

Abstract
The field of glycomics has recently advanced in response to the urgent need for structural characterization and quantification of complex carbohydrates in biologically and medically important applications. The recent success of analytical glycobiology at high sensitivity reflects numerous advances in biomolecular mass spectrometry and its instrumentation, capillary and microchip separation techniques, and microchemical manipulations of carbohydrate reactivity. The multimethodological approach appears to be necessary to gain an in-depth understanding of very complex glycomes in different biological systems.
Keywords: biomolecular mass spectrometry, carbohydrates, permethylation, porous graphitic carbon, sialic acids, capillary electrophoresis
1. INTRODUCTION
The glycomes of various biological systems remain underexplored in terms of important cellular processes and biological recognition. Biosynthesis of structurally complex glycans (oligosaccharides) occurs through a non-template-driven process but nonetheless yields selective structures. However, these structural entities cannot be easily decoded through knowledge of the usual flow of biological information; in other words, glycan biosynthesis is not “hardwired to DNA” (1). Instead, our current knowledge about glycoscience has been accumulated largely through various observations and specialized chemical measurements conducted on complex biological systems. During the past decade, in particular, the field of analytical glycobiology has advanced significantly because of developments and applications of novel methods in separation science and mass spectrometry (MS). Of the many “-omics” technologies, glycomics and glycoproteomics share some methodological approaches with other fields in terms of instrumentation and how they deal with complex analytical data. However, both areas are characterized by chemically and instrumentally unique approaches to carbohydrate research.
Complex carbohydrates are the largest group of biomolecules on Earth. They encompass the structurally diverse subgroups of polysaccharides, proteoglycans, glycolipids, and glycoproteins. Highly sensitive measurements of the glycan constituents of glycoproteins are the primary focus of this review. Such a glycocentric approach is justified due to the increasing importance of oligosaccharide quantitative profiling in biological studies and health-related applications. The growing interest of the scientific community in the structural aspects of carbohydrate constituents arises from their direct participation in biologically important processes, such as their assistance in protein folding, solubility of proteins, and regulation of the pro- or anti-inflammatory abilities of immunoglobulins according to their level of sialylation. The glycomic approach has gained popularity within the biomedical community as the connections between protein glycosylation and practically all major diseases have become recognized. These conditions include inflammatory diseases, various cancers, diabetes, and cardiovascular diseases. Importantly, glycomic measurements can be made both rapidly and at high sensitivity. Such measurements have contributed to biomarker discovery; new tools and incentives have prompted further analytical developments in diagnostic and prognostic measurements.
Traditionally, glycosylation was considered the domain of eukaryotic cells. This view can no longer be supported, given that many studies have implicated glycans (including N-linked structures) in bacteria. Glycosylated structures seem to play important roles in numerous host–pathogen interactions and pathogenicity.
Non-template-directed biosynthesis of glycans by specific glycosyltransferases and exoglycosidases can generate very complex arrays of structurally diverse oligosaccharides. The unique tendency of glycans toward branching, different linkages, and isomerism generally presents formidable analytical challenges. Although certain glycobiological experiments can be difficult, if not intimidating, to perform, substantial advances in glycan separations and mass-spectral measurements have recently been made. Our review summarizes these recent methodological developments and technical innovations toward structural characterization of glycans and their quantification at high sensitivity. We emphasize MS, including its tandem modes and different ionization techniques, as well as new developments in liquid chromatography (LC), capillary electrophoresis (CE), and microchip separations. Glycan array technologies are not covered in this review.
2. CARBOHYDRATE STRUCTURAL ASPECTS AND NOMENCLATURE
Carbohydrates are most frequently attached to the side chains of three amino acids. N-linked glycans are conjugated to the nitrogen atom of an asparagine’s amide, and O-glycans are linked through the hydroxyl groups of serine and threonine residues. N- and O-linked glycans differ in other significant ways. N-linked structures reside within an amino-acid sequence of NXS/T, where N is asparagine, X is any amino acid except for proline, and S/T is serine or threonine. This motif is frequently extended in bacteria to D/EXNXS/T (2), where D/E is aspartic or glutamic acid. N-linked glycans have seldom been observed outside this sequence; examples include recombinant bovine trypsin expressed in maize (3) and recombinant immunoglobulins expressed in CHO cells (4). Conversely, O-linked glycans are not associated with a particular amino-acid sequence. However, in many mucin-like proteins, certain regions of their amino-acid sequences tend to be enriched with serine and threonine residues, and these areas are often heavily O-glycosylated. Several other amino-acid residues can be glycosylated (5), although not nearly as frequently. Among the most prominent of these unusual residues is tryptophan, which may have a mannose (Man) attached at the C2 position of its side chain. This type of glycosylation, referred to as C-mannosylation, has been identified on several proteins, including ribonuclease A (6) and thrombospondin (7). More recently, cysteine was found to be modified by a glucose unit (8) in the glycopeptide sublancin, and this carbohydrate is vital to its antimicrobial properties.
N- and O-glycans are structurally very different and follow different biosynthetic pathways. N-linked glycans have a common core consisting of GlcNAc2Man3 (where GlcNAc stands for N-acetylglucosamine). The final N-linked biosynthetic product is characterized by various monosaccharides extending from the α1,3- and α1,6-branched core Man units and includes Man; galactose (Gal); GlcNAc; fucose (Fuc); and sialic acids, which are usually N-acetylneuraminic acid (NANA) or N-glycolylneuraminic acid (NGNA) in mammalian systems (in plants, xylose monomers are common). On the basis of their monosaccharide composition and unique structural features, N-linked glycans can be divided into three subgroups (Figure 1). The first subgroup, high-Man-type glycans, has only Man units attached to the core, whereas the second subgroup, the complex oligosaccharides, has GlcNAc monosaccharides attached to the core Man units and may be extended with Gal units and lactosamine disaccharides (GlcNAc-Gal disaccharides) and terminated with sialic acids. Members of this second group of carbohydrates often contain Fuc monosaccharides attached to a branch and/or the reducing-end GlcNAc unit. A further variation of complex glycans are the so-called bisecting structures, where a β1,4-linked GlcNAc unit is attached to the β1,4-linked Man of the core. The third subgroup represents a combination of the complex and high-Man classes; its members are known as hybrid glycans. In contrast, O-linked carbohydrates do not have a universal core composition. Essentially, eight unique cores exist (Figure 2), although there are several exceptions (5), including (a) O-linked Fuc found in epidermal growth factor–like repeats and thrombospondin (7) and (b) O-linked Man monosaccharides found in the human and rabbit forms of the protein α-dystroglycan (9, 10). A single O-linked GlcNAc has been found in many cytosolic and nuclear proteins (11) and may be critical for cancer cell growth and proliferation (12). Similarly to the N-linked glycans, the O-glycan cores may be branched and extended with the same monosaccharide building blocks.
Figure 1.
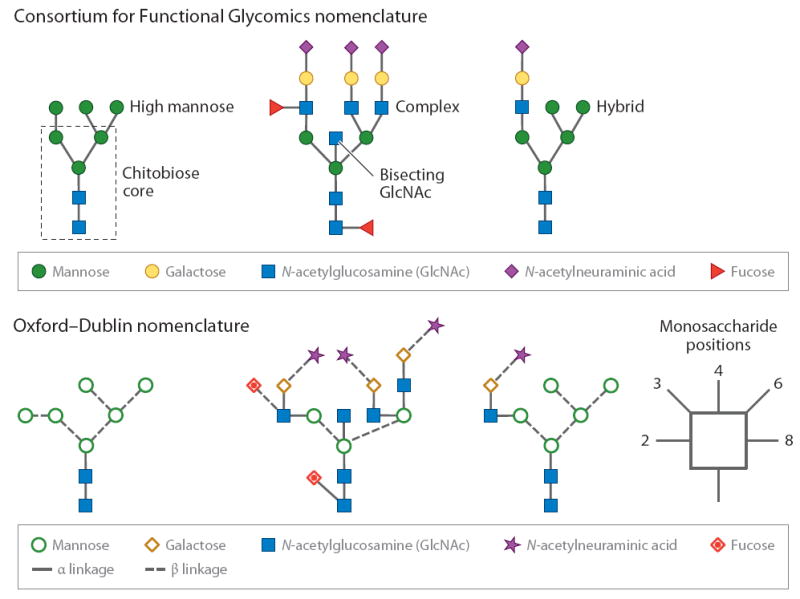
High-mannose-type glycans, complex oligosaccharides, and hybrid structures represented by the Consortium for Functional Glycomics system and the Oxford–Dublin style.
Figure 2.
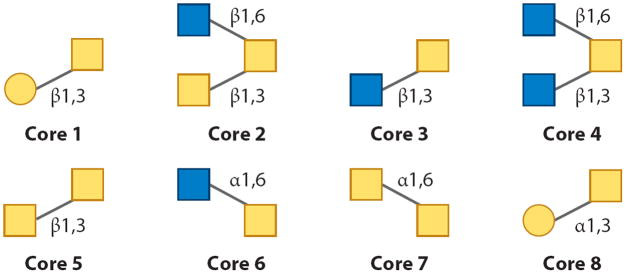
The eight common O-linked glycan cores.
Beyond the myriad of possible structures that can be attached to a protein, the overall complexity of a glycoprotein can be significantly increased by its microheterogeneity. For a given glycoprotein, a single site is rarely occupied by a single carbohydrate. Rather, multiple glycans usually populate a glycosylation site, as exemplified by bovine ribonuclease B. This small glycoprotein has only a single site of glycosylation, yet it is modified by five different high-Man glycans ranging from Man5 to Man9, some of which have different isomeric forms. Further diversity at a potential site of glycosylation is based on the site’s level of occupancy; some sites may remain vacant, others may be only partially occupied, and some may be reversibly modified. It is challenging to perform a comprehensive analytical characterization of a glycoprotein when a thorough structural elucidation of the carbohydrates is required, along with determining the sites of glycosylation, their microheterogeneities, and their levels of occupancy.
Given the broad array of possible carbohydrates, it is necessary to accurately and simply depict their structures. To this end, two main notation schemes (13) are used in the current literature: the Consortium for Functional Glycomics (CFG) nomenclature scheme (http://www.functionalglycomics.org/static/consortium/CFGnomenclature.pdf) and the Oxford–Dublin system (http://www.bioch.ox.ac.uk/glycob). Both recommend the use of different geometric shapes; the CFG system uses the same shape for different monosaccharide isomers. For example, hexoses are represented by circles and the different isomeric structures are indicated by different colors; green represents Man and yellow is used for Gal. Similarly, derivatives of a monosaccharide are represented by the same color; for instance, both glucose and GlcNAc are blue. Because this color scheme may introduce confusion in black-and-white images, the Oxford–Dublin system uses different shapes for each monosaccharide. Linkages are also shown, and the anomeric configuration, either α or β, is indicated by solid or dashed lines, respectively. In the remainder of this review, we refer to Figure 1 for examples using both systems.
3. GLYCAN RELEASE PROCEDURES
Because glycomic studies are focused only on carbohydrates, the release of glycans from the protein backbone is an essential first step. Traditionally, an effective release of glycans, in terms of being quantitative and reproducible, has been an ongoing issue in glycobiology, and these aspects are even more important in current biomedical glycomic studies. Historically, investigators have used chemical release methods, such as hydrazinolysis or classical β-elimination in an alkaline medium, and many technical issues complicating the release procedures have been overcome through enzymatic protocols using peptide-N-glycosidases F and A (PNGase F and PNGase A) for N-linked glycans. Improved microscale chemical release techniques (14-16) for threonine/serine-linked oligosaccharides are still practiced because there is no glycosidase that cleaves all O-linked structures.
