

Abstract
Ciliary neurotrophic factor (CNTF) administration maintains, protects, and promotes the regeneration of both motor neurons (MNs) and skeletal muscle in a wide variety of models. Expression of CNTF receptor α (CNTFRα), an essential CNTF receptor component, is greatly increased in skeletal muscle following neuromuscular insult. Together the data suggest that muscle CNTFRα may contribute to neuromuscular maintenance, protection, and/or regeneration in vivo. To directly address the role of muscle CNTFRα, we selectively-depleted it in vivo by using a “floxed” CNTFRα mouse line and a gene construct (mlc1f-Cre) that drives the expression of Cre specifically in skeletal muscle. The resulting mice were challenged with sciatic nerve crush. Counting of nerve axons and retrograde tracing of MNs indicated that muscle CNTFRα contributes to MN axonal regeneration across the lesion site. Walking track analysis indicated that muscle CNTFRα is also required for normal recovery of motor function. However, the same muscle CNTFRα depletion unexpectedly had no detected effect on the maintenance or regeneration of the muscle itself, even though exogenous CNTF has been shown to affect these functions. Similarly, MN survival and lesion-induced terminal sprouting were unaffected. Therefore, muscle CNTFRα is an interesting new example of a muscle growth factor receptor that, in vivo under physiological conditions, contributes much more to neuronal regeneration than to the maintenance or regeneration of the muscle itself. This novel form of muscle–neuron interaction also has implications in the therapeutic targeting of the neuromuscular system in MN disorders and following nerve injury.
INDEXING TERMS: muscle CNTF receptor α, sciatic, Cre recombinase, mouse, mlc1f-Cre
Ciliary neurotrophic factor receptor α (CNTFRα) is an essential ligand-binding subunit of the CNTF receptor, which is composed of CNTFRα, leukemia inhibitory factor receptor β (LIFRβ) and gp130 (Ip et al., 1992; Davis et al., 1993b). Whereas LIFRβ and gp130 are found in other related receptors, CNTFRα is unique to CNTF receptors and is required for all known forms of CNTF receptor signaling (Davis et al., 1993b; Elson et al., 2000; Derouet et al., 2004).
Exogenous CNTF administration has many protective and regenerative neuromuscular effects. It promotes motor neuron (MN) survival following postnatal axotomy (Sendtner et al., 1990), during development (Oppenheim et al., 1991) and in genetic models of MN disease (Sendtner et al., 1992a; Ikeda et al., 1995). It also increases the rate of axonal regrowth (Sahenk et al., 1994) and the size of neuromuscular junctions (Huang et al., 2002) after peripheral nerve lesion, and maintains axons in genetic models of amyotrophic lateral sclerosis (ALS) (Pun et al., 2006). Moreover, the anatomical and functional atrophy of skeletal muscle observed with disuse (Fraysse et al., 2000), aging (Guillet et al., 1999), MN disease models (Mitsumoto et al., 1994), and peripheral nerve lesion (Helgren et al., 1994) are all reduced by exogenous CNTF administration. These data suggest that endogenous CNTF receptor signaling may protect the neuromuscular system, particularly when it is challenged by traumatic or disease-related insult. This possibility is further supported by data indicating that insults, including peripheral nerve lesion and neuromuscular disease, greatly increase CNTFRα expression in skeletal muscle (Davis et al., 1993a; Helgren et al., 1994; Weis et al., 1998; Poea et al., 2001). Together the data suggest that endogenous skeletal muscle CNTFRα may contribute to neuromuscular regeneration in vivo.
However, the exogenous CNTF in the above studies may have induced pharmacological, not physiological, changes. In addition, this CNTF need not be acting on CNTF receptors given that CNTF, at the concentrations employed, can activate LIF receptors (Saggio et al., 1995) which have been implicated in MN survival (Li et al., 1995). Finally, the cellular site of action is not evident in that in vitro data indicate that CNTFRα is released by denervated muscle and can, if bound to ligand, act on cells expressing LIFRβ and gp130 (like MNs) to enhance CNTF receptor signaling (Davis et al., 1993a). Therefore, binding of exogenous CNTF to muscle-derived CNTFRα may underlie some of its MN effects.
Manipulation of endogenous CNTF receptors in vivo is required to directly address these issues, and in the process, potentially identify targets that may be exploited to treat neuromuscular disorders. Given that CNTFRα is required for all forms of CNTF receptor signaling but is not involved in other signaling, including LIF receptor signaling (Davis et al., 1993b; Elson et al., 2000; Derouet et al., 2004), disruption of the CNTFRα gene is an effective method to identify in vivo functions of endogenous CNTF receptors. Unconditional disruption of the CNTFRα gene in mice leads to perinatal death with reduced MN populations (DeChiara et al., 1995). Although these mice reveal an essential in vivo role for CNTF receptors in the development and/or survival of embryonic MNs, their perinatal death precludes their use to study postnatal functions of CNTF receptors. Moreover, the universal CNTFRα gene disruption in these mice does not allow one to address the functions of CNTF receptors expressed in specific cell types. In the studies reported here, we examine the in vivo functions of muscle CNTFRα following peripheral nerve lesion by selectively disrupting the CNTFRα gene in skeletal muscle with Cre/loxP techniques.
MATERIALS AND METHODS
Mouse lines
The generation and characterization of the floxed CNTFRα (flxCNTFRα) mice have been described previously (Lee et al., 2008). Briefly, exons 3–5 of the CNTFRα gene (“exon1” containing start codon) were flanked by loxP sites (“floxed”) by using previously described methods (Wattler et al., 1999). The mlc1f-Cre mice (Bothe et al., 2000) and YFP16 mice (Feng et al., 2000) were generous gifts from Dr. Steven Burden (New York University Medical School, New York, NY) and Dr. J.R. Sanes (Harvard University, Cambridge, MA), respectively. ROSA26 mice (Soriano, 1999) were obtained from Jackson Laboratories (Bar Harbor, ME).
General design and statistical analysis
The flxCNTFRα+/+ and flxCNTFRα−/− mice were generated by flxCNTFRα+/− × flxCNTFRα+/− breeding. The other gene constructs were bred as heterozygote × wild-type to control for gene dosage. Mlc1f-Cre+/flxCNTFRα−/− mice (i.e., wild-type at the CNTFRα locus) served as primary controls, but were not distinguishable from mlc1f-Cre+/flxCNTFRα +/− mice. All mice (male and female) were backcrossed at least five generations onto a 129SvEvBrd background and genotyped by polymerase chain reaction (PCR) analysis of tail biopsy DNA. All procedures were conducted on 2.5–5-month-old mice. Animal procedures were approved by the University of Cincinnati IACUC committee.
For all procedures, littermate pairs of CNTFRα-depleted and control mice were processed in parallel through the complete procedures including surgery, behavior, and image analysis by individuals kept blind to genotype. This pairing approach controlled for many potential sources of variability including in utero and postnatal rearing environments, variation in genetic background and age, and any variability in surgical and/or anatomical reagents. Therefore, statistical analyses consisted of appropriate paired Student’s t-tests or two-way analysis of variance (ANOVA) tests (depending on the design of the particular experiment). ANOVA tests were following by Bonferroni post hoc tests of individual group differences. In all cases, results of P <0.05 were considered significant.
General anatomical procedures
Cryostat sections (30 μm) were stained with either cresyl violet, standard Xgal histology, or previously described immunohistochemistry procedures (MacLennan et al., 1996). Antibodies (Table 1) recognizing muscle fiber types (see below), neurofilament (Millipore), and Fluoro-Gold (Fluorochrome, Englewood, CO) were visualized through either ABC amplification (Vector, Burlingame, CA) and cyanine-3 tyramide (Perkin Elmer, Oak Brook, IL), or Alexa Fluor-conjugated secondary antibodies (Invitrogen, Carlsbad, CA).
TABLE 1.
Primary Antibodies
| Antibody | Host isotype | Recognizes | lmmunogen | Source | Cat. no. | Lot no. |
|---|---|---|---|---|---|---|
| A4.74 | Mouse IgG1 | Type IIa muscle fibers | Human MyHC | Developmental Studies Hybridoma Bank | – | 2D3 |
| MY-32 | Mouse IgG1 | All type II muscle fibers classes | Rabbit muscle myosin | Sigma | M 4276 | 121K4813 |
| 6H1 | Mouse IgM, kappa light chain | Type IIx muscle fibers | Rabbit muscle myosin | Developmental Studies Hybridoma Bank | – | – |
| 10F5 | Mouse IgM, kappa light chain | Type IIb muscle fibers | Rabbit muscle myosin | Developmental Studies Hybridoma Bank | – | 14J4 |
| WB-MHCs | Mouse IgG1 | Type I muscle fibers | Rabbit muscle myosin | Vector | VP-M667 | – |
| Fluoro-Gold | Rabbit polyclonal | Fluoro-Gold | Fluoro-Gold | Fluorochrome | – | – |
| Neurofilament | Rabbit polyclonal | Neurofilament (NF-M) | Recombinant fusion protein with C-terminal 168 amino acid residues of rat NF-M | Millipore | AB1987 | 19030930 |
CNTFRα RT-PCR
Total RNA was extracted with the Micro-to-Midi total RNA purification system (Invitrogen) according to the manufacturer’s instructions. The RNA was quantified by optical density, and equal amounts of RNA from control and floxed mice were used to synthesize first-strand cDNAs with the SuperScript III kit (Invitrogen). The cDNAs were amplified with TAQ DNA polymerase (Eppendorf, Hamburg, Germany) and the following CNTFRα-specific primers: 5′ GTTCCTGCCTCCATTGAG-CAG 3′ and 5′ GAGCGGCAGCTGAGCACAG 3′. Reactions with the following glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers were used as an extraction/loading control: 5′ AAACCCATCACCATCTTCCA 3′ and 5′ GTGGTTCACACCCATCACAA 3′. The resulting DNAs were separated on an agarose gel with ethidium bromide. The relative intensity of CNTFRα bands (normalized with the intensity of the corresponding GAPDH bands) was determined with MetaMorph software (Molecular Devices, Sunnyvale, CA).
