

Significance
One of the challenging problems in lung cancer is to understand how inflammation pathways influence on the development of lung cancer and to identify early immune mediators. In this study, we functionally dissect the critical events occurring at the interface between endogenously arising lung tumors and the host immune system that determine tumor progression. Our findings greatly advance our knowledge on the function of T helper 17 cells in lung cancer and help understand the mechanisms of inflammatory mediators that promote lung cancer.
Keywords: interleukin 17, lung adenocarcinoma
Abstract
Lung cancer development is associated with extensive pulmonary inflammation. In addition, the linkage between chronic obstructive pulmonary disease (COPD) and lung cancer has been demonstrated in population-based studies. IL-17–producing CD4 helper T cells (Th17 cells) play a critical role in promoting chronic tissue inflammation. Although Th17 cells are found in human COPD and lung cancer, their role is not understood. We have thus used a mouse model of lung cancer, in which an oncogenic form of K-ras (K-rasG12D), frequently found in human lung cancer, is restrictedly expressed in lung epithelial cells [via Clara cell secretory protein (CCSPcre)]. In this model, Th17 and Treg but not Th1 cells were found enriched at the tumor tissues. When CCSPcre/K-rasG12D mice were weekly challenged with a lysate of nontypeable Haemophilus influenza (NTHi), which induces COPD-type inflammation and accelerates the tumor growth, they showed greatly enhanced Th17 cell infiltration in the lung tissues. Lack of IL-17, but not IL-17F, resulted in a significant reduction in lung tumor numbers in CCSPcre/K-rasG12D mice and also those treated with NTHi. Absence of IL-17 not only resulted in reduction of tumor cell proliferation and angiogenesis, but also decreased the expression of proinflammatory mediators and reduced recruitment of myeloid cells. Depletion of Gr-1+CD11b+ myeloid cells in CCSPcre/K-rasG12D mice suppressed tumor growth in lung, indicating Gr-1+CD11b+ myeloid cells recruited by IL-17 play a protumor role. Taken together, our data demonstrate a critical role for Th17 cell-mediated inflammation in lung tumorigenesis and suggest a novel way for prevention and treatment of this disease.
Inflammation plays an important role in tumor development (1, 2). Although targeting inflammation and tumor microenvironment has been considered as a new direction of cancer therapy, the mechanisms underlying cancer-associated inflammation have not been well understood. Lung cancer is a leading cause of death in the world. Accumulating evidence has shown that inflammation is associated with pathogenesis of lung cancer, especially those induced by cigarette smoke (3). The primary risk factor among smokers to develop lung cancer is the presence of chronic obstructive pulmonary disease (COPD) (4), which is characterized by chronic pulmonary inflammation, airway remodeling and destruction of lung parenchyma. Human lung cancers are inflicted with alterations in various subsets of lymphocytes and myeloid cells (5, 6), reminiscent of immune activation during chronic inflammation. Several studies have shown NFκB signaling as a mechanistic link between inflammation and lung cancer using a mouse model of lung adenocarcinoma (7, 8). However, the specific inflammatory cell types or molecules potentiating lung cancer are not understood clearly.
We and others have identified a novel subset of CD4 helper T cells that produce IL-17 and are referred as Th17 cells (9, 10). Th17 cells have been associated with inflammatory diseases such as rheumatoid arthritis, asthma, lupus, and allograft rejection. An important function of IL-17 is to promote tissue inflammation through the up-regulation of proinflammatory cytokines and chemokines (11). Consistently, we have shown that transgenic overexpression of IL-17 in the lungs resulted in chemokine up-regulation and tissue infiltration by leukocytes, although mice treated with neutralizing IL-17–specific antibody were also found to be resistant to the induction of experimental autoimmune encephalomyelitis (9). These and other studies collectively demonstrated that IL-17 and Th17 cells play nonredundant function in promoting inflammation.
Increased frequencies of IL-17 and Th17 cells have been reported in patients with different types of tumors (12), including lung adenocarcinoma (13). The density of intratumoral IL-17–positive cells in primary human nonsmall cell lung cancer was inversely correlated with patient outcome and correlated with smoking status of the patients (14). Th17 cells specific for a common tumor antigen were found in lung cancer patients as part of their spontaneous immune response to the autologous tumor (15). However, the function of Th17 cells and IL-17 in the development of lung cancer remains to be shown. Animal model studies have revealed contrasting roles of IL-17 in various tumors (16). Tumor-promoting effect of IL-17 was shown in some models such as colon cancer (17–20), whereas in others, IL-17 supported anti-tumor immunity, including in B16 melanoma model (21–24). Thus, the role of IL-17 could be complex and tumor-specific.
To properly evaluate the role of IL-17 in inflammation-associated lung cancer, we used a model of oncogenic K-ras mutation expressed only in the lung. Mice expressing K-ras mutation in Clara cells (CCSPcre/K-rasG12D mice) spontaneously develop lung adenocarcinoma (25). In addition, we induced COPD-type lung inflammation by challenging mice with lysates of nontypeable Haemophilus influenza (NTHi). Inflammation driven by NTHi can promote tumor growth in CCSPcre/K-rasG12D mice (25). These experiments collectively indicate a tumorigenic role of IL-17–mediated inflammation in the development of lung cancer.