3.1. Enzymatic Release of N-Linked Structures
The most straightforward and reproducible way to release intact glycans is with PNGase F (17) and PNGase A enzymes. PNGase F is generally effective at releasing N-linked structures; however, in certain living organisms, including plants and insects, many proteins contain α1,3-linked Fuc units that inhibit the activity of this enzyme. For such glycoproteins, PNGase A may be used (18). Both of these enzymes catalyze the cleavage of an asparagine’s side-chain amide bond, convert this residue to an aspartic acid through a deamidation mechanism, and thereby add approximately 0.98 Da to the glycopeptide’s expected mass. Although this mass shift may be used to indicate a site of glycosylation, additional confidence may be gained by performing this reaction in heavy water (19), adding 2.9882 Da to the expected mass. This procedure is still used in large-scale studies (20). However, data from such studies need to be interpreted with caution because natural or experimentally induced deamidation reactions may occur, particularly if an asparagine is followed by a glycine (21).
In an effort to achieve the highest possible digestion efficiency, enzymatic release procedures are frequently performed for up to 24 h. However, for large-scale clinical or industrial studies that require the analysis of many samples, the incubation period must be significantly reduced. One recently reported approach employs ultrahigh-pressure cycling, which subjects proteins to pressures of up to 30 kpsi (22). Even under these extreme conditions, PNGase F remains active, although many glycoproteins become sufficiently denatured to be deglycosylated in as little as 20 min (22). A second method utilizes microwave radiation to assist the enzymatic cleavage; complete removal of the glycans from monoclonal antibody drugs occurs in as little as 10 min, whereas other glycoprotein standards required up to 1 h (23, 24).
Because each commonly used glycanase has an inherent level of specificity, an enzymatic approach that can be applied in any situation is highly desirable and may be possible through the use of Pronase, a mixture of proteolytic enzymes derived from Streptomyces griseus that ultimately digests proteins to yield individual amino acids. The complete digestion of glycoproteins produces single asparagine residues linked to their carbohydrates. The ability of Pronase to free glycans that are resistant to other enzymatic digestion procedures has been demonstrated for N-linked bacterial glycans (25), which are often attached through a bacillosamine monosaccharide. Additionally, Pronase has been used as an alternative to PNGase F enzymes (26) to analyze more common types of N-linked glycans. However, such a procedure requires up to 48 h for complete digestion. To reduce the incubation time to only a few minutes, one may immobilize Pronase on solid supports (27), allowing digestion to be performed online for direct LC-MS analyses.
3.2. O-Glycan Release Procedures
In contrast to the situation with N-linked glycans, a glycanase with a broad specificity toward O-linked structures is not currently available. However, unsubstituted core 1 structures may be freed using O-glycanase (endo-α-N-acetylgalactosaminidase) (28). Therefore, a comprehensive release of O-glycans is typically accomplished through chemical methods; alkaline-based β-elimination has long been one of the most widely used approaches (29). In the classical technique (29), glycans are treated with a mild solution of sodium hydroxide (0.05–0.1 M), and high concentrations of sodium borohydride (1 M) are used to reduce the released glycans as their alditol forms. This process prevents the so-called peeling reactions that damage the structures at their reducing end. Because of the high salt concentrations needed, oligosaccharides released through this procedure require extensive purification before measurement. Consequently, the overall sensitivity is limited, and this approach is only moderately successful for glycans present at trace levels.
Milder release conditions using ammonia-based media have been explored to facilitate effective β-elimination (30). A further adaptation of this approach employs ammonia-borane complex as the reducing agent (14, 15), and its excess can be removed with an appropriate acid. The resulting borane salts are then converted into methyl esters with methanol, which can then be removed under vacuum (15). This procedure eliminated the need for solid-phase extraction and enhanced the overall sensitivity. Similarly to the other β-elimination release methods, the ammonia-borane protocol produces glycan alditols, which are amenable to MS measurements but not optical detection. For use with LC or CE coupled with laser-induced fluorescence (LIF), this procedure has been modified (14) to regenerate a free reducing end.
Other gentle release conditions have been developed through the use of alternative organic bases; one of the most recent procedures employed dimethylamine coupled with microwave radiation (31). This method resulted in a release efficiency of more than 95% in slightly more than 1 h at 70°C and was more efficient than the classical procedure using sodium hydroxide. Additionally, because β-elimination generates dehydroserine and dehydrothreonine, these modified amino acids spontaneously reacted with the dimethylamine and could be used to indicate the site of glycosylation with high confidence, although the efficiency of this reaction was slightly lower than is usually analytically desirable.
As an alternative to the chemical methods, Pronase has been applied to O-linked glycomic studies. Following digestion and subsequent purification, O-glycans linked to their serine or threonine residues were permethylated, and the basic conditions employed induced β-elimination (32). Because the N-linked oligosaccharides remained associated with their asparagines, which underwent their own unique β-elimination reaction (25), this procedure allowed both O- and N-linked glycans to be easily distinguished in the same spectra. A direct comparison between the MS signals showed that samples prepared by the enzymatic/chemical method were typically 10–20 times more intense than those for the samples prepared by the other O-glycan release techniques (Figure 3). When the enzymatic/chemical approach was applied to a 1-μg aliquot of bile salt–stimulated lipase, a large, heavily O-glycosylated protein isolated from human breast milk, 75 total oligosaccharides were identified, of which 40 were unique to this method (32).
Figure 3.

Comparison between different methods of releasing O-linked glycans associated with bovine fetuin. For all carbohydrates, an enzymatic/chemical procedure resulted in the most sensitive matrix-assisted laser desorption/ionization mass spectrometry measurements. Modified from Reference 32.
4. DERIVATIZATIONS FOR IMPROVED MASS-SPECTRAL PERFORMANCE
Analytically, carbohydrates are difficult to measure. Because of the absence of an appropriate chromophore/fluorophore, sensitive UV- or fluorescence-based measurements following a liquid-based separation are challenging to perform. Therefore, for sensitive detection, the carbohydrates must be modified to include an appropriate label. Likewise, carbohydrates are not ideal analytes for MS-based measurements, because their inherent hydrophilicity results in an inefficient desolvation during electrospray ionization (ESI), resulting in suppressed signals. Further complicating MS measurements and hindering the overall measurement sensitivity are the commonly encountered in-source and postsource decay reactions that are particularly prevalent with sialylated or fucosylated oligosaccharides. To assist the MS analyses of these molecules, several structural modifications (derivatizations), many of which were summarized in a recent extensive review (33), can be made to the glycan structure at different sites, including their hydroxyl groups, reducing ends, and sialic acids.
4.1. Permethylation
Permethylation of oligosaccharides has become one of the most popular derivatizations. This reaction modifies a carbohydrate at three locations: (a) The hydroxyl groups become methoxide moieties; (b) the carboxylate of sialic-acid residues becomes esterified; and (c) a methyl group is added to the nitrogen atom of the N-acetyl groups of GlcNAc, GalNAc, and sialic acid. Permethylation offers several advantages, including (a) an improved MS sensitivity of 10 to 20 times that for native glycans; (b) the ability to render acidic structures neutral, permitting the entire glycomic profile to be recorded in the MS positive-ion mode; (c) the ability to enhance cross-ring fragmentation mechanisms during tandem MS analyses, enabling more definitive structural characterization by revealing linkage/branching information; and (d) the production of analytes whose hydrophobicities are sufficient to allow them to be separated by reversed-phase LC, if needed.
The modern approach to permethylation, first developed by Hakomori (34) in the early 1960s, employed dimethylsulfoxide (DMSO) and methyl iodide. Updates to the basic procedure allow the reaction to be accomplished in a slurry of sodium hydroxide (35, 36). However, during the recovery of the permethylated analytes, the analytes are subjected to very basic pH conditions and peeling reactions may occur. These side reactions limit the overall sensitivity of the method, and many important trace-level glycans may not be observed. To improve the overall sensitivity, a solid-phase approach to permethylation that uses discrete sodium hydroxide beads loaded into reactors has been introduced (37, 38). In this approach, the excess sodium hydroxide is easily removed from the carbohydrate-containing solution prior to a liquid/liquid or solid-phase extraction method. Thus, the pH of the solvent system is more controlled, and the peeling reactions seem to be minimized. Although the overall improvements in the subsequent MS recordings were first observed when this reaction was performed in capillaries (37), investigators later developed a higher-throughput method using spin columns with comparable sensitivities (38). The sensitivity of the solid-phase approach may be further enhanced by performing the extraction from the reaction media in an online fashion; doing so maximizes sample transfer and minimizes losses (39).
Although glycans are readily soluble in DMSO and permethylation occurs quickly in this solvent, several problems may arise during the reaction. For chromatographic purposes, it is desirable to analyze the glycans in their alditol states to prevent resolution of the α and β anomeric configurations, which typically produces two peaks for a single analyte during chromatography. Through an interaction with DMSO under basic conditions, the closed-ring structure may be regenerated (36), complicating the mass spectra and compromising detection limits due to the multiple ions being detected for a single analyte. Additionally, a series of +30-Da artifacts can be observed due to the production of iodomethyl methyl ether (40) through a reaction between methyl iodide and DMSO. The ether may react with hydroxyl groups and, again, complicates the overall spectral interpretation. However, both of these reactions are minimized if the permethylation reaction is conducted in an alternate medium, such as N,N-dimethylacetamide (36) or N,N-dimethylformamide (41).
Permethylation reactions have also been successfully applied to phosphorylated and sulfated oligosaccharides. Because both of these moieties are stable throughout the permethylation procedure, the phosphate group becomes singly or doubly esterified (42, 43). Conversely, sulfate groups are not affected by the permethylation procedure and retain their negative charge (44). Therefore, to detect these glycans in the positive-ion mode, a double-permethylation procedure has been developed (44). In this approach, the sulfated glycans were permethylated and the sulfate groups were then removed through a mild treatment with acidified methanol. The samples were again permethylated using deuterated methyl iodide to mark the site of sulfation (44). Additionally, because sialic acids become neutral structures following permethylation, sulfated glycans can be fractionated on the basis of their degree of sulfation by strong anion-exchange chromatography (45). Following desalting, the sulfate group can be chemically removed and the site of sulfation can be indicated using deuterated methyl iodide.
4.2. Modification of Sialic Acids
Sialic acids are an important group of nine-carbon carbohydrates that possess a carboxylic acid group and are usually attached as either α2,3 or α2,6. Sialyl substitutions are involved in many important biological functions, including cell–cell recognition and host–pathogen interactions. Increased levels of α2,6-linked sialylation cause decreased cell–cell interactions, increased adhesion to collagens, enhanced motility, and increased invasiveness of tumor cells. However, sialylated oligosaccharides are difficult to study, particularly under MALDI time-of-flight (TOF) MS conditions, in which the loss of sialic-acid residues is very common (46). To improve the stability of these residues, several different derivatizations have been developed to modify the carboxylate group so that the acidic proton which induces bond cleavage becomes eliminated. An early method applied a simple esterification using methyl iodide performed in DMSO (47). Following this reaction, the sialylated structures were detected as intact species. Sialic-acid esterification offered several secondary advantages, including the ability to measure both acidic and neutral structures simultaneously in the positive-ion mode. Additionally, esterified analytes were not present as a series of multiple metal-ion adducts, which are often associated with a single acidic analyte. To further improve the sensitivity of the subsequent MALDI measurements, investigators have applied the esterification reaction to the products of Pronase digestions of N-linked glycoproteins (48). Not only did the ensuing reaction esterify the carboxylate group of the sialic acids, but a quaternary ammonium moiety was introduced at the N terminus of the asparagine residues and increased the sensitivity of the MS measurements 10-fold in comparison to the native analytes.