Unilateral sciatic nerve crush
Mice were anesthetized with ketamine/xylazine (100 mg/kg). Midthigh skin and muscle were opened to expose the nerve, which was freed from surrounding connective tissue where it passes superficial to the tendon of the obturator internus. The nerve was crushed for 10 seconds with Dumont #5 Biologic Tip forceps (Fine Science Tools, San Mateo, CA). The completeness of lesion produced by the crush procedure was assessed by examination of the tibialis anterior and extensor digitorum longus (EDL) muscles of four YFP16+ mice 4–6 days after the crush. This indicated that whereas the muscles ipsilateral to the lesion contain normal α-bungarotoxin (αBTX)-labeled acetylcholine receptor (AChR) clusters, all are devoid of presynaptic terminals. In contrast, the αBTX-labeled AChR clusters in the contralateral tibialis anterior and EDL muscles all contained presynaptic terminals.
Muscle fiber type analysis
EDL and soleus muscles ipsilateral and contralateral to the nerve crush were harvested 3 weeks post lesion, cut in cross section, and immunohistochemically stained with antibodies specific for different muscle fiber types: type I (WB-MHCs; Vector), total type II (MY-32; Sigma, St. Louis, MO) type IIa (A4.74; Developmental Studies Hybridoma Bank [DSHB], Iowa City, IA), type IIb (10F5; DSHB), and type IIx (6H1; DSHB) fiber types. Images were captured with a Nikon DXM1200 digital camera and analyzed with MetaMorph software.
Motor neuron quantification
Thirty-micron cryostat sections were collected through the complete L5 segment of the lumbar spinal cord, which contains MNs projecting through the sciatic nerve (Janjua and Leong, 1984). The L5 spinal cord segment was located by using the diagrams in Figure 5 of Janjua and Leong (1984). Cresyl violet–stained alpha MNs were counted in every fourth section. Alpha MNs were defined by their characteristic location, large size, irregular shape, heterogeneous cytoplasmic staining, and, in most cases, a visible dark nucleolus in a lighter stained nucleus. To correct for cells potentially split in the Z dimension, all neurons in focus at the top border of the sections were excluded (optical disector; Hyman et al., 1998; Hatton and Von Bartheld, 1999). Counts were multiplied by 4 to estimate total MNs (“fractionator”).
Muscle contractility analysis
We have previously reported the methods used in detail (He et al., 2001; Crawford et al., 2002). In brief, after the resting length of each muscle was measured in situ, the dissected muscles were mounted vertically in a sealed cylindrical chamber fitted with a magnet stirrer. By means of a small plastic ring tied to each tendon of the EDL with surgical silk, muscles were mounted at one end to a fixed stainless steel post. The other end was fixed to the lever of an isometric force transducer (Harvard Apparatus, Dover, MA), and the muscle length was adjusted to produce a passive tension of 5 mN. Muscles were incubated in sterile Krebs solution (in mM): 118 NaCl, 4.7 KCl, 25 NaHCO3, 2.5 CaCl2, 1.2 MgSO4, 1.2 NaH2PO4, 0.026 EDTA, 11 glucose equilibrated with 95% CO2/5% O2) at room temperature (~22°C). Muscles were electrically stimulated via two platinum electrodes positioned along either side of the muscle.
Supramaximal voltage was determined empirically by using a series of short (1.5-second) tetani and subsequently increasing the voltage for each tetani. Similarly, supramaximal frequency of stimulation was determined. Capacitor discharges of equal but alternating polarity (66 Hz at 15 V, 15-ms duration, circuit rate constant, 5 ms) were used for supramaximal stimuli. Experiments consisted of a set (about five) of twitches and a set of tetani with duration of 3–7 seconds. Digital recordings of force were obtained with the BioPac (Goleta, CA) data acquisition system and normalized to cross-sectional area. All recordings were conducted while the observer was blind to genotype. CNTFRα-depleted and control muscles were tested in parallel. The EDL muscle was studied because its smaller dimensions make it unlikely to be oxygen diffusion limited in this in vitro preparation.
Neuromuscular junction analysis
YFP16+ CNTFRα-depleted and YFP16+ control mice were perfused with 4% paraformaldehyde 3 weeks after sciatic nerve crush. Tibialis anterior muscles ipsilateral and contralateral to the nerve crush were dissected, postfixed in 4% paraformaldehyde for 2 hours at 4°C, cryoprotected, and longitudinally sectioned at 100 μm on a cryostat. The sections were preincubated at room temperature with 0.3% Triton X-100, 1% bovine serum albumin (BSA) in phosphate-buffered saline (PBS; pH 7.3) for 1 hour, followed by an overnight 4°C incubation with 1 μg/ml tetramethylrhodamine-conjugated αBTX to label AChR clusters.
To quantify the number of AChR clusters in a particular muscle, all AChR clusters were counted in each section of the muscle and summed to obtain a total.
To quantify the size of AChR clusters, an image was captured of every 60th AChR cluster encountered while counting along the band of AChR clusters in each section. The images were analyzed with MetaMorph software to determine the length of each AChR cluster along the axis of the muscle fiber. An average AChR cluster size was calculated for each muscle, and these values were normalized relative to the value calculated for the intact (unlesioned) side muscles of the control mice. This index of AChR cluster length along the axis of the muscle fiber was chosen because in longitudinal muscle sections the length measured along the axis of the muscle fiber reasonably reflects the overall size of the AChR cluster whether it is present on the side or front face of the muscle fiber relative to the surface of the section (see Fig. 7B for examples). In contrast, measures of the AChR cluster area are much more dependent on AChR cluster position on the muscle fiber such that those clusters present along the side of the fiber appear much smaller in area (see Fig. 7B for examples). In addition, AChR clusters in CNTFRα-depleted mice were indistinguishable from those of controls with regard to shape such that measurement of AChR cluster length reflected relative cluster size.
Figure 7.

Neuromuscular junctions and lesioned-induced terminal sprouting are not affected by depletion of muscle ciliary neurotrophic factor receptor α (CNTFRα). Neuromuscular junctions were visualization in tibialis anterior muscles of muscle CNTFRα-depleted and control mice 3 weeks post lesion through use of the YFP16 gene construct (which drives YFP expression in all motor axons) and rhodamine-conjugated α-bungarotoxin (αBTX) binding (which labels postsynaptic acetylcholine receptor [AChR] clusters). The YFP-labeled axons and terminals (A,C,E) and the corresponding αBTX-labeled, postsynaptic AChR clusters (B,D) were visualized at high resolution with confocal microscopy. Regions boxed in A and B are digitally “magnified” and presented again in C and D respectively. A–D are from muscle contra-lateral to the lesion. E is from muscle ipsilateral to the lesion and illustrates a terminal sprout (arrow) typical of regenerated axons. F: Quantification indicated that both the lesion and CNTFRα depletion had no affect on the total number of AChR clusters per muscle (P >0.05 ; two-way ANOVA). G: The lesion produced a small but statistically significant overall increase in the average size of the AChR clusters (*P <0.05; F =6.95; main effect of lesion in two-way ANOVA [with side relative to lesion treated as a within measure]; Bonferroni post hoc comparisons did not reveal any individual group differences), but CNTFRα depletion did not affect this index (P >0.05 for CNTFRα depletion main effect and interaction terms of same two-way ANOVA described above). H: Finally, the number and distribution of lesioned-induced terminal sprouts were unaffected by CNTFRα depletion (P >0.05 for CNTFRα depletion main effect and interaction terms of two-way ANOVA). Means with SEM are presented. Numbers in or above bars indicate numbers of mice. Legend applies to all graphs. Scale bar =50 μm in B (applies to A,B); 10 μm in C (applies to C,D); 10 μm in E.
The YFP16 gene construct labels all motor axons and their terminals with yellow fluorescent protein (YFP; Feng et al., 2000). Therefore, to quantify terminal sprouts of regenerated axons (no sprouts were observed for terminals on the unlesioned/intact side), we captured images of the terminals in the same neuromuscular junctions (NMJs) sampled for the AChR cluster size analysis described immediately above. For NMJs with no sprouts, the presynaptic terminals align very closely with the postsynaptic AChR clusters as seen for all NMJs in the unlesioned side muscles (e.g., Fig. 7A–D). Terminal sprouts are seen as additional extensions of the terminal emanating from this structure (e.g., Fig. 7E). For each NMJ analyzed, the number of primary fibers/sprouts emanating directly from the terminal areas aligned with the AChR clusters was counted, and the NMJ was qualified according to the number of sprouts it displayed. For each muscle, we calculated the percent of terminals containing zero, one, two, three, or four sprouts (no terminals were observed with more than four sprouts).
Axon quantification
Nerves were harvested 3 weeks post lesion, and the segment containing the crush site and adjacent nerve was dissected, cut in cross section at 30 μm, and immunohistochemically stained for neurofilament. Sections immediately distal and immediately proximal to the lesion (i.e., within 200 μm of the start and end of the lesion) were imaged with a Zeiss 510 LSM confocal microscope and analyzed with MetaMorph software. All axon profiles in all axon bundles of the nerve were counted in three proximal and three distal sections per nerve, and mean proximal and distal values were calculated.
Fluoro-Gold application and analysis
Three weeks after sciatic nerve crush, nerves were transected 7 mm distal to the crush site. The freshly transected proximal stump was immersed in a 5% solution of Fluoro-Gold/saline (Fluorochrome) for 40 minutes prior to religation of the proximal and distal stumps with 9-0 suture. Two weeks later (time for the tracer to be retrogradely transported to MN soma) the mice were perfused, and Fluoro-Gold–positive MNs were immunohistochemically identified and quantified.
Walking track analysis
Mice had nontoxic finger paint smeared on their rear paws and were placed at the end of a 7 (W) × 13 (H) × 70-cm (L) walkway whose floor was lined with white paper. A 13 × 13 × 20-cm box had been previously positioned at the far end of the walkway such that the mice walked down the walkway to enter the dark box (a preferred environment for rodents), leaving footprints in the process. The mice were habituated to the apparatus prior to lesioning so that neophobia did not inhibit the walking behavior during testing.
The distance between the marks left by the first and fifth toes (“1–5”) was measured (while the observer was blind to genotype, as for all other procedures in this report) for feet both ipsilateral (lesioned) and contralateral (unlesioned) to the nerve crush. This was done in accordance with the original publication (De Medinaceli et al., 1982), and subsequent work indicating that this measurement of first to fifth toe spread best reflects the motor dysfunction and subsequent recovery from sciatic nerve damage (Hare et al., 1993).
Wild-type rats and mice typically show little if any recovery in walking track scores during the 2 weeks immediately following sciatic nerve crush, but largely recover during the third week post-lesion (Bervar, 2000; Dijkstra et al., 2000). Therefore, the mice were tested 13, 15, 19, and 21 days after the sciatic nerve crush. At least 10 distinct footprints were measured for each leg of each mouse in each session. Walking track analysis scores were calculated for each mouse in each session from the mean toe spread (1–5) values for each leg by comparing the lesioned (“L”) and unlesioned (“UL”) sides in the following equation: {([1–5L] −[1–5UL]) ÷ [1–5UL]}. Therefore, as the mice recovered motor function following the lesion, the toe spread values of the lesioned side became closer to the values from the unlesioned side, and the overall walking track analysis scores increased.