Results
Th17 Cells Preferentially Accumulate in a Model of Lung Cancer.
Although Th17 cells are found in human COPD and lung cancers, their functional roles have not been understood. To conclusively address this issue, we adopted a mouse model of lung adenocarcinoma (CCSPcre/K-rasG12D) where oncogenic form of K-ras (K-rasG12D) (26) is restrictedly expressed in lung epithelial cells (CCSPcre) (27), hereinafter referred to as CC-LR. CC-LR mice develop lung adenocarcinoma without any potential of metastasis (25). K-ras activation in the bronchiolar epithelium has been associated with a robust inflammatory response (28), therefore providing an ideal spontaneous tumor model to study cancer-associated inflammation regulation. Analysis of bronchoalveolar lavage fluid (BALF) in 14-wk-old CC-LR mice, with visible tumors on lung surface and lung tissue with pulmonary hyperplasia and adenomas, revealed that there is an increased expression of Th17 signature cytokines IL-17, IL-17F, and IL-22 (Fig. 1A). In contrast, IFNγ were not increased in this model. To understand the source of these cytokines in CC-LR mice, we took either BALF cells or mononuclear fraction from tumor-bearing lung and restimulated with phorbol myristate acetate (PMA) and ionomycin for intracellular staining of IL-17 and IFNγ (Fig. 1B). Although IL-17+ cells in CD4 T cells were increased about 10-fold (WT 1.03 ± 0.91%, CC-LR 10.24 ± 6.69%, P = 0.013), IFNγ+ CD4 T cells remained similar between WT and CC-LR mice (WT 6.76 ± 3.89%, CC-LR 6.54 ± 3.58%) (Fig. 1C). Moreover, the level of IFNγ+ CD8 T cells did not change in tumor-bearing mice. One of the major producers of IL-17 in lung is γδ T cell; however, the percentages of γδ T cells producing IL-17 remained similar between WT and CC-LR mice (Fig. 1B). IL-17–positive CD4 T cells were TCRβ+, and IL-17–positive innate lymphoid cells (CD4+CD3−lin−) were not detected in the lungs of CC-LR mice. Foxp3+ regulatory T cells in tumor tissues were also threefold higher than that in normal lungs (WT 8.23 ± 0.40%, CC-LR 27.2 ± 4.5%, P < 0.001). IL-17+/Foxp3+ cells were also increased in tumor tissues (Fig. S1A). An increase in mRNA expression of Il17 and Foxp3 was also detected by RT-PCR (Fig. S1B). Rorc expression was similar between WT and CC-LR mice, whereas Tbx21 expression was reduced in CC-LR mice. Therefore, development of lung cancer is associated with an expansion of Th17 cells and regulatory T cells.
Fig. 1.

Th17 cells are recruited in K-ras–induced lung cancer. (A) Cytokines in BALF by ELISA (n ≥ 5 per group). (B) Representative flow cytometric plots of T cells. Lung mononuclear cells were isolated from 14-wk-old WT or CC-LR mice, stimulated with PMA/Ionomycin and stained with antibodies to CD4, CD8, γδTCR, IL-17, IFN-γ, and Foxp3. (C) Frequency of lung CD4 T cells expressing IL-17, IFN-γ, and Foxp3. Data are shown as mean ± SEM, *P < 0.05, ***P < 0.001 (WT, n = 4; CC-LR, n = 5).
Increased Th17 Cells Associate with Inflammation-Promoted Lung Cancer.
NTHi, found in lower airways of patients with COPD, is associated with exacerbation of COPD (29). Interestingly, chronic exposure to NTHi led to COPD-type lung inflammation characterized with an expanded Th17 cell population in the lung tissues of normal mice (Fig. 2A). NTHi challenge in CC-LR mice further accelerated the production of Th17 related cytokines in comparison with NTHi in WT mice (Fig. 2A). Intracellular cytokine staining of lung mononuclear cells indicated that over 25% of CD4 T cells in lung were stained with anti–IL-17, whereas IFNγ+ Th1 cells among CD4 T cells were decreased upon NTHi challenge (Fig. 2B). Notably, IL-17–deficient mice exhibited not only reduced inflammatory cell infiltration but also a decrease in airway wall thickness after 25 wk of NTHi challenge (Fig. S2).
Fig. 2.

Increased Th17 cells in inflammation-promoted lung cancer. (A) Cytokines in BALF by ELISA (n ≥ 5 per group). (B) Representative flow cytometry plots of T cells. Lung mononuclear cells were isolated from 14-wk-old WT or CC-LR mice which were challenged with NTHi lysate for 4 wk. Isolated cells were stimulated with PMA/Ionomycin and stained with antibodies to CD4, CD8, γδTCR, IL-17, IFN-γ, and Foxp3. (C) Frequency of lung CD4 T cells expressing IL-17, IFN-γ, and Foxp3. Data are shown as mean ± SEM, *P < 0.05, **P < 0.01 (WT + NTHi, n = 6; CC-LR + NTHi, n = 8).