More unusual derivatizations of the carboxylate are possible with an appropriate activating agent to make the carboxylate group on the sialic acids more susceptible to a nucleophilic attack. A particularly useful reagent is 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM) (49), which has been used in an esterification reaction with methanol (50). Interestingly, α2,3-linked sialic acids spontaneously produced lactones (internal cyclic esters), preventing their modification, whereas α2,6-linked sialic acids were activated and ultimately esterified. This differential labeling caused an easily distinguishable 32-Da mass difference between α2,3- and α2,6-linked structures. In another reaction employing DMT-MM, sialic acids were reacted with ammonium ions, causing amidation of the carboxylate (49). The products of this reaction were more suitable analytes under MALDI conditions in comparison to their native analogs; loss of the sialic acids was minimized. Tandem MS analyses of the amidated structures resulted in a more complete set of fragment ions, and due to the lack of salt adducts, the resulting spectra were less complicated. The amidation reaction has been further modified to allow α2,3- and α2,6-linked sialic acids to be distinguished (51). This modification was more suitable for a subsequent permethylation than was the esterification reaction because the lactones are not stable under the basic conditions needed for permethylation and the α2,3-linked sialic acids become esterified. This approach enabled MALDI-based studies of alterations to the linkage ratios of glycans derived from blood serum glycoproteins associated with different states of health (Figure 4). A similar experiment demonstrated the differences in sialyl linkage ratios associated with former smokers with lung cancer, former smokers without the disease, and a control population with no smoking history (52).
Figure 4.

Matrix-assisted laser desorption/ionization mass spectrometry recordings of an amidated and permethylated glycan, showing the differences in sialic-acid linkages in (a) a control individual and (b) a woman diagnosed with late-stage breast cancer. Diamonds pointing to the left indicate α2,3-linked sialic acids, and those pointing to the right indicate α2,6-linked sialic acids. Modified from Reference 51.
In some applications, a nonspecific amidation (one that derivatizes both linkages) may be desirable. Nonspecific amidation can be achieved by reacting the carboxylate groups with acetohydrazide (53). This reaction increased the sensitivity sixfold versus the underivatized sugars; the limit of detection was 1 fmol. However, under tandem MS conditions, the sialic acids appeared to be especially labile, as indicated by an intense ion corresponding to their loss. This loss was attributed to the slightly acidic proton of the amide. Similarly, nonspecific methylami-dation (54) may be accomplished by activating the carboxylate group with the reagent PyAOP [(7-azabenzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate], an activator that is useful in situations wherein steric hindrance effects may prevent complete reaction. Methylamidation also stabilized the sialic-acid units, and no decomposition products were detected during a positive-mode MALDI analysis.
4.3. Modifications at the Reducing End
Another common location for derivatization of a reducing carbohydrate is the reducing end. This part of a glycan molecule is in a dynamic equilibrium between a closed-ring structure and an aldehyde; the latter can easily be modified through reductive amination (55). Consequently, the aldehyde form of a carbohydrate, whose level is enhanced by the acidic buffer conditions, can be conjugated with numerous amines. The resulting Schiff base is subsequently reduced by sodium cyanoborohydride, although 2-picoline-borane complex (56) and sodium triacetoxyborohydride (57) have been proposed to be equally effective and less toxic alternative reagents. Reductive amination has been widely used to introduce a chromophore, such as 2-aminobenzamide (2-AB) or 2-aminobenzoic acid (2-AA), or a fluorophore, such as 8-aminopyrene-1,3,6-trisulfonate (APTS), into the carbohydrate structure. Additionally, such reactions may also improve MS performance by improving the measurement sensitivity and, in some cases, enhancing the tandem MS fragmentation patterns.
Interestingly, several tagging groups previously used to aid UV-based LC detection also enhance MS signal intensities (58); one of the best improvements is associated with 4-aminobenzoic acid 2-(diethylamino)-ethyl ester (ABDEAE). The conjugation of ABDEAE with maltohexaose resulted in an up-to-5,000-fold increase in the measurement sensitivity versus the native carbohydrate, and the limit of detection for the ABDEAE-tagged sugar, obtained via ESI, was 10 fmol (58). The improved ion yield was attributed to the basic functional group on the derivatizing reagent that provided a location for proton attachment. When N-linked glycans were derivatized with this molecule, a diminished but still significant (approximately 50-fold) signal enhancement was observed.
Several other UV or fluorescent tags, such as 2-AB and 2-AA, have been used in both MALDI and ESI studies. Although there was a modest gain in measurement sensitivity [approximately threefold for 2-AB-labeled ovalbumin glycans in comparison to native structures (59)], enhancements in the MALDI tandem MS performance resulted in more diagnostic ions (59), and the levels of cross-ring fragmentation across the innermost GlcNAc unit were increased (60). The improved cross-ring fragmentation mechanisms allowed α1,3-linked core Fuc units to be distinguished from α1,6-linked monosaccharides in the honeybee protein PlA2. The sensitivity may be further improved through the use of the 2-AA label for negative-ion mode studies. Following their derivatization, carbohydrates modified with this label had limits of detection that were approximately fivefold better than those of 2-AB-labeled glycans (61). However, there was an overall loss in tandem MS fragmentation performance, which was probably due to the lack of a mobile proton.
Although many reductive amination protocols are performed in solutions, a modification with 3-aminoquinoline (62) has been reported to occur directly on a MALDI plate. After several experimental parameters, including the organic solvent, pH, inorganic acid, and the concentration of reagent, were optimized, complete derivatization occurred within approximately 1 h. Interestingly, 3-aminoquinoline also functions as an effective MALDI matrix for carbohydrates in both the positive- and negative-ion modes of detection; as little as 1 fmol of a derivatized carbohydrate was detected as a nitrate adduct in the negative-ion mode.
Alternatively, hydrazine-based reactions have also been used to derivatize a carbohydrate’s reducing end; phenylhydrazine improves the sensitivity of ESI-based measurements (63). Among the advantages of this time-honored chemistry, hydrazine labeling reactions are straightforward and require no additional salts. Therefore, the sample-purification steps are simple, and the measurement sensitivities can be further enhanced. Additionally, the loss of sialic-acid groups is minimal. Such tags have also been used to study the effects of their hydrophobicity or hydrophilicity on the carbohydrate ion yield (64, 65). In results that perhaps appear contrary to what may seem intuitive, the best signal enhancements were observed for the more hydrophobic labels, rather than those with a positive charge (65). This finding was attributed to the better overall desolvation characteristics of the hydrophobized glycans.
4.4. Quantitation of Oligosaccharides Through Stable Isotope Labeling
On the basis of the assumption that many human diseases are associated with aberrant glycosylation, comparative profiling of glycans could be a significant starting point for more in-depth studies. Similarly, this approach can be applied to numerous biological investigations of differential systems, for example, in a comparison of the glycosylation patterns in different tissues, chemotaxonomies of different organisms, or phylogenetic trees. In these situations, high precision and accuracy in measuring and comparing glycan abundances become essential, and stable isotopic labeling for MS quantitation becomes an attractive option for direct and simultaneous comparison between multiple differentially labeled samples. Such a comparison may be accomplished through the permethylation reaction through the use of methyl iodide with varying amounts of deuterium; it may be possible to monitor up to four samples simultaneously (66). Importantly, the linearity of this method is acceptable at nearly two orders of magnitude. In an analogous method, a combination of 13C- and deuterium-labeled methyl iodide reagents (13CH3I and 12CDH2) were used to incorporate stable isotopes into glycan structures through permethylation (67, 68). Although this approach may at first appear to be an isobaric method, permethylation using these reagents introduces a mass difference of 0.002922 Da for each site of derivatization. This small mass difference is easily measurable with a high-resolution mass spectrometer (i.e., a Fourier-transform ion cyclotron resonance instrument or an orbitrap).
Isotopically coded tags may also be incorporated into the glycan structure at the reducing end by reductive amination methods or hydrazine-based reactions. These tags include aniline (69-71), 2-aminopyridine (72), 2-AB (73), and 1-phenyl-3-methyl-5-pyrazolone (74). Additional tags, including (13C6 and 12C12) 4-phenethyl-benzohydrazide (75) and a unique set of tetraplexed tags (76, 77), each separated by 4 Da, have been synthesized. Similarly to the SILAC (stable-isotope labeling by the amino acids in a cell culture) method used in proteomic investigations, isotopically labeled glutamine, which is the sole source of nitrogen for the synthesis of GlcNAc, N-acetylgalactosamine, and the sialic acids, has been employed (78) to study cultured mouse embryonic stem cells.
5. TANDEM MASS SPECTROMETRY METHODS
Although MS recordings of glycans can provide information about the glycan’s overall monosaccharide composition, it is also necessary to understand their specific connectivities. At high sensitivity, tandem MS may be the only means to obtain such information through fragmentation. Tandem MS experiments may help reveal fine structural details to distinguish different positional isomers, such as α1,3- versus α1,6-linked Fuc units, that are relevant in many human diseases. Additionally, it is important to place sialic acids on different branches in less than fully sialylated structures, and even to probe more subtle changes in different sialic-acid linkages (α2,3 versus α2,6).
Traditionally, collision-induced dissociation (CID) has been used to fragment glycans under low-energy (a few volts to a few hundred volts) and high-energy (kilovolts) conditions. In this type of fragmentation, analytes are bombarded with a neutral buffer gas, increasing their bond vibrational energies. Once sufficient energy has been imparted to the structure, bond cleavage occurs. The nomenclature system proposed by Domon & Costello (79) is widely used to describe the resulting fragments. Low-energy CID can be performed in ion-trap instruments, and fragmentation occurs between adjacent monosaccharide units. Most frequently, B and Y ions are observed. B ions contain the original structure’s reducing end, whereas Y ions contain its nonreducing end. Similar fragmentation patterns are observed in MALDI postsource decay experiments, wherein certain analytes may undergo spontaneous decomposition. Such bond cleavages are believed to arise from charge-directed mechanisms (80, 81). Complementary fragmentation patterns result if a high-energy CID experiment is performed. These methods, available with modern MALDI tandem TOF instruments, allow access to the higher-energy pathways needed for the so-called cross-ring fragmentation of permethylated (82) and native (83) glycans. The cleavages of two bonds across a single monosaccharide unit, as in A or X ions, can help determine linkage and branching patterns (84).
On the basis of the suspected biomedical importance of glycan isomers, other tandem MS methods for ion-trap instruments have been developed to characterize these isomers. One of these approaches is the sequential MS method. Following an initial CID experiment, a resulting ambiguous ion of interest is isolated and subjected to a subsequent round of fragmentation, and this process is repeated as many times as necessary. Eventually, fragments that are specific to a particular isomer may be detected. In an early application of this technique, a triantennary, tetrasialylated glycan from bovine fetuin was analyzed, and sequential MS showed that the fourth sialic acid resided on an antenna of the α1,6 arm (85). Later, this approach was used to distinguish different ovalbumin glycan isomers and to differentiate isomeric structures of commercially available and humanized immunoglobulin G (IgG) molecules (86). The same approach was also used to characterize glycan isomers in metastatic and nonmetastatic brain cancer cell lines (87). In an application to well-characterized glycans, sequential MS has indicated the presence of unique isomers associated with the high-Man glycans of bovine ribonuclease B (88), although suspected new isomers need to be confirmed through multimethodological analytical approaches (89). Note that the sequential MS approach may be limited to cases in which sufficient amounts of biological material are available.
Because of the relative ease with which glycans form positive ions, through either protonation or cationization with a metal ion, many tandem MS experiments are conducted in positive-ion mode. However, complementary fragmentation patterns arise from negative-ion mode experiments. These patterns can be used to differentiate glycan isomers, including bisecting structures from more highly branched complex carbohydrates (90). In an experiment first reported for ESI, negative ions of neutral glycans were generated and stabilized by an appropriate anion (91-93). For optimal overall MS performance, nitrate ions are the best option because they produce the most intense signals while minimizing in-source decay processes (91). In contrast to the positive-ion mode CID spectra that contain primarily B and Y ions, the fragmentation spectra of negatively charged ions contain numerous A-type cross-ring fragments, which are needed to distinguish different isomers, and C ions (91-93). Figure 5 shows representative negative-ion tandem MS spectra for bi-, tri-, and tetra-antennary glycans, where many of these types of ions were generated. The fragmentation for neutral glycans is efficient and is attributable to the deprotonation of a hydroxyl group; however, sialylated structures, in which the charge is associated with the carboxylate group, require significantly more energy to be fragmented to the same extent (94). Under appropriate negative-ion mode conditions, several fragment ions can be observed with one of the most intense species, the B1 ion, due to the labile nature of the sialic-acid units, which may quench fragmentation throughout the remainder of the molecule. Additionally, cross-ring fragments across the GlcNAc units of the core occur, along with a few glycosidic cleavages. Interestingly, negative-mode fragmentation following esterification of the sialic acids causes more complete dissociation and allows for a more definitive structural determination. Further information that can be obtained from negative-ion tandem MS spectra of acidic glycans may provide clues about the sialic-acid linkages (95). α2,6-Linked sialic acids produce a 0,2A7 fragment, whereas α2,3-linked ones have no such diagnostic ion.