Antibody characterization
The 6H1 and 10F5 muscle fiber type antibodies have been previously characterized and shown to be specific to IIx and IIb fibers, respectively, by both western blot and immunohistochemistry (Lucas et al., 2000). The A4.74 antibody has been characterized by western blot (Hughes et al., 1993) and immunohistochemistry (Smerdu and Soukup, 2008) and found to specifically identify IIa fibers. The WB-MHCs antibody has been characterized by western blot (Klover et al., 2009) and immunohistochemistry (Carson et al., 1998; Klover et al., 2009) and shown to specifically label type I fibers. The My-32 antibody has been characterized by western blot (Okuno et al., 2012) and immunohiostochemistry (Havenith et al., 1990; Rojiani and Cho, 1998) and shown to specifically label type II fibers. In the above studies of these different muscle fiber type–specific antibodies, western blot characterization demonstrated recognition of an appropriate size protein, and immunohistochemistry characterization demonstrated recognition of the appropriate subset of fibers in different muscle types and frequently also compared results with histochemistry-based assays of fiber types. Our studies also indicated that all these muscle fiber type antibodies labeled the fraction of EDL and soleus fibers expected from previous work.
The neurofilament antibody has been characterized by western blot as recognizing the appropriate size protein (Harris et al., 1991) and produces the same immunohisto-chemical staining pattern in the developing chick retina as the antibody RMO270, a mouse monoclonal against neurofilament (NFM; McCabe et al., 1999). Moreover, in the present study, the antibody produced the expected pattern of labeling for axons in the sciatic nerve.
The Fluoro-Gold antibody has been previously characterized in immunohistochemistry by blocking the signal through preadsorption of the antibody with Fluoro-Gold (Lee et al., 2009). In addition, the labeling reported here was seen only in mice loaded with Fluoro-Gold.
Finally, for all immunohistochemistry procedures reported here with the above antibodies, labeling was not observed without primary antibody.
Microscopy
Images were captured with a either a 12-megapixel DXM1200 camera and Nikon E800 microscope or a Zeiss 510 LSM confocal microscope. Image brightness and contrast were optimized with LSM Browser software or Corel Draw software (Mountain View, CA). Images that were compared were identically captured and adjusted.
RESULTS
Muscle-specific CNTFRα depletion
In order to selectively disrupt the CNTFRα gene in skeletal muscle in vivo, we crossed the previously described floxed CNTFRα (flxCNTFRα) mice (Lee et al., 2008) with mice carrying a Cre recombinase (Cre) gene inserted into the myosin light chain 1f locus (mlc1f-Cre) (Bothe et al., 2000), a locus expressed very selectively in skeletal muscle cells (Lyons et al., 1990). Previous Southern blot and reporter gene characterization of the mlc1f-Cre mice indicated that the mice express Cre as designed, in that the mlc1f-Cre leads to excision of floxed sequence in the adult in all skeletal muscles tested, but no excision in brain, liver, heart, or stomach (Bothe et al., 2000). To further characterize this line in the context of the present project, we crossed the mice with ROSA26 reporter mice (Soriano, 1999) to examine Cre activity in the EDL and tibialis anterior (TIB) hindlimb muscles, brain, spinal cord, sciatic nerve, and heart of two 4-month-old ROSA26+/−/mlc1f-Cre+/− mice and an age-matched ROSA26+/−/mlc1f-Cre−/− control. As expected, reporter signal was present only in skeletal muscle of the mlc1f-Cre+ mice, being absent in all other tissues (Fig. 1 and data not shown). Therefore, in agreement with the very selective skeletal muscle activity of the mlc1f promoter (Lyons et al., 1990) and the previous characterization of this mlc1f-Cre mouse (Bothe et al., 2000), the present data indicate that floxed gene excision is highly restricted to skeletal muscle in mlc1f-Cre+ mice, identifying them as a powerful tool for characterizing in vivo functions of muscle-derived CNTFRα, independent of the functions of CNTFRα expressed by other cell types.
Figure 1.

The mlc1f-Cre gene construct leads to selective excision of floxed gene sequence in skeletal muscle. Xgal histology of: (A) longitudinal section of tibialis anterior muscle from a ROSA26 reporter+; mlc1f-Cre+ mouse with blue Xgal reaction product indicating mlc1f-Cre–dependent floxed gene excision; and (B) a ROSA26 reporter +mlc1f-Cre- control; (C) cross section of EDL muscle from a ROSA26 reporter+; mlc1f-Cre+ mouse; (D) transverse spinal cord section from a ROSA26 reporter+; mlc1f-Cre+ mouse showing lack of MN floxed gene excision Scale bar =200 μm in D (applies to A,B,D); 100 μm in C.
We chose to focus on the EDL, soleus, and TIB muscles because these well-characterized muscles: 1) were the subject of previous studies showing the effects of exogenous CNTF treatment on muscle atrophy as well as studies reporting increased muscle CNTFRα expression following denervation (see Introduction); and 2) represent muscles with a range of different fiber types.
We crossed mlc1f-Cre+/−/flxCNTFRα+/− mice with mlc1f-Cre−/−/flxCNTFRα+/− mice and found that approximately 1/8 of the mice generated are mlc1f-Cre+ and homozygous for the floxed CNTFRα gene (mlc1f-Cre+/−/flxCNTFRα+/+; referred to here simply as CNTFRα-depleted mice), indicating that the expression of Cre and the depletion of muscle CNTFRα (see below) do not have any detectable effect on survival. In all the present experiments, these CNTFRα-depleted mice were compared to mlc1f-Cre+, wild-type (mlc1f-Cre+/−/flxCNTFRα−/−) and mlc1f-Cre+, heterozygous floxed (mlc1f-Cre+/−/flxCNTFRα+/−) littermates, with no differences being found between these two control groups.
We then confirmed that CNTFRα expression is depleted in skeletal muscle of the floxed mice. Real-time (RT)-PCR analysis indicated that CNTFRα mRNA levels in muscle are increased by nerve lesion, as previously reported (Helgren et al., 1994), and substantially decreased in the CNTFRα-depleted mice relative to controls (Fig. 2). As expected from the reporter data, the mlc1f-Cre manipulation did not affect spinal cord CNTFRα mRNA expression (floxed spinal cord = 104 ±17.7% of controls, n =3). The decrease, but not complete elimination, in muscle is consistent with previous studies using mlc1f-Cre (Bothe et al., 2000, Wredenberg et al., 2002) and may at least partially reflect the fact that not all cells expressing CNTFRα in muscle tissue are myofibers.
Figure 2.

Combining the mlc1f-Cre gene construct with the floxed ciliary neurotrophic factor receptor α (CNTFRα) gene leads to CNTFRα mRNA depletion in skeletal muscle. CNTFRα mRNA was measured by RT-PCR in EDL muscles ipsilateral (“Lesioned”) and contralateral (“Intact”) to sciatic nerve crush. The muscles were dissected 24 hours post lesion from floxed CNTFRα and litter-mate control mice, all containing the mlc1f-Cre gene construct. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was also measured and used to normalize values. A: Summarized quantitative data. B,C: Representative gels from CNTFRα (B) and GAPDH (C) RT-PCR. As expected from past studies, the lesion led to an increase in muscle CNTFRα mRNA expression (P <0.015; F =10.09; two-way ANOVA). As designed, muscles from floxed CNTFRα mice contained less CNTFRα mRNA than controls (* P <0.002; F =21.90; two-way ANOVA). n =3 mice per condition. The same pattern of results was observed with TIB and soleus muscles (data not shown).
Muscle CNTFRα and body weight
Although most studies of CNTF receptor signaling and weight regulation have focused on the hypothalamus (Lambert et al., 2001; Kokoeva et al., 2005), other work suggests that skeletal muscle CNTF receptors are involved (Watt et al., 2006). Comparison of the age-matched CNTFRα-depleted and control mice did not detect any difference in weight (Fig. 3), suggesting that endogenous muscle CNTF receptor signaling does not play a significant role in regulating weight.
Figure 3.
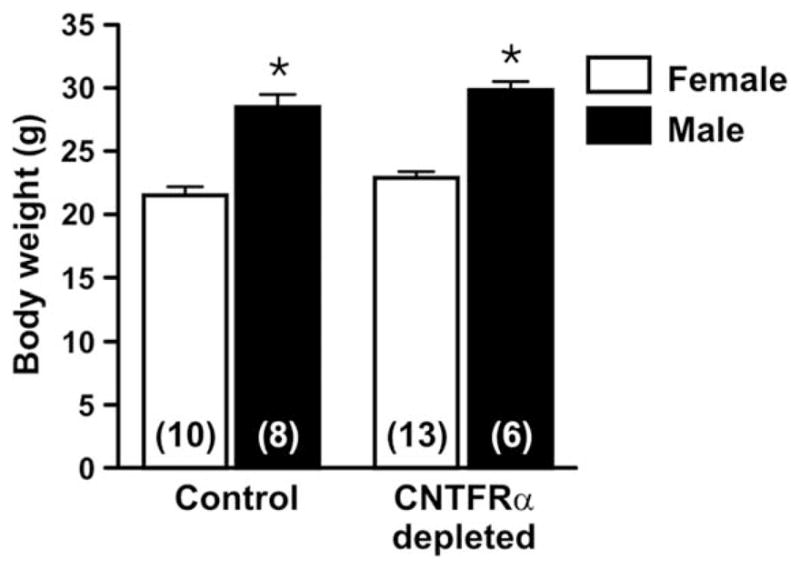
Body weight is not affected by depletion of muscle ciliary neurotrophic factor receptor α (CNTFRα). As expected, male mice weighed significantly more than female mice (*P <0.001; F =92.14; two-way ANOVA). However, CNTFRα-depleted and control mice (age-matched; 3–5 months of age) did not differ (P >0.05; F =3.54; two-way ANOVA). Means with SEM. are presented. Numbers in bars indicate numbers of mice.
Muscle CNTFRα depletion does not affect muscle maintenance or peripheral nerve lesion-induced atrophy of muscle
Naive (unlesioned) muscle CNTFRα-depleted mice did not noticeably differ from control mice in terms of gait, balance, or grip strength (data not shown), thereby suggesting that muscle CNTFRα does not play a significant role in the noninjured neuromuscular system. These results are also consistent with the MN quantification and NMJ analysis presented below for the unlesioned control side of muscle CNTFRα-depleted mice.
