

Abstract
RNA interference (RNAi) is the process of sequence-specific posttranslational gene silencing triggered by double-stranded RNAs (dsRNAs). RNAi is a widely used approach for studying gene function. However, studies have shown that using siRNA can lead to off-target effects when the siRNA contains sufficient sequence identity to non-target mRNA sequences. One of the important steps in designing dsRNA is verification that it has sequence identity to only the target mRNA. In this report, we propose an approach for primary screening dsRNAs for potential off-target effects by using rapid amplification of cDNA ends. This method can be especially useful for model systems using species that have limited availability of sequence data.
Keywords: siRNA, dsRNA, Rapid Amplification of cDNA Ends (RACE)
Introduction
RNA interference (RNAi) is a powerful tool for studying gene function by selectively inhibiting expression of target genes. The RNAi process is mediated by double stranded RNAs (dsRNAs) that are degraded by the RNase III-like protein DICER into small interfering RNA (siRNA) duplexes (Meister and Tuschl 2004). The antisense strand of the siRNA duplex is incorporated into a protein complex called the RNA-induced silencing complex (RISC). Once the RISC complex is formed, it guides the target mRNA for degradation under direction of the antisense strand of the siRNA duplex (Schwarz et al. 2002; Khvorova et al. 2003).
siRNA is a widely used method to silence the expression of genes, but it is critical for the siRNA to be specific. siRNA must selectively knock down the target gene without interfering with the expression or function of other non-target genes or proteins. The most important criterion for siRNA selection is validation that the designed dsRNA is specific to only the target mRNA and fails to have high similarity to other mRNA sequences that would lead to off-target gene silencing (Cullen 2006). Off-target effects can occur when the siRNA contains sufficient sequence identity to a non-target mRNA to induce the inhibition of its mRNA. It has been shown that even an 11-bp homology can support this inhibitory effect (Jackson et al. 2003). For species whose genomes have undergone significant sequencing such as rodents, target specificity of siRNAs can be confirmed by using local alignment algorithms such as the basic local alignment search tool (BLAST; (Altschul et al. 1997)). Moreover, the specificity of siRNAs can be confirmed using DNA microarrays which are ideally suited to provide a global fingerprint of gene regulation to screen for unpredicted off-target effects (Jackson et al. 2003; Semizarov et al. 2003; Birmingham et al. 2006; Anderson et al. 2008). Unfortunately, these approaches are not available for experimental model systems using species whose genomes have yet to be sequenced.
A number of important model organisms have limited sequence information but are amenable to RNAi-mediated transcriptional silencing technology. For instance, in our laboratory, we employ the freshwater pond turtle, Pseudemys scripta elegans, to study mechanisms underlying vertebrate eyeblink classical conditioning using an in vitro brain stem preparation (Zheng and Keifer 2009; Keifer and Zheng 2010). The siRNA approach has been used successfully in this well-established model of learning and memory to understand the function of genes and proteins involved in this form of learning (Keifer et al. 2009). Here, we report a relatively simple technique for primary screening of the specificity of designed siRNAs. This new approach is based on rapid amplification of cDNA ends (RACE) and has broad applications particularly for model systems that have limited availability of genome sequence data. Production of a single RACE product with the predicted size confirms that designed dsRNA is specific only for the target mRNA. The presence of one or more unpredicted 3′ RACE products indicates that the designed dsRNA has significant homology to mRNA sequences other than the targeted mRNA or that there are products of alternative splicing of the target gene.
Materials and Methods
Animal Use and Care
Adult freshwater pond turtles, P. scripta elegans, were obtained from commercial suppliers (Keifer et al. 2009). Protocols involving the use of animals complied with the guidelines of the National Institutes of Health and the Institutional Animal Care and Use Committee.
Design of dsRNAs
dsRNA consisting of 27-mer oligonucleotides with two base 3′ overhangs were designed against turtle glyceraldehyde-3-phosphate dehydrogenase (tGAPDH) mRNA (GenBank number GU108575) using RNAi design software (Integrated DNA Technologies). Designed dsRNAs were checked by BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Since freshwater pond turtle is not sequenced yet, NCBI Transcript Reference Sequences database were used for this analysis. Blast analysis showed that all designed dsRNAs have significant homology to a large number of mRNAs from various organisms.