It has been also shown that NTHi challenge in CC-LR mice resulted in lung cancer promotion (25). We found that in NTHi-exposed CC-LR mice in comparison with untreated CC-LR or NTHi-treated WT mice, percentages of Th17 cells among CD4 population were increased (NTHi WT 31.67 ± 1.45%, NTHi CC-LR 45.25 ± 4.42%, P < 0.05). In NTHi-exposed CC-LR mice, percentage of IFNγ+ CD4 cells was increased in comparison with NTHi-exposed WT mice (NTHi WT 6.40 ± 0.53%, NTHi CC-LR 16.48 ± 2.71, P = 0.01) but similar to untreated CC-LR mice. Percentages of IL-17+ γδ T cells or Foxp3+ regulatory CD4 T cells, however, remained constant between NTHi-exposed WT and NTHi-exposed CC-LR mice (Fig. 2 B and C). These results indicate that NTHi challenge in mice preferentially promotes Th17 cell response and an expansion of Th17 cells is associated with increased tumor growth in the presence of NTHi-induced type of inflammation.
IL-17 Deficiency Inhibited Lung Cancer Development.
The above results suggest a potential role for Th17 cells in lung cancer. To examine this possibility directly, CC-LR mice were crossed with Il17−/− mice. Tumor numbers on the surface of mouse lung were counted at the age of 14 wk. Lack of IL-17 resulted in a ∼75% (52.7 ± 7.1 in CC-LR Il17+/+ vs. 13.2 ± 2.5 in CC-LR Il17−/−, P = 0.003) reduction in lung surface tumor numbers compared with age- and sex-matched CC-LR Il17+/+ mice (Fig. 3A). The mean size of adenomas in CC-LR Il17−/− mice were smaller than that in CC-LR Il17+/+ mice (Fig. 3C and Fig. S3). In contrast, tumor numbers in CC-LR Il17f−/− mice were similar to those in CC-LR Il17f+/+ mice (Fig. 3 A and C). Together, these results indicate that IL-17, but not IL-17F, is required for lung tumor development.
Fig. 3.

IL-17 deficiency reduces lung cancer development. (A) Tumor numbers were counted on the lung surface at 14 wk of age in CC-LR mice that spontaneously develop lung adenocarcinoma, compared with the same mice crossed onto IL-17– or IL-17F–deficient mice, CC-LR Il17−/− or CC-LR Il17f−/−. (B) Total cell number in BALF. Data are shown as mean ± SEM **P < 0.01. ***P < 0.001 (CC-LR, n = 6; CC-LR Il17−/−, n = 7; CC-LR Il17f−/−, n = 5). (C) Representative H&E-stained lung sections from 14-wk-old CC-LR, CC-LR Il17−/− and CC-LR Il17f−/− mice. (D) Tumor numbers were counted on the lung surface at 14 wk of age in CC-LR mice that are exposed to NTHi starting at age 10 wk weekly for 4 wk. (E) Total cell numbers in BALF. Data are shown as means ± SEM (NTHi + CC-LR, n = 5; NTHi + CC-LR Il17−/−, n = 8). *P < 0.05. **P < 0.01.
The majority of recovered cells in BALFs of 14-wk-old tumor-bearing CC-LR mice were macrophages. CC-LR Il17−/− mice showed reduced total white blood cell (WBC) and macrophage numbers in comparison with CC-LR Il17+/+ mice (Fig. 3B). The number of infiltrated lymphocytes and neutrophils were also reduced in CC-LR Il17−/− mice in comparison with CC-LR Il17+/+ mice (Fig. 3B and Table S1). In contrast, we did not observe any difference in the number of infiltrated cells in lung between CC-LR Il17f+/+ and CC-LR Il17f−/− mice (Fig. 3B).
To determine whether IL-17 is involved in tumor promotion by NTHi-induced inflammation, CC-LR Il17−/−mice were challenged with NTHi. In comparison with CC-LR Il17+/+ mice, the numbers of visible tumors on the lung surface of CC-LR Il17−/− mice were reduced by ∼54% (CC-LR Il17+/+; 112 ± 7, CC-LR Il17−/− ; 52 ± 5, P < 0.001) after weekly NTHi exposure for 4 wk from the age of 10 wk (Fig. 3D). CC-LR Il17−/− mice had more hyperplastic early-stage lesions and smaller tumors in comparison with CC-LR Il17+/+ mice. We found the number of neutrophils, macrophages, and lymphocytes in BALF were also reduced in CC-LR Il17−/− mice (Fig. 3E and Table S2). Therefore, our results indicate that IL-17 is critical in inflammation-associated lung adenocarcinoma.
IL-17 Regulates Cancer Cell Proliferation, Angiogenesis, and Production of Proinflammatory Mediators.
To address how IL-17 promotes lung cancer, the proliferation index of tumor cells in situ was quantified by immunohistochemical staining of Ki67 (Fig. 4A). The percentages of Ki67-positive cells among tumor cells were significantly lower in CC-LR Il17−/− mice in comparision to CC-LR Il17+/+ mice (CC-LR Il17+/+ 8.9 ±1.1%, CC-LR Il17−/− 4.8 ± 1.2%, P = 0.011), indicating IL-17 promotes tumor cell proliferation directly or indirectly (Fig. 4B).
Fig. 4.

