipid membrane. Thus, mutation of N39 to alanine serves as a control to evaluate our hypothesis that the identified peripheral basic residues are the primary mediators of Tim4’s additional PS interactions. Mutation of N39 to alanine showed no substantial effect on binding (for clarity, the N39A raw binding data are shown relative to WT Tim4 in Fig. S2).
The absence of a significant loss in free energy for mutations at either R48 or N39 does not conclusively rule out their potential mechanistic importance. It is always possible that mutation of one residue somehow enhances interactions of other residues, causing a compensating effect. Thus, we next evaluated changes in Hill coefficient following alanine mutation to serve as a more definitive diagnostic. Fig. 4D depicts these changes in Hill coefficient on alanine mutation. Notably, alanine substitution at each of the four basic residues did significantly reduce the Hill coefficient from 2.40 (WT) to within a range from 2.02 to 1.57. These results support our hypothesis that these four peripheral basic residues are the mediators of the additional PS-specific interactions. In contrast, the N39 alanine mutant did not appreciably affect the Hill coefficient relative to WT, suggesting that this residue is not capable of forming substantial contacts with PS lipids.
Interestingly, the level of Hill coefficient reduction among the four basic residues was not identical. In particular, alanine mutation of R48 caused a significantly greater reduction in the Hill coefficient than did mutation of either K41 or the chemically identical R27 (R48: h = 1.57 ± 0.09; K41: h = 1.94 ± 0.07; R27: h = 2.02 ± 0.09). This effect occurred despite the fact that a small energetic benefit was observed with the alanine mutant of R48 (Fig. 4B). This is likely because the loss of binding energy associated with the R48 mutation can be compensated for by the strengthening of other interactions (perhaps the opposing K102). Considered in total, our mutational binding evidence suggests it is the combination of the relatively strong, Ca2+-coordinated binding site with the four weaker sites of direct ionic interaction that produces the characteristic “soft” sigmoidal response of Tim4 to increasing PS surface density.
The Tim Proteins Have Different Sensitivities to PS Surface Density.
It has been well established that the key residues that form the Ca2+-coordinated, single-PS binding pocket in Tim4 are strictly conserved in the homologous Tim family members Tim1 and Tim3, both of which also bind PS membranes in a Ca2+-dependent manner (9, 23, 38). Given this conservation, it is likely that the nature of the Ca2+-coordinated PS interaction is quite similar between these three proteins. However, sequence alignments show that although the general structures of the three proteins are quite similar, there nonetheless exist significant differences in the specific types of peripheral residues that surround the central PS binding site (23). Given the importance of the peripheral basic residues for tuning Tim4’s characteristic sensitivity to PS surface density, we hypothesized that these differences in sequence might produce significantly different responses in Tim1 and/or Tim3.
To assess this hypothesis, we performed the PS mole fraction titration with the PS recognition domain of either soluble murine Tim1 or Tim3 (hereafter referred to as Tim1 and Tim3, respectively). Tim1 displayed a much more hyperbolic response relative to Tim4 (Fig. 5A). As such, Tim1 binds significantly stronger to lower PS surface densities relative to Tim4. At higher PS surface densities, the binding strength is essentially identical between the two proteins; this equivalence at high PS percentage is consistent with previous comparative measurements between Tim1 and Tim4 that examined only one, presumably high, PS percentage (30). Also consistent with previous binding measurements, Tim3 bound at weaker levels for all PS percentages examined (30). However, despite these differences in general binding affinity, Fig. 5B shows that Tim3 (h = 1.55 ± 0.10) shares a similar Hill coefficient to that of Tim1 (h = 1.35 ± 0.13). Both these values are significantly less than that of Tim4 (h = 2.40 ± 0.08), confirming that both Tim1 and Tim3 bind with substantially less apparent cooperativity than Tim4. These results further substantiate the importance of the peripheral membrane interacting residues for determining the specific binding characteristics of a particular Tim protein.
Fig. 5.