RNA Isolation
Total RNA from brain tissue of freshwater pond turtles, P. scripta elegans, was extracted using TRIzol reagent (Invitrogen) and purified by the RNeasy Mini kit (Qiagen). During purification, samples were treated by RNase-free DNase (Qiagen).
PCR and RACE
To confirm that AccuPrime Pfx DNA Polymerase (Invitrogen) is able to remove more than one mismatch from the 3′ end of primers and produce 3′ RACE products, we performed amplifications of a 265-bp fragment of pcDNA3.1/V5-His plasmid. Four polymerase chain reactions (PCRs) were performed. All of these reactions were the same except that different sets of forward primers were used. The original forward primer (T7 forward) had no mismatches on the 3′ end. The other three forward primers had one, two or three mismatches on the 3′ ends (T7mis1, T7mis2, T7mis3; Table 1). Ten nanograms of plasmid template was used per reaction. Conditions of the PCRs were the same as for the 3′ RACE. Products of the amplifications were resolved on 1% agarose gels.
Table 1.
Primers used to test AccuPrime Pfx DNA polymerase removal of 3′ end mismatches and produce PCR products during 3′ RACE
| PCRs | Forward primer | Reverse primer (reverse BGH) |
|---|---|---|
| Control | T7 forward TAATACGACTCACTATAGGG | TAGAAGGCACAGTCGAGG |
| 1 Mismatch | T7mis1 TAATACGACTCACTATAGGC | TAGAAGGCACAGTCGAGG |
| 2 Mismatch | T7mis2 TAATACGACTCACTATAGCC | TAGAAGGCACAGTCGAGG |
| 3 Mismatch | T7mis3 TAATACGACTCACTATACCC | TAGAAGGCACAGTCGAGG |
3′ RACE was performed with the FirstChoice RLM-RACE kit (Ambion). Briefly, 1 μg of total RNA was used for reverse transcription by M-MLV Reverse Transcriptase. The sequence of the 3′ RACE adapter was GCGAGCA-CAGAATTAATACGACTCAC TATAGGT12VN. The product from the reverse transcription reaction (2 μl) was used for PCR along with 1 U of AccuPrime Pfx DNA Polymerase (Invitrogen). 3′ RACE was performed according to the manufacturers protocol. The sequence of the 3′ RACE outer primer was GCGAGCACAGAATTAATAC-GACT. The PCR profile consisted of 1 cycle of denaturation (95°C) for 3 min, followed by 35 cycles of denaturation (94°C) for 30 s, annealing for 30 s, and extension (68°C) for 1 min followed by 7 min final extension (68°C). Annealing temperatures of the primers were calculated using the following formula: Tm =81.5°C+ 0.41 (% GC)–675/N–% mismatch. Where N is the primer length in bases and the number of mismatches was 2.
5′ RACE was performed as described by Soutschek and co-authors (2004) with the FirstChoice RLM-RACE kit (Ambion). Briefly, 1 μg of total RNA was ligated to a 5′ RACE adapter without prior treatment. Ligated RNA was used for reverse transcription by M-MLV Reverse Transcriptase with the random Decamers primer. Products of reverse transcriptase reactions were used for nested outer and inner 5′ RACE PCR. The sequence of the gene-specific outer and inner primers was: CTCCACCTCCCC-CAAGCTTCAGATACA and GTAGAGGAATAGGGAA-GACAGATGGAACAG, respectively. 5′ RACE PCR was performed according to the manufacturer′s protocol.