Figure 5.

(a) Bi-, (b) tri-, and (c) tetra-antennary glycans fragmented in the negative-ion mode, resulting in the formation of many cross-ring fragments. Modified from Reference 93.
Negative-ion mode tandem MS analyses have also been performed in MALDI-based experiments, although it is much more challenging to efficiently generate negatively charged ions for neutral structures. However, nitrate adducts (91) are suitable for this purpose under MALDI conditions, and the resulting tandem MS spectra contain numerous cross-ring fragments and C ions, as do the negative-mode ESI tandem MS spectra. Alternatively, glycans tagged with 2-aminobenzamide may be detected in their deprotonated form. The tandem MS spectra of such glycans present extensive 1,3 A cross-ring fragments and are probably caused by the open-ring structure because of reductive amination (96). Importantly, the highly labile Fuc units were stable, and loss of these units was minimal.
6. ION-MOBILITY SPECTROMETRY
In further studies of isomeric glycan structures, several researchers have investigated the use of ion-mobility spectrometry (IMS) coupled to MS; initial studies were performed on the instruments employing a drift tube filled with a neutral buffer gas. In this configuration, ionized gas-phase analytes are subjected to a series of collisions under low–electric field conditions, and ions with the same mass-to-charge (m/z) values are separated according to their unique collisional cross sections—values that are related to the overall shape of the molecule. If their cross sections are sufficiently different, this technique may resolve isomeric structures. Although this type of instrumental configuration is still employed in many research laboratories, the introduction of a commercial instrument (97) using a traveling wave (98) to induce separation allows nearly any laboratory to exploit the advantages of this approach. Regardless of the method used for analyte mobility–based separations, the acquired data seem to be comparable (99).
In 1997, Liu & Clemmer (100) published an early study of carbohydrate isomers with IMS. In a direct infusion approach, the authors used ESI to introduce a solution containing the trisaccharides melezitose, a structure resembling a branched oligosaccharide, and raffinose, a more linear carbohydrate, into a drift tube containing nitrogen at a pressure of ~3 torr at 300° K. Although they observed some separation (the drift times were 2.135 and 2.162 ms for melezitose and raffinose, respectively), further optimization of the experimental and/or instrumentational conditions was required to achieve a more efficient separation.
In a later study, IMS was used to investigate N-linked isomeric structures derived from model glycoproteins (101). In a study of permethylated ovalbumin glycans, three distinct IMS features were repeatedly and reproducibly recorded for a particular m/z value; the sequence was determined to be hexose5-N-acetylhexosamine4. The investigators proposed that these features corresponded to the isomeric structures for this sequence; this hypothesis was further supported by molecular modeling studies in which the lowest-energy theoretical cross sections matched the experimental values within 1.5%.
Further applications of IMS in glycomic studies have involved the analysis of permethylated glycans derived from serum glycoproteins to detect differences among control individuals, individuals diagnosed with cirrhosis, and liver cancer patients (102, 103). Investigators initially suggested that a monosialylated, biantennary structure could act as a marker for liver disease according to supervised principal-components analysis (PCA) (102). A follow-up study obtained better differentiation between the sample cohorts when 10 glycans were used in the supervised PCA. This study demonstrated one of the main advantages of IMS: Glycomic profiles can be collected in 2 min per sample. Although only 17 structures were observed in this case, the possible separation of isomeric structures was beneficial.
A simultaneous glycoproteomic/glycomic method (104) may further improve the throughput in glycomic studies. In this experimental design, glycoprotein standards were first digested with trypsin, followed by PNGase F. The resulting mixtures were analyzed by both MALDI- and ESI-based IMS methods, which yielded slightly different results. The MALDI-based approach led to an amino-acid sequence coverage of 43.5%, and all five high-Man glycans were detected. A better sequence coverage (71.8%) was obtained for ESI-based measurements, although the glycans probably underwent significant in-source fragmentation, and only the products of these reactions were observed.
A similar approach was used to study the N-linked glycans associated with the human immunodeficiency virus protein gp120, which was expressed in different cell lines with various glycosyltransferase inhibitors (105). Perhaps because peptides and oligosaccharides occupied different regions of the IMS drift space (Figure 6a), glycan profiles with high signal-to-noise ratios were acquired directly from the crude digestion mixture (Figure 6b); however, an ESI direct infusion experiment showed only very weak signals associated with the glycans (Figure 6c). Importantly, the IMS profile was very similar to a MALDI-based recording (Figure 6d). Because no sample cleanup was needed, as is often the case with the use of other analytical measurement techniques, this approach has some potential.
Figure 6.

Ion-mobility spectrometry (IMS) analysis of a crude glycan digest (still containing peptides, proteins, etc.) of the human immunodeficiency virus protein gp120. (a) The total drift scope. (b) An electrospray ionization mass spectrometry (ESI MS) spectrum. (c) The glycans after an IMS separation. (d) A matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) profile of this protein. Modified from Reference 105.
7. SEPARATIONS IN GLYCOMICS
Due to the usually high complexity of released glycan pools and the frequently low abundances of some biologically important glycoconjugates, analytical measurements must have high sensitivity. In addition to the inherently sensitive MS techniques, LC, CE, or chip-based electrophoretic methods may also be used to record carbohydrate profiles. A separation technique can greatly benefit MS analyses by reducing the competitive ionization that hinders the detection of low-abundance mixture components. Also, some techniques may cause isomeric structures to resolve, which cannot be easily achieved through MS.
7.1. Liquid Chromatography
Several advances have recently been made in stationary-phase chemistries and technology. These developments are providing new opportunities for carbohydrate analyses at high sensitivity. Given the hydrophilic nature of glycans, separation systems based on hydrophilic interaction chromatography (HILIC) (106) are increasingly being used. This chromatographic mode employs a polar stationary phase; one of the most popular is the TSK gel Amide-80 medium, available from Tosoh Biosciences. In this chromatographic mode, the initial mobile-phase conditions offer lower solvent polarity, such as a high percentage of acetonitrile, and the aqueous content is then gradually increased to elute the retained polar analytes from the column. This method has been applied to several comparative glycomic studies and has revealed differences between glycosylation levels associated with different states of health (107-109), several environmental parameters that influence blood serum glycomic patterns (110), and changes in IgG glycans associated with aging (111).
An alternative to the HILIC stationary phase is porous graphitized carbon (PGC). This medium offers high chemical inertness, stability across a broad pH range, and good performance at elevated temperatures. One of PGC’s most attractive qualities is its high selectivity, which allows for the resolution of the positional isomers of permethylated glycans (112) and various linkage (113) and positional (114) isomers of native glycans. The mechanism(s) of retention with this material is complex and may be based on adsorption through hydrophobic, hydrophilic, and polar interactions (115). Given this diverse array of intermolecular processes, several parameters influence glycan retention. Among them, increasing the mobile-phase ionic strength through the addition of salts causes an increase in the observed retention times and a chromatographic band-broadening; sialylated structures appeared to be particularly sensitive to this parameter (115). Additionally, the retention of these oligosaccharides depends on mobile-phase pH; lower pH values cause increased retention times (115). When compared with that of the other retention mechanisms based on partitioning, the retention of carbohydrates on PGC packings is increased at elevated temperatures (115).
In addition to the advances in stationary-phase chemistries, several recent technological developments have also led to improved carbohydrate separations. Among them is ultrahigh-pressure LC (UPLC), which enables reduced analysis time and increased analyte resolution due to the substantial decrease in the chromatographic particle size. The advantages of reducing the particle diameter have recently been demonstrated through a direct comparison between 3.0-μm and 1.7-μm particles (116) for bovine fetuin glycans labeled with 2-aminobenzamide (Figure 7). In this comparison, not only were the analysis times significantly reduced in UPLC runs, but the higher column efficiencies caused the resolution of several isomers that were not observed with the column packed with the larger particles.
Figure 7.

Glycans derived from bovine fetuin separated by (a) a liquid chromatography column packed with 3-μm particles and (b) an ultrahigh-pressure liquid chromatography (UHPLC) column packed with 1.7-μm particles. Modified from Reference 116.
Another analytical development that has recently become popular is the LC chip (117), which may be interfaced directly to the inlet of an ESI-based mass spectrometer. In this format, a trapping column, switching valve, and LC column are integrated into a single biocompatible unit, while the number of manual connections is reduced. In such a device, higher sensitivities and improved reproducibilities can be obtained. These devices have been used in numerous glycomic studies, including an investigation of permethylated glycans derived from blood serum glycoproteins from control individuals and late-stage breast cancer patients. The separations were accomplished using a reversed-phase system (41). This combination demonstrated that several glycans were differentially present in the different states of health. LC chips with graphite packings are also available and have been used to characterize human milk oligosaccharides (118-120), milk carbohydrates from different primates (121), and glycans present on serum glycoproteins (122). Nearly 200 structures were identified from their exact masses in human serum; these structures included isomeric analytes (123), which were then used to construct a library based on m/z values and retention times to quickly determine the structure of a given glycan. Figure 8 shows chromatograms of only neutral glycans, a mixture of neutral and sialylated carbohydrates, and only sialylated oligosaccharides separated with PGC in the microchip format.
Figure 8.

Base-peak chromatograms of (a) only neutral glycans, (b) a mixture of neutral and sialylated oligosaccharides, and (c) only sialylated carbohydrates derived from human serum glycoproteins. A high-pressure liquid chromatography chip packed with porous graphitic carbon was used in these separations. Abbreviations: Hex, hexose; HexNAc, N-acetylhexosamine; Fuc, fucose; NeuAc, sialic acid. Modified from Reference 122.
A more advanced chip design incorporates an immobilized PNGase F reactor into the analytical scheme (124). Generally, in-solution glycan release procedures are performed for extended periods of time. However, this chip may allow a much higher digestion throughput; an incubation period of only 6 s was needed to achieve an antibody digestion efficiency of approximately 98% (124). Following cleavage, the glycans are trapped on a graphite preconcentrator and subsequently separated by use of a column packed with the same adsorbent.
7.2. Capillary Electrophoresis
The original high-sensitivity glycomic methods can arguably be traced back to CE coupled with LIF detection (125-127), during the time when the modern biomolecular MS instruments and techniques were in the initial stages of development. Although CE-LIF cannot directly identify the monosaccharide sequences of carbohydrates in the same manner that MS and tandem MS methods can, its ability to reliably handle very complex glycan mixtures at high sensitivity and even resolve isomeric glycans still makes CE an attractive approach for glycomic analyses. The advantages of CE-LIF, exemplified by the glycan profile of a monoclonal antibody compared with several glycan standards for peak identification (128), include good peak capacity and resolution of isomers. Although this approach seems effective for proteins with simpler glycomic patterns, it highlights one of the primary difficulties (i.e., solute identification) associated with CE that have seriously hindered its use in biomarker discovery, perhaps more than any other analytical method. In some cases, it may be possible to determine the monosaccharide composition of glycans through the sequential use of exoglycosidases with different specificities, followed by CE analysis (129). This approach is more useful for relatively simple glycoproteins; however, it is generally less effective for biologically complex systems. This complication of CE further stresses the need for a large collection of N- and O-linked glycan standards that can confidently identify the separated peaks for an unknown biological sample.