The Tim proteins have different sensitivities to PS surface density. (A) PS mole fraction titrations at constant total lipid of 600 μM with 170 nM Tim1 (black squares), Tim4 (dark grey circles), or Tim3 (light grey triangles). Each data point is the mean of at least three measurements with error bars denoting 1 SD. Lines through data points are fits to a Hill binding model (Tim1-black; Tim4-dark grey; Tim3-light grey). (B) Comparison of best-fit Hill coefficients between Tim4, Tim1, and Tim3. Error bars signify 1 SD. N.S., differences that do not reach statistical significance (Tim1 relative to Tim3: P = 0.4001); *P < 0.0001.
Discussion
We show that murine Tim4 responds to the surface density of PS on bilayers in vitro with a gently sloped sigmoidal binding curve characterized by a Hill coefficient of 2.40 ± 0.08. We argue that this soft sigmoidicity provides a mechanism for Tim4-bearing phagocytes to distinguish PS exposure on apoptotic versus activated nonapoptotic T cells on the basis of differing PS exposure levels. Specifically, this type of response will cause Tim4 to bind only moderately to cells with intermediate PS surface densities (nonapoptotic, activated T cells) while still maintaining robust binding to cells with relatively high PS surface densities (apoptotic T cells). The soft sigmoidicity also ensures Tim4 binds minimally to cell surfaces with low PS levels (healthy, resting T cells). It is important to note that Tim4 does not directly mediate signaling but is, instead, believed to be a tethering protein that facilitates phagocytosis by confining the target cell to be held in close proximity to the phagocyte (39). Thus, the observed differentiated binding characteristics would likely translate directly to differentiated phagocytic responses by Tim4-bearing phagocytes.
Had Tim4 evolved binding characterized by a significantly higher Hill coefficient, as, for example, seen with Annexin V (h = ∼8) (25) or PKC βII (h = ∼7) (26), the resulting sharp sigmoidal curve would impose an all-or-none response unable to differentially respond to apoptotic versus nonapoptotic activated T cells. At the other extreme, if the binding of Tim4 was closer to that of Tim1 (h = 1.35 ± 0.13) the nearly hyperbolic response would likewise be incapable of mediating the proposed differential recognition because of the much stronger sensitivity to lower PS percentage. Although sharply sigmoidal or hyperbolic binding responses are of course necessary (and present) in a wide array of physiological circumstances, neither would seem capable of satisfying the immunological roles prescribed to Tim4. Thus, it seems logical to conclude that the observed soft sigmoidal response of Tim4 to PS surface density is integral to the protein’s immunological function.
The previous structural characterization of Tim4, as well as that of both Tim1 and Tim3, identified a highly conserved, central PS-binding pocket (23, 30, 38). In all cases, this pocket features a coordinating Ca2+ ion that can form a strong electrostatic interaction with a single, tightly bound PS molecule. We have now identified in Tim4 four peripheral basic residues that are capable of mediating direct ionic interactions with additional PS lipids, albeit each with much weaker individual strength than that of the Ca2+-coordinated site. This heterogeneity of interaction strengths between these putative binding sites has significant implications for the nature of Tim4’s response to increasing PS surface density. Specifically, the relatively lower energetic benefit gained when a PS occupies one of the weaker sites allows the protein to significantly populate bound states in which all five sites are not occupied. By definition, this reduces the apparent cooperativity of the system.
In contrast, if all five Tim4 sites were identically of the Ca2+-coordinated variety, then on membrane binding, each site would have exactly the same strong proclivity for binding an individual PS molecule. Because each site would be experiencing the same surface concentration of PS molecules, they would all fill equally. This would then produce only two significantly populated protein states: either all sites occupied or no sites occupied (when the protein is unbound in solution). Such a scenario constitutes the definition of a perfectly cooperative system in which the Hill coefficient will nearly equal the stoichiometry of the system. Notably, binding measurements of Annexin V, which is believed to have ∼8–12 essentially identical Ca2+-mediated sites of PS binding, yielded Hill coefficients between 7 and 9 (25).