As described above, CNTFRα expression increases in denervated muscle following peripheral nerve lesion. Moreover, many studies using exogenous CNTF administration suggest that endogenous muscle CNTF receptor signaling may protect the neuromuscular system and promote its regeneration following nerve lesion. Therefore, in order to determine the in vivo role(s) played by muscle CNTFRα following nerve lesion, we challenged the muscle CNTFRα-depleted and control mice with unilateral sciatic nerve crush, a well-characterized model in which the lesion produces temporary denervation of muscle followed by regeneration.
The denervated muscle in this model atrophies and then recovers following reinnervation. Exogenous CNTF has been shown to reduce denervation-induced muscle atrophy (Helgren et al., 1994). Comparing muscle weights of CNTFRα-depleted and control mice indicated that CNTFRα-depleted and control muscles are similar in size and atrophy to a similar extent following denervation (Fig. 4). Like muscle weight, muscle fiber cross-sectional area is similar in CNTFRα-depleted and control muscle (data not shown). The depletion of CNTFRα also did not affect the fiber type composition of the muscles ipsilateral and contralateral to the lesion (Fig. 5) nor their in vitro contractile properties (EDL peak tetanic force, normalized to cross-sectional area; Fig. 6).
Figure 4.

Depletion of muscle ciliary neurotrophic factor receptor α (CNTFRα) does not affect muscle weight or nerve lesion induced muscle weight loss. As expected, sciatic nerve crush led to decreased weight of tibialis anterior (A), extensor digitorum longus (EDL; B), and soleus (C) muscles at both 14 and 21 days post lesion (DPL) (*P <0.002; main effect of lesion in two-way ANOVAs with side relative to lesion treated as a within-measure). However, muscle CNTFRα depletion had no effect on muscle weight or the lesion-induced atrophy in any of the muscles at either postlesion interval (all comparisons P >0.05; main effect of CNTFRα depletion and interaction in two-way ANOVAs described above). Means with SEM are presented. Numbers in bars indicate number of mice. Legend in C applies to all panels.
Figure 5.

Depletion of muscle ciliary neurotrophic factor receptor α (CNTFRα) does not affect muscle fiber type composition. Immunohistochemical analysis of muscle fiber types in soleus (A–C) and extensor digitorum longus (EDL; D–G) muscles harvested from CNTFRα-depleted and control mice 3 weeks after nerve crush did not detect any effect of CNTFRα depletion on fiber type composition in muscles either ipsilateral (“Lesioned”) or contralateral (“Intact”) to nerve crush (all comparisons P >0.05; two-way ANOVA terms for main effect of CNTFRα depletion). IIb and IIx fibers were not detected in soleus muscle. Means with SEM are presented. Numbers in bars indicate numbers of mice. Legend applies to all graphs.
Figure 6.
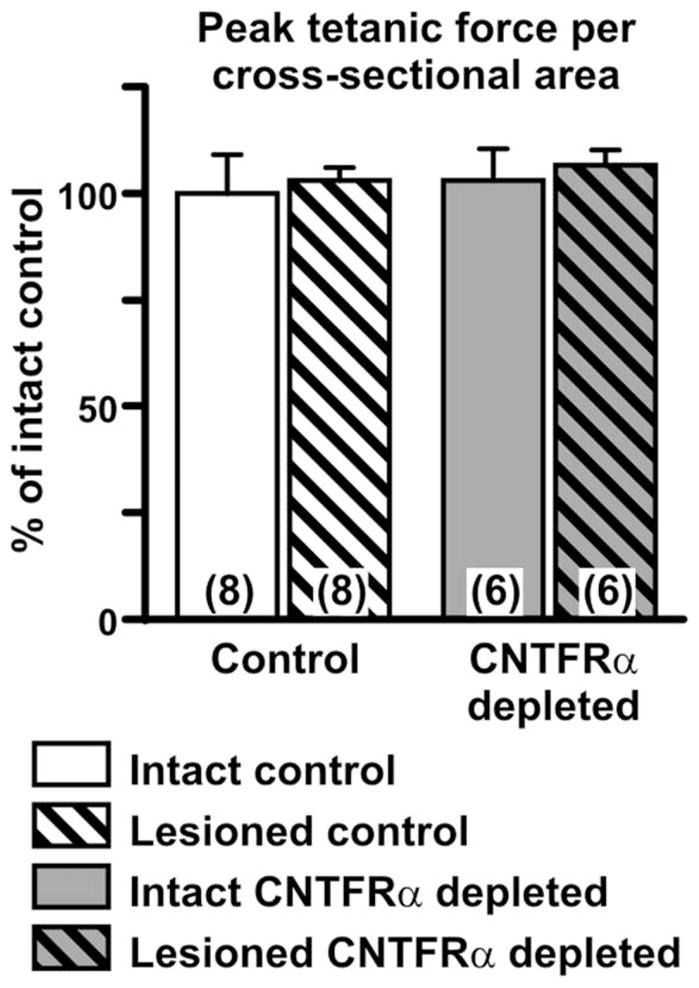
Depletion of muscle ciliary neurotrophic factor receptor α (CNTFRα) does not affect in vitro contractility. EDL muscles ipsilateral (“Lesioned”) and contralateral (“Intact”) to nerve crush were harvested 3 weeks post lesion, and their contractility (peak tetanic force per cross-sectional area) was determined in vitro, as described in the Materials and Methods section. Muscle CNTFRα depletion had no effect on contractility (P >0.05; two-way ANOVA). Numbers in bars indicate numbers of mice. Mean value of the intact controls =438.1 mN/mm2.
Neuromuscular junctions and lesion-induced terminal sprouting are unaffected by muscle CNTFRα depletion
Disruption of critical genes expressed in muscle can lead to changes in postsynaptic AChR clusters (Hosaka et al., 2002). We found no effect of muscle CNTFRα depletion on the number of such structures either ipsilateral or contralateral to the sciatic nerve lesion (Fig. 7F).
Exogenous CNTF administration has been shown to increase the size of AChR clusters following sciatic nerve lesion (Huang et al., 2002) suggesting that endogenous muscle CNTF receptor signaling may regulate AChR cluster size. We found a small, but statistically significant, increase in cluster size following lesion, but no difference between CNTFRα-depleted and control mice, nor any lesion by genotype interaction (Fig. 7G). The CNTFRα depletion also failed to affect the shape of the AChR clusters in that the clusters in CNTFRα-depleted and control mice were indistinguishable and all displayed the normal “pretzel-like” appearance (e.g., Fig. 7B,D).
In order to examine the presynaptic terminals of the NMJs, we utilized the YFP16 gene construct, which drives YFP expression in all MNs and their axons (Feng et al., 2000). This construct was bred into CNTFRα-depleted and control mice that then received the unilateral nerve lesion. NMJs on the intact side of both control and CNTFRα-depleted mice all displayed the normal precise alignment of AChR clusters and terminal arbors with terminal axons corresponding to all parts of the AChR clusters (e.g., Fig. 7C,D). As expected, axons were not observed in ipsilateral muscles following lesion until regenerating axons began to arrive, at approximately 2 weeks post lesion. Although there was variability in the rate of axon return to the AChR clusters, the CNTFRα-depleted and control groups did not differ (data not shown). Moreover, by 3 weeks post lesion, all AChR clusters in both groups were fully reinnervated, in that all parts of all AChR clusters in all mice displayed a matching, fully aligned nerve terminal.
Exogenous CNTF promotes sprouting at axon terminals (Gurney et al., 1992; Kwon and Gurney, 1994; Ikeda et al., 1995; Siegel et al., 2000; Wright and Son, 2007; Wright et al., 2007). Sprouting also occurs from regenerated axon terminals following nerve injury, including sciatic nerve crush (Zhou et al., 2002 and Fig. 7E). Therefore, these data raise the possibility that muscle CNTFRα may regulate the sprouting seen after nerve lesion, either through affecting muscle function and muscle–axon interaction or through the release of muscle CNTFRα (Davis et al., 1993a). We found that the nerve crush led to substantial terminal sprouting in both CNTFRα-depleted and control mice, with most terminals containing at least one sprout and many containing multiple sprouts (Fig. 7H). However, the muscle CNTFRα depletion had no effect on the percentage of terminals with sprouts or the number of sprouts per terminal (Fig. 7H).
Muscle CNTFRα depletion does not decrease motor neuron survival following nerve lesion
Exogenous CNTF administration promotes MN survival during development (Oppenheim et al., 1991), in MN disease models (Sendtner et al., 1992a; Ikeda et al., 1995), and following early postnatal peripheral nerve lesion (Sendtner et al., 1990). Adult peripheral nerve lesion does not lead to MN death, and it is accompanied by an increase in muscle CNTFRα expression (Helgren et al., 1994) and release of soluble CNTFRα that could potentially increase MN CNTF receptor signaling (Davis et al., 1993a). Together, these findings suggest that muscle CNTFRα may be part of an endogenous mechanism that promotes MN survival, most particularly following adult peripheral nerve lesion. Therefore, we quantified sciatic MNs in the muscle CNTFRα-depleted and control mice following sciatic nerve crush. CNTFRα depletion had no effect on MN number on the intact or lesioned sides (Fig. 8).
Figure 8.