IL-17 deficiency reduces inflammation and cancer growth. (A) Representative immunohistochemistry of Ki67 and CD31 staining. (B) Ki67-positive cells or CD31 positive cells per 100 tumor cells at the 14-wk time point. n = 4 per group, 3–7 fields per mouse. (C) Relative expression of mRNA in whole lungs in 14-wk-old CC-LR mice (WT, n = 6; CC-LR Il17+/+, n = 6; CC-LR Il17−/−, n = 5). Data are expressed as fold increase compared with controls. Data represent means ± SEM *P < 0.05. **P < 0.01. (D) Representative flow cytometric plots of BALF or lung cells from WT and tumor-bearing mice. Cells were stained with anti-CD11b and Gr-1 Abs and gated in FSChi SSChi. (E) Absolute number of Gr-1+CD11b+ cells. Data are shown as means ± SEM (n = 5 per group, *P < 0.05).
To examine whether IL-17 increases the in vivo growth of tumors by promoting angiogenesis, we evaluated the vascular density by immunohistochemical examination with CD31 antibody (Fig. 4A). The percentage of CD31-positive cells within the tumors were significantly lower in CC-LR Il17−/− mice in comparision to CC-LR mice (CC-LR Il17+/+ 6.4 ± 1.0%, CC-LR Il17−/− 3.0 ± 1.2%, P = 0.036), indicating that IL-17 promotes angiogenesis in lung adenocarcinoma model (Fig. 4B).
IL-17 is known to stimulate the expression of chemokines and proinflammatory molecules in epithelial cell during inflammation. Therefore, we examined the molecules that are directly regulated by IL-17. Although the expression of Cxcl1 or Csf2 remained similar between CC-LR Il17+/+ and CC-LR Il17−/− mice, Il6, Cxcl2, Ccl2, Il6, Arg1, Csf3, Mmp7, Mmp12, and Mmp13 (Table S3) were up-regulated in tumor-bearing lung and reduced in CC-LR Il17−/− mice in comparison with CC-LR Il17+/+ animals (Fig. 4C). Compensatory T-cell response in CC-LR Il17−/− mice could influence the control of tumor growth; however, IFNγ expression in CD4, CD8 T cells and non-CD4 CD8 T cells were similar in WT and CC-LR Il17−/− mice. Therefore, our data suggest that IL-17 is required for tumorigenesis by inducing proinflammatory molecules during the development of lung cancer.
IL-17 Regulates Gr-1+CD11b+ Myeloid Cells in Lung Cancer.
To understand why CC-LR Il17−/− mice developed fewer adenocarcinomas than CC-LR Il17+/+ mice at 14 wk of age, we examined cellular infiltrates from lung parenchyma and tumors. Although the BALF of WT mice was mostly composed of alveolar macrophages, CC-LR mice showed elevated number of lymphocytes and macrophages and elevated occurrence of a distinctive Gr-1+CD11b+ population, which is significantly reduced in CC-LR Il17−/− mice (Fig. 4D). The gated Gr-1+CD11b+ cells in BALF are mostly Ly6G-positive cells (Fig. S4A). Lung parenchyma of normal mice, however, is comprised of mature and terminally differentiated Gr-1+CD11b+ neutrophils. Cellular analysis of lung parenchyma and tumor showed an accumulation of Gr-1+CD11b+ myeloid cells in CC-LR, in comparison with normal lung. During tumorigenesis, a heterogeneous population of myeloid cells, known as myeloid-derived suppressive cells, is generated (5). Myeloid-derived suppressive cells express the cell-surface markers Gr-1 and CD11b and function to suppress T-cell activity. We found ∼77% reduction in total number of Gr-1+CD11b+ cells among CD45+ fraction of lung mononuclear cells in IL-17–deficient mice (CC-LR Il17+/+, 8.78 ± 2.24; CC-LR Il17−/−, 2.79 ± 1.42; P = 0.027, expressed as ×104 cells) (Fig. 4E).
To evaluate the function of Gr-1+CD11b+ cells, we examined their immune suppressive activity in vitro. Gr-1+CD11b+ cells were isolated from tumors of CC-LR mice and from lung parenchyma of WT mice. When Gr-1+CD11b+ cells were cultured with total splenocytes in the presence of αCD3 and αCD28, Gr-1+CD11b+ cells isolated from tumors exhibited suppressive activity to T-cell proliferation and cytokine production (Fig. S4B). This finding is in agreement with a previous study using CC10 promoter-driven SV40 TAg transgene model of lung adenocarcinoma (30). This result suggests that CC-LR Il17−/− may have increased cytotoxic CD8 T activity due to reduced Gr-1+CD11b+ cells. However, when CD8 T cells in CC-LR Il17−/− were depleted using anti-CD8 antibody for 2 wk, we did not observe altered tumor development in CC-LR Il17−/− mice (Fig. 5). In addition, we observed that CC-LR mice that were depleted of CD8 T cells exhibited similar tumor burden as isotype control antibody-treated mice (Fig. 5). Therefore, the cytotoxic activity of CD8 T cells is not attributable to reduced tumor growth in CC-LR mice lacking IL-17, at least in a period of 14 wk we observed.
Fig. 5.