Thus, it appears that Tim4 has achieved its evolutionarily required binding response, using a hierarchy of PS interaction strengths. The Ca2+-coordinated PS interaction ensures a strong, PS-specific affinity, whereas the peripheral basic residues collectively serve to tune the apparent cooperativity of the system to the optimal level. It is noteworthy that the four peripheral basic residues do not contribute equally to PS specificity, and that this may be correlated with whether the residues are biased toward interactions with anionic carboxyl (PS only) or phosphate (common to both PC and PS) groups. This organization may further suggest some degree of PS specificity for interactions that are biased toward anionic carboxyl residues (e.g., R48 and K102), given that all other physiological anionic residues of any abundance have only phosphate anionic moieties (e.g., phosphatidic acid or phosphatidylinositol head groups). In contrast, residues such as K41 and R27 that appear to be biased toward phosphate interactions would not necessarily be expected to bear the same “PS specificity” and might be able to interact with other anionic lipids with strengths rivaling those of PS interactions.
Notably, the peripheral basic residues that are biased toward anionic carboxyl moieties (R48 and K102) are strictly conserved between mouse and human Tim4. In contrast, the phosphate-biased residues (R27 and K41) are only loosely conserved in humans, with the functional contributions of R27 potentially replaced by H25, and those contributions of K41 replaced by K39. Thus, although the total number of interfacial basic residues is conserved between mouse and human, only the residues positioned such that they will predominately interact with PS carboxyl moieties are strictly conserved. This conservation further highlights the potential importance of residues K102 and R48 for determining the characteristic PS sensitivity of Tim4.
It is also important to note that none of the Tim4-related hypotheses developed here in any way preclude the presence or importance of other factors in determining the phagocytic fate of a PS-exposing cell. As previously mentioned, it has been shown that PS exposure alone is not sufficient in all cases to elicit phagocytic removal by macrophages (10). This result does not, however, prevent Tim4-bearing phagocytes from using the level of PS exposure as a determinant of the appropriate level of cellular removal when conditions are suitable for phagocytic removal (i.e., when all necessary factors are present).
Our observation that both Tim1 and Tim3 display nearly hyperbolic responses to increasing PS surface density suggests that unlike Tim4, these proteins would be relatively insensitive to the differences in PS surface density between apoptotic and nonapoptotic T cells, and that this may be an important feature for their respective physiological functions. As an example, it has now been shown that Tim1 can use apoptotic cells exposing PS as a nonphagocytic signal to produce proliferation in natural killer T cells (11). Thus, it seems plausible that the as-yet-unidentified target of Tim1 for facilitating proliferation, when expressed on conventional T cells, may also be membrane PS, exposed either by the activated T cells themselves or possibly on the antigen-presenting cells. At the very least, these differences in the recognition properties of the three Tim proteins highlight the need for future functional studies to assess the immunological significance of these recognition differences.
Although the dynamic nature of lipid membrane systems presents significant barriers to atomic-level studies, this work demonstrates that our multidisciplinary approach can effectively surmount these barriers. In the case of Tim4, the resultant binding model reveals a previously unappreciated sensitivity to a parameter known to vary under different physiological instances of PS exposure (i.e., PS surface density). Given the structural and functional diversity now attributed to the growing list of PS receptors, it seems reasonable, in light of our results, that there may exist an equally diverse range of sensitivities to the defining properties of the PS-exposing membrane (properties such as PS surface density, membrane composition, or membrane fluidity). This would suggest that these membrane properties might have a critical role to play in PS recognition. Furthermore, it is possible that in some cases, the PS may serve more as an anchor to allow a receptor to interrogate the nature of the surrounding membrane as a determinant of the appropriate immunological response. Although much remains to be done to assess these hypotheses, this study of Tim4 establishes that all PS-containing membranes are not necessarily equal with respect to immune recognition.
Experimental Procedures
Materials.
Stocks of Hepes, CaCl2, NaCl, and EGTA were obtained from Fisher Scientific. Lipid stocks of POPC, POPS, SOPC, and SOPS were obtained from Avanti Polar Lipids and used without further purification. CHCl3 for use with lipids was HPLC-grade and was obtained from Fisher Scientific, as were stocks of MeOH and acetone for cleaning.
