

Abstract
Background:
There is insufficient data on the clinical and radiological features of neuromyelitis optica (NMO) and neuromyelitis optica spectrum disorders (NMOSD) from India.
Objective:
The objective of the following study is to examine the clinico-radiological features of NMO and NMOSD in an Indian cohort.
Materials and Methods:
This retrospective study included 44 consecutive patients who (1) satisfied the 2006 Wingerchuk criteria for NMO (16 seropositive and 7 seronegative); or (2) had isolated or recurrent optic neuritis (ON) with seropositivity (n = 4); or (3) had isolated or recurrent myelitis with seropositivity (n = 17).
Results:
The female:male ratio was 7.8:1 with median age of onset 26.5 (range 8-72). Annualized relapse rate (ARR) was comparable across all groups (F [3, 40] = 0.938 and P = 0.431). Various presentations other than ON and myelitis were noted. All 40 patients with myelitis had spinal cord lesions involving ≥3 vertebral segments during the course of the disease. Cervicomedullary involvement was seen in 32.5% (13/40) patients. Brain magnetic resonance imaging was available for 40 patients; eight of these (20%) had brain lesions in locations described in multiple sclerosis (MS), 27.5% (11/40) had lesions at sites unusual for MS and 52.5% (21/40) had normal brain imaging.
Conclusion:
NMO and NMOSD patients in this cohort have comparable ARR regardless of clinical presentation, supporting the emerging trend of treating all patients with immunotherapeutic agents at an early stage. Varied presentations seen in NMO and NMOSD highlight the need for a high index of suspicion for NMO in demyelinating episodes not classical for MS.
Keywords: Myelitis, neuromyelitis optica, neuromyelitis optica spectrum disorders, optic neuritis
Introduction
Neuromyelitis optica (NMO) is an inflammatory demyelinating, necrotizing disease of the central nervous system, with a predilection for the optic nerves and spinal cord. Clinical, radiological and immunopathological characteristics distinguish it from multiple sclerosis (MS). Classical NMO is defined by the revised diagnostic criteria set out by Wingerchuk et al. in 2006, which requires the presence of optic neuritis (ON) and myelitis, plus any two of: (a) Brain magnetic resonance imaging (MRI) not satisfying the McDonald criteria; (b) contiguous spinal cord MRI T2 lesions spanning three or more vertebral segments; (c) positive serology for NMO-immunoglobulin G (IgG) (anti-aquaporin-4 antibody).[1] Since the discovery of NMO-IgG as the pathogenic marker for NMO, various limited forms of NMO have also been recognized based on NMO-IgG seropositivity;[2] these constitute the neuromyelitis optica spectrum disorders (NMOSD).
Various studies on NMO from the pre-MRI era from India report that NMO comprised 9-24% of the demyelinating diseases in India.[3] However, very few studies have examined NMO in India after the adoption of the 2006 revised Wingerchuk criteria and there is insufficient data on the clinical and radiological features of NMO in India. In addition, there is very little data available on NMOSD in India. The objective of the present study was to examine clinical and radiological features of NMO and NMOSD, in an Indian cohort.
Materials and Methods
During the study period from January 2010 to April 2013, 460 patients with demyelinating diseases were seen by us at the outpatient and inpatient department of a tertiary care hospital. These included new and follow-up cases. Of this population, 44 consecutive patients were included in this retrospective study, fulfilling the following criteria:
Patients satisfying the revised (2006) Wingerchuk diagnostic criteria for NMO (seropositive NMO and seronegative NMO) or
Patients with isolated or recurrent ON and NMO-IgG seropositivity (seropositive ON) or
Patients with isolated or recurrent myelitis and NMO-IgG seropositivity (seropositive myelitis).
Patients with a confirmed diagnosis of MS, acute disseminated encephalomyelitis, seronegative transverse myelitis and seronegative ON were excluded.
Patients included in the study were divided into four groups on the basis of clinical presentation and NMO-IgG seropositivity:
NMO-IgG positive NMO (Seropositive NMO)
NMO-IgG negative NMO (Seronegative NMO)
NMO-IgG positive ON (Seropositive ON)
NMO-IgG positive myelitis (Seropositive myelitis).