Crucial to the high-sensitivity measurements in CE-LIF is the choice of an appropriate fluorescent tag. Although many different labels were explored early in the development of this technique, the use of APTS as a tagging group by Guttman and colleagues (130, 131) has been nearly universally adopted. However, the search for fluorescent tags with improved analytical attributes continues; recent examples include 4-fluoro-7-nitro-2,1,3-benzoxdiazole (132) and the rhodamine 110 dye, which has a large fluorescence quantum yield (133). Because the use of each of these tags results in carbohydrate derivatives with slightly different overall structures, the buffer compositions and methods must be somewhat modified to achieve separations with the highest possible resolution.
Although CE-LIF was initially practiced mainly in academic laboratories, it has recently become more popular in the biopharmaceutical industry, where new therapeutics, including monoclonal antibodies and glycoprotein-based vaccines, need to be assessed for quality control. CE-LIF is often used to provide quantitative glycomic profiles to support product efficacy and minimize immunogenicity effects (134, 135). In contrast to clinical biomarker discovery studies, most applications in the biotechnology industry do not require the high level of sensitivity at which glycan identity is often known. However, both clinical investigations and biopharmaceutical analyses demand high sample throughput, which CE readily achieves. This ability is exemplified by a recent CE-based instrument used to evaluate glycan profiles of patients with different liver diseases (136, 137), a situation in which it may be useful to analyze many samples rapidly. Figure 9 shows that an optimized series of sample treatments, beginning with a 3-μl aliquot serum, leading to APTS labeling, and ending with CE-fluorescence glycan profiling, can be accomplished with an instrumentally modified DNA sequencer.
Figure 9.
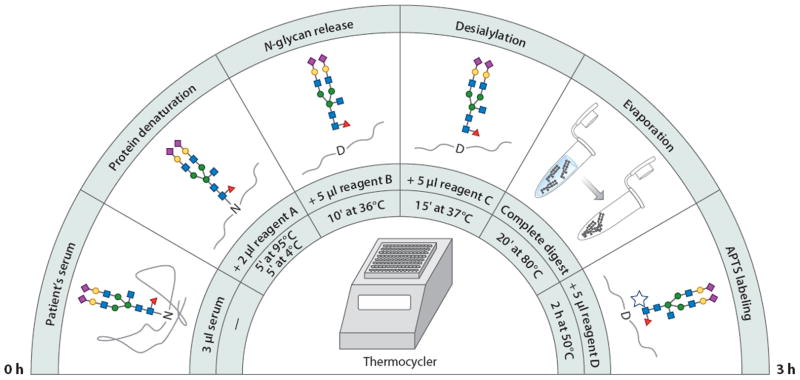
An optimized work flow for the preparation of glycans prior to a capillary electrophoretic analysis. Abbreviation: APTS, 8-aminopyrene-1,3,6-trisulfonate. Modified from Reference 137.
Although electrophoretic separations of glycans of biomedical interest have traditionally been performed in capillary-based instruments, chip-based systems have recently become more widely used (138-142). This format offers significant reduction in analysis times (i.e., higher throughput) and more reproducible separations. Figure 10 shows an early example of this trend (139). In contrast to the separation performed in a capillary, which required a run time of more than 30 min, the CE chip using a spiral channel design was completed in only 2.8 min at a comparable, or better, separation efficiency. Recently, more advanced chip designs, including serpentine channels, have produced separations of even greater efficiency (143).
Figure 10.

A chip-based electropherogram of N-linked glycans from the serum of a patient with late-stage breast cancer. Modified from Reference 139.
The major remaining problem with CE analysis of glycans is its limited compatibility with MS, although progress in the coupling of these two techniques for the analysis of glycoconjugates has been made and recently reviewed (142, 144, 145). The best separation efficiencies for capillary zone electrophoresis have been achieved for only minute sample quantities in both capillary- and chip-based formats. This observation implies that most on-column preconcentration solutions (e.g., stacking or solute trapping) may not be particularly helpful for enhancing signal intensities in different CE-MS combinations. However, an elaborate instrumental setup consisting of CE-LIF coupled to negative-ion mode ESI MS has been used to successfully analyze recombinant monoclonal antibody glycans tagged with APTS (146).
Additional advances in CE-MS analyses of glycoconjugates will further advance the field of analytical glycobiology. The best CE separations to date have been achieved with buffer media compositions and polymeric additives that are largely incompatible with typical MS conditions. It is difficult to predict whether the needed improvements will come from the development of different derivatization schemes and separation conditions or from innovations in CE-MS interfacing technologies and MS designs. Nevertheless, the results produced so far have been encouraging, showing significant potential for this hybrid method.
8. CONCLUSIONS
The multilateral importance of glycosylated structures in biology and biomedical research demands further advances in analytical methods and instrumentation. The structural and functional attributes of glycans present complex and challenging problems to investigators in glycobiology; these challenges could be clarified through more precise structural assignments and more reliable quantification in glycomic profiling. Since our laboratory reviewed the overall area of analytical glycobiology (including high-sensitivity determinations at both the protein and oligosaccharide levels) a decade ago (147), substantial progress has been made in carbohydrate derivatization; capillary separations, including capillary LC with its HILIC and PGC modes; CE; and especially MS and its various new technologies and tandem modes. Separations in capillaries are now giving way to microchip formats. All of these areas now emphasize substantial needs for glycan standards. Today’s analytical glycobiology represents a unique combination of microscale chemistry and instrumental methods. As the frontiers of glycoscience continue to attract attention due to their substantial scientific importance (in, for example, developmental biology, immunology, disease biomarkers, and modern biopharmaceuticals), appreciation for glycomic methodologies is likely to increase, as will the proliferation of new analytical tools.
Acknowledgments
The work described in this review was supported by grant number R01GM024349 from the National Institute of General Medical Sciences and grant number UO1CA128535 from the National Cancer Institute, US Department of Health and Human Services.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Hart GW, Copeland RJ. Glycomics hits the big time. Cell. 2010;143:672–76. doi: 10.1016/j.cell.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nita-Lazar M, Wacker M, Schegg B, Amber S, Aebi M. The N-X-S/T consensus sequence is required but not sufficient for bacterial N-linked protein glycosylation. Glycobiology. 2005;15:361–67. doi: 10.1093/glycob/cwi019. [DOI] [PubMed] [Google Scholar]
- 3.Zhang H, Huang RY, Jalili PR, Irungu JW, Nicol GR, et al. Improved mass spectrometric characterization of protein glycosylation reveals unusual glycosylation of maize-derived bovine trypsin. Anal Chem. 2010;82:10095–101. doi: 10.1021/ac1020722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valliere-Douglass JF, Eakin CM, Wallace A, Ketchem RR, Wang W, et al. Glutamine-linked and non-consensus asparagine–linked oligosaccharides present in human recombinant antibodies define novel protein glycosylation motifs. J Biol Chem. 2010;285:16012–22. doi: 10.1074/jbc.M109.096412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol. 2012;13:448–62. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hofsteenge J, Müller DR, de Beer T, Löffler A, Richter WJ, Vliegenthart JF. New type of linkage between a carbohydrate and a protein: C-glycosylation of a specific tryptophan residue in human RNase Us. Biochemistry. 1994;33:13524–30. doi: 10.1021/bi00250a003. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez de Peredo A, Klein D, Macek B, Hess D, Peter-Katalinic J, Hofsteenge J. C-mannosylation and o-fucosylation of thrombospondin type 1 repeats. Mol Cell Proteomics. 2002;1:11–18. doi: 10.1074/mcp.m100011-mcp200. [DOI] [PubMed] [Google Scholar]
- 8.Oman TJ, Boettcher JM, Wang H, Okalibe XN, van der Donk WA. Sublancin is not a lantibiotic but an S-linked glycopeptide. Nat Chem Biol. 2011;7:78–80. doi: 10.1038/nchembio.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nilsson J, Larson G, Grahn A. Characterization of site-specific O-glycan structures within the mucin-like domain of α-dystroglycan from human skeletal muscle. Glycobiology. 2010;20:1160–69. doi: 10.1093/glycob/cwq082. [DOI] [PubMed] [Google Scholar]
- 10.Stalnaker SH, Hashmi S, Lim JM, Aoki K, Porterfield M, et al. Site mapping and characterization of O-glycan structures on α-dystroglycan isolated from rabbit skeletal muscle. J Biol Chem. 2010;285:24882–91. doi: 10.1074/jbc.M110.126474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hart GW. Glycosylation. Curr Opin Cell Biol. 1992;4:1017–23. doi: 10.1016/0955-0674(92)90134-x. [DOI] [PubMed] [Google Scholar]
- 12.Yi W, Clark PM, Mason DE, Keenan MC, Hill C, et al. Phosphofructokinase 1 glycosylation regulates cell growth and metabolism. Science. 2012;337:975–80. doi: 10.1126/science.1222278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harvey DJ, Merry AH, Royle L, Campbell MP, Dwek RA, Rudd PM. Proposal for a standard system for drawing structural diagrams of N- and O-linked carbohydrates and related compounds. Proteomics. 2009;9:3796–801. doi: 10.1002/pmic.200900096. [DOI] [PubMed] [Google Scholar]
- 14.Huang Y, Mechref Y, Novotny MV. Microscale nonreductive release of O-linked glycans for subsequent analysis through MALDI mass spectrometry and capillary electrophoresis. Anal Chem. 2001;73:6063–69. doi: 10.1021/ac015534c. [DOI] [PubMed] [Google Scholar]
- 15.Huang Y, Konse T, Mechref Y. Matrix-assisted laser desorption/ionization mass spectrometry compatible β-elimination of O-linked oligosaccharides. Rapid Commun Mass Spectrom. 2002;16:1199–204. doi: 10.1002/rcm.701. [DOI] [PubMed] [Google Scholar]
- 16.Miura Y, Kato K, Takegawa Y, Kurogochi M, Furukawa J, et al. Glycoblotting-assisted O-glycomics: Ammonium carbamate allows for highly efficient O-glycan release from glycoproteins. Anal Chem. 2010;82:10021–29. doi: 10.1021/ac101599p. [DOI] [PubMed] [Google Scholar]
- 17.Plummer TH, Jr, Elder JH, Alexander S, Phelan AW, Tarentino AL. Demonstration of peptide: N-glycosidase F activity in endo-β-N-acetylglucosaminidase F preparations. J Biol Chem. 1984;259:10700–4. [PubMed] [Google Scholar]
- 18.Tretter V, Altmann F, Marz L. Peptide-N4-(N-acetyl-β-glucosaminyl)asparagine amidase F cannot release glycans with fucose attached a(1→3) to the asparagine-linked N-acetylglucosamine residue. Eur J Biochem. 1991;199:647–52. doi: 10.1111/j.1432-1033.1991.tb16166.x. [DOI] [PubMed] [Google Scholar]
- 19.Kuster B, Mann M. 18O-labeling of N-glycosylation sites to improve the identification of gelseparated glycoproteins using peptide mass mapping and database searching. Anal Chem. 1999;71:1431–40. doi: 10.1021/ac981012u. [DOI] [PubMed] [Google Scholar]