Protein Production.
The cDNA of Tim4, Tim1, and Tim3 were a generous gift from Shigekazu Nagata (Kyoto University, Kyoto). The cDNA corresponding to the murine form of the PS recognition domains (IgV domain) of Tim4 [residues D4-A112; Protein Data Bank code 3BIB (23)], Tim1 [residues D2-E112; Protein Data Bank code 2OR8 (38)], and Tim3 [residues G6-K112; Protein Data Bank code 3KAA (30)] were cloned in frame with a C-terminal 6× His tag into the pAcGP67A vector (BD Biosciences). Recombinant Tim4, Tim1, and Tim3 were all produced in Hi5 cells via baculovirus transfection, as previously described (40), with the exception that a higher salt buffer was used for purification (10 mM Hepes at pH 7.2 plus 450 mM NaCl buffer). The higher concentration of NaCl buffer was critical for maintaining protein solubility during the purification process. After purification, protein was stored at 4 °C and was stable for several months. All mutant proteins were purified via the same procedures and showed identical elution profiles over FPLC and nearly equivalent protein yields, suggesting minimal disruption to the underlying protein structure.
Lipid Vesicle Preparation.
LUVs were prepared by extrusion method. Briefly, POPC/POPS powder lipids (Avanti Polar Lipids) were dissolved in CHCl3 to the desired concentration and then dried under nitrogen flow. The lipids were then dried further under vacuum for 1 h. Next, a buffer of 10 mM Hepes at pH 7.2 plus 150 mM NaCl was added to the lipids and vortexed for 1 h at 40 °C. After vortex, 1 mM CaCl2 was added, and the lipid solutions were subjected to five freeze thaw cycles, using a dry ice/ethanol slurry. Finally, the lipids were extruded 21 times using 100-nm-diameter filters (Avanti Polar Lipids). Lipid size and polydispersity were characterized by dynamic light scattering (Zen3600 Malvern Nano Zetasizer). Lipid concentration was then quantified using a standard phosphate analysis, with all reagents and protocol from Avanti Polar Lipids.
Tryptophan Shift Binding Assay.
The crystal structure of the PS recognition domain of murine Tim4 revealed a solvent-exposed tryptophan at residue 97 (W97) that is believed to bury in the membrane during binding (confirmed by our structural studies) (23). In the presence of 7:3 POPC:POPS LUVs at 600 μM total lipid concentration, the fluorescent spectra of soluble Tim4 is red-shifted ∼10 nm, and the quantum yield is increased by a factor of ∼2.9; both changes are consistent with the burial of a tryptophan in a nonpolar environment. Therefore, we used the Tim4 spectral shift as a readout for binding to LUVs. To determine the fraction of Tim4 bound, we fit individual measured spectrum to a linear combination of “unbound” and “bound” spectrum, using custom Matlab code, in which each standard spectrum was an average of at least 3 measurements. For lipid titration experiments, no lipid (unbound) and 300 μM total lipid 7:3 POPC:POPS were used as the standards for fitting of intermediate spectrum. For the PS mole fraction titration, the standards were 600 μM pure POPC (unbound) or 600 μM total lipid 7:3 POPC:POPS (bound). Individual standards were generated for each protein (Tim4, Tim1, Tim3, and all Tim4 mutants). The unbound spectrum of each of the Tim4 mutants was essentially identical to that of WT, again suggesting little disruption to the underlying protein structure. Fluorescence measurements were made on a Fluorolog-3 spectrofluorometer (Horiba), using incident beam intensity to normalize the collected signal. The cuvette used was a 10-mm path-length Quartz cuvette from Starna Cells Inc. Protein was excited with 280 nm light and collected in a range from 300 to 420 nm. Each measurement was performed at least in triplicate, using degassed buffer. Measurements were done at least 2 min after initial mixing of the sample, and then a repeat measurement was performed ∼3 min after the initial measurement to ensure equilibrium had been reached.