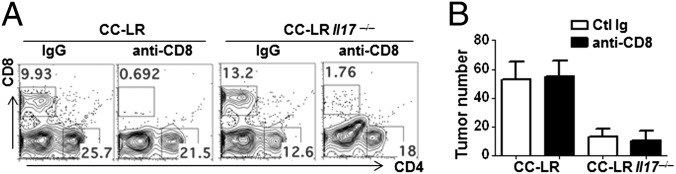
CD8 T-cell depletion did not restore tumor growth. (A) Flow cytometric analyses of CD8 T cells in BALF and lung. Lung mononuclear cells were stained for CD4 and CD8 after 2 wk of either rat IgG or anti-CD8 antibody treatment. (B) Tumor burden after anti-CD8 treatment (n = 5–6 per group). Data represent means ± SEM.
To investigate the role of tumor-associated Gr-1+CD11b+ cells in the development of lung adenocarcinoma and establish whether IL-17 mediated cellular changes in tumor are in agreement with the immune responses mediated by Gr-1+CD11b+, we depleted Gr-1+ cells by i.p. injection of the RB6.8C5 monoclonal antibody in tumor-bearing mice. Depletion of Gr-1+ cells was confirmed by flow cytometric analysis of BALF and lung (Fig. 6A). Depletion of Gr-1+CD11b+ cells in CC-LR mice resulted in suppression of lung tumor growth (isotype control group, 51.1 ± 1.90, RB6.8C5 treated group, 27.33 ± 6.76, P = 0.042) (Fig. 6B). Tumor cell proliferation (isotype control group, 16.96 ± 2.63%; RB6.8C5-treated group, 8.49 ± 1.92%, P = 0.013) and tumor microvessel density (isotype control group, 10.5 ± 1.5%; RB6.8C5-treated group, 2.24 ± 1.23%, P = 0.003) were also significantly reduced in Gr-1 antibody-treated group (Fig. 6C). In addition, inflammatory molecules induced upon tumor growth were significantly suppressed upon Gr-1 antibody injection (Fig. 6D). Based on the above data, Gr-1+ cells depletion in CC-LR mice considerably recapitulates the phenotype of CC-LR Il17−/− mice. The expression level of IL-17 upon Gr-1+ cell depletion was reduced but the expression level of Foxp3 or IFNγ remained similar (Fig. 6D). We also found that administration of a neutralizing antibody to IL-17A in vivo significantly reduced tumor count as well as Gr-1+CD11b+ cell recruitment (Fig. S5). This result not only highlights the potential use of IL-17 blockade in lung cancer patients, but also rules out possible microbiota difference in WT and CC-LR Il17−/− mice accounting for the tumor growth differences. Collectively, our data demonstrated that Gr-1+CD11b+ myeloid cells recruited during primary lung tumor development promote tumor growth and IL-17 orchestrates their recruitment.
Fig. 6.

Gr-1+ cell depletion reduces the growth of lung adenocarcinoma. CC-LR mice were treated every 3 d for 2 wk with anti–Gr-1 or isotype control antibody. (A) Flow cytometric analyses of Gr-1+CD11b+ in BALF and lung. Cells were gated in FSChi SSChi. (B) Tumor burden after anti–Gr-1 treatment (control Ig, n = 5; anti–Gr-1, n = 8). (C) Representative sections of lung adenocarcinoma immunostained with Ki67 or CD31. Ki67-positive cells or CD31 positive cells per 100 tumor cells (circle, control Ig; square, anti–Gr-1). (D) Relative expression of mRNA in whole lungs after anti–Gr-1 treatment in CC-LR mice. Data are expressed as fold increase compared with controls. Data represent means ± SEM *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Understanding the developmental process and function of immune response during tumorigenesis is a challenging problem. One of important factors to consider is to use a disease model that resembles the development of tumors in human. The most common molecular changes identified in human lung cancer are K-ras mutations (31). K-ras mutation restrictively expressed in mouse lung epithelial cells led to cellular hyperplasia, adenoma, and eventually adenocarcinoma, resembling human lung cancer development (25, 28). Therefore, this system allows immune response to be shaped against endogenous arising lung tumor and reflects tumor microenvironment during the initiation and early stages of pulmonary adenocarcinoma.
Th17 cells and its signature cytokine, IL-17, have been detected in different types of human cancers (12). Despite relatively well defined role of Th17 cells in promoting inflammation in autoimmune disease, their function in tumor development has been obscure. In this study, we made an intriguing observation that activating K-ras mutation in lung epithelial cells promotes the recruitment of Th17 cells in tumor tissue. More importantly, our study revealed an indispensable role of IL-17 in tumorigenesis during the early stages of pulmonary adenocarcinoma. Our conclusion is in agreement with the recent studies using spontaneous or chemically induced colorectal cancer models (17, 19, 20) and prostate cancer model with Pten deficiency (32).