Patients came to the department mostly as referrals and therefore were seen by us at variable intervals from the onset of the disease and from the time of treatment for acute episodes. Clinical and radiological features of these patients were analyzed. Single-variable ANOVA was used as a test of significance where required.
NMO (anti-aquaporin 4) antibody testing was done by indirect immunofluorescence using the Euroimmun kit (Luebeck, Germany), a visual fluorescence-observation cell-based assay that incorporated fixed HEK293 cells transfected singly with either human AQP4-M1 or M23 isoform.[4] Testing was carried out at Metropolis Healthcare Limited, Mumbai. The study was approved by the institutional ethics committee.
Results
Of the 44 patients included in the study, 23 patients satisfied the revised (2006) Wingerchuk et al. diagnostic criteria for NMO (16 seropositive and seven seronegative). 21 patients satisfied the criteria for NMOSD,[2] among which four had recurrent ON (all seropositive) and 17 had isolated or recurrent myelitis (all seropositive). The female:male ratio for the whole cohort was 7.8:1. The female:male ratio for the ON onset group was 3.5:1 whereas all myelitis onset patients were females. The median age of onset was 26.5 (range 8-72). There was no significant difference in the age of onset of NMO patients presenting with either ON or myelitis (F [1, 16] = 0.536 and P = 0.475). The median duration of follow-up was 3 years (ranges from 4 months to 30 years).
The median number of relapses was three (range 1-15). The mean annualized relapse rate (ARR) for the whole cohort was 0.89 ± 0.62 (range 0.14-3.03). There was no significant difference in ARR across the four groups (F [3, 40] = 0.938 and P = 0.431). The initial presentation was myelitis in 59% (26/44) patients, ON in 30% (13/44) and simultaneous ON and myelitis in 7% (3/44) [Graph 1]. There was no significant difference in ARR of NMO patients presenting with either ON or myelitis (F [1, 16] = 0.604 and P = 0.448). Apart from the relapses of isolated ON, isolated myelitis and simultaneous ON + myelitis, various other presentations were noted with brainstem and cerebellar involvement, with or without ON or myelitis. The clinical parameters of the four groups are described in Table 1.
Graph 1.
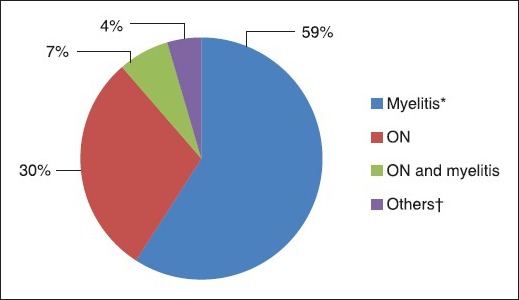
Initial presentations (*Hiccups and vomiting in 4, †Diplopia + vomiting 1; Myelitis + ON + diplopia + ataxia 1)
Table 1.
Clinical characteristics of the four study groups

MRI parameters
Among the 40 patients having clinical evidence of myelitis, 77.5% (31/40) of the patients had cervicodorsal cord involvement (15/16 seropositive NMO, 4/7 seronegative NMO and 12/17 seropositive myelitis). All patients having clinical evidence of myelitis (40/40) had spinal cord lesions involving three or more vertebral segments during the course of the disease. One seropositive NMO patient and one seropositive myelitis patient presented initially with short segment myelitis (two vertebral lengths). Both subsequently had relapses of myelitis involving more than three vertebral lengths. Cervicomedullary junction (CMJ) involvement was seen in 32.5% (13/40) of the patients in these three groups (6 seropositive NMO, one seronegative NMO and six seropositive myelitis).
Of the 40 patients for whom brain MRI was available, 20% (8/40) had brain lesions at locations described in MS (5 seropositive NMO and 3 seropositive myelitis). Two of these eight patients were tested for cerebrospinal fluid (CSF) oligoclonal bands and it was negative in both. 27.5% (11/40) patients had brain lesions at sites unusual for MS (four seropositive NMO, two seronegative NMO and five seropositive myelitis). Brain MRI was normal for 52.5% (21/40) patients and was not available for four seropositive myelitis patients. Optic nerve imaging was not consistently available and was therefore not analyzed.