Custom Matlab code was used to fit measured spectra as a linear combination of the bound and unbound spectra to yield protein bound fraction values. These protein bound fraction values as a function of either PS concentration or PS mole fraction were fit in GraphPad Prism software, using nonlinear least squares fitting. The [PS] titrations were fit using a single site-binding model with fit parameters of Kd and Bmax. PS mole fraction titrations were fit to a Hill model with fit parameters of Kd, Bmax, and h (Hill coefficient). Statistical significance was evaluated in GraphPad Prism, using an extra sum-of-squares F test to compare relevant fit parameters. All evaluated P values are provided in the figure legends.
X-Ray Reflectivity.
X-ray reflectivity measurements were all performed at ChemMatCARS sector 15ID at the Advanced Photon Source of Argonne National Lab. A custom mini Teflon trough was machined to fit the existing X-ray reflectivity setup at 15ID. The trough has dimensions of ∼60 × 80 × 5 mm, for a total volume of ∼30 mL when filled to a positive meniscus. Surface pressure was measured using a Nima Technology surface pressure sensor. Lipids were deposited to a measured surface pressure roughly equivalent to that of a bilayer (∼25 mN/m at an area per lipid of ∼65 Å2/molecule) (34) and allowed to relax 30 min before scanning. Subsequently, ∼300 μL Tim4 protein at ∼100 μM was injected through a side port on the trough to a final concentration of ∼1 μM. The system was then stirred with a small stir bar (introduced before lipid deposition) for ∼1 h to allow the protein to equilibrate over the full surface. All systems used freshly prepared buffer of 10 mM Hepes at pH 7.2, 1 mM CaCl2, and 150 mM NaCl2 (Fisher Scientific) that was degassed for at least 1 h before setup.
Data collection.
Data were collected on a liquid surface reflectometer covering a Qz range of 0.018–0.55 Å−1, with beam intensity limited via inclusion of aluminum absorbers to the minimal values possible to reduce the possibility of radiation-induced lipid film damage while preserving acceptable signal to noise. Data were highly reproducible within a given film preparation, and in situ monitoring of surface pressure gave no indication of film damage resulting from oxidation. Samples were contained within a hermetically sealed box under helium, with O2 levels maintained below ∼1% (vol/vol), both to ensure little chance of lipid tail oxidation and to avoid atmospheric scatter of X-rays. Each lipid film was scanned one to two times before protein injection and at least three times after protein injection, each at distinctly separate locations covering the whole film to ensure homogeneity of protein coverage. After EGTA injections, an additional two scans were typically performed. The film was frequently translated both within and between complete reflectivity scans to ensure fresh patches of the lipid film were examined.
The wavelength of the X-ray beam used was 1.24 Å, and data were collected on an area detector (Pilatus 100K). The best-fit protein orientation X-ray data presented are representative of triplicate measurements of a single film. The extracted best-fit orientation is an average of all three measurements. More than 15 additional measurements of this system were made on five different film preparations, exploring a range of initial surface pressures from 20 to 30 mN/m and subphase protein concentrations from 250 nM to 3 μM. Altering these conditions did not produce any significant changes in the best-fit protein orientation but did, in some cases, lower the confidence interval between good and bad fits because of weakened signal to noise. The data presented here represent the highest-quality data with optimized conditions for best protein coverage and highest resolution in terms of fit quality between different protein orientations.
Analysis.
For our lipid-only control systems, we used the standard two-box model to characterize an averaged structure of the lipid head and tail group regions. This model is characterized by four parameters: the lengths of each of the head and tail group regions, as well as their respective average electron densities. To account for thermal fluctuations of the lipids, we convoluted the intrinsic electron density profile with a Gaussian distribution. The width of the Gaussian was estimated to be 3.4 Å on the basis of fitting. We performed iterative fitting of all free-fit parameters for a range of sigma values between 2.7 and 4.5 Å. A sigma value of 3.4 Å minimized fitting error while maintaining physically reasonable parameters (values of sigma between ∼3.5 and 4.0 give equally good fits, but the head group thickness rapidly drops to nonphysical fit values of ∼1 Å). The roughness of 3.4 Å is similar to, although slightly larger than, the prediction given by capillary wave theory (31). This value appeared ideal in both protein and lipid systems and was therefore used for all subsequent fittings.