The mechanisms underlying protumorigenic role of IL-17, however, were not completely understood. In our study, IL-17 accelerated cancer development at least in part by recruiting myeloid cells and promoting inflammation. A recent study demonstrated that IL-17 promotes tumor resistance to VEGF inhibition due to its ability of Gr-1+CD11b+ cell mobilization and recruitment (33). Il17rc−/− (deficiency in IL-17 and IL-17F) mice crossed with deletion of PTEN in prostate cells showed reduced prostate adenocarcinoma due to reduction in MMP7. MMP7 was induced directly by IL-17 in prostate cells in this study (32). Because IL-17 can induce IL-6 from tumor and stromal cells, STAT3 activation by IL-6 was proposed as one of mechanisms for protumorigenic role of IL-17 (22). In our study, we also observed reduced pSTAT3 in tumors of CC-LR Il17−/− and anti–IL-17 antibody treated CC-LR mice in comparison with CC-LR WT (Fig. S6).
In contrast, antitumorigenic role of IL-17 has been reported in several cancer models. In a colon cancer model adopting truncated form of adenomatous polyposis coli (APC), Il17−/− × APCΔ468 mice developed more invasive cancer (34), even though fewer polyps and reduced Gr-1+CD11b+ myeloid cells were found in Il17−/− × APCΔ468 mice. In other metastasis accompanying models, IL-17 has shown to promote the activation of tumor-specific CD8+ T cells that help to eradicate tumor (21, 23, 35). Despite the increased tumor burden in Il17−/− mice, tumor lung of Il17−/− mice expressed reduced Gr-1+CD11b+ myeloid cells in comparison with WT (23). In transplantation models, CD8+ T cell response initiated upon tumor injection is a critical determinant of tumor elimination, which may outweigh other cellular effects mediated by IL-17. Therefore, the role of IL-17 in tumor development is likely to be dependent on the local tumor microenvironments and the stage of tumor development.
At present, it is unclear which pathways or molecules are responsible downstream of IL-17 that promote the recruitment of myeloid cells. A recent report indicates CXCL1/2 mediates mammary tumor growth and lung metastasis by recruiting CD11b+Ly6G+ granulocytic MDSC population to the tumor site (36). Alveolar epithelial cells transformed by oncogenic K-ras are known to express high level of CXCL1 and CXCL2 (37). Also, oncogenic K-ras–induced GM-CSF is capable of inducing proliferation and differentiation of c-kit+ Lineage– precursors into functional MDSC (38, 39). Because IL-17 can directly act on lung epithelial cells to induce CXCL2 and G-CSF, reduced expression of these molecules in the lungs of IL-17–deficient mice could contribute to decrease in Gr-1+CD11b+ cells recruitment. Even though Gr-1+CD11b+ cells in tumor lung were capable of suppressing CD8 T-cell activity ex vivo, depletion of CD8 T cells did not lead to enhanced tumor growth in our study. Gr-1+ CD11b+ cells may directly promote angiogenesis or promote regulatory T cells to enhance tumor in CC-LR mice (5, 40, 41).
Th17 cells can be readily found in a steady state in intestine but efficiently generated and recruited to tissues upon inflammation. Analysis of human lung cells from emphysema patients revealed infiltration of Th17 cells (42). We and others have previously used an experimental model of smoke exposure and demonstrated IL-17–deficient mice are resistant to smoke-induced airway inflammation and emphysema (43, 44). Tobacco smoke accounts for the most common causes of lung cancer. Therefore, these studies suggest the link between inflammation in lung caused by smoking, promotion of lung cancer, and involvement of IL-17 and Th17 cells. In our study, CC-LR mice were challenged with NTHi to induce COPD-like inflammation in the lung. Th17 cells were major adaptive immune cells generated in the lung during inflammation, and IL-17 played a pivotal role in promoting tumor in inflammation-accelerated lung cancer model.
Currently, we do not know how lung cancer microenvironment in K-ras mutant mice directs Th17 cell responses. K-ras activation in the bronchiolar epithelium has been associated with promotion of inflammatory response (28). More recent data indicate K-ras activation in pancreas leads to perturbation of multiple metabolic pathways (45). It is possible that inflammatory molecules or metabolites upon K-ras activation in lung epithelium influence the polarization and recruitment of Th17 cells in lung. Also, it is not known whether Th17 cells in CC-LR mice develop in antigen-specific pathway and recognize self or tumor antigen. In NTHi-challenged CC-LR mice, recall response indicates the generation of NTHi-specific T cells. In contrast, we did not detect IL-17 or IFNγ response to tumor lysates. In APCΔ468 mice, microbial translocation upon epithelial barrier loss directed the generation of Th17 cells, suggesting the possibility of microbiota-specific Th17 cells in colon cancer (19). CC-LR mice represent the early stage of lung cancer; it would be interesting to define the antigens in this model, which could also differ from the antigens of established tumors.