Cell Culture
Turtle brain primary cell cultures were produced as described (Lu et al. 1999) with some modification. Brain tissue was collected and incubated at 4°C for 30 min in antibiotic incubation medium (AIM) consisting of 2× RPMI 1640 and 1,000 μg streptomycin, 1,000 IU penicillin, and 250 μg gentamicin/ml. Following two additional washes in AIM, tissue was minced and digested with 0.25% trypsin at 30°C in a 5% CO2 incubator for 15 min. Dissociated cells were then washed once with RPMI 1640 medium, and approximately 100,000–200,000 cells were transferred to each well of a 6-well plate. Cell cultures were maintained in RPMI 1640 medium supplemented with 20% heat-inactivated fetal bovine serum and antibiotics (200 U penicillin, 200 μg streptomycin, 50 μg gentamicin/ml) at 25°C in a 5% CO2 incubator.
dsRNA Transfection
Turtle brain primary cells were forward transfected with tGAPDH dsRNAs #6, #8, #12, and #17 or with scrambled dsRNAs #12 and #17 (Table 2) using Lipofectamine RNAiMAX transfection reagent (Invitrogen). Final concentrations of dsRNAs were 5, 10, 20, 50, and 100 nM. Lipofectamine RNAiMAX transfection reagent alone was added in one well as a control to determine any effect of the transfection reagent itself. The levels of GAPDH and actin proteins were examined 25 h later by Western blotting. All experiments were repeated three times using different cultures.
Table 2.
Designed dsRNA and primers used for 3′ RACE
| dsRNA | Sense strand | Antisense strand | Primer | Predicted size of 3′ RACE |
|---|---|---|---|---|
| GAPDH #6 | CUAUGUUUGUGAUGGGUGUCAACCA | UGGUUGACACCCAUCACAAACAUAGGU | ACCTATGTTTGTGATGGGTGTCAACCA | 900 |
| GAPDH #8 | GUGCCAAGAGGGUUGUCAUCUCUGC | GCAGAGAUGACAACCCUCUUGGCACCU | AGGTGCCAAGAGGGTTGTCATCTCTGC | 942 |
| GAPDH #12 | GGACCAGGUUGUCUCCUGUGACUTC | GAAGUCACAGGAGACAACCUGGUCCUC | GAGGACCAGGTTGTCTCCTGTGACTTC | 450 |
| GAPDH #17 | CAGGUGCAUGGAUCAGCCUCUGCCT | AGGCAGAGGCUGAUCCAUGCACCUGGC | GCCAGGTGCATGGATCAGCCTCTGCCT | 275 |
| Scrambled #12 | GCGUGCCGGAUAUGAUGUCGUUCAC | AGGUGAACGACAUCAUAUCCGGCACGC | GCGTGCCGGATATGATGTCGTTCACCT | Not predicted |
| Scrambled #17 | GCCGAUCCGCGACUCCGCUGUAUAG | ACCUAUACAGCGGAGUCGCGGAUCGGC | GCCGATCCGCGACTCCGCTGTATAGGT | Not predicted |
Western Blotting
Whole-cell extracts were prepared in RIPA lysis buffer (20 mM Tris, pH 7.4, 50 mM NaCl, 5 mM ethylene diaminetetraacetic acid, 1% Nonidet P-40, 0.1% SDS, 5 mM NaF, 1 mM PMSF, 1 mM Na3VO4, 1 μM leupeptin, and 0.3 μM aprotinin). Protein content was quantified and normalized using the Bradford method, and proteins were separated on SDS–PAGE. Proteins were blotted onto PVDF transfer membranes (Millipore). Primary antibodies against GAPDH (Santa Cruz Biotechnology; sc-166574) were used, and loading controls were performed using primary antibodies to beta actin (Millipore; catalog# 04–1116). Proteins were detected by the ECL-Plus chemiluminescence system (Amersham Pharmacia). Immunoreactive signals were captured on Kodak X-omatic AR film and quantified by computer-assisted densitometry. After being washed in phosphate-buffered saline, blots were incubated with Alexa Fluor 680 secondary antibodies (Molecular Probes). Proteins were visualized and quantified using the Odyssey infrared imaging system and software (Li-Cor Bioscience). Optical densities of the bands were determined relative to background levels. Quantification of the level of GAPDH protein was determined relative to actin and compared to the control sample. Data were analyzed using StatView software by using a one-way ANOVA.