To determine the precise orientation of Tim4 bound to a lipid monolayer, we used a recently developed extension of the two-box model that uses existing protein crystal structure information (32, 33). The analysis performed here follows closely that of these previous studies and uses essentially the same custom C code for calculating electron density profile and performing iterative fitting (done in the Cplot program, available at www.certif.com/content/cplot/). The best-fit orientation was first determined from all possible rotations. Then a statistical F test was used to assess the quality of all other orientations relative to the best fit. A 95% confidence window was used to establish the orientations that were indistinguishable in terms of fit quality. Calculated values of reduced χ2 for best fits were typically in the range of 7–15, which is consistent with previous measurements on similar systems (32, 33).
MD Simulations.
All-atom MD simulations on the membrane-docked Tim4 system were performed using explicit water, lipids, and ions in NAMD 2.6 with the CHARMM27 force field and TIP3P water model. The simulated system was generated using the CHARMM-GUI Membrane Builder (36, 37) (available at www.charmm-gui.org/?doc=input/membrane) with the best-fit Tim4 orientation and included 136 lipids (70 POPC in the nonprotein interacting leaflet and 48 POPC-20 POPS in the leaflet where the protein was bound; the two fewer lipids were to accommodate the cross-sectional area taken up by partial insertion of Tim4, as calculated by the Membrane Builder). NaCl concentration was set to 150 mM and then adjusted as necessary by the VMD Autoionize plugin to neutralize the charge of the system. Periodic boundary conditions were used, and a 15-Å water cushion was included beyond both the protein and membrane structure to prevent interactions between periodic cells. A constant number, pressure, area, and temperature ensemble with P = 1 atm, A = 4605 Å2, and T = 303.15 K was used to maintain appropriate system parameters, most notably the area per lipid molecule, which was set at ∼65 Å per molecule to match both the X-ray experimental value and an approximate value for 7:3 POPC:POPS bilayer. The system was equilibrated following the standard method produced by the CHARMM-GUI Membrane Builder, with 1 ns of production performed as the final step of equilibration. Unrestrained simulation was then performed for 100 ns, and data analysis was done using both VMD (custom TCL code) and Matlab.
Supplementary Material
Acknowledgments
We thank Dr. Shigekazu Nagata and his lab (Kyoto University, Kyoto) for generously providing cDNA for the Tim proteins used in this study. We also specifically acknowledge Dr. Adam Hammond for many fruitful discussions during the preparation of the manuscript, as well as Dr. Lorenzo Pesce (computational studies: Beagle supercomputer) and Dr. Elena Solomaha (biochemical studies: University of Chicago Biophysics Core) for their help with data collection. This research was supported by National Institutes of Health Grant R01AI073922 (to E.J.A.) and also through resources provided by the Computation Institute and the Biological Sciences Division of the University of Chicago and Argonne National Laboratory under Grant S10 RR029030-01. Funding support was also provided by the National Science Foundation through Grants MCB-0920316 (to K.Y.C.L), CHE-0910825 (to M.L.S), and MCB-0920261 (to B.R.). K.Y.C.L. acknowledges support from The University of Chicago Materials Research Science and Engineering Center (DMR 0820054). ChemMatCARS Sector 15-ID is principally supported by the National Science Foundation/Department of Energy (DOE) under Grant NSF/CHE-0822838. Use of the Advanced Photon Source was supported by the DOE Office of Science, Office of Basic Energy Sciences, under Contract DE-AC02-06CH11357. This work was also partially funded by the Chicago Biomedical Consortium with support from the Searle Funds at the Chicago Community Trust.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320174111/-/DCSupplemental.