Laboratory parameters
NMO antibody testing was done at the time of a relapse in 45.45% (20/44) patients (three of these patients were seronegative); in 54.55% (24/44) patients, NMO antibody testing was done at variable intervals after a relapse (four of these patients were seronegative). Only one of the 20 patients for whom testing for CSF oligoclonal bands was done tested positive. This patient presented with bilateral ON and had a normal brain MRI with positive NMO-IgG. The median CSF leukocyte count was 10 (range 2-940) in the 28 patients for whom CSF analysis was done; four patients had >50 leukocytes in CSF. Three out of these four patients had lymphocytic predominance while one patient had neutrophilic predominance. The median CSF protein level was 40.5 (range 16-790).
Treatment
59.09% (26/44) patients (75% [12/16] seropositive NMO, 42.8% [3/7] seronegative NMO, 50% [2/4] seropositive ON, 52.9% [9/17] seropositive myelitis) received various immunotherapeutic agents to prevent relapses. Azathioprine + prednisolone (12/26) and rituximab (6/26) were most commonly used as initial therapy. Other agents used as initial therapy were mycophenolate with or without prednisolone, cyclophosphamide, methotrexate, mitoxantrone and interferon beta-1a (Rebif®). In one patient interferon beta-1 a (Rebif®) was used initially as this patient was thought to have opticospinal MS before NMO antibody testing became available. It was discontinued when this patient tested positive for NMO IgG. In eight patients, second immunotherapeutic agent was used, rituximab being the most common (7/8).
Discussion
NMO is an inflammatory demyelinating, necrotizing disorder affecting optic nerves and spinal cord. Identification of NMO IgG as the pathogenic biomarker has contributed to widening the spectrum of NMO to include limited forms and atypical clinical presentations. A strong female preponderance has been reported for NMO in various studies in different patient populations.[5,6,7] The present study also found a high female:male ratio of 7.8:1.
Although the first episode of NMO can occur at any age from childhood to old age, in most reported series the average age at onset is in the 4th decade.[8] In the present series, the median age at onset was 26.5, with a wide range of 8-72. A study of British and Japanese NMO spectrum patients found that the mean age of patients presenting with ON was significantly lower than the mean age of patients presenting with long-segment myelitis.[6] This was supported by a large study of Japanese NMO-IgG positive patients, which found that ON was more predominant in younger subgroups while spinal involvement was more predominant in the older subgroups.[7] In the present study, however, there was no significant difference in the age of onset of patients presenting with ON and myelitis; the smaller sample size of this study may be a factor in this finding.
Classically, NMO has been known to present with isolated or simultaneous ON and myelitis. In the present series, myelitis was the presenting feature in 59% patients, ON in 30% and simultaneous ON and myelitis in 7%. Apart from ON and myelitis, the present study also noted other clinical presentations involving brainstem and cerebellum [Graph 1]. Brainstem presentations such as intractable hiccups, nausea and vomiting, vertigo, ataxia and various bulbar symptoms have been well-documented in NMO and NMOSD.[9] Four patients in the present study presented with intractable hiccups and vomiting and all four had CMJ involvement. Various hypothalamic presentations such as narcolepsy, syndrome of inappropriate anti-diuretic hormone secretion, anorexia or hyperphagia, as well as certain cerebral presentations such as posterior reversible encephalopathy syndrome, impairment of consciousness and seizures have also been described.[9]
Brain MRI in NMO and NMOSD was initially thought to be either normal or to show lesions atypical for MS. However, brain lesions have now been reported in 60-79% of patients with NMO and NMOSD.[10] In the present study, 47.5% patients demonstrated brain lesions. Certain characteristic brain lesions, such as confluent hemispheric lesions, longitudinally extensive corticospinal tract lesions, lesions in areas of high aquaporin-4 expression like hypothalamus and periependymal regions and central and dorsal brainstem lesions with CMJ involvement, have been described.[9,10] CMJ involvement was seen in an appreciable number (32.5%) of patients with myelitis in the present study. A small study on NMO and NMOSD lesions on 7 T MRI reported that NMO lesions are less often perivascular, rarely show characteristic hypointense rim unlike MS lesions and less frequently involve cortex as compared to MS.[11] These results will need to be confirmed in larger studies.