Results
Rationale for Using RACE to Test Specificity of siRNAs
RACE is a technique used to obtain the full-length sequence of an mRNA transcript of interest and results in the production of a cDNA copy-produced through reverse transcription followed by PCR amplification of the cDNA copies (Fig. 1). 3′ RACE PCR uses the natural polyA tail at the 3′ end of eukaryotic mRNAs for priming during reverse transcription. The cDNAs are typically generated using an oligo-dT-adaptor primer that complements the polyA stretch and adds a special adaptor sequence to the 5′ end of each cDNA. The cDNA is then subjected to PCR using antisense 3′ RACE primers that are complimentary to the anchored and sense gene-specific primer (GSP). The GSP primer is complimentary to the known region of mRNA.
Fig. 1.
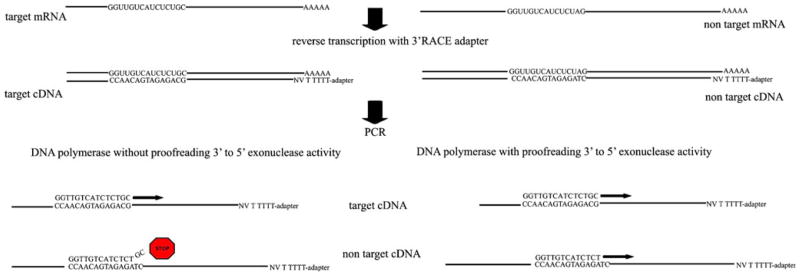
Scheme of 3′ RACE. 3′ RACE uses the natural polyA tail at the 3′ end of eukaryotic mRNAs for priming during reverse transcription. cDNAs will be generated using an oligo-dT-adaptor primer that complements the polyA stretch and adds a special adaptor sequence to the 5′ end of each cDNA. The cDNA is then subjected to PCR using antisense 3′ RACE primers that are complimentary to the anchored and sense GSP. However, Taq DNA polymerase is not able to perform amplification, if GSP has mismatches on the 3′ end. Using DNA polymerase with proofreading 3′ to 5′ exonuclease activity helps to avoid this problem
To confirm that a designed dsRNA is specific to the target mRNA, we generated GSP primers complementary to the region targeted by the dsRNA. These primers were used as GSP primers for 3′ RACE. Here, we used a 265-bp fragment of pcDNA3.1/V5-His plasmid for initial testing of our protocol. If the sequence of the gene of interest is known, as is the case here, one can predict the size of the product of 3′ RACE. When the dsRNA-specific primer is complimentary to other off-target mRNAs, except for one or more mismatches on the 3′ end of primer, Taq DNA polymerase is not able to perform amplification. Thus, it is impossible to predict if there are other off-target mRNAs affected by the dsRNA (Fig. 1). Using AccuPrime Pfx DNA polymerase helps to avoid this problem. This polymerase has proofreading 3′ to 5′ exonuclease activity that provides higher fidelity than Pfu DNA polymerase (Cline et al. 1996). The results of PCRs show that AccuPrime Pfx DNA polymerase is able to produce PCR products even with primers that have three mismatches on the 3′ end (Fig. 2). For the experiment shown in Fig. 2, the original forward primer (T7 forward) had no mismatches on the 3′ end (Fig. 2, lane 1). The other three forward primers had one, two, or three mismatches on the 3′ ends (Fig. 2, lanes 2–4; T7mis1, T7mis2, T7mis3, respectively, Table 1).
Fig. 2.