- 20.Zielinska DF, Gnad F, Wisniewski JR, Mann M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell. 2010;141:897–907. doi: 10.1016/j.cell.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 21.Palmisano G, Melo-Braga MN, Engholm-Keller K, Parker BL, Larsen MR. Chemical deamidation: a common pitfall in large-scale N-linked glycoproteomic mass spectrometry-based analyses. J Proteome Res. 2012;11:1949–57. doi: 10.1021/pr2011268. [DOI] [PubMed] [Google Scholar]
- 22.Szabo Z, Guttman A, Karger BL. Rapid release of N-linked glycans from glycoproteins by pressurecycling technology. Anal Chem. 2010;82:2588–93. doi: 10.1021/ac100098e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandoval WN, Arellano F, Arnott D, Raab H, Vandlen R, Lill JR. Rapid removal of N-linked oligosaccharides using microwave assisted enzyme catalyzed deglycosylation. Int J Mass Spectrom. 2007;259:117–23. [Google Scholar]
- 24.Tzeng YK, Chang CC, Huang CN, Wu CC, Han CC, Chang HC. Facile MALDI-MS analysis of neutral glycans in NaOH-doped matrixes: microwave-assisted deglycosylation and one-step purification with diamond nanoparticles. Anal Chem. 2008;80:6809–14. doi: 10.1021/ac801137g. [DOI] [PubMed] [Google Scholar]
- 25.Liu X, McNally DJ, Nothaft H, Szymanski CM, Brisson JR, Li J. Mass spectrometry–based glycomics strategy for exploring N-linked glycosylation in eukaryotes and bacteria. Anal Chem. 2006;78:6081–87. doi: 10.1021/ac060516m. [DOI] [PubMed] [Google Scholar]
- 26.An HJ, Peavy TR, Hedrick JL, Lebrilla CB. Determination of N-glycosylation sites and site heterogeneity in glycoproteins. Anal Chem. 2003;75:5628–37. doi: 10.1021/ac034414x. [DOI] [PubMed] [Google Scholar]
- 27.Temporini C, Perani E, Calleri E, Dolcini L, Lubda D, et al. Pronase-immobilized enzyme reactor: an approach for automation in glycoprotein analysis by LC/LC-ESI/MS. Anal Chem. 2007;79:355–63. doi: 10.1021/ac0611519. [DOI] [PubMed] [Google Scholar]
- 28.Iwase H, Hotta K. Release of O-linked glycoprotein glycans by endo-α-N-acetylgalactosaminidase. Methods Mol Biol. 1993;14:151–59. doi: 10.1385/0-89603-226-4:151. [DOI] [PubMed] [Google Scholar]
- 29.Carlson DM. Structures and immunochemical properties of oligosaccharides isolated from pig submaxillary mucins. J Biol Chem. 1968;243:616–26. [PubMed] [Google Scholar]
- 30.Rademaker JG, Haverkamp J, Thomas-Oates J. Determination of glycosylation sites in O-linked glycopeptides: a sensitive mass spectrometric protocol. Org Mass Spectrom. 1993;28:1536–41. [Google Scholar]
- 31.Maniatis S, Zhou H, Reinhold V. Rapid de-O-glycosylation concomitant with peptide labeling using microwave radiation and an alkyl amine base. Anal Chem. 2010;82:2421–25. doi: 10.1021/ac902734w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goetz JA, Novotny MV, Mechref Y. Enzymatic/chemical release of O-glycans allowing MS analysis at high sensitivity. Anal Chem. 2009;81:9546–52. doi: 10.1021/ac901363h. [DOI] [PubMed] [Google Scholar]
- 33.Harvey DJ. Derivatization of carbohydrates for analysis by chromatography, electrophoresis and mass spectrometry. J Chromatogr B. 2011;879:1196–225. doi: 10.1016/j.jchromb.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 34.Hakomori S. A rapid permethylation of glycolipid and polysaccharide catalyzed by methylsulfinyl carbanion in dimethyl sulfoxide. J Biochem. 1964;55:205–8. [PubMed] [Google Scholar]
- 35.Ciucanu I, Kerek F. A simple and rapid method for the permethylation of carbohydrates. Carbohydr Res. 1984;131:209–17. [Google Scholar]
- 36.Ciucanu I, Costello CE. Elimination of oxidative degradation during the per-O-methylation of carbohydrates. J Am Chem Soc. 2003;125:16213–19. doi: 10.1021/ja035660t. [DOI] [PubMed] [Google Scholar]
- 37.Kang P, Mechref Y, Klouckova I, Novotny MV. Solid-phase permethylation of glycans for mass spectrometric analysis. Rapid Commun Mass Spectrom. 2005;19:3421–28. doi: 10.1002/rcm.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang P, Mechref Y, Novotny MV. High-throughput solid-phase permethylation of glycans prior to mass spectrometry. Rapid Commun Mass Spectrom. 2008;22:721–34. doi: 10.1002/rcm.3395. [DOI] [PubMed] [Google Scholar]
- 39.Desantos-Garcia JL, Khalil SI, Hussein A, Hu Y, Mechref Y. Enhanced sensitivity of LC-MS analysis of permethylated N-glycans through online purification. Electrophoresis. 2011;32:3516–25. doi: 10.1002/elps.201100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robinson S, Routledge A, Thomas-Oates J. Characterisation and proposed origin of mass spectrometric ions observed 30 Th above the ionised molecules of per-O-methylated carbohydrates. Rapid Commun Mass Spectrom. 2005;19:3681–88. doi: 10.1002/rcm.2246. [DOI] [PubMed] [Google Scholar]
- 41.Alley WR, Jr, Madera M, Mechref Y, Novotny MV. Chip-based reversed-phase liquid chromatography–mass spectrometry of permethylated N-linked glycans: a potential methodology for cancer-biomarker discovery. Anal Chem. 2010;82:5095–106. doi: 10.1021/ac100131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McConville MJ, Thomas-Oates JE, Ferguson MA, Homans SW. Structure of the lipophosphoglycan from Leishmania major. J Biol Chem. 1990;265:19611–23. [PubMed] [Google Scholar]
- 43.Yu SY, Wu SW, Hsiao HH, Khoo KH. Enabling techniques and strategic workflow for sulfoglycomics based on mass spectrometry mapping and sequencing of permethylated sulfated glycans. Glycobiology. 2009;19:1136–49. doi: 10.1093/glycob/cwp113. [DOI] [PubMed] [Google Scholar]
- 44.Lei M, Mechref Y, Novotny MV. Structural analysis of sulfated glycans by sequential double permethylation using methyl iodide and deuteromethyl iodide. J Am Soc Mass Spectrom. 2009;20:1660–71. doi: 10.1016/j.jasms.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Lei M, Novotny MV, Mechref Y. Sequential enrichment of sulfated glycans by strong anion-exchange chromatography prior to mass spectrometric measurements. J Am Soc Mass Spectrom. 2010;21:348–57. doi: 10.1016/j.jasms.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 46.Talbo G, Mann M. Aspects of the sequencing of carbohydrates and oligonucleotides by matrix-assisted laser desorption/ionization post-source decay. Rapid Commun Mass Spectrom. 1996;10:100–3. doi: 10.1002/(SICI)1097-0231(19960115)10:1<100::AID-RCM402>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 47.Powell AK, Harvey DJ. Stabilization of sialic acids in N-linked oligosaccharides and gangliosides for analysis by positive ion matrix-assisted laser desorption/ionization mass spectrometry. Rapid Commun Mass Spectrom. 1996;10:1027–32. doi: 10.1002/(SICI)1097-0231(19960715)10:9<1027::AID-RCM634>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Li X, Chan K, Zou W, Pribil P, et al. “One-pot” methylation in glycomics application: esterification of sialic acids and permanent charge construction. Anal Chem. 2007;79:3894–900. doi: 10.1021/ac070091j. [DOI] [PubMed] [Google Scholar]
- 49.Sekiya S, Wada Y, Tanaka K. Derivatization for stabilizing sialic acids in MALDI-MS. Anal Chem. 2005;77:4962–68. doi: 10.1021/ac050287o. [DOI] [PubMed] [Google Scholar]
- 50.Wheeler SF, Domann P, Harvey DJ. Derivatization of sialic acids for stabilization in matrix-assisted laser desorption/ionization mass spectrometry and concomitant differentiation of α(2→3) and α(2→6) isomers. Rapid Commun Mass Spectrom. 2009;23:303–12. doi: 10.1002/rcm.3867. [DOI] [PubMed] [Google Scholar]
- 51.Alley WR, Jr, Novotny MV. Glycomic analysis of sialic acid linkages in glycans derived from blood serum glycoproteins. J Proteome Res. 2010;9:3062–72. doi: 10.1021/pr901210r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vasseur JA, Goetz JA, Alley WR, Jr, Novotny MV. Smoking- and lung cancer–induced changes in N-glycosylation of blood serum proteins. Glycobiology. 2012;22:1684–708. doi: 10.1093/glycob/cws108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Toyoda M, Ito H, Matsuno YK, Narimatsu H, Kameyama A. Quantitative derivatization of sialic acids for the detection of sialoglycans by MALDI MS. Anal Chem. 2008;80:5211–18. doi: 10.1021/ac800457a. [DOI] [PubMed] [Google Scholar]
- 54.Liu X, Qiu H, Lee RK, Chen W, Li J. Methylamidation for sialoglycomics by MALDI-MS: a facile derivatization strategy for both α2,3- and α2,6-linked sialic acids. Anal Chem. 2010;82:8300–6. doi: 10.1021/ac101831t. [DOI] [PubMed] [Google Scholar]
- 55.Bigge JC, Patel TP, Bruce JA, Goulding PN, Charles SM, Parekh RB. Nonselective and efficient fluorescent labeling of glycans using 2-amino benzamide and anthranilic acid. Anal Biochem. 1995;230:229–38. doi: 10.1006/abio.1995.1468. [DOI] [PubMed] [Google Scholar]
- 56.Ruhaak LR, Steenvoorden E, Koeleman CA, Deelder AM, Wuhrer M. 2-Picoline-borane: a non-toxic reducing agent for oligosaccharide labeling by reductive amination. Proteomics. 2010;10:2330–36. doi: 10.1002/pmic.200900804. [DOI] [PubMed] [Google Scholar]
- 57.Dalpathado DS, Jiang H, Kater MA, Desaire H. Reductive amination of carbohydrates using NaBH(OAc)3. Anal Bioanal Chem. 2005;381:1130–37. doi: 10.1007/s00216-004-3028-9. [DOI] [PubMed] [Google Scholar]
- 58.Yoshino K, Takao T, Murata H, Shimonishi Y. Use of the derivatizing agent 4-aminobenzoic acid 2-(diethylamino)ethyl ester for high-sensitivity detection of oligosaccharides by electrospray ionization mass spectrometry. Anal Chem. 1995;67:4028–31. doi: 10.1021/ac00117a034. [DOI] [PubMed] [Google Scholar]
- 59.Lattova E, Snovida S, Perreault H, Krokhin O. Influence of the labeling group on ionization and fragmentation of carbohydrates in mass spectrometry. J Am Soc Mass Spectrom. 2005;16:683–96. doi: 10.1016/j.jasms.2005.01.021. [DOI] [PubMed] [Google Scholar]
- 60.Maslen S, Sadowski P, Adam A, Lilley K, Stephens E. Differentiation of isomeric N-glycan structures by normal-phase liquid chromatography–MALDI-TOF/TOF tandem mass spectrometry. Anal Chem. 2006;78:8491–98. doi: 10.1021/ac0614137. [DOI] [PubMed] [Google Scholar]
- 61.Harvey DJ. Collision-induced fragmentation of negative ions from N-linked glycans derivatized with 2-aminobenzoic acid. J Mass Spectrom. 2005;40:642–53. doi: 10.1002/jms.836. [DOI] [PubMed] [Google Scholar]
- 62.Rohmer M, Meyer B, Mank M, Stahl B, Bahr U, Karas M. 3-Aminoquinoline acting as matrix and derivatizing agent for MALDI MS analysis of oligosaccharides. Anal Chem. 2010;82:3719–26. doi: 10.1021/ac1001096. [DOI] [PubMed] [Google Scholar]
- 63.Lattova E, Perreault H. Labelling saccharides with phenylhydrazine for electrospray and matrix-assisted laser desorption/ionization mass spectrometry. J Chromatogr B. 2003;793:167–79. doi: 10.1016/s1570-0232(03)00374-x. [DOI] [PubMed] [Google Scholar]