References
- 1.Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: Progress and conundrums. J Exp Med. 2010;207(9):1807–1817. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zwaal RFA, Comfurius P, Bevers EM. Surface exposure of phosphatidylserine in pathological cells. Cell Mol Life Sci. 2005;62(9):971–988. doi: 10.1007/s00018-005-4527-3. [DOI] [PubMed] [Google Scholar]
- 3.Fischer K, et al. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood. 2006;108(13):4094–4101. doi: 10.1182/blood-2006-03-011742. [DOI] [PubMed] [Google Scholar]
- 4.Dillon SR, Constantinescu A, Schlissel MS. Annexin V binds to positively selected B cells. J Immunol. 2001;166(1):58–71. doi: 10.4049/jimmunol.166.1.58. [DOI] [PubMed] [Google Scholar]
- 5.Dillon SR, Mancini M, Rosen A, Schlissel MS. Annexin V binds to viable B cells and colocalizes with a marker of lipid rafts upon B cell receptor activation. J Immunol. 2000;164(3):1322–1332. doi: 10.4049/jimmunol.164.3.1322. [DOI] [PubMed] [Google Scholar]
- 6.Elliott JI, et al. Phosphatidylserine exposure in B lymphocytes: A role for lipid packing. Blood. 2006;108(5):1611–1617. doi: 10.1182/blood-2005-11-012328. [DOI] [PubMed] [Google Scholar]
- 7.Elliott JI, et al. Membrane phosphatidylserine distribution as a non-apoptotic signalling mechanism in lymphocytes. Nat Cell Biol. 2005;7(8):808–816. doi: 10.1038/ncb1279. [DOI] [PubMed] [Google Scholar]
- 8.Bratton DL, Henson PM. Apoptotic cell recognition: Will the real phosphatidylserine receptor(s) please stand up? Curr Biol. 2008;18(2):R76–R79. doi: 10.1016/j.cub.2007.11.024. [DOI] [PubMed] [Google Scholar]
- 9.Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235(1):172–189. doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Segawa K, Suzuki J, Nagata S. Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci USA. 2011;108(48):19246–19251. doi: 10.1073/pnas.1114799108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee HH, et al. Apoptotic cells activate NKT cells through T cell Ig-like mucin-like-1 resulting in airway hyperreactivity. J Immunol. 2010;185(9):5225–5235. doi: 10.4049/jimmunol.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown S, et al. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature. 2002;418(6894):200–203. doi: 10.1038/nature00811. [DOI] [PubMed] [Google Scholar]
- 13.Gardai SJ, Bratton DL, Ogden CA, Henson PM. Recognition ligands on apoptotic cells: A perspective. J Leukoc Biol. 2006;79(5):896–903. doi: 10.1189/jlb.1005550. [DOI] [PubMed] [Google Scholar]
- 14.Gardai SJ, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123(2):321–334. doi: 10.1016/j.cell.2005.08.032. [DOI] [PubMed] [Google Scholar]
- 15.Albacker LA, et al. TIM-4, a receptor for phosphatidylserine, controls adaptive immunity by regulating the removal of antigen-specific T cells. J Immunol. 2010;185(11):6839–6849. doi: 10.4049/jimmunol.1001360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Albacker LA, et al. TIM-4, expressed by medullary macrophages, regulates respiratory tolerance by mediating phagocytosis of antigen-specific T cells. Mucosal Immunol. 2013;6(3):580–590. doi: 10.1038/mi.2012.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rodriguez-Manzanet R, DeKruyff R, Kuchroo VK, Umetsu DT. The costimulatory role of TIM molecules. Immunol Rev. 2009;229(1):259–270. doi: 10.1111/j.1600-065X.2009.00772.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meyers JH, Sabatos CA, Chakravarti S, Kuchroo VK. The TIM gene family regulates autoimmune and allergic diseases. Trends Mol Med. 2005;11(8):362–369. doi: 10.1016/j.molmed.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 19.Umetsu SE, et al. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat Immunol. 2005;6(5):447–454. doi: 10.1038/ni1186. [DOI] [PubMed] [Google Scholar]
- 20.Kobayashi N, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27(6):927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyanishi M, et al. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450(7168):435–439. doi: 10.1038/nature06307. [DOI] [PubMed] [Google Scholar]