Agarose gel electrophoresis of PCR products of a 265-bp fragment of pcDNA3.1/V5-His plasmid using the Pfx DNA polymerase. PCR was performed using primers from Table 1. Lane 1 T7 forward primer+reverse primer; lane 2 forward primer with one mismatch on the 3′ end+reverse primer; lane 3 forward primer with two mismatches on the 3′ end+reverse primer; lane 4 forward primer with three mismatches on the 3′ end+reverse primer
Design and Selection of dsRNAs by 3′ RACE
There are certain requirements for use of 3′ RACE primers: temperature of melting must be higher than 60°C, placing more than three G or C residues in the last five positions on the 3′ end must be avoided, and primers with G as the 3′-terminal base should not be used. Several dsRNAs were previously designed against the pond turtle, P. scripta elegans, tGAPDH mRNA (GenBank accession: FJ514828; Sabirzhanov et al. 2007). In the present analysis, six primers specific to these dsRNA sequences and which suit 3′ RACE requirements were synthesized (Table 2). Using these primers specific to tGAPDH dsRNAs, 3′ RACE reactions were performed as shown in Fig. 3. Primers #12 and #17 each produced a single PCR product with the predicted MW for the targeted tGAPDH sequence (Table 2; Fig. 3a, lanes 3 and 4) indicating that dsRNAs #12 and #17 are complementary to and selectively inhibited to expression of tGAPDH. However, primer #6 produced two PCR products, one band with the predicted size of 900 bp and another nonspecific band at 450 bp (Fig. 3a, Lane 1). Primer #8 produced three PCR products. Only one of these bands had the predicted size, 942 bp, and the two others were not specific for tGAPDH (Fig. 3a, Lane 2). These products from 3′ RACE were sequenced. Previously, we found that tGAPDH has an 83%, 82%, and 81% sequence identity to chick, rat, and human GAPDH, respectively. Analysis of the sequences generated by 3′ RACE by Nucleotide BLAST in the Reference mRNA Sequences database showed that primers #12 and #17 produced GAPDH-specific products. Primer #6 resulted in one GAPDH specific and one nonspecific band. No significant similarity to any known genes was found for this nonspecific band by BLAST. Primer #8 resulted in one GAPDH specific band and two nonspecific bands. No significant similarity was found for the 800-bp band; however, the 750-bp band showed 71% similarity to Drosophila mojavensis GI20756 (Dmoj\GI20756) mRNA (NCBI Reference Sequence: XM_002005907.1). These results demonstrate that dsRNAs #6 and #8 against tGAPDH have significant identity to off-target mRNAs and thus would inhibit expression of other genes (Cullen 2006; Jackson et al. 2003).
Fig. 3.

a Agarose gel electrophoresis of products of 3′ RACE with primers specific to tGAPDH dsRNAs. 3′ RACE was performed with primers (Table 2) specific to tGAPDH dsRNA #6 (lane 1), #8 (lane 2), #12 (lane 3), and #17 (lane 4). b Agarose gel electrophoresis of products of 3′ RACE with primers specific to scrambled dsRNA #12 (lane 1) and #17 (lane 2)
Most likely, dsRNAs #6 and #8 will knock down the target gene (GAPDH). However, there is a possibility that these dsRNAs will also induce the inhibition of non-target gene.
Scrambled dsRNAs and primers specific for dsRNAs #12 and #17 were designed and synthesized by Integrated DNA Technologies (Table 2). Target specificity of these scrambled dsRNAs was also examined by 3′ RACE. No PCR products were observed as expected (Fig. 3b).
Suppression of tGAPDH Protein by 3′ RACE-Selected dsRNAs
Turtle brain primary cell cultures were transfected with different concentrations of anti-tGAPDH dsRNAs. Western blot analysis showed that treatment of cells with anti-tGAPDH dsRNA #12 and #17 significantly reduced tGAPDH protein levels compared to controls that were treated with transfection agent alone (Figs. 4a and 5a). Treatment with low concentrations of tGAPDH dsRNA resulted in a 30% reduction in GAPDH protein at only 5 nM for dsRNA #12 (P<0.001) and to about 50% or less for both dsRNA #12 and #17 at 20 nM (P<0.0001). Turtle brain primary cell cultures were also transfected by different concentrations of scrambled anti-tGAPDH dsRNA #12 and #17. Treatment of cells had no significant effect on expression of tGAPDH protein levels (Figs. 4b, 5b; #12, P=0.97; #17, P=0.46).
Fig. 4.

Western blot analysis of the level of GAPDH and actin after treatment by different concentration of a tGAPDH dsRNAs #12 and #17 or b by scrambled dsRNAs #12 and #17
Fig. 5.