- 64.Walker SH, Lilley LM, Enamorado MF, Comins DL, Muddiman DC. Hydrophobic derivatization of N-linked glycans for increased ion abundance in electrospray ionization mass spectrometry. J Am Soc Mass Spectrom. 2011;22:1309–17. doi: 10.1007/s13361-011-0140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Walker SH, Papas BN, Comins DL, Muddiman DC. Interplay of permanent charge and hydrophobicity in the electrospray ionization of glycans. Anal Chem. 2010;82:6636–42. doi: 10.1021/ac101227a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang P, Mechref Y, Kyselova Z, Goetz JA, Novotny MV. Comparative glycomic mapping through quantitative permethylation and stable-isotope labeling. Anal Chem. 2007;79:6064–73. doi: 10.1021/ac062098r. [DOI] [PubMed] [Google Scholar]
- 67.Alvarez-Manilla G, Warren NL, Abney T, Atwood JA, 3rd, Azadi P, et al. Tools for glycomics: relative quantitation of glycans by isotopic permethylation using 13CH3I. Glycobiology. 2007;17:677–87. doi: 10.1093/glycob/cwm033. [DOI] [PubMed] [Google Scholar]
- 68.Atwood JA, 3rd, Cheng L, Alvarez-Manilla G, Warren NL, York WS, Orlando R. Quantitation by isobaric labeling: applications to glycomics. J Proteome Res. 2008;7:367–74. doi: 10.1021/pr070476i. [DOI] [PubMed] [Google Scholar]
- 69.Lawrence R, Olson SK, Steele RE, Wang L, Warrior R, et al. Evolutionary differences in glycosaminoglycan fine structure detected by quantitative glycan reductive isotope labeling. J Biol Chem. 2008;283:33674–84. doi: 10.1074/jbc.M804288200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ridlova G, Mortimer JC, Maslen SL, Dupree P, Stephens E. Oligosaccharide relative quantitation using isotope tagging and normal-phase liquid chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 2008;22:2723–30. doi: 10.1002/rcm.3665. [DOI] [PubMed] [Google Scholar]
- 71.Xia B, Feasley CL, Sachdev GP, Smith DF, Cummings RD. Glycan reductive isotope labeling for quantitative glycomics. Anal Biochem. 2009;387:162–70. doi: 10.1016/j.ab.2009.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hashii N, Kawasaki N, Itoh S, Nakajima Y, Kawanishi T, Yamaguchi T. Alteration of N-glycosylation in the kidney in a mouse model of systemic lupus erythematosus: relative quantification of N-glycans using an isotope-tagging method. Immunology. 2009;126:336–45. doi: 10.1111/j.1365-2567.2008.02898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Prien JM, Prater BD, Qin Q, Cockrill SL. Mass spectrometric–based stable isotopic 2-aminobenzoic acid glycan mapping for rapid glycan screening of biotherapeutics. Anal Chem. 2010;82:1498–508. doi: 10.1021/ac902617t. [DOI] [PubMed] [Google Scholar]
- 74.Zhang P, Zhang Y, Xue X, Wang C, Wang Z, Huang L. Relative quantitation of glycans using stable isotopic labels 1-(d0/d5) phenyl-3-methyl-5-pyrazolone by mass spectrometry. Anal Biochem. 2011;418:1–9. doi: 10.1016/j.ab.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 75.Walker SH, Budhathoki-Uprety J, Novak BM, Muddiman DC. Stable-isotope labeled hydrophobic hydrazide reagents for the relative quantification of N-linked glycans by electrospray ionization mass spectrometry. Anal Chem. 2011;83:6738–45. doi: 10.1021/ac201376q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bowman MJ, Zaia J. Tags for the stable isotopic labeling of carbohydrates and quantitative analysis by mass spectrometry. Anal Chem. 2007;79:5777–84. doi: 10.1021/ac070581b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bowman MJ, Zaia J. Comparative glycomics using a tetraplex stable-isotope coded tag. Anal Chem. 2010;82:3023–31. doi: 10.1021/ac100108w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Orlando R, Lim JM, Atwood JA, 3rd, Angel PM, Fang M, et al. IDAWG: metabolic incorporation of stable isotope labels for quantitative glycomics of cultured cells. J Proteome Res. 2009;8:3816–23. doi: 10.1021/pr8010028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Domon B, Costello CE. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/ MS spectra of glycoconjugates. Glycoconj J. 1988;5:397–409. [Google Scholar]
- 80.Cancilla MT, Wong AW, Voss LR, Lebrilla CB. Fragmentation reactions in the mass spectrometry analysis of neutral oligosaccharides. Anal Chem. 1999;71:3206–18. doi: 10.1021/ac9813484. [DOI] [PubMed] [Google Scholar]
- 81.Harvey DJ. Collision-induced fragmentation of underivatized N-linked carbohydrates ionized by electrospray. J Mass Spectrom. 2000;35:1178–90. doi: 10.1002/1096-9888(200010)35:10<1178::AID-JMS46>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 82.Mechref Y, Novotny MV, Krishnan C. Structural characterization of oligosaccharides using MALDI-TOF/TOF tandem mass spectrometry. Anal Chem. 2003;75:4895–903. doi: 10.1021/ac0341968. [DOI] [PubMed] [Google Scholar]
- 83.Stephens E, Maslen SL, Green LG, Williams DH. Fragmentation characteristics of neutral N-linked glycans using a MALDI-TOF/TOF tandem mass spectrometer. Anal Chem. 2004;76:2343–54. doi: 10.1021/ac030333p. [DOI] [PubMed] [Google Scholar]
- 84.Mechref Y, Kang P, Novotny MV. Differentiating structural isomers of sialylated glycans by matrix-assisted laser desorption/ionization time-of-flight/time-of-flight tandem mass spectrometry. Rapid Commun Mass Spectrom. 2006;20:1381–89. doi: 10.1002/rcm.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sheeley DM, Reinhold VN. Structural characterization of carbohydrate sequence, linkage, and branching in a quadrupole ion trap mass spectrometer: neutral oligosaccharides and N-linked glycans. Anal Chem. 1998;70:3053–59. doi: 10.1021/ac9713058. [DOI] [PubMed] [Google Scholar]
- 86.Ashline DJ, Lapadula AJ, Liu YH, Lin M, Grace M, et al. Carbohydrate structural isomers analyzed by sequential mass spectrometry. Anal Chem. 2007;79:3830–42. doi: 10.1021/ac062383a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Prien JM, Huysentruyt LC, Ashline DJ, Lapadula AJ, Seyfried TN, Reinhold VN. Differentiating N-linked glycan structural isomers in metastatic and nonmetastatic tumor cells using sequential mass spectrometry. Glycobiology. 2008;18:353–66. doi: 10.1093/glycob/cwn010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Prien JM, Ashline DJ, Lapadula AJ, Zhang H, Reinhold VN. The high-mannose glycans from bovine ribonuclease B isomer characterization by ion trap MS. J Am Soc Mass Spectrom. 2009;20:539–56. doi: 10.1016/j.jasms.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Prien JM, Prater BD, Cockrill SL. A multi-method approach toward de novo glycan characterization: a Man-5 case study. Glycobiology. 2010;20:629–47. doi: 10.1093/glycob/cwq012. [DOI] [PubMed] [Google Scholar]
- 90.Harvey DJ, Crispin M, Scanlan C, Singer BB, Lucka L, et al. Differentiation between isomeric triantennary N-linked glycans by negative ion tandem mass spectrometry and confirmation of glycans containing galactose attached to the bisecting (β1-4-GlcNAc) residue in N-glycans from IgG. Rapid Commun Mass Spectrom. 2008;22:1047–52. doi: 10.1002/rcm.3470. [DOI] [PubMed] [Google Scholar]
- 91.Harvey DJ. Fragmentation of negative ions from carbohydrates. Part 1. Use of nitrate and other anionic adducts for the production of negative ion electrospray spectra from N-linked carbohydrates. J Am Soc Mass Spectrom. 2005;16:622–30. doi: 10.1016/j.jasms.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 92.Harvey DJ. Fragmentation of negative ions from carbohydrates. Part 2. Fragmentation of high-mannose N-linked glycans. J Am Soc Mass Spectrom. 2005;16:631–46. doi: 10.1016/j.jasms.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 93.Harvey DJ. Fragmentation of negative ions from carbohydrates. Part 3. Fragmentation of hybrid and complex N-linked glycans. J Am Soc Mass Spectrom. 2005;16:647–59. doi: 10.1016/j.jasms.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 94.Seymour JL, Costello CE, Zaia J. The influence of sialylation on glycan negative ion dissociation and energetics. J Am Soc Mass Spectrom. 2006;17:844–54. doi: 10.1016/j.jasms.2006.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Harvey DJ, Rudd PM. Fragmentation of negative ions from N-linked carbohydrates. Part 5. Anionic N-linked glycans. Int J Mass Spectrom. 2011;305:120–30. [Google Scholar]
- 96.Wuhrer M, Deelder AM. Negative-mode MALDI-TOF/TOF-MS of oligosaccharides labeled with 2-aminobenzamide. Anal Chem. 2005;77:6954–59. doi: 10.1021/ac051117e. [DOI] [PubMed] [Google Scholar]
- 97.Pringle SD, Giles K, Wildgoose JL, Williams JP, Slade SE, et al. An investigation of the mobility separation of some peptide and protein ions using a new hybrid quadrupole/travelling wave IMS/TOF instrument. Int J Mass Spectrom. 2007;261:1–12. [Google Scholar]
- 98.Giles K, Pringle SD, Worthington KR, Little D, Wildgoose JL, Bateman RH. Applications of a travelling wave–based radio-frequency-only stacked ring ion guide. Rapid Commun Mass Spectrom. 2004;18:2401–14. doi: 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- 99.Williams JP, Grabenauer M, Carpenter CJ, Holland RJ, Wormald MR, et al. Characterization of simple isomeric oligosaccharides and the rapid separation of glycan mixtures by ion mobility mass spectrometry. Int J Mass Spectrom. 2010;298:119–27. [Google Scholar]
- 100.Liu Y, Clemmer DE. Characterizing oligosaccharides using injected ion mobility/mass spectrometry. Anal Chem. 1997;69:2504–9. doi: 10.1021/ac9701344. [DOI] [PubMed] [Google Scholar]
- 101.Plasencia MD, Isailovic D, Merenbloom SI, Mechref Y, Novotny MV, Clemmer DE. Resolving and assigning N-linked glycan structural isomers from ovalbumin by IMS-MS. J Am Soc Mass Spectrom. 2008;19:1706–15. doi: 10.1016/j.jasms.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Isailovic D, Kurulugama RT, Plasencia MD, Stokes ST, Kyselova Z, et al. Profiling of human serum glycans associated with liver cancer and cirrhosis by IMS-MS. J Proteome Res. 2008;7:1109–17. doi: 10.1021/pr700702r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Isailovic D, Plasencia MD, Gaye MM, Stokes ST, Kurulugama RT, et al. Delineating diseases by IMS-MS profiling of serum N-linked glycans. J Proteome Res. 2012;11:576–85. doi: 10.1021/pr200777u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fenn LS, McLean JA. Simultaneous glycoproteomics on the basis of structure using ion mobility–mass spectrometry. Mol Biosyst. 2009;5:1298–302. doi: 10.1039/b909745g. [DOI] [PubMed] [Google Scholar]
- 105.Harvey DJ, Sobott F, Crispin M, Wrobel A, Bonomelli C, et al. Ion mobility mass spectrometry for extracting spectra of N-glycans directly from incubation mixtures following glycan release: application to glycans from engineered glycoforms of intact, folded HIV gp120. J Am Soc Mass Spectrom. 2011;22:568–81. doi: 10.1007/s13361-010-0053-0. [DOI] [PubMed] [Google Scholar]