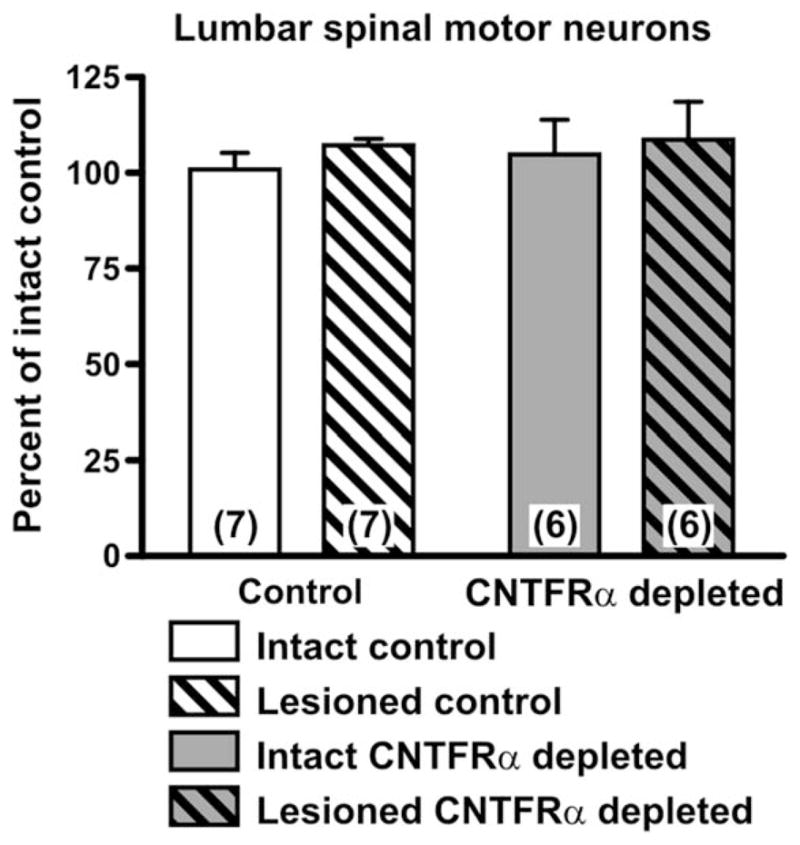
Motor neuron survival is not affected by muscle ciliary neurotrophic factor receptor α (CNTFRα) depletion. Lumbar spinal motor neurons were counted in spinal cords harvested from muscle CNTFRα-depleted and control mice 3 weeks after sciatic nerve crush. Neither the lesion nor the CNTFRα depletion influenced the number of motor neurons, nor was there any interaction (P >0.05 for all two-way ANOVA terms). Means with SEM are presented. Numbers in bars indicate numbers of mice.
Muscle CNTFRα plays an essential role in axonal regeneration following peripheral nerve lesion
The increase in muscle CNTFRα expression and release following nerve lesion also raises the possibility that muscle CNTFRα, even though not essential for MN survival, may contribute to axonal regeneration. Moreover, adult CNTF administration increases the rate of axonal regrowth following peripheral nerve lesion (Sahenk et al., 1994) and maintains MN axons in genetic models of ALS (Pun et al., 2006).
Our examination of NMJs (see above) indicated that muscle CNTFRα depletion does not produce a detectable change in the rate or extent of reinnervation following nerve lesion, with complete reinnervation occurring in both CNTFRα-depleted and control mice. However, axonal branching anywhere distal to the lesion could lead to rapid and complete reinnervation of NMJs even if the receptor depletion leads to a reduction in the number of regenerating axons. Therefore, to first determine whether muscle CNTFRα is essential for normal regeneration of axons across the lesion site, we quantified axons immediately proximal and distal to the lesion site in CNTFRα-depleted and control mice 3 weeks after lesion, when essentially all normally regenerating axons should have extended across the lesion site, with most reaching target tissues.
As expected, we found that in control mice the number of distal axons was equivalent to the number of proximal axons (distal =97.0 ±6.0% of proximal; n =10). In contrast, the number of distal axons was only 81.1 ±7.3% of proximal axons in CNTFRα-depleted mice (n =10; Fig. 9). Comparing the CNTFRα-depleted mice with littermate controls that were lesioned, assayed, and analyzed in parallel confirmed that the ratio of distal to proximal axons was significantly lower in the CNTFRα-depleted mice (P =0.02; 10 pair; t =2.81; paired t-test).
Figure 9.
Axonal regeneration across the lesion site is impaired by muscle CNTFRα depletion. Examples of cross sections from nerve segments harvested 3 weeks post lesion from a control mouse (A–E) and a littermate muscle CNTFRα-depleted mouse (F–J), which were lesioned and processed in parallel. Neurofilament-stained (to label axons) sections are presented proximal to distal (left to right) showing sections proximal to the lesion site (A,F), sections from the lesion site (B,C,G,H) with the typical expansion of axon growth in the proximal part of the lesion (B,G), and sections distal to the lesion site (D,E,I,J). Distal to the lesion the nerves from the muscle CNTFRα-depleted mice displayed fewer axons, and in some cases, like the one shown here, appeared “collapsed” relative to controls with less clearly separated axon bundles. Sections presented above each other originate from the same position relative to respective crush sites. Quantification of the data indicated that while controls displayed the same number of distal and proximal axons by this postlesion interval (distal =97.0 ±6.0% of proximal; n =10), this ratio was significantly decreased in muscle CNTFRα-depleted littermates that were lesioned, assayed, and analyzed in parallel (distal =81.1 ±7.3% of proximal; n =10; P =0.02; 10 pair; t =2.81; paired t-test). Scale bar =75 μm in J (applies to A–J).
Muscle CNTFRα contributes to motor neuron axon regeneration following peripheral nerve lesion
The above result indicates that muscle CNTFRα is required for normal axon regeneration following the lesion, in that without it there is a decreased/abnormal amount of axons extending across the lesion site. The reduction in distal axons in the CNTFRα-depleted mice could result from a decrease in the number of lesioned neurons that respond by extending a regenerating axon at least across the crush site by 3 weeks post lesion. However, the result is also theoretically consistent with a reduction in axonal branching at the lesion site, if any such branching were muscle CNTFRα-dependent. To distinguish between these possibilities, we loaded axons 7 mm distal to the lesion site with the retrograde tracer Fluoro-Gold and quantified the number of subsequently labeled MNs. The spinal cords of the muscle CNTFRα-depleted mice contained 81.5 ±4.5% as many labeled MNs as their paired littermate controls run in parallel (P <0.015; 6 pair; t =3.65; paired t-test; Fig. 10). Therefore, at 3 weeks post lesion, when MNs have normally regenerated axons back to the muscle, approximately 20% fewer of the MNs in the muscle CNTFRα-depleted mice have regenerated axons even as far as 7 mm distal to the lesion site.
Figure 10.

Depletion of muscle ciliary neurotrophic factor receptor α (CNTFRα) reduces motor neuron axonal regeneration following nerve lesion. Axons distal to the crush site in CNTFRα-depleted and control mice were loaded with the retrograde tracer Fluoro-Gold 3 weeks post lesion. Spinal cords harvested 2 weeks later were processed for Fluoro-Gold immunohistochemistry to identify motor neurons with axons that regenerated to the loading site. Control (A) and muscle CNTFRα-depleted (B) examples are shown here with the lesioned and tracer loaded sides on the right. A graph summarizing all the data is presented as an insert in B. The muscle CNTFRα-depleted mice displayed significantly fewer labeled motor neurons than their littermate control pairs processed in parallel (*P <0.015; 6 pair; t =3.65; paired t-test). Broken lines designate approximate borders of spinal cord gray matter. Scale bar =100 μm in B (applies to A,B).
Muscle CNTFRα is essential for normal recovery of motor function following peripheral nerve lesion
In order to assess the potential contribution of muscle CNTFRα to motor function recovery following nerve lesion, we compared the muscle CNTFRα-depleted and control mice with “walking track,” or “footprint” analysis, the most widely used behavioral method to quantify the recovery of motor function following sciatic nerve lesion (De Medinaceli et al., 1982; Bain et al., 1988; Wang et al., 1997) (see Materials and Methods section). Wild-type rats and mice typically show little if any recovery in walking track scores during the 2 weeks immediately following sciatic nerve crush, but largely recover during the third week post lesion (Bervar, 2000; Dijkstra et al., 2000). As animals regain motor function, the footprints left by their foot ipsilateral to the lesion progressively take on the characteristics of the normal footprints of the contralateral control side. In particular, recovery from sciatic nerve lesion has been shown to be best reflected by the “toe spread” between the first and fifth toes such that the lesion reduces this value ipsilateral to the lesion and with recovery the toe spread increases with time to more closely match that of the uninjured side (Hare et al., 1993).
In our study the control mice displayed a recovery consistent with this literature (Fig. 11). In significant contrast, the muscle CNTFRα-depleted mice recovered function to a substantially lesser extent (Fig. 11; P <0.003; two-way ANOVA; main effect of CNTFRα depletion). Less powerful post hoc individual group comparisons, appropriately designed to protect against cumulative type I error, were unable to identify differences at individual time points. Consequently, the data do not allow for more refined conclusions regarding the relative time courses of motor function recovery in the muscle CNTFRα-depleted and control mice. However, given the highly significant overall effect of CNTFRα depletion on motor function recovery (ANOVA; see above), it is clear that endogenous muscle CNTFRα is required for normal motor function recovery following sciatic nerve crush because with CNTFRα depletion motor function is clearly impaired.
Figure 11.

Depletion of muscle ciliary neurotrophic factor receptor α (CNTFRα) impairs recovery of motor function following nerve lesion. Walking track analysis revealed that the muscle CNTFRα-depleted mice display impaired motor function recovery following sciatic nerve crush, relative to controls (P <0.003; F =9.71; main effect of muscle CNTFRα depletion in two-way ANOVA; less powerful post hoc individual group comparisons, appropriately designed to protect against cumulative type I error, were unable to detect differences on individual days). “Walking track analysis scores” compare lesioned and control sides within animals such that values increase as the lesioned side leg recovers function to more closely resemble the function of the unlesioned side (see Materials and Methods). Animals were tested in parallel by investigators blind to genotype. Means with SEM bars are presented. Numbers of control mice tested: 13 days post lesion (DPL) 9; 15DPL, 11; 19DPL, 11; 21DPL, 9. Number of muscle CNTFRα-depleted mice tested: 13DPL, 7; 15DPL, 7; 19DPL, 6; 21DPL, 6.
DISCUSSION
We report that selective in vivo depletion of skeletal muscle CNTFRα decreases MN axonal regeneration and the recovery of motor function following sciatic nerve crush. Surprisingly, the same manipulation has no detectable effect on the aspects of muscle maintenance and regeneration previously shown to respond to exogenous CNTF administration. These results suggest that either the exogenous CNTF in the previous studies acted on sites other than muscle CNTF receptors or it elicited pharmacological effects not seen under physiological conditions.
The depletion of skeletal muscle CNTFRα produced a significant but modest effect on axon regeneration, indicating that, not surprisingly, other proteins participate in axon regeneration, and that the system likely has some redundancy in vivo. Some of this redundancy may well involve CNTFRα expressed by the regenerating MNs themselves. Future work combining MN and muscle-specific CNTFRα gene disruption in vivo will be required to definitively address this issue. Regardless, 3 weeks after sciatic nerve crush, when most normal MNs have regenerated axons back to the muscle, the muscle CNTFRα-depleted mice displayed a decrease of approximately 20% relative to controls in the number of MNs regenerating axons even as far as the Fluoro-Gold loading site, just 7 mm distal to the crush site. Therefore, the data clearly indicate that muscle CNTFRα is required for normal axonal regeneration, in that without it the regeneration is decreased/abnormal. However, the present data do not distinguish between the possibility that a distinct subclass of MNs, which constitute approximately 20% of all MNs, absolutely require muscle CNTFRα for axonal regeneration and the possibility of redundancy in the system such that multiple proteins contribute to axonal regeneration and partially compensate for the loss of a single protein from muscle.