The rubric of ‘NMO spectrum disorders’[2] included the following clinical presentations associated with NMO-IgG seropositivity: Idiopathic single or recurrent attacks of longitudinally extensive transverse myelitis (LETM), recurrent or simultaneous bilateral ON, Asian opticospinal MS, ON or LETM associated with systemic autoimmune diseases and ON or LETM associated with brain lesions typical of NMO. Recently Lana-Peixato and Callegaro[9] suggested that NMO spectrum should include either NMO-IgG seropositivity or NMO-typical brain lesions plus one of the following: Single, recurrent or simultaneous bilateral ON, longitudinally extensive myelitis (≥3 vertebral segments), or recurrent brainstem, hypothalamic or cerebral symptoms. This definition does not include NMO-IgG seropositivity as an absolute requirement for the diagnosis of NMOSD. Although these suggested criteria may be more sensitive, the specificity may be compromised by removing the absolute requirement for NMO-IgG seropositivity. More recently, Sato et al.[12] have also proposed a widening of the criteria for NMO spectrum to include even single episodes of brain/brainstem presentations; however, these criteria retain the absolute requirement for NMO-IgG seropositivity. These criteria need further validation and testing.
Comparisons have been made in the ARR for patients with seropositive NMO, seronegative NMO and NMOSD. ARR was not significantly different between definite NMO and limited forms of NMO in two Japanese series.[7,13] A French study compared ARR between seropositive and seronegative NMO and found no significant difference between the two groups,[14] which is in contrast to other studies that have reported a higher relapse rate in seropositive forms of NMO.[14] A Japanese study also found no significant difference in ARR of seropositive and seronegative NMO patients.[7] A recent study from Mayo clinic also did not find any significant difference in the interval to relapse, attack severity, disability outcome or ARR among seropositive and seronegative NMO patients.[15] Our study found that there was no significant difference in ARR across the four study groups. There was also no significant difference in ARR among NMO patients presenting with either ON or myelitis. A large study of Japanese NMO-IgG positive patients also found no significant difference in ARR of patients presenting with either ON or myelitis.[7] Similar relapse rates across NMO (regardless of seropositivity status) and NMOSD further support the case for early treatment of all these disorders.
There are no randomized controlled trials concerning treatment of NMO or NMOSD. High-dose intravenous (IV) steroids have been widely used for acute relapses; plasma exchange has been recommended for steroid-unresponsive relapses.[16] There is no adequate evidence for use of Intravenous immunoglobulin (IVIg) in acute relapses.
Traditional relapse-preventing therapies used for MS, such as interferons and natalizumab,[17] are not effective for prevention of NMO relapses; interferons have actually been shown to increase ARR and neurological disability in patients with NMO/NMOSD.[18,19] Only class IV evidence is available regarding the use of relapse-preventing therapies in NMO/NMOSD. 2010 European Federation of Neurological Societies/Peripheral Nerve Society (EFNS/PNS) guidelines for treatment of NMO considered azathioprine with or without prednisolone and rituximab as the first line therapies.[16] In the present series, azathioprine with prednisolone and rituximab were the most frequently used initial therapies. Monthly pulse cyclophosphamide, mitoxantrone, mycophenolate mofetil and methotrexate have been suggested as second line therapies.[16] Rituximab has also been used as a second-line therapy in treatment-refractory cases and has been seen to reduce relapse rates.[20] Cyclosporine A has been used in a small case series for patients with NMO/NMOSD.[21] There is insufficient evidence in the form of small case series for intermittent plasma exchange and IVIg for relapse prevention.[16] There is no consensus over treatment of NMOSD; EFNS/PNS guidelines considered NMO-IgG seropositivity and severity or failure to recover from the acute attacks as the deciding factors for treatment of NMOSD.[16]
Recently, eculizumab, a humanized monoclonal antibody against the complement protein C5, has shown promise in reducing relapse frequency and stabilizing or improving neurological disability in patients with aggressive NMO/NMOSD, in an open-label pilot study.[22] Tocilizumab, a humanized anti-IL-6 antibody, has also been shown to decrease ARR for rituximab-refractory NMO in a small case series.[23]
A recent work by Tradtrantip et al. has focused on targeted therapy for NMO based on blocking of pathogenic NMO-IgG to its target antigen, aquaporin-4. Experimental approaches devised by them have examined introduction of blocking antibodies,[24] as well as inactivation of NMO-IgG by deglycosylation: this might be accomplished by therapeutic apheresis using surface-immobilized bacterial endoglycosidase S.[25]
This retrospective study is limited by the small sample size and variable follow-up in each group. In addition, the unequal numbers of patients in each group preclude more robust statistical analysis. Patients included in the study came to us primarily as referrals and therefore investigations and treatment were not as per a single defined protocol. The variable proportion of treated patients in the four study groups may have contributed to the comparable ARR seen across all groups. Furthermore as this is a hospital based study, results cannot be generalized. Further studies and long-term follow-up with a larger patient cohort are needed to better describe the clinical profile of NMO and NMOSD in India.