Quantitative analysis of the level of GAPDH and actin proteins after treatment by a tGAPDH dsRNAs #12 and #17 or b by scrambled dsRNAs #12 and #17 (n=3/group). Error bars indicate standard deviation. Asterisk indicates significant differences from the controls which were treated with tranfection agent alone
Treatment with tGAPDH dsRNAs Induces siRNA-Directed Cleavage of GAPDH mRNA
To show that the reduction in the level of tGAPDH protein was due to siRNA-directed cleavage of tGAPDH mRNA, we characterized specific mRNA cleavage products using a modified 5′ RACE method. This method was successfully utilized to demonstrate microRNA (miRNA)-directed mRNA cleavage in plants and mouse (Soutschek et al. 2004; Llave et al. 2002; Yekta et al. 2004). Total RNA from turtle brain primary cell cultures transfected with 100 nM anti-tGAPDH dsRNA #12, #17, or from control samples, was isolated followed by the 5′ RACE procedure. PCR products with the predicted size were observed for samples transfected by anti-tGAPDH dsRNA #12 and #17 (Fig. 6). The identity of the PCR products was confirmed by sequencing. Analysis of sequences showed that cleavage of tGAPDH mRNA occurred after position 902 and 1,083 bp, ten and 14 nucleotides downstream of the 5′ end of the tGAPDH dsRNA #12 and #17 antisense strand, respectively. The amount of PCR product from samples treated by anti-tGAPDH dsRNA #17 was considerably lower than that from samples treated by anti-tGAPDH dsRNA #12. This observation corresponds with the Western blot data. No PCR products were detected from control samples. These data demonstrate that treatment of turtle brain primary cell cultures by tGAPDH dsRNA #12 and #17 results in reduced levels of tGAPDH protein by “predicted” RNAi mechanism of action.
Fig. 6.

Agarose gel electrophoresis of 5′ RACE products. 5′ RACE was performed with a control sample (lane 1) and samples transfected by 100 nM anti-tGAPDH dsRNA #12 (lane 2) or #17 (lane 3)
Discussion
Confirming the ability to selectively knockdown a target gene without interfering with the expression or function of other genes or proteins is an essential component of experiments using siRNA-mediated gene silencing. There are two common ways to confirm target specificity of designed dsRNAs: (1) dsRNA specificity can be predicted by local alignment algorithms such as BLAST (Altschul et al. 1997) or the Smith–Waterman local alignment algorithm (Smith and Waterman 1981), and (2) dsRNA target specificity can be characterized by application of microarray profiling (Jackson et al. 2003; Semizarov et al. 2003; Birmingham et al. 2006; Anderson et al. 2008). However, both of these approaches are not suitable for species in which there is limited genome sequence data. It has been also shown that detection of specific RNA cleavage products generated by RISC-mediated hydrolysis of target mRNA using the 5′ RACE PCR method is a good indicator for confirming the effectiveness of dsRNAs against a target mRNA (Soutschek et al. 2004; Llave et al. 2002; Yekta et al. 2004). This method, however, is not able to predict whether the dsRNA targets only one mRNA sequence.
Here, we report development of a method that allows for screening of dsRNA sequences to determine whether they have significant sequence identity to non-target mRNAs using 3′ RACE (Cullen 2006; Jackson et al. 2003). Since the RACE strategy has certain requirements for primers (high Tm and C/G content), it is impossible to guarantee the complete elimination of all dsRNAs that may cross-react with non-target mRNAs because some of the designed dsRNAs, and primers will not fit these requirements. However, this technique can be employed as a useful initial step for dsRNA screening to eliminate dsRNAs that are likely to produce off-target effects.
Microarray profiling is the most direct method for demonstrating that a designed dsRNA selectively inhibits expression of the target gene and does not produce off-target effects. However, even microarray profiling will not confirm that the designed dsRNA affects only the target gene and not products of alternative splicing of the same gene of interest. Regardless, this approach is not suitable for model systems using species in which there is limited genome sequence data. The strategy presented in this study is also applicable for screening designed primers and probes for species that have limited available sequence information. Moreover, this method is also suitable for confirmation that the designed dsRNA affects only certain mRNAs but not products of alternative splicing. This is especially important because 40–80% of mammalian pre-mRNAs are alternatively spliced, and different products of alternative splicing may have different functions (Modrek and Lee 2002; Johnson et al. 2003).