Muscle-specific manipulation studies have identified muscle-derived factors that regulate the differentiation and stabilization of motor nerve terminals (Nguyen et al., 2000; Yumoto et al., 2012). There is also evidence that as yet unidentified muscle-derived factors may influence axon growth in terminal branching decisions following nerve lesion (Robinson and Madison, 2009). However, to the best of our knowledge, the present data are the first to identify a specific endogenous muscle protein, muscle CNTFRα, that contributes to motor axon regeneration across the nerve lesion site. Application of exogenous MDP77, an unrelated protein largely, but not exclusively, expressed in skeletal muscle, has been shown to increase motor axon regeneration across the site of sciatic nerve lesion (Itoh et al., 2005). Along with the present results, these findings suggest that the interaction of skeletal muscle and regenerating motor axons at the lesion site may involve a variety of proteins. With the advent of genetic methods to selectively disrupt genes in skeletal muscle, exploration for additional muscle proteins involved in this interaction in vivo should become straightforward.
The muscle CNTFRα-depleted mice also display a decrease in motor recovery as determined by walking track analysis. The decrease in axonal regeneration may be at least partially responsible for the impaired motor recovery detected by this sensitive test. The decrease in motor recovery could reflect the reduced number of MN soma that successfully regenerate axons back to the muscle. It could also reflect the detrimental effects of the apparent collateral branching/sprouting that occurs to occupy the NMJs that would otherwise have been innervated by the nonregenerating MNs. However, other as yet unidentified mechanisms may also contribute. Regardless, the present data reveal an essential in vivo role for muscle CNTFRα in the normal recovery of motor function following peripheral nerve lesion.
There are several possible mechanisms by which muscle CNTFRα could play its role in axonal regeneration following nerve lesion. In vitro data indicate that soluble CNTFRα is released from muscle following denervation (Davis et al., 1993a). If it combines with a CNTF receptor ligand, such as the CNTF that has been shown to be released at the lesion site from Schwann cells following the same injury (Sendtner et al., 1992b), it can act on other cell types that express the other receptor components, LIFRβ and gp130 (such as MNs), to enhance CNTF receptor signaling (Davis et al., 1993a). Therefore, if the increase in muscle CNTFRα expression seen following sciatic nerve lesion (Helgren et al., 1994) leads to muscle CNTFRα reaching the regenerating axons at the lesion site and/or cell bodies of the MNs, it would be expected to enhance MN CNTF receptor signaling, and potentially increase MN axonal regeneration. Alternatively, or in addition, the increase in muscle CNTFRα may promote CNTF receptor signaling in denervated muscle, which could lead to an increased release of one or more unidentified factors that act on MNs to promote their regeneration. Although the separation between the midthigh nerve lesion site and the denervated muscles of the lower limb would at first seem to argue against the above possible mechanisms, the rapid and dramatic increase in CNTFRα mRNA expression in denervated muscle that is sustained for at least 10 days post lesion (Helgren et al., 1994) suggests that CNTFRα and/or CNTFRα-dependent factors released from the denervated muscles could be involved. In addition, evidence suggests that, although denervation increases the release of muscle CNTFRα, the CNTFRα is also released from intact muscle at a slower rate (Davis et al., 1993a). Therefore, muscles adjacent to the nerve may continuously supply the nerve with this soluble CNTFRα, even in the absence of nerve injury, such that sufficient quantities are immediately available to contribute to axonal regeneration following nerve injury. Finally, the Schwann cell CNTF released at the injury site may act on CNTF receptors in this adjacent muscle to induce the local release of a factor or factors that promote axonal regeneration.
As noted above, the number of retrogradely labeled MNs was decreased by approximately 20% in the muscle CNTFRα-depleted mice. The number of total sciatic nerve axons distal to the lesion site was similarly affected. Given that MN axons make up a small minority of total sciatic nerve axons at the lesion site (Swett et al., 1991), a decrease of approximately 20% in MN axons cannot account for the approximate 20% decrease in total axons. Therefore, the results suggest that muscle CNTFRα also contributes to the regeneration of at least some sensory axons. Unfortunately, we were unable to obtain reliable, quantifiable retrograde labeling of dorsal root ganglion neurons or identify workable markers for sensory axons in order to confirm this.
We targeted CNTFRα because CNTFRα disruption is the most comprehensive approach to determining the in vivo functions of endogenous CNTF receptor signaling. Thus, CNTFRα is essential for all known forms of CNTF receptor signaling, regardless of which ligands and signaling pathways are involved (Davis et al., 1993b; Elson et al., 2000; Derouet et al., 2004). Consequently, the present data do not address which individual ligand(s) or pathways participate in the muscle CNTFRα-dependent signaling that promotes MN regeneration and recovery of motor function. As noted above, CNTF may contribute to MN regeneration given that it is released from Schwann cells at the site of peripheral nerve lesion (Sendtner et al., 1992b) and may combine with CNTFRα released from the denervated muscle (Davis et al., 1993a). A previous study showing decreased motor recovery following sciatic nerve crush in unconditional CNTF knockout mice (Yao et al., 1999) suggests that endogenous CNTF may be involved, but it does not address the source of the CNTF or the CNTFRα. The cardiotrophin-like cytokine/cytokine-like factor 1 (CLC/CLF) CNTF receptor ligand, which plays a substantial role in embryonic MN survival (Forger et al., 2003; Zou et al., 2009), may also participate. Exogenous CLC has been found to protect retinal ganglion cells after optic nerve crush (Schuettauf et al., 2005). Although this neuroprotection was observed with a central nervous system lesion, the study raises the possibility that endogenous CLC may be involved in the muscle CNTFRα-dependent signaling underlying the effects reported here. Studies are needed to determine whether CLC is expressed in the adult neuromuscular system. Of course, the ligands may act together. Regardless, conditional disruption of individual ligands will be necessary to definitively answer these questions.
The conditional gene disruption technique utilized here produced a substantial but not total depletion of muscle CNTFRα. This result is consistent with other work done with the same mlc1f-Cre gene construct (Bothe et al., 2000, Wredenberg et al., 2002), and it has two important implications for interpretation of the present data. First, it is still theoretically possible that muscle CNTFRα contributes to muscle regeneration, such that even a small amount of muscle CNTFRα is sufficient to provide maximal effect. Similarly, muscle CNTFRα may play a role in the other functions that were unaffected in the present studies. However, it seems most likely that if muscle CNTFRα were to play a significant role in any of these processes, its substantial depletion would be expected to produce a detectable effect. The second important implication regarding the degree of muscle CNTFRα depletion pertains to the interpretation of the effects seen on neuronal regeneration. It should be noted that muscle CNTFRα may play a larger role in these functions than that detected here, in that total removal of all muscle CNTFRα may produce larger effects. Regardless, the present results surprisingly demonstrate that muscle CNTFRα plays a more important role in the regeneration of neurons than the regeneration of the muscle itself.
Exogenous CNTF administration has been shown by several groups to elicit terminal sprouting of MN axons (Gurney et al., 1992; Kwon and Gurney, 1994; Ikeda et al., 1995; Tarabal et al., 1996; Siegel et al., 2000; Wright and Son, 2007; Wright et al., 2007). Conflicting reports from CNTF knockout studies suggest that endogenous CNTF is (Siegel et al., 2000) or is not (Wright and Son, 2007) involved in MN terminal sprouting observed after nerve lesion. The present results suggest that muscle CNTFRα does not significantly contribute to terminal sprouting following sciatic nerve crush. However, the data do not rule out the possibility that MN CNTFRα is essential for this response, through an interaction with either CNTF or CLC/CLF.
Work with genetic models of ALS indicates that exogenous CNTF administration can protect MN axons from this genetic insult (Pun et al., 2006). Several lines of evidence suggest that loss of MN axons is a critical event leading to ALS symptoms (Gould et al., 2006; Pun et al., 2006; Dadon-Nachum et al., 2011). However, clinical ALS trials with systemic CNTF injections were stopped due to unacceptable side effects (ALS CNTF Treatment Study Group, 1995, 1996; Miller et al., 1996), indicating that any therapeutic manipulation of CNTF signaling will need to be more specifically targeted. Therefore, the present data indicating that endogenous muscle CNTFRα-dependent signaling contributes to MN axon regeneration following a different insult (nerve lesion) raises the possibility that muscle CNTFRα should be considered as a potential specific target in the treatment of ALS, whether this involves interventions designed to increase muscle CNTFRα expression or other approaches. Similarly, the present data suggest that targeting muscle CNTFRα may prove beneficial in the treatment of peripheral nerve injury.
Acknowledgments
Grant sponsor: National Institutes of Health; Grant numbers: NS35224 and NS052700 (to A.J.M.).
We thank Dr. S. Burden for the mlc1f-Cre mice and for helpful comments on the manuscript, Dr. J.R. Sanes for the YFP16 mice, Peggy Bowman, Tatiana Radzyukevich, and Dan Shelly for technical assistance, and Glenn Doerman for assistance with the figures.
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
ROLE OF AUTHORS
All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: AJM. Acquisition of data: NL, RPS, KML, RR, DST, COM, RJZ, MKB, RJP, and AJM. Analysis and interpretation of data NL, RPS, KML, RR, DST, COM, RJZ, MKB, RJP, and AJM. Drafting of the manuscript: AJM. Manuscript review: NL, RPS, KML, RR, DST, COM, RJZ, MKB, and RJP.