Conclusion
NMO and NMOSD patients in this cohort have significant female preponderance and median age of onset in the third decade with comparable ARR regardless of clinical presentation, supporting the emerging trend of treating all patients with immunotherapeutic agents at an early stage. Varied presentations seen in NMO and its spectrum highlight the need for a high index of suspicion for NMO in demyelinating episodes not classical for MS.
Acknowledgments
We are grateful to Dr. B. K. Goyal, Dean, Bombay Hospital Medical Institute of Medical Sciences and Dr. D. P. Vyas, Medical Director, Bombay Hospital Medical Institute of Medical Sciences for allowing us to use the medical records. Dr. Nisha Ahmad, Metropolis Healthcare Limited, Mumbai provided invaluable assistance in carrying out neuromyelitis optica-IgG testing.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared
References
- 1.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66:1485–9. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- 2.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6:805–15. doi: 10.1016/S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- 3.Jacob A, Boggild M. Neuromyelitis optica. Ann Indian Acad Neurol. 2007;10:231–9. doi: 10.4103/0972-2327.58277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, et al. Serologic diagnosis of NMO: A multicenter comparison of aquaporin-4-IgG assays. Neurology. 2012;78:665–71. doi: 10.1212/WNL.0b013e318248dec1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collongues N, Marignier R, Zéphir H, Papeix C, Blanc F, Ritleng C, et al. Neuromyelitis optica in France: A multicenter study of 125 patients. Neurology. 2010;74:736–42. doi: 10.1212/WNL.0b013e3181d31e35. [DOI] [PubMed] [Google Scholar]
- 6.Kitley J, Leite MI, Nakashima I, Waters P, McNeillis B, Brown R, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain. 2012;135:1834–49. doi: 10.1093/brain/aws109. [DOI] [PubMed] [Google Scholar]
- 7.Nagaishi A, Takagi M, Umemura A, Tanaka M, Kitagawa Y, Matsui M, et al. Clinical features of neuromyelitis optica in a large Japanese cohort: Comparison between phenotypes. J Neurol Neurosurg Psychiatry. 2011;82:1360–4. doi: 10.1136/jnnp-2011-300403. [DOI] [PubMed] [Google Scholar]
- 8.Jacob A, McKeon A, Nakashima I, Sato DK, Elsone L, Fujihara K, et al. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2013;84:922–30. doi: 10.1136/jnnp-2012-302310. [DOI] [PubMed] [Google Scholar]
- 9.Lana-Peixoto MA, Callegaro D. The expanded spectrum of neuromyelitis optica: Evidences for a new definition. Arq Neuropsiquiatr. 2012;70:807–13. doi: 10.1590/s0004-282x2012001000010. [DOI] [PubMed] [Google Scholar]
- 10.Sato D, Fujihara K. Atypical presentations of neuromyelitis optica. Arq Neuropsiquiatr. 2011;69:824–8. doi: 10.1590/s0004-282x2011000600019. [DOI] [PubMed] [Google Scholar]
- 11.Sinnecker T, Dörr J, Pfueller CF, Harms L, Ruprecht K, Jarius S, et al. Distinct lesion morphology at 7-T MRI differentiates neuromyelitis optica from multiple sclerosis. Neurology. 2012;79:708–14. doi: 10.1212/WNL.0b013e3182648bc8. [DOI] [PubMed] [Google Scholar]
- 12.Sato DK, Nakashima I, Takahashi T, Misu T, Waters P, Kuroda H, et al. Aquaporin-4 antibody-positive cases beyond current diagnostic criteria for NMO spectrum disorders. Neurology. 2013;80:2210–6. doi: 10.1212/WNL.0b013e318296ea08. [DOI] [PubMed] [Google Scholar]