- 106.Alpert AJ. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J Chromatogr. 1990;499:177–96. doi: 10.1016/s0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- 107.Saldova R, Royle L, Radcliffe CM, Abd Hamid UM, Evans R, et al. Ovarian cancer is associated with changes in glycosylation in both acute-phase proteins and IgG. Glycobiology. 2007;17:1344–56. doi: 10.1093/glycob/cwm100. [DOI] [PubMed] [Google Scholar]
- 108.Abd Hamid UM, Royle L, Saldova R, Radcliffe CM, Harvey DJ, et al. A strategy to reveal potential glycan markers from serum glycoproteins associated with breast cancer progression. Glycobiology. 2008;18:1105–18. doi: 10.1093/glycob/cwn095. [DOI] [PubMed] [Google Scholar]
- 109.Arnold JN, Saldova R, Galligan MC, Murphy TB, Mimura-Kimura Y, et al. Novel glycan biomarkers for the detection of lung cancer. J Proteome Res. 2011;10:1755–64. doi: 10.1021/pr101034t. [DOI] [PubMed] [Google Scholar]
- 110.Knezevic A, Gornik O, Polasek O, Pucic M, Redzic I, et al. Effects of aging, body mass index, plasma lipid profiles, and smoking on human plasma N-glycans. Glycobiology. 2010;20:959–69. doi: 10.1093/glycob/cwq051. [DOI] [PubMed] [Google Scholar]
- 111.Pucic M, Knezevic A, Vidic J, Adamczyk B, Novokmet M, et al. High throughput isolation and glycosylation analysis of IgG variability and heritability of the IgG glycome in three isolated human populations. Mol Cell Proteomics. 2011;10 doi: 10.1074/mcp.M111.010090. 010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Costello CE, Contado-Miller JM, Cipollo JF. A glycomics platform for the analysis of permethylated oligosaccharide alditols. J Am Soc Mass Spectrom. 2007;18:1799–812. doi: 10.1016/j.jasms.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pabst M, Bondili JS, Stadlmann J, Mach L, Altmann F. Mass + retention time = structure: a strategy for the analysis of N-glycans by carbon LC-ESI-MS and its application to fibrin N-glycans. Anal Chem. 2007;79:5051–57. doi: 10.1021/ac070363i. [DOI] [PubMed] [Google Scholar]
- 114.Pabst M, Grass J, Toegel S, Liebminger E, Strasser R, Altmann F. Isomeric analysis of oligomannosidic N-glycans and their dolichol-linked precursors. Glycobiology. 2012;22:389–99. doi: 10.1093/glycob/cwr138. [DOI] [PubMed] [Google Scholar]
- 115.Pabst M, Altmann F. Influence of electrosorption, solvent, temperature, and ion polarity on the performance of LC-ESI-MS using graphitic carbon for acidic oligosaccharides. Anal Chem. 2008;80:7534–42. doi: 10.1021/ac801024r. [DOI] [PubMed] [Google Scholar]
- 116.Ahn J, Bones J, Yu YQ, Rudd PM, Gilar M. Separation of 2-aminobenzamide labeled glycans using hydrophilic interaction chromatography columns packed with 1.7 micron sorbent. J Chromatogr B. 2010;878:403–8. doi: 10.1016/j.jchromb.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 117.Yin H, Killeen K, Brennen R, Sobek D, Werlich M, van de Goor T. Microfluidic chip for peptide analysis with an integrated HPLC column, sample enrichment column, and nanoelectrospray tip. Anal Chem. 2005;77:527–33. doi: 10.1021/ac049068d. [DOI] [PubMed] [Google Scholar]
- 118.Wu S, Tao N, German JB, Grimm R, Lebrilla CB. Development of an annotated library of neutral human milk oligosaccharides. J Proteome Res. 2010;9:4138–51. doi: 10.1021/pr100362f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wu S, Grimm R, German JB, Lebrilla CB. Annotation and structural analysis of sialylated human milk oligosaccharides. J Proteome Res. 2011;10:856–68. doi: 10.1021/pr101006u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dallas DC, Martin WF, Strum JS, Zivkovic AM, Smilowitz JT, et al. N-linked glycan profiling of mature human milk by high-performance microfluidic chip liquid chromatography time-of-flight tandem mass spectrometry. J Agric Food Chem. 2011;59:4255–63. doi: 10.1021/jf104681p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tao N, Wu S, Kim J, An HJ, Hinde K, et al. Evolutionary glycomics: characterization of milk oligosaccharides in primates. J Proteome Res. 2011;10:1548–57. doi: 10.1021/pr1009367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chu CS, Ninonuevo MR, Clowers BH, Perkins PD, An HJ, et al. Profile of native N-linked glycan structures from human serum using high performance liquid chromatography on a microfluidic chip and time-of-flight mass spectrometry. Proteomics. 2009;9:1939–51. doi: 10.1002/pmic.200800249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hua S, An HJ, Ozcan S, Ro GS, Soares S, et al. Comprehensive native glycan profiling with isomer separation and quantitation for the discovery of cancer biomarkers. Analyst. 2011;136:3663–71. doi: 10.1039/c1an15093f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bynum MA, Yin H, Felts K, Lee YM, Monell CR, Killeen K. Characterization of IgG N-glycans employing a microfluidic chip that integrates glycan cleavage, sample purification, LC separation, and MS detection. Anal Chem. 2009;81:8818–25. doi: 10.1021/ac901326u. [DOI] [PubMed] [Google Scholar]
- 125.Liu JP, Shirota O, Wiesler D, Novotny M. Ultrasensitive fluorometric detection of carbohydrates as derivatives in mixtures separated by capillary electrophoresis. Proc Natl Acad Sci USA. 1991;88:2302–6. doi: 10.1073/pnas.88.6.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu JP, Shirota O, Novotny M. Separation of fluorescent oligosaccharide derivatives by microcolumn techniques based on electrophoresis and liquid chromatography. J Chromatogr. 1991;559:223–35. doi: 10.1016/0021-9673(91)80073-p. [DOI] [PubMed] [Google Scholar]
- 127.Liu J, Dolnik V, Hsieh YZ, Novotny M. Experimental evaluation of the separation efficiency in capillary electrophoresis using open tubular and gel-filled columns. Anal Chem. 1992;64:1328–36. doi: 10.1021/ac00037a006. [DOI] [PubMed] [Google Scholar]
- 128.Mechref Y, Muzikar J, Novotny MV. Comprehensive assessment of N-glycans derived from a murine monoclonal antibody: a case for multimethodological approach. Electrophoresis. 2005;26:2034–46. doi: 10.1002/elps.200410345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Guttman A. Multistructure sequencing of N-linked fetuin glycans by capillary gel electrophoresis and enzyme matrix digestion. Electrophoresis. 1997;18:1136–41. doi: 10.1002/elps.1150180719. [DOI] [PubMed] [Google Scholar]
- 130.Guttman A. High-resolution carbohydrate profiling by capillary gel electrophoresis. Nature. 1996;380:461–62. doi: 10.1038/380461a0. [DOI] [PubMed] [Google Scholar]
- 131.Guttman A, Chen FT, Evangelista RA. Separation of 1-aminopyrene-3,6,8-trisulfonate-labeled asparagine-linked fetuin glycans by capillary gel electrophoresis. Electrophoresis. 1996;17:412–17. doi: 10.1002/elps.1150170221. [DOI] [PubMed] [Google Scholar]
- 132.Oyama T, Yodohsi M, Yamane A, Kakehi K, Hayakawa T, Suzuki S. Rapid and sensitive analyses of glycoprotein-derived oligosaccharides by liquid chromatography and laser-induced fluorometric detection capillary electrophoresis. J Chromatogr B. 2011;879:2928–34. doi: 10.1016/j.jchromb.2011.08.026. [DOI] [PubMed] [Google Scholar]
- 133.Ijiri S, Todoroki K, Yoshida H, Yoshitake T, Nohta H, Yamaguchi M. Highly sensitive capillary electrophoresis analysis of N-linked oligosaccharides in glycoproteins following fluorescence derivatization with rhodamine 110 and laser-induced fluorescence detection. Electrophoresis. 2011;32:3499–509. doi: 10.1002/elps.201100258. [DOI] [PubMed] [Google Scholar]
- 134.Schwarzer J, Rapp E, Reichl U. N-glycan analysis by CGE-LIF: profiling influenza A virus hemagglutinin N-glycosylation during vaccine production. Electrophoresis. 2008;29:4203–14. doi: 10.1002/elps.200800042. [DOI] [PubMed] [Google Scholar]
- 135.Szabo Z, Guttman A, Bones J, Karger BL. Rapid high-resolution characterization of functionally important monoclonal antibody N-glycans by capillary electrophoresis. Anal Chem. 2011;83:5329–36. doi: 10.1021/ac2007587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vanderschaeghe D, Laroy W, Sablon E, Halfon P, Van Hecke A, et al. GlycoFibroTest is a highly performant liver fibrosis biomarker derived from DNA sequencer–based serum protein glycomics. Mol Cell Proteomics. 2009;8:986–94. doi: 10.1074/mcp.M800470-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Vanderschaeghe D, Szekrenyes A, Wenz C, Gassmann M, Naik N, et al. High-throughput profiling of the serum N-glycome on capillary electrophoresis microfluidics systems: toward clinical implementation of GlycoHepatoTest. Anal Chem. 2010;82:7408–15. doi: 10.1021/ac101560a. [DOI] [PubMed] [Google Scholar]
- 138.Zamfir AD, Lion N, Vukelic Z, Bindila L, Rossier J, et al. Thin chip microsprayer system coupled to quadrupole time-of-flight mass spectrometer for glycoconjugate analysis. Lab Chip. 2005;5:298–307. doi: 10.1039/b413282c. [DOI] [PubMed] [Google Scholar]
- 139.Zhuang Z, Starkey JA, Mechref Y, Novotny MV, Jacobson SC. Electrophoretic analysis of N-glycans on microfluidic devices. Anal Chem. 2007;79:7170–75. doi: 10.1021/ac071261v. [DOI] [PubMed] [Google Scholar]
- 140.Smejkal P, Szekrenyes A, Ryvolova M, Foret F, Guttman A, et al. Chip-based CE for rapid separation of 8-aminopyrene-1,3,6-trisulfonic acid (APTS) derivatized glycans. Electrophoresis. 2010;31:3783–86. doi: 10.1002/elps.201000457. [DOI] [PubMed] [Google Scholar]
- 141.Primack J, Flynn GC, Pan H. A high-throughput microchip-based glycan screening assay for antibody cell culture samples. Electrophoresis. 2011;32:1129–32. doi: 10.1002/elps.201000619. [DOI] [PubMed] [Google Scholar]
- 142.Cortes DF, Kabulski JL, Lazar AC, Lazar IM. Recent advances in the MS analysis of glycoproteins: capillary and microfluidic workflows. Electrophoresis. 2011;32:14–29. doi: 10.1002/elps.201000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhuang Z, Mitra I, Hussein A, Novotny MV, Mechref Y, Jacobson SC. Microchip electrophoresis of N-glycans on serpentine separation channels with asymmetrically tapered turns. Electrophoresis. 2011;32:246–53. doi: 10.1002/elps.201000461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Amon S, Zamfir AD, Rizzi A. Glycosylation analysis of glycoproteins and proteoglycans using capillary electrophoresis–mass spectrometry strategies. Electrophoresis. 2008;29:2485–507. doi: 10.1002/elps.200800105. [DOI] [PubMed] [Google Scholar]
- 145.Mechref Y, Novotny MV. Glycomic analysis by capillary electrophoresis–mass spectrometry. Mass Spectrom Rev. 2009;28:207–22. doi: 10.1002/mas.20196. [DOI] [PubMed] [Google Scholar]
- 146.Gennaro LA, Salas-Solano O. On-line CE-LIF-MS technology for the direct characterization of N-linked glycans from therapeutic antibodies. Anal Chem. 2008;80:3838–45. doi: 10.1021/ac800152h. [DOI] [PubMed] [Google Scholar]
- 147.Mechref Y, Novotny MV. Structural investigations of glycoconjugates at high sensitivity. Chem Rev. 2002;102:321–69. doi: 10.1021/cr0103017. [DOI] [PubMed] [Google Scholar]