Acknowledgments
We thank Drs. Zhaoqing Zheng for contributing the specimens of turtle brain and Pat Ronan for helpful comments on the manuscript. This study is supported by NIH grants NS051187 and P20 RR015567 which is designated as a Center of Biomedical Research Excellence (COBRE) to J.K.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EM, Birmingham A, Baskerville S, Reynolds A, Maksimova E, Leake D, Fedorov Y, Karpilow J, Khvorova A. Experimental validation of the importance of seed complement frequency to siRNA specificity. RNA. 2008;14:853–861. doi: 10.1261/rna.704708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham A, Anderson EM, Reynolds A, Ilsley-Tyree D, Leake D, Fedorov Y, Baskerville S, Maksimova E, Robinson K, Karpilow J, Marshall WS, Khvorova A. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat Methods. 2006;3:199–204. doi: 10.1038/nmeth854. [DOI] [PubMed] [Google Scholar]
- Cline J, Braman JC, Hogrefe HH. PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res. 1996;24:3546–3551. doi: 10.1093/nar/24.18.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Enhancing and confirming the specificity of RNAi experiments. Nat Meth. 2006;3:677–681. doi: 10.1038/nmeth913. [DOI] [PubMed] [Google Scholar]
- Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- Keifer J, Zheng Z. AMPA receptor trafficking and learning. Eur J Neurosci. 2010;32:269–277. doi: 10.1111/j.1460-9568.2010.07339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keifer J, Sabirzhanov BE, Zheng Z, Li W, Clark TG. Cleavage of proBDNF to BDNF by a tolloid-like metalloproteinase is required for acquisition of in vitro eyeblink classical conditioning. J Neurosci. 2009;29:14956–14964. doi: 10.1523/JNEUROSCI.3649-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvorova A, Reynolds A, Jayasena SD. Functional siRNAs and miRNAs exhibit strand bias. Cell. 2003;115:209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- Llave C, Xie Z, Kasschau KD, Carrington JC. Cleavage of Scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002;20:2053–2056. doi: 10.1126/science.1076311. [DOI] [PubMed] [Google Scholar]
- Lu Y, Nerurkar VR, Aguirre AA, Work TM, Balazs GH, Yanagihara R. Establishment and characterization of 13 cell lines from a green turtle (Chelonia mydas) with fibropapillomas. In Vitro Cell Dev Biol Anim. 1999;35:389–393. doi: 10.1007/s11626-999-0113-6. [DOI] [PubMed] [Google Scholar]
- Meister G, Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- Modrek B, Lee C. A genomic view of alternative splicing. Nat Genet. 2002;30:13–19. doi: 10.1038/ng0102-13. [DOI] [PubMed] [Google Scholar]
- Sabirzhanov BE, Keifer J, Clark TG. Characterization of a novel reptilian tolloid-like gene in the pond turtlePseudemys scripta elegans. Brain Res. 2007;1154:22–30. doi: 10.1016/j.brainres.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DS, Hutvágner G, Haley B, Zamore PD. Evidence that siRNAs function as guides, not primers, in the Drosophila and human RNAi pathways. Mol Cell. 2002;10:537–548. doi: 10.1016/s1097-2765(02)00651-2. [DOI] [PubMed] [Google Scholar]
- Semizarov D, Frost L, Sarthy A, Kroeger P, Halbert DN, Fesik SW. Specificity of short interfering RNA determined through gene expression signatures. Proc Natl Acad Sci USA. 2003;100:6347–6352. doi: 10.1073/pnas.1131959100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, John M, Kesavan V, Lavine G, Pandey RK, Racie T, Rajeev KG, Röhl I, Toudjarska I, Wang G, Wuschko S, Bumcrot D, Koteliansky V, Limmer S, Manoharan M, Vornlocher HP. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- Yekta S, Shih IH, Bartel DP. MicroRNA-directed cleavage of HOXB8 mRNA. Science. 2004;304:594–596. doi: 10.1126/science.1097434. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Keifer J. PKA has a critical role in synaptic delivery of GluR1- and GluR4-containing AMPARs during initial stages of acquisition of in vitro classical conditioning. J Neurophysiol. 2009;101:2539–2549. doi: 10.1152/jn.91282.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]