LITERATURE CITED
- ALS CNTF Treatment Study (ACTS) Phase I–II Study Group. A phase I study of recombinant human ciliary neurotrophic factor (rHCNTF) in patients with amyotrophic lateral sclerosis. Clin Neuropharmacol. 1995;18:515–532. doi: 10.1097/00002826-199512000-00004. [DOI] [PubMed] [Google Scholar]
- ALS CNTF Treatment Study Group. A double-blind placebo-controlled clinical trial of subcutaneous recombinant human ciliary neurotrophic factor (rHCNTF) in amyotrophic lateral sclerosis. Neurology. 1996;46:1244–1249. doi: 10.1212/wnl.46.5.1244. [DOI] [PubMed] [Google Scholar]
- Bain JR, Mackinnon SE, Hunter DA. Functional evaluation of complete sciatic, peroneal, and posterior tibial nerve lesions in the rat. Plast Reconstr Surg. 1988;82:1052–1066. doi: 10.1097/00006534-198901000-00024. [DOI] [PubMed] [Google Scholar]
- Bervar M. Video analysis of standing—an alternative footprint analysis to assess functional loss following injury to the rat sciatic nerve. J Neurosci Methods. 2000;102:109–116. doi: 10.1016/s0165-0270(00)00281-8. [DOI] [PubMed] [Google Scholar]
- Bothe GWM, Haspel JA, Smith CL, Wiener HH, Burden SJ. Selective expression of Cre recombinase in skeletal muscle fibers. Genesis. 2000;26:165–166. [PubMed] [Google Scholar]
- Carson NE, Gu J, Ianuzzo CD. Detection of myosin heavy chain in skeletal muscles using monoclonal antibodies on formalin fixed, paraffin embedded tissue sections. J Histotechnol. 1998;21:19–24. [Google Scholar]
- Crawford K, Flick R, Close L, Shelly D, Paul RJ, Bove K, Kumar A, Lessard J. Mice lacking skeletal muscle actin show reduced muscle strength and growth deficits and die during the neonatal period. Mol Cell Biol. 2002;22:5887–5896. doi: 10.1128/MCB.22.16.5887-5896.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadon-Nachum M, Melamed E, Offen D. The “dying back” phenomenon of motor neurons in ALS. J Mol Neurosci. 2011;43:470–477. doi: 10.1007/s12031-010-9467-1. [DOI] [PubMed] [Google Scholar]
- Davis S, Aldrich TH, Ip NY, Stahl N, Scherer S, Farruggella T, DiStefano PS, Curtis R, Panayotatos N, Gascan H, Chevalier S, Yancopoulos GD. Released form of CNTF receptor α component is a soluble mediator of CNTF responses. Science. 1993a;259:1736–1739. doi: 10.1126/science.7681218. [DOI] [PubMed] [Google Scholar]
- Davis S, Aldrich TH, Stahl N, Pan L, Taga T, Kishimoto T, Ip NY, Yancopoulos GD. LIFRβ and gp130 as heterodimerizing signal transducers of the tripartite CNTF receptor. Science. 1993b;260:1805–1808. doi: 10.1126/science.8390097. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Vejsada R, Poueymirou WT, Acheson A, Suri C, Conover JC, Friedman B, McClain J, Pan L, Stahl N, Ip NY, Kato A, Yancopoulos GD. Mice lacking the CNTF receptor, unlike mice lacking CNTF, exhibit profound motor neuron deficits at birth. Cell. 1995;83:313–322. doi: 10.1016/0092-8674(95)90172-8. [DOI] [PubMed] [Google Scholar]
- De Medinaceli L, Freed WJ, Wyatt RJ. An index of the functional condition of rat sciatic nerve based on measurements made from walking tracks. Exp Neurol. 1982;77:634–643. doi: 10.1016/0014-4886(82)90234-5. [DOI] [PubMed] [Google Scholar]
- Derouet D, Rousseau F, Alfonsi F, Froger J, Hermann J, Barbier F, Perret D, Diveu C, Guillet C, Preisser L, Dumont A, Barbado M, Morel A, deLapeyriere O, Gascan H, Chevalier S. Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proc Natl Acad Sci U S A. 2004;101:4827–4832. doi: 10.1073/pnas.0306178101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra JR, Meek MF, Robinson PH, Gramsbergen A. Methods to evaluate functional nerve recovery in adult rats: walking track analysis, video analysis and the withdrawal reflex. J Neurosci Methods. 2000;96:89–96. doi: 10.1016/s0165-0270(99)00174-0. [DOI] [PubMed] [Google Scholar]
- Elson GCA, Lelievre E, Guillet C, Chevalier S, Plun-Favreau H, Froger J, Suard I, Benoit de Coignac A, Delneste Y, Bonnefoy J-Y, Gauchat J-F, Gascan H. CLF associates with CLC to form a functional heteromeric ligand for the CNTF receptor complex. Nat Neurosci. 2000;3:867–872. doi: 10.1038/78765. [DOI] [PubMed] [Google Scholar]
- Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- Forger NG, Prevette D, deLapeyriere O, de Bovis B, Wang S, Bartlett P, Oppenheim RW. Cardiotrophin-like cytokine/cytokine-like factor 1 is an essential trophic factor for lumbar and facial motoneurons in vivo. J Neurosci. 2003;23:8854–8858. doi: 10.1523/JNEUROSCI.23-26-08854.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraysse B, Guillet C, Huchet-Cadiou C, Conte Camerino D, Gascan H, Leoty C. Ciliary neurotrophic factor prevents unweighting-induced functional changes in rat soleus muscle. J Appl Physiol. 2000;88:1623–1630. doi: 10.1152/jappl.2000.88.5.1623. [DOI] [PubMed] [Google Scholar]
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillet C, Auguste P, Mayo W, Kreher P, Gascan H. Ciliary neurotrophic factor is a regulator of muscular strength in aging. J Neurosci. 1999;19:1257–1262. doi: 10.1523/JNEUROSCI.19-04-01257.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Yamamoto H, Kwon Y. Induction of motor neuron sprouting in vivo by ciliary neurotrophic factor and basic fibroblast growth factor. J Neurosci. 1992;12:3241–3247. doi: 10.1523/JNEUROSCI.12-08-03241.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hare GM, Evans PJ, Mackinnon SE, Best TJ, Midha R, Szalai JP, Hunter DA. Walking track analysis: utilization of individual footprint parameters. Ann Plast Surg. 1993;30:147–153. doi: 10.1097/00000637-199302000-00009. [DOI] [PubMed] [Google Scholar]
- Harris J, Ayyub C, Shaw G. A molecular dissection of the carboxyterminal tails of the major neurofilament subunits NF-M and NF-H. J Neurosci Res. 1991;30:47–62. doi: 10.1002/jnr.490300107. [DOI] [PubMed] [Google Scholar]
- Hatton WJ, Von Bartheld CS. Analysis of cell death in the trochlear nucleus of the chick embryo: calibration of the optical dissector counting method reveals systematic bias. J Comp Neurol. 1999;409:169–186. [PubMed] [Google Scholar]
- Havenith MG, Visser R, Schrijvers-van Schendel JM, Bosman FT. Muscle fiber typing in routinely processed skeletal muscle with monoclonal antibodies. Histochemistry. 1990;93:497–499. doi: 10.1007/BF00266407. [DOI] [PubMed] [Google Scholar]
- He S, Shelly DA, Moseley AE, James PF, James JH, Paul RJ, Lingrel JB. The alpha(1)- and alpha(2)-isoforms of Na-K-ATPase play different roles in skeletal muscle contractility. Am J Physiol Regul Integr Comp Physiol. 2001;281:R917–R925. doi: 10.1152/ajpregu.2001.281.3.R917. [DOI] [PubMed] [Google Scholar]
- Helgren ME, Squinto SP, Davis HL, Parry DJ, Boulton TG, Heck CS, Zhu Y, Yancopoulos GD, Lindsay RM, DiStefano PS. Trophic effect of ciliary neurotrophic factor on denervated skeletal muscle. Cell. 1994;76:493–504. doi: 10.1016/0092-8674(94)90113-9. [DOI] [PubMed] [Google Scholar]
- Hosaka Y, Yokota T, Miyagoe-Suzuki Y, Yuasa K, Imamura M, Matsuda R, Ikemoto T, Kameya S, Takeda S. α1-Syntrophin-deficient skeletal muscle exhibits hypertrophy and aberrant formation of neuromuscular junctions during regeneration. J Cell Biol. 2002;158:1097–1107. doi: 10.1083/jcb.200204076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Wang F, Hong G, Wan S, Kang H. Protective effects of ciliary neurotrophic factor on denervated skeletal muscle. J Tongji Med Univ. 2002;22:148–151. doi: 10.1007/BF02857680. [DOI] [PubMed] [Google Scholar]
- Hughes SM, Cho M, Karsch-Mizrachi I, Travis M, Silberstein L, Leinwand LA, Blau HM. Three slow myosin heavy chains sequentially expressed in developing mammalian skeletal muscle. Dev Biol. 1993;158:183–199. doi: 10.1006/dbio.1993.1178. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Gomez-Isla T, Irizarry MC. Stereology: A practical primer for neuropathology. J Neuropathol Exp Neurol. 1998;57:305–310. doi: 10.1097/00005072-199804000-00001. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Wong V, Holmlund TH, Greene T, Cedarbaum JM, Lindsay RM, Mitsumoto H. Histometric effects of ciliary neurotrophic factor in wobbler mouse motor neuron disease. Ann Neurol. 1995;37:47–54. doi: 10.1002/ana.410370110. [DOI] [PubMed] [Google Scholar]
- Ip NY, Yancopoulos GD. Ciliary neurotrophic factor and its receptor complex. Prog Growth Factor Res. 1992;4:139–155. doi: 10.1016/0955-2235(92)90028-g. [DOI] [PubMed] [Google Scholar]
- Itoh S, Fujimori KE, Uyeda A, Matsuda A, Kobayashi H, Shinomiya K, Tanaka J, Taguchi T. Long-term effects of muscle-derived protein with molecular mass of 77 kDa (MDP77) on nerve regeneration. J Neurosci Res. 2005;81:730–738. doi: 10.1002/jnr.20582. [DOI] [PubMed] [Google Scholar]
- Janjua MZ, Leong SK. Organization of neurons forming the femoral, sciatic, common peroneal and tibial nerves in rats and monkeys. Brain Res. 1984;310:311–323. doi: 10.1016/0006-8993(84)90154-9. [DOI] [PubMed] [Google Scholar]
- Klover P, Chen W, Zhu B-M, Hennighausen L. Skeletal muscle growth and fiber composition in mice are regulated through the transcription factors STAT5a/b: linking growth hormone to the androgen receptor. FASEB J. 2009;23:3140–3148. doi: 10.1096/fj.08-128215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokoeva MV, Yin H, Flier JS. Neurogenesis in the hypothalamus of adult mice: potential role in energy balance. Science. 2005;310:679–683. doi: 10.1126/science.1115360. [DOI] [PubMed] [Google Scholar]
- Kwon YW, Gurney ME. Systemic injections of ciliary neurotrophic factor induce sprouting by adult motor neurons. Neuroreport. 1994;5:789–792. doi: 10.1097/00001756-199403000-00013. [DOI] [PubMed] [Google Scholar]