In summary, we have shown that oncogene-driven and inflammation-promoted lung adenocarcinoma in mice is associated with increased Th17 cells in the tumor tissue. Our study also demonstrates, to our knowledge, for the first time that inflammation occurs in the lung can accelerate lung cancer through Th17 cells and IL-17 upon oncogenic activation. Because the lung is vulnerable to many airborne infections and environmental insults that lead to various degrees of inflammation, it is important to identify the cellular and molecular pathways of inflammatory diseases, such as COPD, that predispose individuals to lung cancer. Currently, no screening strategies exist that can identify patients with chronic lung inflammation with the potential to develop lung cancer. A large cohort study measuring the level of Th17 cells and IL-17 in patients with chronic lung inflammation and different stages of lung cancer with history of inflammatory diseases may provide causal link indicative of individuals with lung inflammation at higher risk of developing lung cancer. This link would further help setting the basis for future translational studies targeting Th17 cells or IL-17. Although inhibition of IL-17 using neutralizing antibody or Th17 cells development by targeting IL-23 have shown their promising results treating autoimmune diseases in clinical trials, the potential benefits and risks of inhibiting Th17 cells or IL-17 for cancer therapy have not been investigated. Our study establishes that targeting Th17 cells or IL-17 in the early stage of lung cancer and in COPD patients with Th17 cell response may be a promising strategy for controlling inflammation that promotes tumorigenesis.
Methods
Mice.
All of the mice were housed in the MD Anderson cancer center animal facility, and all experiments were performed in accordance with relevant institutional and national guidelines and regulations approved by the Animal Studies Committee. CCSPCre (27), LSL–K-rasG12D (26), Il17−/−, and Il17f−/− mice (46) have been described. C57BL/6 mice were obtained from the Jackson Laboratory and crossed with CC-LR mice. CC-LR WT, CC-LR Il17−/−, and CC-LR Il17f−/− mice were kept in separate cages.
Assessment of Lung Tumor Burden.
Lung surface tumor numbers were counted, and then BALF was obtained by collecting 1 mL of PBS through a tracheostomy cannula. The lungs were prepared for histologic analysis first by perfusion with PBS, and then inflation with 10% phosphate-buffered formalin (pH 7. 4) at a 20-cm pressure of H2O. Total leukocyte count was determined using a hemacytometer, and cell populations were determined by cytocentrifugation and Wright–Giemsa staining. BALF was centrifuged at 1,250 × g for 10 min, and supernatants were collected for ELISA.
NTHi Lysate Aerosol Exposure.
A lysate of NTHi strain 12 was prepared as described (25). CC-LR mice were exposed to NTHi starting at 10 wk of age for 4 wk.
Isolation of Lung Resident Mononuclear Cells.
Lungs were harvested after perfusion with PBS. Lungs were first inflated with 0.1 mg/mL collagenase IV and DNase1 for 15 min at 37 °C. Single cell suspensions were prepared by mechanical dissociation of lung tissue through a 70-μm nylon mesh. Lung cells were suspended in PBS and layered on LSM Lymphocyte Separation (Medium MP Biomedical). Cells were centrifuged at room temperature for 20 min at 900 × g. Mononuclear cells were harvested from the gradient interphase.
Depletion of Gr-1 Cells or CD8 T Cells.
For Gr-1 depletion study, 12-wk-old mice were injected with 150 μg of anti–Gr-1 antibody (Clone: RB6-8C5; Bioxcell) or isotype control Ab (Rat IgG2a) i.p. two times per week for 2 wk. Depletion of the cells was monitored by FACS analysis of blood. For CD8 T-cell depletion study, the same experimental schedule as above was performed using anti-CD8 antibody (Clone: 2.43; BioXCell). Additional information is provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Drs. Tyler Jacks for the LSL-K-rasG12D, Francesco DeMayo for the CCSP-cre mice, Jonathan Kurie for the LKR13 cells, and members of the C.D. laboratory for their help and discussion. The work is supported by research grants from the National Institutes of Health (to C.D.), Cancer Prevention and Research Institute of Texas (to C.D. and S.H.C.), and Research Scholar Grant (RSG-11-115-01-CNE) from American Cancer Society (to S.J.M.). C.D. is the Olga and Harry Wiess Distinguished University Chair in Cancer Research of the University of Texas MD Anderson Cancer Center.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1319051111/-/DCSupplemental.