- 22.Rodriguez-Manzanet R, et al. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc Natl Acad Sci USA. 2010;107(19):8706–8711. doi: 10.1073/pnas.0910359107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Santiago C, et al. Structures of T cell immunoglobulin mucin protein 4 show a metal-Ion-dependent ligand binding site where phosphatidylserine binds. Immunity. 2007;27(6):941–951. doi: 10.1016/j.immuni.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frasch SC, et al. Regulation of phospholipid scramblase activity during apoptosis and cell activation by protein kinase Cdelta. J Biol Chem. 2000;275(30):23065–23073. doi: 10.1074/jbc.M003116200. [DOI] [PubMed] [Google Scholar]
- 25.Tait JF, Gibson DF, Smith C. Measurement of the affinity and cooperativity of annexin V-membrane binding under conditions of low membrane occupancy. Anal Biochem. 2004;329(1):112–119. doi: 10.1016/j.ab.2004.02.043. [DOI] [PubMed] [Google Scholar]
- 26.Mosior M, Newton AC. Mechanism of the apparent cooperativity in the interaction of protein kinase C with phosphatidylserine. Biochemistry. 1998;37(49):17271–17279. doi: 10.1021/bi981344t. [DOI] [PubMed] [Google Scholar]
- 27.Weiss JN. The Hill equation revisited: Uses and misuses. FASEB J. 1997;11(11):835–841. [PubMed] [Google Scholar]
- 28.Mosior M, McLaughlin S. Electrostatics and reduction of dimensionality produce apparent cooperativity when basic peptides bind to acidic lipids in membranes. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1992;1105(1):185–187. doi: 10.1016/0005-2736(92)90178-o. [DOI] [PubMed] [Google Scholar]
- 29.Sizing ID, et al. Epitope-dependent effect of anti-murine TIM-1 monoclonal antibodies on T cell activity and lung immune responses. J Immunol. 2007;178(4):2249–2261. doi: 10.4049/jimmunol.178.4.2249. [DOI] [PubMed] [Google Scholar]
- 30.DeKruyff RH, et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J Immunol. 2010;184(4):1918–1930. doi: 10.4049/jimmunol.0903059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pershan PS, Schlossman M. Liquid Surfaces and Interfaces: Synchotron X-ray Methods. Cambridge: Cambridge University Press; 2012. [Google Scholar]
- 32.Chen C-H, et al. Configuration of PKCalpha-C2 domain bound to mixed SOPC/SOPS lipid monolayers. Biophys J. 2009;97(10):2794–2802. doi: 10.1016/j.bpj.2009.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Málková Š, et al. X-ray reflectivity studies of cPLA2α-C2 domains adsorbed onto Langmuir monolayers of SOPC. Biophys J. 2005;89(3):1861–1873. doi: 10.1529/biophysj.105.061515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ly HV, Longo ML. The influence of short-chain alcohols on interfacial tension, mechanical properties, area/molecule, and permeability of fluid lipid bilayers. Biophys J. 2004;87(2):1013–1033. doi: 10.1529/biophysj.103.034280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stace CL, Ktistakis NT. Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim Biophys Acta. 2006;1761(8):913–926. doi: 10.1016/j.bbalip.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 36.Jo S, Kim T, Im W. Automated builder and database of protein/membrane complexes for molecular dynamics simulations. PLoS ONE. 2007;2(9):e880. doi: 10.1371/journal.pone.0000880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jo S, Lim JB, Klauda JB, Im W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys J. 2009;97(1):50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santiago C, et al. Structures of T Cell immunoglobulin mucin receptors 1 and 2 reveal mechanisms for regulation of immune responses by the TIM receptor family. Immunity. 2007;26(3):299–310. doi: 10.1016/j.immuni.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park D, Hochreiter-Hufford A, Ravichandran KS. The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr Biol. 2009;19(4):346–351. doi: 10.1016/j.cub.2009.01.042. [DOI] [PubMed] [Google Scholar]
- 40.Scharf L, et al. The 2.5 Å structure of CD1c in complex with a mycobacterial lipid reveals an open groove ideally suited for diverse antigen presentation. Immunity. 2010;33(6):853–862. doi: 10.1016/j.immuni.2010.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.