- 13.Yanagawa K, Kawachi I, Toyoshima Y, Yokoseki A, Arakawa M, Hasegawa A, et al. Pathologic and immunologic profiles of a limited form of neuromyelitis optica with myelitis. Neurology. 2009;73:1628–37. doi: 10.1212/WNL.0b013e3181c1deb9. [DOI] [PubMed] [Google Scholar]
- 14.Marignier R, Bernard-Valnet R, Giraudon P, Collongues N, Papeix C, Zéphir H, et al. Aquaporin-4 antibody-negative neuromyelitis optica: Distinct assay sensitivity-dependent entity. Neurology. 2013;80:2194–200. doi: 10.1212/WNL.0b013e318296e917. [DOI] [PubMed] [Google Scholar]
- 15.Jiao Y, Fryer JP, Lennon VA, Jenkins SM, Quek AM, Smith CY, et al. Updated estimate of AQP4-IgG serostatus and disability outcome in neuromyelitis optica. Neurology. 2013;81:1197–204. doi: 10.1212/WNL.0b013e3182a6cb5c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sellner J, Boggild M, Clanet M, Hintzen RQ, Illes Z, Montalban X, et al. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur J Neurol. 2010;17:1019–32. doi: 10.1111/j.1468-1331.2010.03066.x. [DOI] [PubMed] [Google Scholar]
- 17.Kleiter I, Hellwig K, Berthele A, Kümpfel T, Linker RA, Hartung HP, et al. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch Neurol. 2012;69:239–45. doi: 10.1001/archneurol.2011.216. [DOI] [PubMed] [Google Scholar]
- 18.Jarernsook B, Siritho S, Prayoonwiwat N. Efficacy and safety of beta-interferon in Thai patients with demyelinating diseases. Mult Scler. 2013;19:585–92. doi: 10.1177/1352458512459290. [DOI] [PubMed] [Google Scholar]
- 19.Kim SH, Kim W, Li XF, Jung IJ, Kim HJ. Does interferon beta treatment exacerbate neuromyelitis optica spectrum disorder? Mult Scler. 2012;18:1480–3. doi: 10.1177/1352458512439439. [DOI] [PubMed] [Google Scholar]
- 20.Jacob A, Weinshenker BG, Violich I, McLinskey N, Krupp L, Fox RJ, et al. Treatment of neuromyelitis optica with rituximab: Retrospective analysis of 25 patients. Arch Neurol. 2008;65:1443–8. doi: 10.1001/archneur.65.11.noc80069. [DOI] [PubMed] [Google Scholar]
- 21.Kageyama T, Komori M, Miyamoto K, Ozaki A, Suenaga T, Takahashi R, et al. Combination of cyclosporine A with corticosteroids is effective for the treatment of neuromyelitis optica. J Neurol. 2013;260:627–34. doi: 10.1007/s00415-012-6692-2. [DOI] [PubMed] [Google Scholar]
- 22.Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, et al. Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: An open-label pilot study. Lancet Neurol. 2013;12:554–62. doi: 10.1016/S1474-4422(13)70076-0. [DOI] [PubMed] [Google Scholar]
- 23.Ayzenberg I, Kleiter I, Schröder A, Hellwig K, Chan A, Yamamura T, et al. Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol. 2013;70:394–7. doi: 10.1001/jamaneurol.2013.1246. [DOI] [PubMed] [Google Scholar]
- 24.Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC, et al. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol. 2012;71:314–22. doi: 10.1002/ana.22657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tradtrantip L, Ratelade J, Zhang H, Verkman AS. Enzymatic deglycosylation converts pathogenic neuromyelitis optica anti-aquaporin-4 immunoglobulin G into therapeutic antibody. Ann Neurol. 2013;73:77–85. doi: 10.1002/ana.23741. [DOI] [PMC free article] [PubMed] [Google Scholar]