References
- 1.Elinav E, et al. Inflammation-induced cancer: Crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer. 2013;13(11):759–771. doi: 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- 2.Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spitz MR, et al. A risk model for prediction of lung cancer. J Natl Cancer Inst. 2007;99(9):715–726. doi: 10.1093/jnci/djk153. [DOI] [PubMed] [Google Scholar]
- 4.Celli BR. Chronic obstructive pulmonary disease and lung cancer: Common pathogenesis, shared clinical challenges. Proc Am Thorac Soc. 2012;9(2):74–79. doi: 10.1513/pats.201107-039MS. [DOI] [PubMed] [Google Scholar]
- 5.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fridman WH, Pages F, Sautes-Fridman C, Galon J. The immune contexture in human tumours: Impact on clinical outcome. Nat Rev Cancer. 2012;12(4):298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 7.Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco smoke promotes lung tumorigenesis by triggering IKKbeta- and JNK1-dependent inflammation. Cancer Cell. 2010;17(1):89–97. doi: 10.1016/j.ccr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meylan E, et al. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature. 2009;462(7269):104–107. doi: 10.1038/nature08462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Park H, et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6(11):1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrington LE, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6(11):1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 11.Kolls JK, Linden A. Interleukin-17 family members and inflammation. Immunity. 2004;21(4):467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- 12.Kryczek I, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood. 2009;114(6):1141–1149. doi: 10.1182/blood-2009-03-208249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reppert S, et al. A role for T-bet-mediated tumour immune surveillance in anti-IL-17A treatment of lung cancer. Nat Commun. 2011;2:600. doi: 10.1038/ncomms1609. [DOI] [PubMed] [Google Scholar]
- 14.Chen X, et al. Increased IL-17-producing cells correlate with poor survival and lymphangiogenesis in NSCLC patients. Lung Cancer. 2010;69(3):348–354. doi: 10.1016/j.lungcan.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 15.Hamai A, et al. Human T(H)17 immune cells specific for the tumor antigen MAGE-A3 convert to IFN-gamma-secreting cells as they differentiate into effector T cells in vivo. Cancer Res. 2012;72(5):1059–1063. doi: 10.1158/0008-5472.CAN-11-3432. [DOI] [PubMed] [Google Scholar]
- 16.Martin-Orozco N, Dong C. The IL-17/IL-23 axis of inflammation in cancer: Friend or foe? Curr Opin Investig Drugs. 2009;10(6):543–549. [PubMed] [Google Scholar]
- 17.Chae WJ, et al. Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci USA. 2010;107(12):5540–5544. doi: 10.1073/pnas.0912675107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu S, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med. 2009;15(9):1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grivennikov SI, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491(7423):254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tong Z, et al. A protective role by interleukin-17F in colon tumorigenesis. PLoS ONE. 2012;7(4):e34959. doi: 10.1371/journal.pone.0034959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kryczek I, Wei S, Szeliga W, Vatan L, Zou W. Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood. 2009;114(2):357–359. doi: 10.1182/blood-2008-09-177360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, et al. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206(7):1457–1464. doi: 10.1084/jem.20090207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin-Orozco N, et al. T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity. 2009;31(5):787–798. doi: 10.1016/j.immuni.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muranski P, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112(2):362–373. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moghaddam SJ, et al. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease-like airway inflammation in a K-ras-induced mouse model. Am J Respir Cell Mol Biol. 2009;40(4):443–453. doi: 10.1165/rcmb.2008-0198OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jackson EL, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15(24):3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li H, et al. Cre-mediated recombination in mouse Clara cells. Genesis. 2008;46(6):300–307. doi: 10.1002/dvg.20396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji H, et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene. 2006;25(14):2105–2112. doi: 10.1038/sj.onc.1209237. [DOI] [PubMed] [Google Scholar]
- 29.Bandi V, et al. Nontypeable Haemophilus influenzae in the lower respiratory tract of patients with chronic bronchitis. Am J Respir Crit Care Med. 2001;164(11):2114–2119. doi: 10.1164/ajrccm.164.11.2104093. [DOI] [PubMed] [Google Scholar]
- 30.Corzo CA, et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J Exp Med. 2010;207(11):2439–2453. doi: 10.1084/jem.20100587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359(13):1367–1380. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Q, et al. Interleukin-17 promotes formation and growth of prostate adenocarcinoma in mouse models. Cancer Res. 2012;72(10):2589–2599. doi: 10.1158/0008-5472.CAN-11-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maniati E, Hagemann T. IL-17 mediates resistance to anti-VEGF therapy. Nat Med. 2013;19(9):1092–1094. doi: 10.1038/nm.3333. [DOI] [PubMed] [Google Scholar]
- 34.Blatner NR, et al. Expression of RORgammat marks a pathogenic regulatory T cell subset in human colon cancer. Sci Transl Med. 2012;4(164):ra159. doi: 10.1126/scitranslmed.3004566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Benchetrit F, et al. Interleukin-17 inhibits tumor cell growth by means of a T-cell-dependent mechanism. Blood. 2002;99(6):2114–2121. doi: 10.1182/blood.v99.6.2114. [DOI] [PubMed] [Google Scholar]
- 36.Acharyya S, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150(1):165–178. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wislez M, et al. High expression of ligands for chemokine receptor CXCR2 in alveolar epithelial neoplasia induced by oncogenic kras. Cancer Res. 2006;66(8):4198–4207. doi: 10.1158/0008-5472.CAN-05-3842. [DOI] [PubMed] [Google Scholar]
- 38.Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21(6):836–847. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bayne LJ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21(6):822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murdoch C, Muthana M, Coffelt SB, Lewis CE. The role of myeloid cells in the promotion of tumour angiogenesis. Nat Rev Cancer. 2008;8(8):618–631. doi: 10.1038/nrc2444. [DOI] [PubMed] [Google Scholar]
- 41.Gong L, et al. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol Cancer. 2013;12(1):154. doi: 10.1186/1476-4598-12-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shan M, et al. Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med. 2009;1(4):ra10. doi: 10.1126/scitranlsmed.3000154. [DOI] [PubMed] [Google Scholar]
- 43.Shan M, et al. Cigarette smoke induction of osteopontin (SPP1) mediates T(H)17 inflammation in human and experimental emphysema. Sci Transl Med. 2012;4(117):ra119. doi: 10.1126/scitranslmed.3003041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen K, et al. IL-17RA is required for CCL2 expression, macrophage recruitment, and emphysema in response to cigarette smoke. PLoS ONE. 2011;6(5):e20333. doi: 10.1371/journal.pone.0020333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ying H, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149(3):656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang XXO, et al. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205(5):1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.