- Lambert PD, Anderson KD, Sleeman MW, Wong V, Tan J, Hijarunguru A, Corcoran TL, Murray JD, Thabet KE, Yancopoulos GD, Wiegand SJ. Ciliary neurotrophic factor activates leptin-like pathways and reduces body fat, without cachexia or rebound weight gain, even in leptin-resistant obesity. Proc Natl Acad Sci U S A. 2001;98:4652–4657. doi: 10.1073/pnas.061034298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee N, Robitz R, Zurbrugg RJ, Karpman AM, Mahler AM, Cronier SA, Vesey R, Spearry RP, Zolotukhin S, MacLennan AJ. Conditional, genetic disruption of ciliary neurotrophic factor receptors reveals a role in adult motor neuron survival. Eur J Neurosci. 2008;27:2830–2837. doi: 10.1111/j.1460-9568.2008.06298.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S-B, Beak SK, Park SH, Waterhouse BD, Lee HS. Collateral projection from the locus coeruleus to whisker-related sensory and motor brain regions of the rat. J Comp Neurol. 2009;514:387–402. doi: 10.1002/cne.22012. [DOI] [PubMed] [Google Scholar]
- Li M, Sendtner M, Smith A. Essential function of LIF receptor in motor neurons. Nature. 1995;378:724–727. doi: 10.1038/378724a0. [DOI] [PubMed] [Google Scholar]
- Lucas CA, Kang LHD, Hoh JFY. Monospecific antibodies against the three mammalian fast limb myosin heavy chains. Biochem Biophys Res Comm. 2000;272:303–308. doi: 10.1006/bbrc.2000.2768. [DOI] [PubMed] [Google Scholar]
- Lyons GE, Ontell M, Cox R, Sassoon D, Buckingham M. The expression of myosin genes in developing skeletal muscle in the mouse embryo. J Cell Biol. 1990;111:1465–1476. doi: 10.1083/jcb.111.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan AJ, Vinson EN, Marks L, McLaurin DL, Pfeifer M, Lee N. Immunohistochemical localization of ciliary neurotrophic factor receptor α expression in the rat nervous system. J Neurosci. 1996;16:621–630. doi: 10.1523/JNEUROSCI.16-02-00621.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe KL, Gunther EC, Reh TA. The development of the pattern of retinal ganglion cells in the chick retina: mechanisms that control differentiation. Development. 1999;126:5713–5724. doi: 10.1242/dev.126.24.5713. [DOI] [PubMed] [Google Scholar]
- Miller RG, Bryan WW, Dietz MA, Munsat TL, Petajan JH, Smith SA, Goodpasture JC. Toxicity and tolerability of recombinant human ciliary neurotrophic factor in patients with amyotrophic lateral sclerosis. Neurology. 1996;47:1329–1331. doi: 10.1212/wnl.47.5.1329. [DOI] [PubMed] [Google Scholar]
- Mitsumoto H, Ikeda K, Holmlund T, Greene T, Cedarbaum JM, Wong V, Lindsay RM. The effects of ciliary neurotrophic factor on motor dysfunction in wobbler mouse motor neuron disease. Ann Neurol. 1994;36:142–148. doi: 10.1002/ana.410360205. [DOI] [PubMed] [Google Scholar]
- Nguyen QT, Son Y-J, Sanes JR, Lichtman JW. Nerve terminals form but fail to mature when postsynaptic differentiation is blocked: in vivo analysis using mammalian nerve-muscle chimeras. J Neurosci. 2000;20:6077–6086. doi: 10.1523/JNEUROSCI.20-16-06077.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno H, Kishimoto KN, Hatori M, Itoi E. 1α,25-Dihydroxyvitamin D3 enhances fast-myosin heavy chain expression in differentiated C2C12 myoblasts. Cell Biol Int. 2012;36:441–447. doi: 10.1042/CBI20100782. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Prevette D, Qin-Wei Y, Collins F, MacDonald J. Control of embryonic motoneuron survival in vivo by ciliary neurotrophic factor. Science. 1991;251:1616–1618. doi: 10.1126/science.2011743. [DOI] [PubMed] [Google Scholar]
- Poea S, Guyon T, Lavasseur P, Berrih-Aknin S. Expression of ciliary neurotrophic factor receptor in myasthenia gravis. J Neuroimmunol. 2001;120:180–189. doi: 10.1016/s0165-5728(01)00423-4. [DOI] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–19. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Robinson GA, Madison RD. Influence of terminal nerve branch size on motor neuron regeneration accuracy. Exp Neurol. 2009;215:228–235. doi: 10.1016/j.expneurol.2008.10.002. [DOI] [PubMed] [Google Scholar]
- Rojiani AM, Cho ES. Neuropathologic applications of immunohistochemical fiber typing in the non-neoplastic muscle biopsy. Mod Pathol. 1998;11:334–338. [PubMed] [Google Scholar]
- Saggio I, Gloaguen I, Poiana G, Laufer R. CNTF variants with increased biological potency and receptor selectivity define a functional site of receptor interaction. EMBO J. 1995;14:3045–3054. doi: 10.1002/j.1460-2075.1995.tb07307.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahenk Z, Seharaseyon J, Mendell JR. CNTF potentiates peripheral nerve regeneration. Brain Res. 1994;655:246–250. doi: 10.1016/0006-8993(94)91621-7. [DOI] [PubMed] [Google Scholar]
- Schuettauf F, Zurakowski D, Quinto K, Varde MA, Besch D, Laties A, Anderson R, Wen R. Neuroprotective effects of cardiotropin-like cytokine on retinal ganglion cells. Graefes Arch Clin Exp Ophthalmol. 2005;243:1036–1042. doi: 10.1007/s00417-005-1152-7. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Kreutzberg GW, Thoenen H. Ciliary neurotrophic factor prevents the degeneration of motor neurons after axotomy. Nature. 1990;345:440–441. doi: 10.1038/345440a0. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Schmalbruch H, Stockli KA, Carroll P, Kreutzberg GW, Thoenen H. Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy. Nature. 1992a;358:502–504. doi: 10.1038/358502a0. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Stockli KA, Thoenen H. Synthesis and localization of ciliary neurotrophic factor in the sciatic nerve of the adult rat after lesion and during regeneration. J Cell Biol. 1992b;118:139–148. doi: 10.1083/jcb.118.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel SG, Patton B, English AW. Ciliary neurotrophic factor is required for motoneuron sprouting. Exp Neurol. 2000;166:205–212. doi: 10.1006/exnr.2000.7528. [DOI] [PubMed] [Google Scholar]
- Smerdu V, Soukup T. Demonstration of myosin heavy chain isoforms in rat and humans: the specificity of seven available monoclonal antibodies used in immunohistochemical and immunoblotting methods. Eur J Histochem. 2008;52:179–190. doi: 10.4081/1210. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Swett JE, Torigoe Y, Elie VR, Bourassa CM, Miller PG. Sensory neurons of the rat sciatic nerve. Exp Neurol. 1991;114:82–103. doi: 10.1016/0014-4886(91)90087-s. [DOI] [PubMed] [Google Scholar]
- Tarabal O, Caldero J, Esquerda JE. Intramuscular nerve sprouting induced by CNTF is associated with increases in CGRP content in mouse motor nerve terminals. Neurosci Lett. 1996;219:60–64. doi: 10.1016/s0304-3940(96)13174-8. [DOI] [PubMed] [Google Scholar]
- Wang M-S, Zeleny-Pooley M, Gold BG. Comparative dose-dependence study of FK506 and cyclosporin A on the rate of axonal regeneration in the rat sciatic nerve. J Pharm Exp Therap. 1997;282:1084–1093. [PubMed] [Google Scholar]
- Watt MJ, Dzamko N, Thomas WG, Rose-John S, Ernst M, Carling D, Kemp BE, Febbraio MA, Steinberg GR. CNTF reverses obesity-induced insulin resistance by activating skeletal muscle AMPK. Nature Med. 2006;12:541–548. doi: 10.1038/nm1383. [DOI] [PubMed] [Google Scholar]
- Wattler S, Kelly M, Nehls M. Construction of gene targeting vectors from lKOS genomic libraries. BioTechniques. 1999;26:1150–1160. doi: 10.2144/99266rr02. [DOI] [PubMed] [Google Scholar]
- Weis J, Lie DC, Ragoss U, Zuchner SL, Schroder JM, Karpati G, Farruggella T, Stahl N, Yancopoulos GD, DiStefano PS. Increased expression of CNTF receptor α in denervated human skeletal muscle. J Neuropathol Exp Neurol. 1998;57:850–857. doi: 10.1097/00005072-199809000-00006. [DOI] [PubMed] [Google Scholar]
- Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westereblad H, Larsson N-G. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci U S A. 2002;99:15066–15071. doi: 10.1073/pnas.232591499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MC, Son Y-J. Ciliary neurotrophic factor is not required for terminal sprouting and compensatory reinnervation of neuromuscular synapses: reevaluation of CNTF null mice. Exp Neurol. 2007;205:437–448. doi: 10.1016/j.expneurol.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright MC, Cho W-J, Son Y-J. Distinct patterns of motor nerve terminal sprouting induced by ciliary neurotrophic factor vs. botulinum toxin. J Comp Neurol. 2007;504:1–16. doi: 10.1002/cne.21439. [DOI] [PubMed] [Google Scholar]
- Yao M, Moir MS, Wang MZ, To MP, Terris DJ. Peripheral nerve regeneration in CNTF knockout mice. Laryngoscope. 1999;109:1263–1268. doi: 10.1097/00005537-199908000-00015. [DOI] [PubMed] [Google Scholar]
- Yumoto N, Kim N, Burden SJ. Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature. 2012;489:438–442. doi: 10.1038/nature11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CJ, Kawabuchi M, Wang S, Liu WT, Hirata K. Age differences in morphological patterns of axonal sprouting and multiple innervation of neuromuscular junctions during muscle reinnervation following nerve crush injury. Ann Anat. 2002;184:461–472. doi: 10.1016/S0940-9602(02)80080-2. [DOI] [PubMed] [Google Scholar]
- Zou X, Bolon B, Pretorius JK, Kurahara C, McCabe J, Christiansen KA, Sun N, Duryea D, Foreman O, Senaldi G, Itano AA, Siu G. Neonatal death in mice lacking cardiotropin-like cytokine is associated with multifocal neuronal hypoplasia. Vet Pathol. 2009;46:514–519. doi: 10.1354/vp.08-VP-0239-B-BC. [DOI] [PubMed] [Google Scholar]